

Abstract
Candida albicans is commonly found in mixed infections with Pseudomonas aeruginosa, especially in the lungs of cystic fibrosis (CF) patients. Both of these opportunistic pathogens are able to form resistant biofilms and frequently infect immunocompromised individuals. The interaction between these two pathogens, which includes physical interaction as well as secreted factors, is mainly antagonistic. In addition, research suggests considerable interaction with their host, especially with immunomodulatory lipid mediators, termed eicosanoids. Candida albicans and Pseudomonas aeruginosa are both able to utilize arachidonic acid (AA), liberated from the host cells during infection, to form eicosanoids. The production of these eicosanoids, such as Prostaglandin E2, by the host and the pathogens may affect the dynamics of polymicrobial infection and the outcome of infections. It is of considerable importance to elucidate the role of host-produced, as well as pathogen-produced eicosanoids in polymicrobial infection. This review will focus on in vitro as well as in vivo interaction between C. albicans and P. aeruginosa, paying special attention to the role of eicosanoids in the cross-talk between host and the pathogens.
Keywords: Candida albicans, co-infection, eicosanoid, interaction, prostaglandin, Pseudomonas aeruginosa
Introduction
Recently it has become increasingly evident that microorganisms are not only found as free floating cells, but exist as surface associated, structured and cooperative consortia, called biofilms (Douglas, 2003; Hentzer et al., 2003; Burmølle et al., 2006; Harriott and Noverr, 2011). In addition, these communities are embedded in an extracellular matrix of self-produced polymeric material. In these interactive organizations for microorganisms secreted factors and physical proximity enable metabolic interactions (Diaz et al., 2014). This often involves interkingdom interactions necessary for ecological balance and survival of certain species (Rinzan, 2009).
Pseudomonas aeruginosa is a Gram-negative, aerobic rod colonizing a remarkable assortment of niches, including aquatic environments, terrestrial environments and eukaryotic organisms (Pier, 1985; Tan et al., 1999). It is an opportunistic pathogen, frequently isolated from healthy humans as part of the human microbiota and is commonly found in mixed infections with the yeast, Candida albicans (Kaleli et al., 2006). Candida albicans is found as part of the normal microbiota of the skin, gastrointestinal tract and female genital tract (Morales and Hogan, 2010) and is a major cause of opportunistic infections ranging from superficial to fatal systemic infections (Sandven, 2000). Fungal infections have become increasingly troublesome in the past decades, especially in immunocompromised patients and in the hospital setting, with C. albicans being the most frequently isolated fungal pathogen and the most commonly isolated bloodstream pathogen (Rinzan, 2009). Selective pressure of nutrient limitation and competition between bacteria and fungi regulate the colonization of potential pathogenic microorganisms such as C. albicans and P. aeruginosa, with a disruption in this equilibrium resulting in infection by opportunistic pathogens (Calderone and Fonzi, 2001).
These two microorganisms have tendencies to form polymicrobial biofilms and as such play extensive roles in nosocomial infections, infection in immunocompromised individuals and especially in cystic fibrosis (CF) patients (El-Azizi et al., 2004; Bianchi et al., 2008; McAlester et al., 2008). This review, therefore, aims to evaluate the complex cross-kingdom relationship of these two pathogens and the impressive interaction and communication between them as well as the collateral damage to hosts caught in the cross-fire. Additionally, special attention will be given to the known immunomodulatory lipids produced by both of these microorganisms as well as the host and the role this may play during infection.
Pathogenesis of Pseudomonas aeruginosa
Pseudomonas aeruginosa possesses numerous virulence factors including exotoxin A, proteases and lipases, released by a type II secretion system (Xcp regulon), as well as exotoxins exoS, T, U, and Y, secreted into host cells by a type III secretion system (Hogardt et al., 2004). Interestingly, it was found that P. aeruginosa possesses two type II secretory pathways, previously not seen in one organism (Ball et al., 2002). Additionally, pyoverdine, rhamnolipids, lipopolysaccharide (LPS) and pili also form part of this formidable pathogen's virulence arsenal (Gilligan, 1991; Méar et al., 2013). A study by Bianchi et al. (2008) showed that P. aeruginosa impairs the engulfment of apoptotic cells through the action of yet another virulence factor, the phenazine, pyocyanin (PYO) (Gibson et al., 2009). Interestingly, it has been shown that multiple drug resistant strains of P. aeruginosa show decreased production of PYO, and thus have a reduction in virulence (Fuse et al., 2013). As previously mentioned, P. aeruginosa forms biofilms, and a universal model for the formation of P. aeruginosa biofilm formation was suggested (O'Toole et al., 2000; Klausen et al., 2003). According to this model, P. aeruginosa cells move by means of flagella to an adequate surface and movement along this surface is accomplished through type IV-pili. Cells aggregate, leading to microcolony formation. During maturation, large mushroom-shaped structures are formed. Klausen et al. (2003) proposed an alternate model, with evidence indicating that flagella do not play a role in the attachment of P. aeruginosa cells. The formation of P. aeruginosa biofilms are, however, highly dependent on the carbon source. Additionally, the circumstances during growth, such as flow vs. stationary growth, might elicit large morphological changes.
In addition to the previously mentioned factors, the resistance of P. aeruginosa to antimicrobial agents is key to its pathogenic capabilities. Various mechanisms for antibiotic resistance in P. aeruginosa biofilms have been proposed (Drenkard, 2003). These include the reduced transport of antimicrobial agents in the biofilm due to extracellular matrix and accompanied nutrient and oxygen limitation of cells deeply embedded in the biofilm. This causes a decrease in metabolic activity of the cells. Antibiotic resistant persisters embedded in the biofilm structure, stress responses of the cells, efflux pumps and quorum sensing among cells may all contribute to the increased resistance observed in bacterial biofilms. Evidence also suggest that a protein, PvrR, regulates susceptibility and resistance phenotypes of P. aeruginosa (Drenkard and Ausubel, 2002; Benamara et al., 2011). An impact on lipid composition is also speculated due to differential protein expression in biofilms. In this regard, Benamara et al. (2011) examined the effect of biofilm formation on inner membrane lipid composition in P. aeruginosa that indicated a reduced amount of uneven numbered phospholipids. In addition, an increase in long chain phosphatidylethanolamines was observed, suggesting an increase in bilayer lipid stability and a decrease in membrane fluidity.
Pathogenesis of Candida albicans
Candida albicans is a dimorphic yeast, meaning that both yeast and hyphal morphology is shown, with a tendency to form drug resistant biofilms (Ramage et al., 2001). The ability of this microorganism to switch between the planktonic single yeast cell and hyphal morphologies has a major influence on its virulence (Andes et al., 2004; Pierce, 2005; Bruzual et al., 2007; Brand et al., 2008; Gil-Bona et al., 2015). In addition to this morphological plasticity, the aggressiveness of C. albicans colonization is due to a collection of other virulence factors. These include adhesins (biomolecules that enable binding to host cells or host cell ligands), lipolytic and proteolytic enzymes and phenotypic switching (white to opaque switching) (Calderone and Fonzi, 2001). Interaction of C. albicans with the host is largely accomplished by contact with the C. albicans cell surface and subsequent biofilm formation (Gow and Hube, 2012). Ramage et al. (2001) investigated the formation of C. albicans biofilms through visualization of the biofilms at various stages of development and monitoring the metabolic activity of the cells. Through scanning electron microscopy (SEM) and confocal scanning laser microscopy (CSLM) a dense network of hyphae and yeast cells in a matrix of exopolymeric material was visualized in the matured biofilm. This study also tested the effect of antifungal drugs on Candida biofilms and planktonic C. albicans cells. It was found that cells in the biofilm had a 250-fold increase in resistance against fluconazole. Dumitru et al. (2004) argued that this increased resistance might be due to the hypoxic conditions found in biofilms, with anaerobically grown C. albicans also showing resistance against certain antifungal agents. Interestingly, a study by Kuhn et al. (2002) evaluated a range of antifungal agents against Candida and found that sub-inhibitory concentrations of certain antifungals elicited alterations in biofilm formation.
Interaction between Pseudomonas aeruginosa and Candida albicans In vitro
Physical/direct interaction
Several studies were conducted to evaluate the interaction of P. aeruginosa and C. albicans. The antagonistic interaction of C. albicans and P. aeruginosa was examined by Brand et al. (2008) who showed that P. aeruginosa cells kill C. albicans hyphal cells, but not C. albicans yeast cells. The deadly effect on C. albicans is thought to be due to PYO, which alters the cell wall of C. albicans (Kerr et al., 1999). Further research into this interaction provided evidence that there is a difference in P. aeruginosa mediated C. albicans killing among different morphotypes of C. albicans (Rinzan, 2009). Increased susceptibility to the killing effect of P. aeruginosa was seen with filamentous cells compared to planktonic counterparts, similar to the study by Brand et al. (2008), as well as a reversion of germ tube formation in the presence of P. aeruginosa. Further analysis of this interaction indicated that attachment of P. aeruginosa and C. albicans killing is mediated by lectin-carbohydrate interaction, type IV pili as well as mannans. The authors also speculated on the possible involvement of O-linked mannans in the survival of C. albicans yeast cells during combined incubation, as was proposed previously (Brand et al., 2008; Rinzan, 2009). In Figure 1, scanning electron micrographs of a dual species biofilm with C. albicans and P. aeruginosa is seen showing extensive colonization of C. albicans cells by P. aeruginosa.
Figure 1.
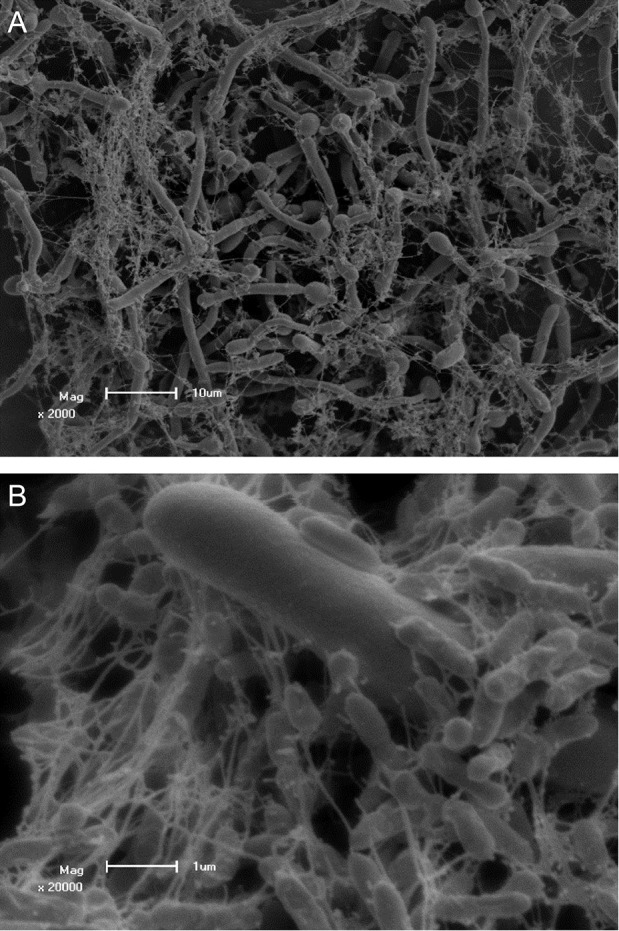
Scanning electron micrographs of Candida albicans colonized by Pseudomonas aeruginosa PAO1 showing adhesion to C. albicans hyphae. Scale bars represent (A) 10 μm and (B) 1 μm. Co-culture of C. albicans and P. aeruginosa was performed and visualized by Ruan Fourie.
Indirect interaction
Role of Pseudomonas aeruginosa quorum sensing molecules during In vitro interaction
The interaction of C. albicans and P. aeruginosa is mediated by quorum sensing molecules (QSM), produced by both organisms (Cugini et al., 2007). The bulk of Gram-negative bacterial quorum sensing systems utilize N-acyl homoserine lactones (AHL) that bind and activate their respective transcriptional activators (R protein) to induce expression of target genes (de Kievit and Iglewski, 2000). When adequate population density is reached by bacterial cells, AHL concentrations are high enough to induce these transcriptional changes.
Two AHL-dependent QS systems were identified in P. aeruginosa, namely the las and rhl systems (de Kievit and Iglewski, 2000). 3-oxododecanoyl-L-homoserine lactone (3-oxo-HSL) is an autoinducer with its production directed by LasI autoinducer synthase (las QS system). The production of another autoinducer, butanoylhomoserine lactone, is similarly regulated by RhlI autoinducer synthase (rhl QS system). These bind and activate their respective transcriptional activators LasR and RhlR (Passador et al., 1993; Pearson et al., 1995). These QSMs may regulate up to 10% of the genome of P. aeruginosa depending on culture conditions (Hentzer et al., 2003; Wagner et al., 2003). The structures of these autoinducers are shown in Figure 2.
Figure 2.
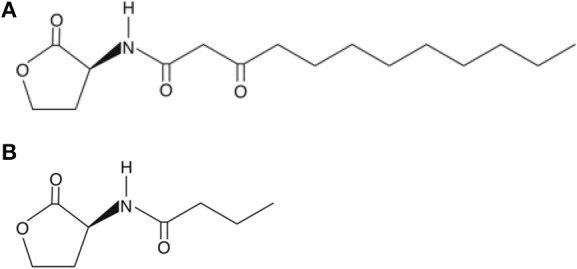
Structures of (A) 3-oxododecanoyl-L-homoserine lactone at the top and (B) butanoylhomoserine lactone underneath.
The QSM, 3-oxo-HSL, was studied for its effect on cell adherence in polymicrobial biofilms of P. aeruginosa and C. albicans (Ovchinnikova et al., 2012). The study showed that mutant P. aeruginosa strains lacking the lasI gene for the LasI autoinducer synthase was unable to adhere to C. albicans hyphae, while a P. aeruginosa strain without the mutation was able to adhere to C. albicans cells. The study suggested that 3-oxo-HSL is needed for the adherence of P. aeruginosa cells to C. albicans hyphae, because 3-oxo-HSL is needed for the production of surface adherence proteins on P. aeruginosa cells. A study by McAlester et al. (2008) showed that if cell free supernatant from high 3-oxo-HSL producing P. aeruginosa strains is added to C. albicans cultures, the yeast to hyphal switch is inhibited. Pseudomonas aeruginosa strains that produced low amounts of 3-oxo-HSL did not inhibit the yeast to hyphal switch when the supernatants of their cultures were added to C. albicans cultures, suggesting that 3-oxo-HSL affects yeast morphology in a dose dependent manner. To ensure that the 3-oxo-HSL was the cause of the inhibition of morphological switch, pure 3-oxo-HSL was added to a C. albicans culture with the same results obtained. The reaction of C. albicans toward 3-oxo-HSL may lead to the dispersal of C. albicans cells in the presence of P. aeruginosa (Morales and Hogan, 2010; Ovchinnikova et al., 2012). These studies thus show that AHLs are not only important for bacterial communication, but are responsible for considerable interaction with other microorganisms such as C. albicans.
In addition to AHLs, a QS signal, 2-heptyl-3-hydroxyl-4-quinolone signal or Pseudomonas quinolone signal (PQS), was later identified and this molecule is released in the late exponential phase (Pesci et al., 1999; Lépine et al., 2003). The production of PQS can be induced by the LasI/R system and inhibited by the RhlI/R system (De Sordi and Mühlschlegel, 2009). Strikingly, PQS was shown to have both a damaging effect on P. aeruginosa through a pro-oxidative effect, as well as an anti-inflammatory effect (Haussler and Becker, 2008). The authors speculate that this contradictory effect drives survival of the fittest through selection of phenotypic variants able to survive in stressful conditions and molding populations sufficiently adapted. It also modulates swarming motility of P. aeruginosa (Déziel et al., 2004; Ha et al., 2011). In addition to PQS, its immediate precursor, 2-heptyl-4-quinolone (HHQ), has been shown to repress C. albicans biofilm formation (Reen et al., 2011).
PQS (Figure 3A) induces the formation of several virulence factors, including phenazine compounds (Phelan et al., 2014). Of these, PYO or methyl-1-hydroxyphenazine (Figure 3B), is the best studied. Pyocyanin is a chloroform soluble compound with a blue color (Cox, 1986). It has been shown to be a QSM produced in the early stationary phase (Hernandez et al., 2004; Price-Whelan et al., 2007) and a study by Dietrich et al. (2006) suggested that PYO is a terminal signaling molecule that controls its own cycling. It plays a major role in maintaining NADH/NAD+ ratio stability in P. aeruginosa cells when they encounter oxygen limiting conditions due to the limited fermentation capability of Pseudomonas (Dietrich et al., 2006). Pyocyanin can then act as an alternate terminal electron acceptor and decrease the NADH/NAD+ ratio in the stationary phase of growth. It can then later be reoxidized by oxygen when it becomes available and this could be a mechanism for the production of reactive oxygen species (ROS). Phenazines, including PYO, phenazine-1-carboxylic acid, 1-hydroxyphenazine and phenazine-1-carboxamide play extensive roles in the interaction between Pseudomonas species and eukaryotes, including fungal microorganisms (Kaleli et al., 2006; Phelan et al., 2014).
Figure 3.
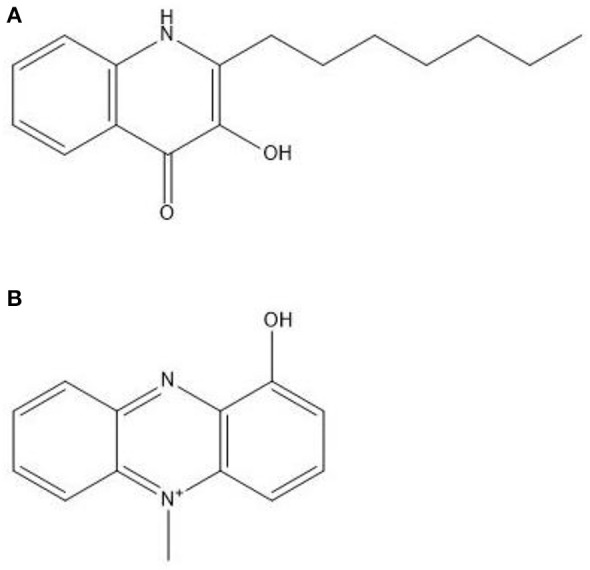
Structures of (A) Pseudomonas quinolone signal at the top and (B) pyocyanin underneath.
In addition, PYO has antimicrobial activity against a wide range of cells including a bactericidal effect against a wide variety of bacterial species with Gram-positive species being more susceptible than Gram-negative species (Hassan and Fridovich, 1980). Interestingly, Pseudomonas species seem to be resistant to this bactericidal effect (Baron and Rowe, 1981). It is also toxic to eukaryotic cells (O'Malley et al., 2003). The mechanism of this effect is due to the ability of this compound to undergo non-enzymatic redox cycling intracellularly, resulting in the generation of ROS (Gloyne et al., 2011). Another effect of PYO is the reduction in cyclic adenosine monophosphate (cAMP) (Kerr et al., 1999). This enables PYO to inhibit the shift from yeast to hyphal morphology in C. albicans because the yeast-mycelium transition is promoted by increased levels of intracellular cAMP. Gibson et al. (2009) observed a red pigment with the co-incubation of P. aeruginosa and C. albicans produced during close proximity of the yeast and bacterial cells. This pigment was localized in fungal cells. The authors speculate that C. albicans enzymes participate in the formation of this product intracellularly. The precursor of this red pigment was identified as 5-methylphenazine-1-carboxylic acid (5-MPCA) through the use of P. aeruginosa strains with disruptions in the phenazine biosynthesis pathway. The presence of the red pigmented compound was linked to significant repression of C. albicans viability. In addition, a recent study indicated that the phenazines, phenazine-1-ol, phenazine-1-carboxylic acid and phenazine-1-carboxamide, has a synergistic effect with three antifungals: fluconazole, itraconazole and clotrimazole against Candida species (Kumar et al., 2014). This then suggests that the presence of phenazine producing organisms such as Pseudomonas can drastically alter the treatment of simultaneous fungal infection.
The role of Candida albicans quorum sensing molecules during In vitro interaction
Candida albicans has also been shown to produce QSMs (Hornby et al., 2001). The QSM, farnesol (Figure 4), inhibits germ tube formation and also caused a morphological shift from predominant mycelial state to predominant yeast morphology, indicating the effect as 3-oxo-HSL. The effect on the morphology of C. albicans is thought to be due to inhibition of the Ras1-controlled pathway involved in hyphal growth (Morales and Hogan, 2010). Recently, farnesol was identified to attract macrophages in hosts (Hargarten et al., 2015). The authors speculate that engulfment and movement of these immune cells then aid in dissemination as macrophages are killed by C. albicans after engulfment. Farnesol induces the generation of ROS which could play a role in the competition of C. albicans with bacteria. Resistance of C. albicans to oxidative stress has also been shown to be linked, in part, to farnesol (Westwater et al., 2005). A study by Cugini et al. (2007) indicated that farnesol inhibits P. aeruginosa PQS and a subsequent virulence factor, PYO, whose production is controlled by PQS, in a dose dependent manner. Interestingly, there was no effect on the overall growth of P. aeruginosa. Additionally, when P. aeruginosa was co-cultured with C. albicans, reduction in PQS and PYO produced by P. aeruginosa was also observed, suggesting that high enough concentrations of farnesol is produced by C. albicans to exert an effect on P. aeruginosa. Later, the same research group found that C. albicans and its secreted factors increase PQS and butanoylhomoserine lactone in lasR defective mutants of P. aeruginosa, with a downstream increase in phenazine production (Cugini et al., 2010). The authors speculated that oxidative stress may trigger downstream quorum sensing pathways.
Figure 4.
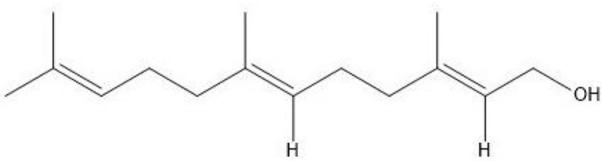
Structure of farnesol.
As seen with proteomic analysis by Jones-Dozier (2008), a possible decrease in virulence of P. aeruginosa is evident when exposed to farnesol, due to alterations in protein expression of P. aeruginosa subjected to this compound. In addition to decreasing PQS and PYO production, farnesol inhibits swarming motility in P. aeruginosa (McAlester et al., 2008). Rhamnolipids, a class of glycolipids, play a role in swarming motility and has been implicated as playing part in the development of ventilator associated pneumonia (VAP) (Köhler et al., 2010). Due to the fact that rhamnolipid production is partly regulated by PQS, the mechanism of decreased swarming motility may be due to the reduction in PQS production by farnesol.
Other factors influencing In vitro interaction
Iron availability
In a study by Purschke et al. (2012), C. albicans exhibited a lower metabolic activity in mixed biofilms with P. aeruginosa when compared to single species biofilms. Secretome analysis of the proteins of the single vs. mixed species biofilms revealed an overall increase of secreted proteins of mixed species biofilms of C. albicans and P. aeruginosa compared to the single species counterparts. This increase was largely found to be due to increased secreted proteins by P. aeruginosa. Interestingly, a large proportion of the increased protein production was attributed to a siderophore, pyoverdine, specific to Pseudomonas. This increase in pyoverdine was thought to be due to the increased iron utilization by the two species in the mixed biofilm. This was confirmed by the addition of iron, which abolished the production of pyoverdine. The authors speculated sequestration of available iron by pyoverdine results in decreased availability to C. albicans, although C. albicans is able to utilize iron bound to certain other microbial siderophores. Recent evidence suggests that this phenomenon may not be of importance during in vivo interaction (Lopez-Medina et al., 2015). In this study, C. albicans secreted factors significantly reduced pyoverdine and another siderophore, pyochelin, expression by P. aeruginosa during gastrointestinal colonization in a murine model. This decrease of expression by P. aeruginosa was linked to diminished virulence of P. aeruginosa with growth unaffected. The authors suspect the heterogeneity of the biofilms between in vivo and in vitro studies may cause the differential results.
Recently, Trejo-Hernández et al. (2014) found that hypoxia influences the ability of P. aeruginosa to inhibit C. albicans filamentation in vitro compared to aerobic conditions. This was attributed to decreased AHL produced by P. aeruginosa in the presence of C. albicans. Previously, it was shown that hypoxic conditions promote filamentation in C. albicans and reduces farnesol production and response to it (Dumitru et al., 2004). Additionally, the authors also speculated that competition for iron may also be greater during hypoxia (Synnott et al., 2010; Trejo-Hernández et al., 2014). Therefore, both the interaction of P. aeruginosa with C. albicans, the concentration of oxygen and iron competition influences the production of AHLs (Trejo-Hernández et al., 2014). The authors also found that proteins known to play roles in iron uptake in P. aeruginosa through siderophores were upregulated in mixed biofilms, confirming previous observations. Additionally, iron supplementation increased the growth of P. aeruginosa in single and mixed biofilms, with this effect not seen with C. albicans. This increase in growth of the bacterium may increase the destruction of the fungal population. Lamont et al. (2002) indicated that pyoverdine may act as a signaling molecule to modulate other virulence factors including exotoxin A and pyoverdine itself. Because pyoverdine production is increased in mixed biofilms, the virulence of P. aeruginosa might also be upregulated in mixed biofilms (Trejo-Hernández et al., 2014). Additionally, PQS as well as products of the PQS system including rhamnolipids and PYO, were upregulated. A significant increase in P. aeruginosa mutability frequency was seen with a large number of antibiotic resistant mutant phenotypes arising over time. The authors speculate that the decreased catalase activity observed in mixed biofilms may result in increased oxidative stress, concomitantly increasing mutability. In the case of C. albicans, the same trend was seen with hypermutability arising with a high frequency of antimicrobial resistant phenotypes, possibly attributed to the increased oxidative stress caused by PYO produced by P. aeruginosa. Additionally, C. albicans iron dependant processes, including aerobic respiration, were downregulated. Glycolytic enzyme activity in C. albicans was also altered, with PYO possibly attributing to this and leading to other pathways for energy utilization. To confirm the increased virulence of C. albicans and P. aeruginosa in mixed biofilms, the authors used a rat infection model. Candida albicans was shown to promote pathogenicity of P. aeruginosa. Therefore, the ability of these pathogens to compete for iron may alter population dynamics and influence the nature of the interaction.
Bacterial cell wall components
In addition to various secreted factors produced by P. aeruginosa in polymicrobial interaction (Holcombe et al., 2010), bacterial LPS were shown to have adverse effects on Candida spp. biofilms (Bandara et al., 2010). The same group later confirmed these results by evaluating the effect of P. aeruginosa LPS on C. albicans (Bandara et al., 2013). The study suggested a decrease of C. albicans filamentation and biofilm metabolic activity, including glycolysis, and growth with the addition of high concentrations of P. aeruginosa LPS. In addition to this, peptidoglycan was shown to trigger filamentation in C. albicans (Xu et al., 2008).
Ethanol
Chen et al. (2014) evaluated the effect of C. albicans produced ethanol on P. aeruginosa and found that ethanol stimulated adhesion and biofilm formation of P. aeruginosa. In addition, swarming motility by P. aeruginosa decreased and a stimulation of phenazine derivatization and production of 5-MPCA by P. aeruginosa in the presence of ethanol was observed. The authors speculate that there is a positive feedback loop where C. albicans ethanol production increases P. aeruginosa 5-MPCA production and biofilm formation. In turn, 5-MPCA stimulates ethanol production in C. albicans (Morales et al., 2013).
Extracellular DNA
A recent study also identified extracellular DNA as a large factor in biofilm formation by C. albicans (Sapaar et al., 2014). Low amounts of extracellular DNA (1.0 μg/mL) was shown to promote biofilm formation and increase biofilm stability, whereas higher concentrations (10 μg/mL) hampered the formation of biofilms by C. albicans as well as the stability of the biofilms. The study also indicated that the source of the extracellular DNA, whether it is C. albicans, or from bacterial sources such as P. aeruginosa, does not matter. This increase in biofilm formation by C. albicans due to extracellular DNA may increase the virulence of the fungus. Evidence also suggest that the concentration of extracellular DNA can reach 4 mg/mL in CF patient sputum samples, raising the question if this facet of interaction might have clinical relevance (Sapaar et al., 2014).
A summary of several facets of interaction between C. albicans and P. aeruginosa can be seen in Figure 5.
Figure 5.

Illustration of competition between Candida albicans and Pseudomonas aeruginosa. Pseudomonas aeruginosa attaches to C. albicans hyphae and kills hyphal cells through secreted hydrolytic enzymes such as hemolytic phospholipase C (PlcH) and phenazines such as pyocyanin and 5-methylphenazine-1-carboxylic acid (5-MPCA). 3-oxo-homoserine lactone produced by P. aeruginosa and phenazines inhibit filamentation by C. albicans, similar to farnesol, produced by C. albicans. Pseudomonas aeruginosa lipopolysaccharide (LPS) inhibits C. albicans filamentation. Ethanol production is increased by the fungus, inhibiting the motility of P. aeruginosa (adapted from Lindsay and Hogan, 2014).
Interaction between Pseudomonas aeruginosa and Candida albicans In vivo
A high number of C. albicans nosocomial infections are polymicrobial with P. aeruginosa a frequent co-isolate in blood stream infections and pneumonia (Lindsay and Hogan, 2014). Kerr (1994) was the first to describe the anticandidal activity of P. aeruginosa in vivo. The study evaluated lung infection of three surgery patients postoperatively with inhibition of C. albicans growth seen after P. aeruginosa colonization. This inhibition was confirmed with the regrowth of C. albicans seen after eradication of P. aeruginosa, even with fluconazole treatment. Additional in vitro susceptibility experiments confirmed the suppression of Candida growth by P. aeruginosa. Gupta et al. (2005) evaluated 300 burn patients over 2 years and found repression of Candida spp. in the presence of P. aeruginosa. Several studies also indicate that prior colonization of Candida may promote susceptibility of the host to P. aeruginosa infection (Roux et al., 2009; Hamet et al., 2012; Xu et al., 2014). Nseir et al. (2007) reported that antifungal treatment during Candida spp. tracheobronchial colonization may be associated with reduced risk for P. aeruginosa colonization. The case is strengthened by Azoulay et al. (2006), who reported a possible link between Candida colonization of the respiratory tract and an increased risk for Pseudomonas VAP. Roux et al. (2013) reported that the Th1-Th17 immune response, associated with Candida colonization, caused a decrease in P. aeruginosa phagocytosis by alveolar macrophages, the primary innate immune response against bacterial invasion. The adaptive Th1-Th17 response evoked by C. albicans invasion, is characterized by an increase in interferon ɤ(IFNɤ), as well as an increase in interleukin-17 (IL-17) and decrease in IL-2. The authors suggest that the increase in IFNɤ, associated with the Th1-Th17 response, is responsible for the inhibition of bacterial phagocytosis, through inhibiting expression of scavenger receptors on alveolar macrophages.
Remarkably, contradictory results to the notion that P. aeruginosa infection is more aggressive after prior C. albicans colonization, was provided by Ader et al. (2011). In a murine model, C. albicans short term colonization prior to P. aeruginosa colonization caused a reduction in P. aeruginosa bacterial load compared to the absence of C. albicans colonization (Ader et al., 2011). Additionally, a reduction in P. aeruginosa induced lung injury was observed with the prior colonization of C. albicans. Interestingly, this effect was reversed with treatment by the antifungal caspofungin during C. albicans colonization. Candida albicans initiates alveolar innate immunity in a murine model, protecting the host against subsequent P. aeruginosa infection (Mear et al., 2014). The authors showed that prior C. albicans infection induces interleukin-17 (IL-17) and IL-22 secretion through innate lymphoid cell recruitment. The cytokines produced, induce the production of antimicrobial peptides as well as the mobilization of phagocytic cells. In a murine gut model, C. albicans secreted factors inhibited expression of siderophores as well as cytotoxic molecules by P. aeruginosa, reducing the virulence of the bacteria (Lopez-Medina et al., 2015). Due to this, increased survival of the host was observed during co-incubation of P. aeruginosa with C. albicans. Interestingly, Neely et al. (1986) demonstrated increased mortality in a murine model when C. albicans infection was preceded by P. aeruginosa. This reciprocal effect may also be due to alterations in innate immune response, as Faure et al. (2014) reported that the P. aeruginosa type III secretion system induced IL-18 secretion causing substantial neutrophil recruitment and host cell damage, and decreased IL-17 secretion in a mouse model, possibly leading to the reduced clearance of pathogens.
The co-infection of P. aeruginosa and C. albicans has been well documented in cystic fibrosis (CF) patients. CF is one of the most commonly encountered autosomal recessive disorders found, with the occurrence varying in race (Andersen, 1938). The disease is caused by a genetic disorder where a mutation exists in the CF transmembrane conductance regulator (CFTR) gene (Delhaes et al., 2012). CF is a disease with two pathophysiological properties, namely pancreatic insufficiency with malnutrition and also airway infections due to extremely viscous secretions (Andersen, 1938). The increased viscosity of lung secretions is thought to be due to the increased sodium absorption of the respiratory epithelium and the defective regulation of chloride ion secretion (Gilligan, 1991). This is thought to be the reason why CF patients have comparatively dehydrated surface liquid which leads to defective mucociliary clearance. The thick bronchial mucus traps viral particles, fungal spores and bacteria and provides a suitable environment for the fungal spores and bacteria to grow, causing infection (Delhaes et al., 2012). Ninety percent of deaths in CF are due to pulmonary dysfunction and in effect, chronic airway infection (Gilligan, 1991). A study by Güngör et al. (2013) evaluated the most prevalent fungal colonization in Turkish CF patients. The most prevalent fungal microorganisms isolated from these CF patients, were shown to be C. albicans at 62.5% (30 patients). In addition, fungal-bacterial co-colonization in the CF patients was shown to be 98.2% in these C. albicans infections. The most frequent bacterial co-colonizer of CF patients with C. albicans infections was found to be Staphylococcus aureus (53.57%), with P. aeruginosa at 48.21%, Staphylococcus maltophilia at 16.07% and Haemophilus influenza at 5.97%. Other similar studies have also implicated S. aureus and P. aeruginosa as the most prevalent bacterial species isolated (Valenza et al., 2008; Williamson et al., 2011). Several studies addressed the correlation between C. albicans and P. aeruginosa in CF infection, with a recent study suggesting a significant correlation (Conrad and Bailey, 2015). Discrepancies may arise due to method of analysis and the population analyzed.
Production and role of oxylipins during infection
Lipids have crucial cellular significance, forming membranes, as well as acting as cellular signals (reviewed by Tehlivets et al., 2007). The latter is of great importance in multicellular eukaryotic organisms. Although lipids as signaling molecules have been well studied in mammals and plants, their roles in fungi are not as well characterized.
Arachidonic acid (AA) or 5,8,11,14-eicosatetraenoic acid (Figure 6A), a major constituent of the mammalian phospholipids, together with its wide range of metabolites (termed eicosanoids), have substantial roles as lipid signals (Chilton et al., 1996), including modulation of the innate immune response (Rodríguez et al., 2014). As the constituent of cellular membranes, it is predominantly found in the sn2 position of phospholipids with incorporation into inflammatory cells through CoA-dependant acyl transferases.
Figure 6.

Structures of (A) arachidonic acid (AA) at the top, followed by (B) 3-hydroxyeicosatetraenoic acid (3-HETE); (C) 15-HETE; (D) Prostaglandin E2 at the bottom.
The metabolism of AA to form a plethora of lipid mediators involve various pathways in vertebrates, including the action of cyclooxygenases (COX), lipoxygenases (LOX), monooxygenases (CYP450), and non-enzymatic (NE) pathways. Cyclooxygenases, or prostaglandin endoperoxide synthases, are enzymes catalyzing the insertion of two oxygen atoms in AA (Marnett et al., 1999). In mammalian cells, two isoforms exist, namely, COX-1, which is constitutively expressed, and COX-2, which is inducible. The initial reaction of AA oxidation, mediated by COX, yields prostaglandin G2 (PGG2) (Rodríguez et al., 2014). Through peroxidase activity, PGG2 is reduced to prostaglandin H2 (PGH2), which serves as a precursor for various other immunomodulatory compounds, including prostaglandins as well as thromboxanes. For example, further action by Prostaglandin E synthase converts PGH2 to Prostaglandin E2 (PGE2) (Figure 6D).
Lipoxygenases are a large group of dioxygenases that catalyze oxygen insertion into polyunsaturated fatty acids in animals, plants as well as microorganisms (Kuhn and O'Donnell, 2006; Brodhun and Feussner, 2011). The reaction of oxygenation consists of various steps, starting with hydrogen abstraction, followed by radical rearrangement and the insertion of oxygen. In addition to oxygenation and hydroperoxidation, LOX also catalyze the synthesis of leukotrienes, lipoxins and hepoxilins through the combination of various enzymatic activities (Kuhn and O'Donnell, 2006; Dennis and Norris, 2015). Various isoforms of LOX exist with different stereospecificity and activities, for example 12/15-LOX catalyze oxygenation and hydroperoxidation of PUFAs at the 12 or 15 carbon position. Various studies suggest that the 12/15-LOX can be induced by interleukin-4 (IL4) and IL13, which are important in the Th2 response. Additionally, 12/15-LOX is inhibited by interferon ɤ. The low expression of 12/15-LOX has been linked to pro-inflammatory responses and has been implicated in airway functions. Prostaglandin E2 has also been argued to induce the expression of 12/15-LOX in neutrophils through cAMP elevation. Various products of LOX have also been implicated in anti-inflammatory responses in neutrophils. Many LOX products, including hydroperoxy-eicosatetraenoic acids (HPETEs) and hydroxy-eicosatetraenoic acids (HETEs) are intermediate products leading to the formation of lipoxins and leukotrienes (Dennis and Norris, 2015). The interaction of COX- and LOX-derived lipid mediators as well as the combination of these two pathways leads to the modulation of the inflammatory response (Dennis and Norris, 2015). The non-steroidal anti-inflammatory drug (NSAID), acetylsalicylic acid (ASA) (also known as aspirin), was shown to acetylate COX isozymes leading to the formation of 15(R)-HETE, which acts as LOX substrate for the formation of lipoxins (Serhan, 2002). These lipoxins are potent anti-inflammatory molecules, inhibiting neutrophil recruitment and leukotriene formation. In addition to these pathways, cytochrome P450s are also responsible for the formation of epoxyeicosatetraenoic acids (EETs) from AA, with concurrent modification to diHETEs, playing differential effects on the host.
The effects of eicosanoids on mammalian cells are dependent on the type of target tissue and the physiological state of these tissues (Dennis and Norris, 2015). Considerable research is being done to determine the eicosanoids that play a role in host protection against pathogens during infection as they can enhance the clearance of pathogens. For this review, the focus will fall on lipid mediators in terms of C. albicans and P. aeruginosa infection.
Role of mammalian oxylipins during Pseudomonas aeruginosa infection
Several studies have addressed the effect of various invading pathogens on the production of PGE2 to gain a better understanding of the immunological aspects of infection. In mammalian cells, PGE2 is formed through the metabolism of AA by the action of cyclooxygenase (COX-1 or COX-2) to first form PGH2, followed by PGE synthases to form PGE2 (Murakami et al., 2003). Immune cells are the main source of PGE2, although this compound is also produced by various other cell types (Kalinski, 2012; Agard et al., 2013). It elicits a response in mammalian cells through activation of four receptors, designated EP1–EP4, with the effect dependent on the receptor activated.
Pseudomonas aeruginosa pulmonary infection is associated with an overproduction of PGE2 and concurrent decrease in phagocytosis by alveolar macrophages (Ballinger et al., 2006; Agard et al., 2013). This increase in PGE2 is due to the large amount of AA released during P. aeruginosa infection, mediated by ExoU, an intracellular phospholipase (König et al., 1996; Saliba et al., 2005; Sadikot et al., 2007; Agard et al., 2013). As such it plays a crucial role in initial infection and infiltration by P. aeruginosa. The absence of ExoU in P. aeruginosa was linked to diminished severity of infection and PGE2 production. The importance of PGE2 during P. aeruginosa infection was seen when a COX-2 inhibitor was employed, resulting in a decrease in severity of infection by this pathogen (Sadikot et al., 2007). Several other virulence factors also elicit changes in PGE2 levels. The QSM 3-oxo-HSL produced by P. aeruginosa was shown to induce COX-2 and, therefore, PGE2 production in human lung fibroblasts (Smith et al., 2002). Similarly, P. aeruginosa PYO and LPS increased the release of PGE2 in urothelial cells in a concentration dependent manner (McDermott et al., 2013).
Role of Pseudomonas aeruginosa oxylipins during infection
A number of microorganisms including bacteria are able to form eicosanoids (Lamacka and Sajbidor, 1995). Although the presence of LOX in plants and animals has long been known, their presence in lower eukaryotes and prokaryotes has only recently been established, with P. aeruginosa one of the few bacteria with typical LOX genes. Pseudomonas aeruginosa has been found to possess a secretable 15-LOX, homologous to mammalian LOX, producing 15-HETE (Figure 6C) which is similar to host 15-HETE and elicits anti-inflammatory effects on the host, through acting as a substrate for lipoxin formation (Serhan, 2002; Vance et al., 2004). The formation of these lipoxins may alter the severity of infection through inhibiting neutrophil recruitment and generation of leukotrienes (Serhan, 2002). In addition to 15-HETE, P. aeruginosa is able to produce a range of products including dihydroxy unsaturated fatty acids such as 7,10-dihydroxy-8(E)-octadecenoic acid (DOD) (Hou, 2008). In addition, the production of prostaglandins and prostaglandin-analog compounds have been identified in P. aeruginosa (Lamacka and Sajbidor, 1995), however, the effect of these compounds during infection has not been addressed.
Role of mammalian oxylipins during Candida albicans infection
The alteration of immune response is not unique to bacteria, but plays a significant role as a virulence factor during C. albicans infection. Candida albicans is able to metabolize AA, liberated from host cells by host as well as yeast-derived phospholipase A2 (Castro et al., 1994; Filler et al., 1994; Brash, 2001; Niewerth and Korting, 2001; Theiss et al., 2006; Parti et al., 2010). In addition, C. albicans can induce significant production of PGE2 by mammalian cells (Filler et al., 1994; Deva et al., 2001). Candida albicans mannans, forming part of the C. albicans cell wall, can directly induce PGE2 production by mammalian cells (Smeekens et al., 2010). In addition, the induction of the release of AA and the eicosanoid metabolites from alveolar macrophages has been shown to be partly regulated by mannose and β-glucan receptors interacting with fungal cell wall components and have also been shown to inhibit phagocytosis during C. albicans infection (Castro et al., 1994).
Eradication of infection by the immune system is highly dependent on the balance of Th1 and Th2 responses (Romani, 2000). The Th1 response is associated with the removal of pathogens through phagocytosis, in contrast to the hampering of this effect during the Th2 response. Prostaglandin E2 is able to modulate this balance and frequently promotes colonization of pathogens such as C. albicans, as well as causing tissue eosinophilia. Prostaglandin E2 production in response to C. albicans invasion also induces the protective Th-17 response in mammalian systems (Smeekens et al., 2010).
Role of Candida albicans oxylipins during infection
AA can be used as a carbon source by C. albicans (Deva et al., 2000). The authors also identified a HETE produced by C. albicans from AA namely 3,18-diHETE, that is associated with hyphal forms and may play a role in adhesion during infection (Deva et al., 2000; Ells et al., 2012). Additionally, the authors showed that most AA is metabolized by non-mitochondrial pathways, concurrent with the speculation that fatty acids are metabolized by peroxisomal β-oxidation in yeasts. Acetylsalicylic acid-sensitive 3-HETE (Figure 6B), produced by C. albicans was also speculated to be linked to C. albicans morphogenesis (Deva et al., 2001). 3-HETE can also serve as substrate for COX-2 in mammalian cells to form 3-hydroxyeicosanoids due to the similarity to AA (Ciccoli et al., 2005). Further analysis of one of these metabolites, 3-OH-PGE2, indicated a similar or even more robust effect on mammalian cells eliciting a pro-inflammatory response.
The anti-inflammatory lipid Resolvin E1, which Candida albicans is able to produce from eicosapentaenoic acid, also plays a role in the interspecies signaling (Haas-Stapleton et al., 2007). This compound was shown to protect C. albicans against host immunity at low doses, however, at higher concentration, this protective effect is lost. This is thought to facilitate the commensal carriage of C. albicans as part of the human microbiome.
An important aspect of the determination of C. albicans virulence was the identification of a PGE2 cross reactive compound produced by C. albicans from exogenous AA (Noverr et al., 2001). This compound was later identified as PGE2, identical to the host product (Erb-Downward and Noverr, 2007). This was especially surprising as C. albicans does not possess homologs to COX present in mammalian cells responsible for the formation of PGE2. The involvement of various enzymes or homologous enzymes was speculated in the production of PGE2. A multicopper oxidase homolog (Fet3), fatty acid desaturase homolog (Ole2) and cytochrome P450s has been shown to be involved in the production of PGE2 by C. albicans (Erb-Downward and Noverr, 2007; Ells et al., 2011; Krause et al., 2015). Biofilm formation in C. albicans as well as germ tube formation has been shown to be enhanced by the addition of synthetic PGE2. In addition, various COX-inhibitors such as ASA drastically affect the formation of biofilms and germ tubes (Alem and Douglas, 2004). Interestingly, the simultaneous addition of PGE2 abolished the inhibitory effect of ASA on C. albicans biofilm formation. In a later study by the same researchers, a comparison of PGE2 production by C. albicans planktonic cells and biofilms showed a significant increase in PGE2 production of C. albicans biofilms (Alem and Douglas, 2005). In addition, various COX-inhibitors, namely ASA, diclofenac and etodolac, significantly decreased the production of PGE2 by C. albicans biofilms. These findings suggest a possible link between prostaglandin production and biofilm formation. Deva et al. (2001) reported significant decrease in yeast to hyphal transition by C. albicans with the addition of ASA. This morphological effect was attributed to the possible suppression of 3(R)-hydroxy-oxylipins.
Figure 7 indicates the interaction of C. albicans and P. aeruginosa with host cells with the focus on eicosanoid production. In the figure, arrows indicate the production or increase of an eicosanoid, whereas a question mark indicates an unknown effect or interaction.
Figure 7.

Interaction of Candida albicans and Pseudomonas aeruginosa with host cells highlighting the production of eicosanoids. Arrows indicate production/increase. Question marks indicate uncharacterized interactions (AA, Arachidonic acid; PLA2, Phospholipase A2; PGs, Prostaglandins; PGE2, Prostaglandin E2; ExoU, Exotoxin U; PYO, pyocyanin; LPS, lipopolysaccharide; 3-HETE, 3-hydroxyeicosatetraenoic acid; 3-oxo-HLS, 3-oxododecanoyl-L-homoserine lactone; 15-LOX, 15-Lipoxygenase).
Considering the information regarding the direct and indirect interaction of C. albicans and P. aeruginosa, as well as the information regarding the role of eicosanoids during single species infection, several questions can be asked relating to polymicrobial infection with C. albicans and P. aeruginosa.
Role of oxylipins in polymicrobial infection
Although information is available regarding the eicosanoids produced, and their effect during single species infection, very little is known regarding the effect of microbially-produced eicosanoids on co-infecting pathogens, as well as on the host.
In a study by Peters and Noverr (2013), the role of eicosanoids during polymicrobial infection in another microbial interaction became evident. In this study, a murine model was used to elucidate the effect of polymicrobial interactions using C. albicans and Staphylococcus aureus. Through their work, it was evident that polymicrobial infections with these organisms resulted in a significant increase in morbidity and mortality during murine peritoneal infection, with this effect not seen with single species infection. In addition, disease progression and microbial load in infected mice was significantly higher, compared to monomicrobial infections. The authors also detected a significant increase in pro-inflammatory chemokines released, as well as recruitment of polymorphonucleocytes. To determine the effect of the pro-inflammatory response on infection dynamics, indomethacin was used. Indomethacin, a non-selective COX inhibitor, similar to ASA, caused a significant reduction in morbidity and mortality in polymicrobial infection, as well as a reduction in cytokine release. Indomethacin also caused a significant reduction in microbial load during co-infection. This effect was not seen with monomicrobial infection. In vitro experiments showed that this effect was not due to inhibition of growth of C. albicans and S. aureus. The authors also observed a significant increase in PGE2 release by the host cells in response to polymicrobial infection with indomethacin also causing a significant reduction in PGE2. The authors suggested that PGE2 may play a role in the non-protective pro-inflammatory response during polymicrobial infection, as PGE2 induces the release of several cytokines. To further determine the effect of PGE2 in co-infection, mice were co-infected with C. albicans and S. aureus and treated with indomethacin as well as PGE2. Under this circumstance, high morbidity and mortality was observed in the mice, although they were treated with indomethacin. In the absence of infection, with only the administration of indomethacin and PGE2, no mortality was observed. Further, the administration of PGE2 caused a significant increase in microbial burden during co-infection. During this study, the production of eicosanoids, such as PGE2, by C. albicans itself was not addressed. Interestingly, Krause et al. (2015) indicated that C. albicans PGE2 production significantly enhances S. aureus biofilm growth in vitro. The authors also found that co-incubation of C. albicans with S. aureus did not elicit an increase in PGE2 production by C. albicans. Nothing is known yet regarding the eicosanoid production of C. albicans and P. aeruginosa during co-infection.
Conclusions
In addition to the ample evidence supporting the interaction of C. albicans and P. aeruginosa, not only with their host, but also with each other, it is evident that the interaction is multifaceted. Various virulence factors such as morphogenesis, hypermutabililty and secreted factors (including lipid mediators) affect and damage hosts to facilitate rapid and aggressive colonization and infection. Any disequilibrium in host defenses, such as in CF, immune disorders, and breaching of host barriers, is rapidly exploited by these opportunistic pathogens.
On their own, C. albicans and P. aeruginosa pose a risk to compromised hosts. Recent studies, however, illuminate that the interaction of these microorganisms are antagonistic with substantial damage caused to the host during the chemical war they play part in. Host immunity also plays a large role in damage to its own tissue due to radical generation by the innate immune response and various lipid mediators. This is even more evident in the work by Peters and Noverr (2013). It is also evident that various secreted factors of C. albicans (including farnesol and various hydrolytic enzymes) and P. aeruginosa (including AHL's, PQS, PYO and various peptides) form a large amount of radicals and can elicit oxidative damage to each other, as well as the host.
Although the role of lipid mediators between single pathogens and hosts has been studied, a gap in knowledge still exists regarding the effect of lipid mediators in polymicrobial infection of C. albicans and P. aeruginosa. Polymicrobial infection by C. albicans and S. aureus, however, highlight the importance of lipid mediators, such as PGE2, in infection by multiple microbes. Due to the fact that C. albicans and P. aeruginosa are both able to produce significant amounts of prostaglandins and other eicosanoids from exogenous AA, this could affect the dynamics of this co-infection as well as host survival during infection. This warrants investigation to fully understand the interaction of C. albicans and P. aeruginosa in terms of eicosanoids, as well as the effect of these eicosanoids in the host during co-infection.
Author contributions
RF compiled the information, co-wrote the manuscript and approved the final version submitted. RE, CW, OS, JA provided scholarly input in placing the literature into context, edited the manuscript and approved the final version submitted. CP provided scholarly input in placing the literature into context, co-wrote the manuscript and approved the final version submitted
Funding
The financial assistance of the National Research Foundation (NRF) (Grant: CPRR 93485) toward this research is hereby acknowledged. Opinions expressed and conclusions arrived at, are those of the author and are not necessarily to be attributed to the NRF.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. The reviewer AH and handling Editor declared a common affiliation and the handling Editor states that the process nevertheless met the standards of a fair and objective review.
References
- Ader F., Jawhara S., Nseir S., Kipnis E., Faure K., Vuotto F., et al. (2011). Short term Candida albicans colonization reduces Pseudomonas aeruginosa-related lung injury and bacterial burden in a murine model. Crit. Care 15:R150. 10.1186/cc10276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard M., Asakrah S., Morici L. A. (2013). PGE2 suppression of innate immunity during mucosal bacterial infection. Front. Cell. Infect. Microbiol. 3:45. 10.3389/fcimb.2013.00045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alem M. A. S., Douglas L. J. (2004). Effects of aspirin and other nonsteroidal anti-inflammatory drugs on biofilms and planktonic cells of Candida albicans. Antimicrob. Agents. Chemother. 48, 41–47. 10.1128/AAC.48.1.41-47.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alem M. A. S., Douglas L. J. (2005). Prostaglandin production during growth of Candida albicans biofilms. J. Med. Microbiol. 54, 1001–1005. 10.1099/jmm.0.46172-0 [DOI] [PubMed] [Google Scholar]
- Andersen D. H. (1938). Cystic fibrosis of the pancreas and its relation to celiac disease: a clinical and pathologic study. Am. J. Dis. Child. 56, 344–399. 10.1001/archpedi.1938.01980140114013 [DOI] [Google Scholar]
- Andes D., Nett J., Oschel P., Albrecht R., Marchillo K., Pitula H. (2004). Development and characterization of an in vivo central venous catheter Candida albicans biofilm model. Infect. Immun. 72, 6023–6031. 10.1128/IAI.72.10.6023-6031.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay E., Timsit J.-F., Tafflet M., de Lassence A., Darmon M., Zahar J.-R., et al. (2006). Candida colonization of the respiratory tract and subsequent Pseudomonas ventilator-associated pneumonia. Chest 129, 110–117. 10.1378/chest.129.1.110 [DOI] [PubMed] [Google Scholar]
- Ball G., Durand É., Lazdunski A., Filloux A. (2002). A novel type II secretion system in Pseudomonas aeruginosa. Mol. Cell. Biochem. 43, 475–485. 10.1046/j.1365-2958.2002.02759.x [DOI] [PubMed] [Google Scholar]
- Ballinger M. N., Aronoff D. M., McMillan T. R., Cooke K. R., Olkiewicz K., Toews G. B., et al. (2006). Critical role of prostaglandin E2 overproduction in impaired pulmonary host response following bone marrow transplantation. J. Immunol. 177, 5499–5508. 10.4049/jimmunol.177.8.5499 [DOI] [PubMed] [Google Scholar]
- Bandara H. M. H. N., K Cheung B. P., Watt R. M., Jin L. J., Samaranayake L. P. (2013). Pseudomonas aeruginosa lipopolysaccharide inhibits Candida albicans hyphae formation and alters gene expression during biofilm development. Mol. Oral. Microbiol. 28, 54–69. 10.1111/omi.12006 [DOI] [PubMed] [Google Scholar]
- Bandara H. M. H. N., Lam O. L. T., Watt R. M., Jin L. J., Samaranayake L. P. (2010). Bacterial lipopolysaccharides variably modulate in vitro biofilm formation of Candida species. J. Med. Microbiol. 59, 1225–1234. 10.1099/jmm.0.021832-0 [DOI] [PubMed] [Google Scholar]
- Baron S. S., Rowe J. J. (1981). Antibiotic action of pyocyanin. Antimicrob. Agents Chemother. 20, 814–820. 10.1128/AAC.20.6.814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benamara H., Rihouey C., Jouenne T., Alexandre S. (2011). Impact of the biofilm mode of growth on the inner membrane phospholipid composition and lipid domains in Pseudomonas aeruginosa. Biochim. Biophys. Acta 1808, 98–105. 10.1016/j.bbamem.2010.09.004 [DOI] [PubMed] [Google Scholar]
- Bianchi S. M., Prince L. R., McPhillips K., Allen L., Marriott H. M., Taylor G. W., et al. (2008). Impairment of apoptotic cell engulfment by pyocyanin, a toxic metabolite of Pseudomonas aeruginosa. Am. J. Respir. Crit. Care. Med. 177, 35–43. 10.1164/rccm.200612-1804OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand A., Barnes J. D., Mackenzie K. S., Odds F. C., Gow N. A. R. (2008). Cell wall glycans and soluble factors determine the interactions between the hyphae of Candida albicans and Pseudomonas aeruginosa. FEMS Microbiol. Lett. 287, 48–55. 10.1111/j.1574-6968.2008.01301.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash A. R. (2001). Arachidonic acid as a bioactive molecule. J. Clin. Investig. 107, 1339–1345. 10.1172/JCI13210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodhun F., Feussner I. (2011). Oxylipins in fungi. FEBS 278, 1047–1063. 10.1111/j.1742-4658.2011.08027.x [DOI] [PubMed] [Google Scholar]
- Bruzual I., Riggle P., Hadley S., Kumamoto C. A. (2007). Biofilm formation by fluconazole-resistant Candida albicans strains is inhibited by fluconazole. J. Antimicrob. Chemother. 59, 441–450. 10.1093/jac/dkl521 [DOI] [PubMed] [Google Scholar]
- Burmølle M., Webb J. S., Rao D., Hansen L. H., Sørensen S. J., Kjelleberg S. (2006). Enhanced biofilm formation and increased resistance to antimicrobial agents and bacterial invasion are caused by synergistic interactions in multispecies biofilms. Appl. Environ. Microbiol. 72, 3916–3923. 10.1128/AEM.03022-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone R. A., Fonzi W. A. (2001). Virulence factors of Candida albicans. Trends. Microbiol. 9, 327–335. 10.1016/S0966-842X(01)02094-7 [DOI] [PubMed] [Google Scholar]
- Castro M., Ralston N. V. C., Morgenthaler T. I., Rohrbach M. S., Limper A. H. (1994). Candida albicans stimulates arachidonic acid liberation from alveolar macrophages through alpha-mannan and beta-glucan cell wall components. Infect. Immun. 62, 3138–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A. I., Dolben E. F., Okegbe C., Harty C. E., Golub Y., Thao S., et al. (2014). Candida albicans ethanol stimulates Pseudomonas aeruginosa WspR-controlled biofilm formation as part of a cyclic relationship involving phenazines. PLoS Pathog. 10:e1004480. 10.1371/journal.ppat.1004480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilton F. H., Fonteh A. N., Surette M. E., Triggiani M., Winkler J. D. (1996). Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1299, 1–15. 10.1016/0005-2760(95)00169-7 [DOI] [PubMed] [Google Scholar]
- Ciccoli R., Sahi S., Singh S., Prakash H., Zafiriou M.-P., Ishdorj G., et al. (2005). Oxygenation by COX-2 (cyclo-oxygenase-2) of 3-HETE (3-hydroxyeicosatetraenoic acid), a fungal mimetic of arachidonic acid, produces a cascade of novel bioactive 3-hydroxyeicosanoids. Biochem. J. 390, 737–747. 10.1042/BJ20041995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad D. J., Bailey B. A. (2015). Multidimensional clinical phenotyping of an adult cystic fibrosis patient population. PLoS ONE 10:e0122705. 10.1371/journal.pone.0122705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox C. D. (1986). Role of pyocyanin in the acquisition of iron from transferrin. Infect. Immun. 52, 263–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cugini C., Calfee M. W., Farrow J. M., III, Morales D. K., Pesci E. C., Hogan D. A. (2007). Farnesol, a common sesquiterpene, inhibits PQS production in Pseudomonas aeruginosa. Mol. Microbiol. 65, 896–906. 10.1111/j.1365-2958.2007.05840.x [DOI] [PubMed] [Google Scholar]
- Cugini C., Morales D. K., Hogan D. A. (2010). Candida albicans-produced farnesol stimulates Pseudomonas quinolone signal production in LasR-defective Pseudomonas aeruginosa strains. Microbiology 156, 3096–3107. 10.1099/mic.0.037911-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kievit T. R., Iglewski B. H. (2000). Bacterial quorum sensing in pathogenic relationships. Infect. Immun. 68, 4839–4849. 10.1128/IAI.68.9.4839-4849.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhaes L., Monchy S., Fréalle E., Hubans C., Salleron J., Leroy S., et al. (2012). The airway microbiota in cystic fibrosis: a complex fungal and bacterial community-implications for therapeutic management. PLoS ONE 7:e36313. 10.1371/journal.pone.0036313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis E. A., Norris P. C. (2015). Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 15, 511–523. 10.1038/nri3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sordi L., Mühlschlegel F. A. (2009). Quorum sensing and fungal-bacterial interactions in Candida albicans: a communicative network regulating microbial coexistence and virulence. FEMS Yeast Res. 9, 990–999. 10.1111/j.1567-1364.2009.00573.x [DOI] [PubMed] [Google Scholar]
- Deva R., Ciccoli R., Kock L., Nigam S. (2001). Involvement of aspirin-sensitive oxylipins in vulvovaginal candidiasis. FEMS Microbiol. Lett. 198, 37–43. 10.1111/j.1574-6968.2001.tb10616.x [DOI] [PubMed] [Google Scholar]
- Deva R., Ciccoli R., Schewe T., Kock J. L. F., Nigam S. (2000). Arachidonic acid stimulates cell growth and forms a novel oxygenated metabolite in Candida albicans. Biochim. Biophys. Acta 1486, 299–311. 10.1016/S1388-1981(00)00073-1 [DOI] [PubMed] [Google Scholar]
- Déziel E., Lépine F., Milot S., He J., Mindrinos M. N., Tompkins R. G., et al. (2004). Analysis of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines (HAQs) reveals a role for 4-hydroxy-2-heptylquinoline in cell-to-cell communication. Proc. Natl. Acad. Sci. U.S.A. 101, 1339–1344. 10.1073/pnas.0307694100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz P. I., Strausbaugh L. D., Dongari-Bagtzoglou A. (2014). Fungal-bacterial interactions and their relevance to oral health: linking the clinic and the bench. Front. Cell. Infect. Microbiol. 4:101. 10.3389/fcimb.2014.00101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich L. E. P., Price-Whelan A., Petersen A., Whiteley M., Newman D. K. (2006). The phenazine pyocyanin is a terminal signalling factor in the quorum sensing network of Pseudomonas aeruginosa. Mol. Microbiol. 61, 1308–1321. 10.1111/j.1365-2958.2006.05306.x [DOI] [PubMed] [Google Scholar]
- Douglas L. J. (2003). Candida biofilms and their role in infection. Trends Microbiol. 11, 30–36. 10.1016/S0966-842X(02)00002-1 [DOI] [PubMed] [Google Scholar]
- Drenkard E. (2003). Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 5, 1213–1219. 10.1016/j.micinf.2003.08.009 [DOI] [PubMed] [Google Scholar]
- Drenkard E., Ausubel F. M. (2002). Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 416, 740–743. 10.1038/416740a [DOI] [PubMed] [Google Scholar]
- Dumitru R., Hornby J. M., Nickerson K. W. (2004). Defined anaerobic growth medium for studying Candida albicans basic biology and resistance to eight antifungal drugs. Antimicrob. Agents Chemother. 48, 2350–2354. 10.1128/AAC.48.7.2350-2354.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Azizi M. A., Starks S. E., Khardori N. (2004). Interactions of Candida albicans with other Candida spp. and bacteria in the biofilms. J. Appl. Microbiol. 96, 1067–1073. 10.1111/j.1365-2672.2004.02213.x [DOI] [PubMed] [Google Scholar]
- Ells R., Kock J. L. F., Albertyn J., Kemp G., Pohl C. H. (2011). Effect of inhibitors of arachidonic acid metabolism on prostaglandin E2 production by Candida albicans and Candida dubliniensis biofilms. Med. Microbiol. Immunol. 200, 23–28. 10.1007/s00430-010-0169-7 [DOI] [PubMed] [Google Scholar]
- Ells R., Kock J. L. F., Albertyn J., Pohl C. H. (2012). Arachidonic acid metabolites in pathogenic yeasts. Lipids Health Dis. 11:100. 10.1186/1476-511X-11-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erb-Downward J. R., Noverr M. C. (2007). Characterization of prostaglandin E2 production by Candida albicans. Infect. Immun. 75, 3498–3505. 10.1128/IAI.00232-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure E., Mear J.-B., Faure K., Normand S., Couturier-Maillard A., Grandjean T., et al. (2014). Pseudomonas aeruginosa type-3 secretion system dampens host defense by exploiting the NLRC4-coupled inflammasome. Am. J. Respir. Crit. Care. Med. 189, 799–811. 10.1164/rccm.201307-1358OC [DOI] [PubMed] [Google Scholar]
- Filler S. G., Ibe B. O., Ibrahim A. S., Ghannoum M. A., Rai J. U., Edwards J. E. (1994). Mechanisms by which Candida albicans induces endothelial cell prostaglandin synthesis. Infect. Immun. 62, 1064–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuse K., Fujimura S., Kikuchi T., Gomi K., Iida Y., Nukiwa T., et al. (2013). Reduction of virulence factor pyocyanin production in multidrug-resistant Pseudomonas aeruginosa. J. Infect. Chemother. Off. J. Japan. Soc. Chemother. 19, 82–88. 10.1007/s10156-012-0457-9 [DOI] [PubMed] [Google Scholar]
- Gibson J., Sood A., Hogan D. A. (2009). Pseudomonas aeruginosa-Candida albicans interactions: localization and fungal toxicity of a phenazine derivative. Appl. Environ. Microbiol. 75, 504–513. 10.1128/AEM.01037-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Bona A., Parra-Giraldo C. M., Hernáez M. L., Reales-Calderon J. A., Solis N. V., Filler S. G., et al. (2015). Candida albicans cell shaving uncovers new proteins involved in cell wall integrity, yeast to hypha transition, stress response and host–pathogen interaction. J. Prot. 127, 340–351. 10.1016/j.jprot.2015.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilligan P. H. (1991). Microbiology of airway disease in patients with cystic fibrosis. Clin. Microbiol. 4, 35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyne L. S., Grant G. D., Perkins A. V., Powell K. L., McDermott C. M., Johnson P. V., et al. (2011). Pyocyanin-induced toxicity in A549 respiratory cells is causally linked to oxidative stress. Toxicol. Vitr. 25, 1353–1358. 10.1016/j.tiv.2011.05.004 [DOI] [PubMed] [Google Scholar]
- Gow N. A. R., Hube B. (2012). Importance of the Candida albicans cell wall during commensalism and infection. Curr. Opin. Microbiol. 15, 406–412. 10.1016/j.mib.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Güngör Ö., Tamay Z., Güler N., Erturan Z. (2013). Frequency of fungi in respiratory samples from Turkish cystic fibrosis patients. Mycoses 56, 123–129. 10.1111/j.1439-0507.2012.02221.x [DOI] [PubMed] [Google Scholar]
- Gupta N., Haque A., Mukhopadhyay G., Narayan R. P., Prasad R. (2005). Interactions between bacteria and Candida in the burn wound. Burns 31, 375–378. 10.1016/j.burns.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Ha D., Merritt J. H., Hampton T. H., Hodgkinson J. T., Janecek M., Spring D. R., et al. (2011). 2-Heptyl-4-Quinolone, a precursor of the Pseudomonas quinolone signal molecule, modulates swarming motility in Pseudomonas aeruginosa. J. Bacteriol. 193, 6770–6780. 10.1128/JB.05929-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas-Stapleton E. J., Lu Y., Hong S., Arita M., Favoreto S., Nigam S., et al. (2007). Candida albicans modulates host defense by biosynthesizing the pro-resolving mediator resolvin E1. PLoS ONE 2:e1316. 10.1371/journal.pone.0001316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamet M., Pavon A., Dalle F., Pechinot A., Prin S., Quenot J.-P., et al. (2012). Candida spp. airway colonization could promote antibiotic-resistant bacteria selection in patients with suspected ventilator-associated pneumonia. Intensive Care Med. 38, 1272–1279. 10.1007/s00134-012-2584-2 [DOI] [PubMed] [Google Scholar]
- Hargarten J. C., Moore T. C., Petro T. M., Nickerson K. W., Atkin A. L. (2015). Candida albicans quorum sensing molecules stimulate mouse macrophage migration. Infect. Immun. 83, 3857–3864. 10.1128/IAI.00886-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriott M. M., Noverr M. C. (2011). Importance of Candida-bacterial polymicrobial biofilms in disease. Trends Microbiol. 19, 557–563. 10.1016/j.tim.2011.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan H. M., Fridovich I. (1980). Mechanism of the antibiotic action of pyocyanine. J. Bacteriol. 141, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler S., Becker T. (2008). The Pseudomonas Quinolone Signal (PQS) balances life and death in Pseudomonas aeruginosa populations. PLoS Pathog. 4:e1000166. 10.1371/journal.ppat.1000166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentzer M., Wu H., Andersen J. B., Riedel K., Rasmussen T. B., Bagge N., et al. (2003). Attenuation of Pseudomonas aeruginosa virulence by quorum sensing inhibitors. EMBO J. 22, 3803–3815. 10.1093/emboj/cdg366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez M. E., Kappler A., Newman D. K. (2004). Phenazines and other redox-active antibiotics promote microbial mineral reduction. Appl. Environ. Microbiol. 70, 921–928. 10.1128/AEM.70.2.921-928.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogardt M., Roeder M., Schreff A. M., Eberl L., Heesemann J. (2004). Expression of Pseudomonas aeruginosa exoS is controlled by quorum sensing and RpoS. Microbiology 150, 843–851. 10.1099/mic.0.26703-0 [DOI] [PubMed] [Google Scholar]
- Holcombe L. J., McAlester G., Munro C. A., Enjalbert B., Brown A. J. P., Gow N. A. R., et al. (2010). Pseudomonas aeruginosa secreted factors impair biofilm development in Candida albicans. Microbiology 156, 1476–1486. 10.1099/mic.0.037549-0 [DOI] [PubMed] [Google Scholar]
- Hornby J. M., Jensen E. C., Lisec A. D., Tasto J. J., Jahnke B., Shoemaker R., et al. (2001). Quorum sensing in the dimorphic fungus Candida albicans is mediated by farnesol. Appl. Environ. Microbiol. 67, 2982–2992. 10.1128/AEM.67.7.2982-2992.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou C. T. (2008). New bioactive fatty acids. Asia. Pac. J. Clin. Nutr. 17, 192–195. [PubMed] [Google Scholar]
- Jones-Dozier S. L. (2008). Proteomic Analysis of the Response of Pseudomonas aeruginosa PAO1 to the Cell to Cell Signaling Molecule Trans, Trans-Farnesol of Candida albicans. Department of Biology, Georgia State University, Atlanta, USA. [Google Scholar]
- Kaleli I., Cevahir N., Demir M., Yildirim U., Sahin R. (2006). Anticandidal activity of Pseudomonas aeruginosa strains isolated from clinical specimens. Mycoses 50, 74–78. 10.1111/j.1439-0507.2006.01322.x [DOI] [PubMed] [Google Scholar]
- Kalinski P. (2012). Regulation of immune responses by prostaglandin E2. J. Immunol. 188, 21–28. 10.4049/jimmunol.1101029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. R. (1994). Suppression of fungal growth exhibited by Pseudomonas aeruginosa. J. Clin. Microbiol. 32, 525–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr J. R., Taylor G. W., Rutman A., Hoiby N., Cole P. J., Wilson R. (1999). Pseudomonas aeruginosa pyocyanin and 1-hydroxyphenazine inhibit fungal growth. J. Clin. Pathol. 52, 385–387. 10.1136/jcp.52.5.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klausen M., Heydorn A., Ragas P., Lambertsen L., Aaes-Jørgensen A., Molin S., et al. (2003). Biofilm formation by Pseudomonas aeruginosa wild type, flagella and type IV pili mutants. Mol. Microbiol. 48, 1511–1524. 10.1046/j.1365-2958.2003.03525.x [DOI] [PubMed] [Google Scholar]
- Köhler T., Guanella R., Carlet J., van Delden C. (2010). Quorum sensing-dependent virulence during Pseudomonas aeruginosa colonisation and pneumonia in mechanically ventilated patients. Thorax 65, 703–710. 10.1136/thx.2009.133082 [DOI] [PubMed] [Google Scholar]
- König B., Jaeger K. E., Sage A. E., Vasil M. L., König W. (1996). Role of Pseudomonas aeruginosa lipase in inflammatory mediator release from human inflammatory effector cells (platelets, granulocytes, and monocytes). Infect. Immun. 64, 3252–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause J., Geginat G., Tammer I. (2015). Prostaglandin E2 from Candida albicans stimulates the growth of Staphylococcus aureus in mixed biofilms. PLoS ONE 10:e0135404. 10.1371/journal.pone.0135404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn D. M., George T., Chandra J., Mukherjee P. K., Ghannoum M. A. (2002). Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob. Agents Chemother. 46, 1773–1780. 10.1128/AAC.46.6.1773-1780.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H., O'Donnell V. B. (2006). Inflammation and immune regulation by 12/15-lipoxygenase. Prog. Lipid. Res. 45, 334–356. 10.1016/j.plipres.2006.02.003 [DOI] [PubMed] [Google Scholar]
- Kumar S. N., Nisha G. V., Sudaresan A., Venugopal V. V., Sree Kumar M. M., Lankalapalli R. S., et al. (2014). Synergistic activity of phenazines isolated from Pseudomonas aeruginosa in combination with azoles against Candida species. Med. Mycol. 52, 480–488. 10.1093/mmy/myu01 [DOI] [PubMed] [Google Scholar]
- Lamacka M., Sajbidor J. (1995). The occurrence of prostaglandins and related compounds in lower organisms. Prostaglandins Leukot. Essiential Fat. acids 52, 357–364. 10.1016/0952-3278(95)90062-4 [DOI] [PubMed] [Google Scholar]
- Lamont I. L., Beare P. A., Ochsner U., Vasil A. I., Vasil M. L. (2002). Siderophore-mediated signaling regulates virulence factor production in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 99, 7072–7077. 10.1073/pnas.092016999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lépine F., Déziel E., Milot S., Rahme L. G. (2003). A stable isotope dilution assay for the quantification of the Pseudomonas quinolone signal in Pseudomonas aeruginosa cultures. Biochim. Biophys. Acta 1622, 36–41. 10.1016/S0304-4165(03)00103-X [DOI] [PubMed] [Google Scholar]
- Lindsay A. K., Hogan D. A. (2014). Candida albicans: molecular interactions with Pseudomonas aeruginosa and Staphylococcus aureus. Fungal Biol. Rev. 28, 85–96. 10.1016/j.fbr.2014.10.002 [DOI] [Google Scholar]
- Lopez-Medina E., Fan D., Coughlin L. A., Ho E. X., Lamont I. L., Reimmann C., et al. (2015). Candida albicans inhibits Pseudomonas aeruginosa virulence through suppression of pyochelin and pyoverdine biosynthesis. PLoS Pathog. 11:e1005129. 10.1371/journal.ppat.1005129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnett L. J., Rowlinson S. W., Goodwin D. C., Kalgutkar A. S., Lanzo C. A. (1999). Arachidonic acid oxygenation by COX-1 and COX-2: mechanisms of catalysis and inhibition. J. Biol. Chem. 274, 22903–22906. 10.1074/jbc.274.33.22903 [DOI] [PubMed] [Google Scholar]
- McAlester G., O'Gara F., Morrissey J. P. (2008). Signal-mediated interactions between Pseudomonas aeruginosa and Candida albicans. J. Med. Microbiol. 57, 563–569. 10.1099/jmm.0.47705-0 [DOI] [PubMed] [Google Scholar]
- McDermott C., Chess-Williams R., Mills K. A., Kang S. H., Farr S. E., Grant G. D., et al. (2013). Alterations in acetylcholine, PGE2 and IL6 release from urothelial cells following treatment with pyocyanin and lipopolysaccharide. Toxicol. Vitr. 27, 1693–1698. 10.1016/j.tiv.2013.04.015 [DOI] [PubMed] [Google Scholar]
- Mear J. B., Gosset P., Kipnis E., Faure E., Dessein R., Jawhara S., et al. (2014). Candida albicans airway exposure primes the lung innate immune response against Pseudomonas aeruginosa infection through innate lymphoid cell recruitment and interleukin-22-associated mucosal response. Infect. Immun. 82, 306–315. 10.1128/IAI.01085-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méar J.-B., Kipnis E., Faure E., Dessein R., Schurtz G., Faure K., et al. (2013). Candida albicans and Pseudomonas aeruginosa interactions: more than an opportunistic criminal association? Médecine Mal. Infect. 43, 146–151. 10.1016/j.medmal.2013.02.005 [DOI] [PubMed] [Google Scholar]
- Morales D. K., Grahl N., Okegbe C., Dietrich L. E. P., Jacobs N. J., Hogan D. A. (2013). Control of Candida albicans metabolism and biofilm formation by Pseudomonas aeruginosa phenazines. MBio 4:e00526–12. 10.1128/mBio.00526-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales D. K., Hogan D. A. (2010). Candida albicans interactions with bacteria in the context of human health and disease. PLoS Pathog. 6:e1000886. 10.1371/journal.ppat.1000886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M., Nakashima K., Kamei D., Masuda S., Ishikawa Y., Ishii T., et al. (2003). Cellular prostaglandin E2 production by membrane-bound prostaglandin E synthase-2 via both cyclooxygenases-1 and -2. J. Biol. Chem. 278, 37937–37947. 10.1074/jbc.M305108200 [DOI] [PubMed] [Google Scholar]
- Neely A. N., Law E. J., Holder I. A. (1986). Increased susceptibility to lethal Candida Infections in burned mice preinfected with Pseudomonas aeruginosa or pretreated with proteolytic enzymes. Infect. Immun. 52, 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niewerth M., Korting H. C. (2001). Phospholipases of Candida albicans. Mycoses 367, 361–367. 10.1046/j.1439-0507.2001.00685.x [DOI] [PubMed] [Google Scholar]
- Noverr M. C., Phare S. M., Toews G. B., Coffey M. J., Huffnagle G. B. (2001). Pathogenic yeasts Cryptococcus neoformans and Candida albicans produce immunomodulatory prostaglandins. Infect. Immun. 69, 2957–2963. 10.1128/IAI.69.5.2957-2963.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nseir S., Jozefowicz E., Cavestri B., Sendid B., Di Pompeo C., Dewavrin F., et al. (2007). Impact of antifungal treatment on Candida-Pseudomonas interaction: a preliminary retrospective case-control study. Intensive Care Med. 33, 137–142. 10.1007/s00134-006-0422-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley Y. Q., Abdalla M. Y., McCormick M. L., Reszka K. J., Denning G. M., Britigan B. E. (2003). Subcellular localization of Pseudomonas pyocyanin cytotoxicity in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 284, L420–L430. 10.1152/ajplung.00316.2002 [DOI] [PubMed] [Google Scholar]
- O'Toole G., Kaplan H. B., Kolter R. (2000). Biofilm formation as microbial development. Annu. Rev. Microbiol. 54, 49–79. 10.1146/annurev.micro.54.1.49 [DOI] [PubMed] [Google Scholar]
- Ovchinnikova E. S., Krom B. P., van der Mei H. C., Busscher H. J. (2012). Force microscopic and thermodynamic analysis of the adhesion between Pseudomonas aeruginosa and Candida albicans. Soft Matter 8, 6454–6461. 10.1039/c2sm25100k [DOI] [Google Scholar]
- Parti R. P., Loper R., Brown G. D., Gordon S., Taylor P. R., Bonventre J. V., et al. (2010). Cytosolic phospholipase A2 activation by Candida albicans in alveolar macrophages: role of dectin-1. Am. J. Respir. Cell. Mol. Biol. 42, 415–423. 10.1165/rcmb.2009-0110OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passador L., Cook J. M., Gambello M. J., Rust L., Iglewski B. H. (1993). Expression of Pseudomonas aeruginosa virulence genes requires cell-to-cell communication. Science 260, 1127–1130. 10.1126/science.8493556 [DOI] [PubMed] [Google Scholar]
- Pearson J. P., Passador L., Iglewski B. H., Greenberg E. P. (1995). A second N-acylhomoserine lactone signal produced by Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 92, 1490–1494. 10.1073/pnas.92.5.1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesci E. C., Milbank J. B. J., Pearson J. P., McKnight S., Kende A. S., Greenberg E. P., et al. (1999). Quinolone signaling in the cell-to-cell communication system of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 96, 11229–11234. 10.1073/pnas.96.20.11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters B. M., Noverr M. C. (2013). Candida albicans-Staphylococcus aureus polymicrobial peritonitis modulates host innate immunity. Infect. Immun. 81, 2178–2189. 10.1128/IAI.00265-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan V. V., Moree W. J., Aguilar J., Cornett D. S., Koumoutsi A., Noble S. M., et al. (2014). Impact of a transposon insertion in phzF2 on the specialized metabolite production and interkingdom interactions of Pseudomonas aeruginosa. J. Bacteriol. 196, 1683–1693. 10.1128/JB.01258-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pier G. B. (1985). Pulmonary disease associated with Pseudomonas aeruginosa in cystic fibrosis: current status of the host-bacterium interaction. J. Infect. Dis. 151, 575–580. 10.1093/infdis/151.4.575 [DOI] [PubMed] [Google Scholar]
- Pierce G. E. (2005). Pseudomonas aeruginosa, Candida albicans, and device-related nosocomial infections: implications, trends, and potential approaches for control. J. Ind. Microbiol. Biotechnol. 32, 309–318. 10.1007/s10295-005-0225-2 [DOI] [PubMed] [Google Scholar]
- Price-Whelan A., Dietrich L. E. P., Newman D. K. (2007). Pyocyanin alters redox homeostasis and carbon flux through central metabolic pathways in Pseudomonas aeruginosa PA14. J. Bacteriol. 189, 6372–6381. 10.1128/JB.00505-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke F. G., Hiller E., Trick I., Rupp S. (2012). Flexible survival strategies of Pseudomonas aeruginosa in biofilms result in increased fitness compared with Candida albicans. Mol. Cell. Proteomics 11, 1652–1669. 10.1074/mcp.M112.017673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramage G., VandeWalle K., Wickes B. L., López-Ribot J. L. (2001). Characteristics of biofilm formation by Candida albicans. Rev. Iberoam. Micol. 18, 163–170. [PubMed] [Google Scholar]
- Reen F. J., Mooij M. J., Holcombe L. J., McSweeney C. M., McGlacken G. P., Morrissey J. P., et al. (2011). The Pseudomonas quinolone signal (PQS), and its precursor HHQ, modulate interspecies and interkingdom behaviour. FEMS Microbiol. Ecol. 77, 413–428. 10.1111/j.1574-6941.2011.01121.x [DOI] [PubMed] [Google Scholar]
- Rinzan F. F. (2009). Pseudomonas Aeruginosa-Candida albicans Interactions from Ecological and Molecular Perspectives. Department of Biology, Georgia State University, Atlanta, USA. [Google Scholar]
- Rodríguez M., Domingo E., Municio C., Alvarez Y., Hugo E., Fernández N., et al. (2014). Polarization of the innate immune response by prostaglandin E2: a puzzle of receptors and signals. Mol. Pharmacol. 85, 187–197. 10.1124/mol.113.089573 [DOI] [PubMed] [Google Scholar]
- Romani L. (2000). Innate and adaptive immunity in Candida albicans infections and saprophytism. J. Leukoc. Biol. 68, 175–179. [PubMed] [Google Scholar]
- Roux D., Gaudry S., Dreyfuss D., El-Benna J., de Prost N., Denamur E., et al. (2009). Candida albicans impairs macrophage function and facilitates Pseudomonas aeruginosa pneumonia in rat. Crit. Care Med. 37, 1062–1067. 10.1097/CCM.0b013e31819629d2 [DOI] [PubMed] [Google Scholar]
- Roux D., Gaudry S., Khoy-Ear L., Aloulou M., Phillips-Houlbracq M., Bex J., et al. (2013). Airway fungal colonization compromises the immune system allowing bacterial pneumonia to prevail. Crit. Care Med. 41, 191–199. 10.1097/ccm.0b013e31828a25d6 [DOI] [PubMed] [Google Scholar]
- Sadikot R. T., Zeng H., Azim A. C., Joo M., Dey S. K., Breyer R. M., et al. (2007). Bacterial clearance of Pseudomonas aeruginosa is enhanced by the inhibition of COX-2. Eur. J. Immunol. 37, 1001–1009. 10.1002/eji.200636636 [DOI] [PubMed] [Google Scholar]
- Saliba A. M., Nascimento D. O., Silva M. C. A., Assis M. C., Gayer C. R. M., Raymond B., et al. (2005). Eicosanoid-mediated proinflammatory activity of Pseudomonas aeruginosa ExoU. Cell. Microbiol. 7, 1811–1822. 10.1111/j.1462-5822.2005.00635.x [DOI] [PubMed] [Google Scholar]
- Sandven P. (2000). Epidemiology of candidemia. Rev. Iberoam. Micol. 17, 73–81. [PubMed] [Google Scholar]
- Sapaar B., Nur A., Hirota K., Yumoto H., Murakami K., Amoh T., et al. (2014). Effects of extracellular DNA from Candida albicans and pneumonia-related pathogens on Candida biofilm formation and hyphal transformation. J. Appl. Microbiol. 116, 1531–1542. 10.1111/jam.12483 [DOI] [PubMed] [Google Scholar]
- Serhan C. N. (2002). Lipoxins and aspirin-triggered 15-epi-lipoxin biosynthesis: an update and role in anti-inflammation and pro-resolution. Prostaglandins Other Lipid. Mediat. 68–69, 433–455. 10.1016/S0090-6980(02)00047-3 [DOI] [PubMed] [Google Scholar]
- Smeekens S. P., van de Veerdonk F. L., van der Meer J. W. M., Kullberg B. J., Joosten L. A. B., Netea M. G. (2010). The Candida Th17 response is dependent on mannan- and beta-glucan-induced prostaglandin E2. Int. Immunol. 22, 889–895. 10.1093/intimm/dxq442 [DOI] [PubMed] [Google Scholar]
- Smith R. S., Harris S. G., Phipps R., Iglewski B. (2002). The Pseudomonas aeruginosa quorum-sensing molecule N-(3-Oxododecanoyl) homoserine lactone contributes to virulence and induces inflammation in vivo. J. Bacteriol. 184, 1132–1139. 10.1128/jb.184.4.1132-1139.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synnott J. M., Guida A., Mulhern-Haughey S., Higgins D. G., Butler G. (2010). Regulation of the hypoxic response in Candida albicans. Eukaryot. Cell 9, 1734–1746. 10.1128/EC.00159-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M. -W., Mahajan-Miklos S., Ausubel F. M. (1999). Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc. Natl. Acad. Sci. U.S.A. 96, 715–720. 10.1073/pnas.96.2.715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tehlivets O., Scheuringer K., Kohlwein S. D. (2007). Fatty acid synthesis and elongation in yeast. Biochim. Biophys. Acta 1771, 255–270. 10.1016/j.bbalip.2006.07.004 [DOI] [PubMed] [Google Scholar]
- Theiss S., Ishdorj G., Brenot A., Kretschmar M., Lan C.-Y., Nichterlein T., et al. (2006). Inactivation of the phospholipase B gene PLB5 in wild-type Candida albicans reduces cell-associated phospholipase A2 activity and attenuates virulence. Int. J. Med. Microbiol. 296, 405–420. 10.1016/j.ijmm.2006.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trejo-Hernández A., Andrade-Domínguez A., Hernández M., Encarnación S. (2014). Interspecies competition triggers virulence and mutability in Candida albicans – Pseudomonas aeruginosa mixed biofilms. ISME J. 8, 1974–1988. 10.1038/ismej.2014.53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza G., Tappe D., Turnwald D., Frosch M., König C., Hebestreit H., et al. (2008). Prevalence and antimicrobial susceptibility of microorganisms isolated from sputa of patients with cystic fibrosis. J. Cyst. Fibros. 7, 123–127. 10.1016/j.jcf.2007.06.006 [DOI] [PubMed] [Google Scholar]
- Vance R. E., Hong S., Gronert K., Serhan C. N., Mekalanos J. J. (2004). The opportunistic pathogen Pseudomonas aeruginosa carries a secretable arachidonate 15-lipoxygenase. Proc. Natl. Acad. Sci. U.S.A. 101, 2135–2139. 10.1073/pnas.0307308101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner V. E., Bushnell D., Passador L., Brooks A. I., Iglewski B. H. (2003). Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185, 2080–2095. 10.1128/JB.185.7.2080-2095.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwater C., Balish E., Schofield D. A. (2005). Candida albicans-conditioned medium protects yeast cells from oxidative stress: a possible link between quorum sensing and oxidative stress resistance. Eukaryot. Cell 4, 1654–1661. 10.1128/EC.4.10.1654-1661.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson D. R., Albert M., Perreault M. M., Delisle M.-S., Muscedere J., Rotstein C., et al. (2011). The relationship between Candida species cultured from the respiratory tract and systemic inflammation in critically ill patients with ventilator-associated pneumonia. Can. J. Anesth. 58, 275–284. 10.1007/s12630-010-9439-5 [DOI] [PubMed] [Google Scholar]
- Xu L., Wang F., Shen Y., Hou H., Liu W., Liu C., et al. (2014). Pseudomonas aeruginosa inhibits the growth of pathogenic fungi: in vitro and in vivo studies. Exp. Ther. Med. 7, 1516–1520. 10.3892/etm.2014.1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Teck Ho Lee R., Fang H., Wang Y., Li R., Zou H., et al. (2008). Bacterial peptidoglycan triggers Candida albicans hyphal growth by directly activating the adenylyl cyclase Cyr1p. Cell Host Microbe 4, 28–39. 10.1016/j.chom.2008.05.014 [DOI] [PubMed] [Google Scholar]