

Abstract
Cystic fibrosis (CF) is characterized by chronic infection and inflammation of the airways. In vitro culture of select bacterial species from respiratory specimens has been used to guide antimicrobial therapy in CF for the past few decades. More recently, DNA sequence-based, culture-independent approaches have been used to assess CF airway microbiology, although the role that these methods will (or should) have in routine microbiologic analysis of CF respiratory specimens is unclear. We performed DNA sequence analyses to detect bacterial species in 945 CF sputum samples that had been previously analyzed by selective CF culture. We determined the concordance of results based on culture and sequence analysis, highlighting the comparison of the results for the most prevalent genera. Although overall prevalence rates were comparable between the two methods, results varied by genus. While sequence analysis was more likely to detect Achromobacter, Stenotrophomonas, and Burkholderia, it was less likely to detect Staphylococcus. Streptococcus spp. were rarely reported in culture results but were the most frequently detected species by sequence analysis. A variety of obligate and facultative anaerobic species, not reported by culture, was also detected with high prevalence by sequence analysis. Sequence analysis indicated that in a considerable proportion of samples, taxa not reported by selective culture constituted a relatively high proportion of the total bacterial load, suggesting that routine CF culture may underrepresent significant segments of the bacterial communities inhabiting CF airways.
INTRODUCTION
A hallmark of cystic fibrosis (CF) is chronic bacterial infection of the respiratory tract. Infection and the associated inflammation contribute to progressive lung damage, which ultimately results in respiratory failure, the leading cause of death in CF. For the past few decades, an important aspect of CF care has been the use of intensive antimicrobial therapy. Although therapy in younger patients is often provided in an attempt to eradicate initial infection, antibiotics in older patients are typically prescribed to suppress chronic infection or to treat the intermittent exacerbations of respiratory symptoms characteristic of CF. In vitro culture of CF respiratory specimens (most often expectorated sputum) has been used to guide the choice of antibiotics and has focused on the selective recovery of a relatively small suite of bacterial species believed to contribute to lung disease in CF. Historically, this set included Staphylococcus aureus, Haemophilus influenzae, and Pseudomonas aeruginosa (1). Since the 1990s, the list of CF species of interest has expanded to include a small number of other opportunistic bacterial pathogens, including Achromobacter xylosoxidans, Stenotrophomonas maltophilia, and the Burkholderia cepacia complex, which also have been associated with disease progression in CF (2).
During the past decade, the microbiology of CF respiratory tract infection has been further explored through the use of methods that detect the presence of bacterial species without relying on microbial growth in culture. Such culture-independent approaches, enabled by rapid advances in DNA sequencing technology, differ fundamentally from traditional culture-based clinical microbiology in several important ways. While culture adroitly exploits phenotypic differences to selectively recover, isolate, and identify a relatively small set of specific bacterial species of interest (i.e., species believed to be pathogenic in CF), culture-independent methods do not consider the clinical relevance of the species being detected and do not rely on microbial growth in vitro. Consequently, the breadth of microbial species detected in a biological specimen by DNA sequence analysis is far greater than that expected to be reported by standard culture approaches. Another important feature of DNA sequence-based analyses is that they are typically capable of providing estimations of the relative abundances of the species detected.
Culture-independent, DNA sequence-based analyses have heightened awareness that CF airway infection is most often polymicrobial. In addition to the traditional CF pathogens recovered in standard CF culture, sequence-based analyses have consistently detected a broad array of bacterial species in CF respiratory samples. While some of these species are not routinely sought in the culture of CF specimens (e.g., anaerobic species), others may not be considered clinically significant (e.g., discarded as oral flora) (3). Although the role that these latter species may play in contributing to CF lung disease remains unclear, recent work suggests that the oral cavity and upper airway serve as important sources of species inhabiting the lower airways (4–6).
Cross-sectional and longitudinal studies have also shown that changes in the structure of airway communities may be associated with progression of lung disease or exacerbations of respiratory symptoms (7–12). Thus, analyses that provide a depiction of community structure may become an expectation of CF caregivers in the not-too-distant future (13). As DNA sequence analysis continues to become less expensive and more accessible, the role of culture-independent analysis of CF respiratory specimens in patient management will warrant further discussion. How the results of sequence-based analyses compare to the results obtained from selective CF culture will be an important consideration in these discussions.
In this study, we retrospectively analyzed a large set of CF sputum samples that had been assessed by DNA sequence analysis and by routine CF selective culture in a single CF care center. We compared the results obtained by the two methods to better understand the ways that these methods are complementary and the ways that they are distinct.
MATERIALS AND METHODS
Patients, sputum specimens, and medical record review.
Sputum sample collection and medical record review were approved by the University of Michigan Institutional Review Board. Sputum specimens, collected during the course of routine medical care, were processed by the University of Michigan Health System clinical microbiology laboratory using standard protocols for selective culturing of CF respiratory specimens (14). Specimens were routinely cultured onto several solid media, including chocolate agar supplemented with cefsulodin, sheep's blood agar, mannitol salt agar, MacConkey agar, and Burkholderia cepacia selective agar. Beginning in 2008, chocolate agar supplemented with bacitracin was used. Anaerobic culture was not routinely used during this period to culture CF specimens. After processing, the remaining specimen was stored at −80°C in 0.5-ml aliquots. Culture results for each sputum specimen were obtained from the clinical microbiology laboratory database. Results indicating enteric Gram-negative rod (n = 41), fermenting Gram-negative rod (n = 3), Gram-negative rod (n = 1), or nonfermenting Gram-negative rod (n = 4) were not included in the analysis.
DNA extraction and sequence analysis.
DNA was prepared from frozen sputum samples as previously described (8). Briefly, samples were treated with Sputolysin (EMD Chemicals, Gibbstown, NJ) and were subjected to bead beating before DNA was extracted using a MagNA Pure nucleic acid purification platform (Roche Diagnostics Corp., Indianapolis, IN). Pyrosequencing of the V3 to V5 hypervariable region of the bacterial 16S ribosomal subunit (16S rRNA) gene was performed by the Human Genome Sequencing Center at Baylor College of Medicine using Roche 454-based sequencing protocols developed for the Human Microbiome Project (http://www.hmpdacc.org/resources/tools_protocols.php) as previously described (8). The mothur (v.1.29) software package was used to process sequences as described elsewhere (15). The total number of reads for each sample was randomly subsampled to 547, the smallest number of reads obtained in the sample set, to control for differences in sequencing depth before alpha diversity measures, richness, and evenness were calculated. DNA sequencing reads were assigned to operational taxonomic units (OTUs) using a 3% dissimilarity cutoff, which roughly correlates to the species level. OTUs were binned into genera by aligning the DNA sequences to the Ribosomal Database Project (16) training set containing 9,665 bacterial and 384 archaeal 16S rRNA gene sequences. To identify OTUs that were unclassified at the genus level, a representative sequence was generated that contained the highest homology to all other sequences in the OTU. These representative sequences were identified by using a BLAST search against the NCBI nucleotide collection (nr/nt) database and reporting the best match.
RESULTS
Sample set.
In total, 945 sputum samples collected over an 11-year period (2000 to 2011) from 132 persons with CF and previously assessed by routine culture were analyzed. The number of sputum samples per patient ranged from 1 to 30 (average, 7 samples per patient), and the time frame for sample collection per patient ranged from <1 year to 10 years (average, 3 years). The age distribution of patients at the time of sample collection is shown in the supplemental data (see Fig. S1 in the supplemental material).
Bacteria identified/detected.
Selective culture had previously identified bacterial species belonging to 15 genera among the 945 sputum samples. Six of these genera (Pseudomonas, Staphylococcus, Haemophilus, Achromobacter, Stenotrophomonas, and Burkholderia) may be considered traditional CF pathogens (2). The numbers of samples that were culture positive for each of these six genera are shown in Table 1. Although typically not considered important CF pathogens, Streptococcus spp. were reported to have been recovered in several samples as well. The eight remaining genera identified by culture were each represented by smaller numbers of positive samples: Pandoraea (7 samples reported as culture positive), Alcaligenes (6), Sphingobacterium (4), Acinetobacter (2), Chryseobacterium (2), and one culture-positive sample each for Mycobacterium, Citrobacter, and Rhizobium. Sequence analysis detected 618 OTUs among the 945 samples; these were binned into 393 genera. The mean Good's coverage was 0.99 (range, 0.962 to 1). The numbers of samples in which each of the six traditional CF pathogens and Streptococcus spp. were detected by sequence analysis are also shown in Table 1.
TABLE 1.
Prevalence of bacterial genera in 945 CF sputum samples as determined by selective culture and DNA sequence analysis
| Bacterial genus | No. identified by selective culture (%) | No. detected by DNA sequence analysis (%) |
|---|---|---|
| Pseudomonas | 723 (76.5) | 755 (79.9) |
| Staphylococcus | 491 (52.0) | 398 (42.1) |
| Haemophilus | 45 (4.8) | 351 (37.2) |
| Achromobacter | 118 (12.5) | 134 (14.2) |
| Stenotrophomonas | 111 (11.8) | 155 (16.4) |
| Burkholderia | 109 (11.5) | 137 (14.5) |
| Streptococcus | 15 (1.6) | 838 (88.7) |
Concordance between culture and sequencing results.
With the exceptions of Haemophilus and Streptococcus, the proportions of samples positive for these genera based on selective culture were similar to those obtained by sequence analysis (Table 1). Closer examination of the consistency of results between the two methods, however, revealed less concordance than expected based on these summary data (Fig. 1). For example, although only a 3.4% overall difference in detection of Pseudomonas in samples was observed between methods, this result included 4.0% of samples in which Pseudomonas was only detected by culture and 7.4% in which Pseudomonas was only detected by sequence analysis. Similar or greater disparities in prevalence by detection method were also observed for the other genera (Fig. 1).
FIG 1.

Concordance of culture-based and sequencing-based detection of bacteria. Euler diagrams (31) showing concordance between culture-based and sequencing-based detection of bacteria in CF sputum samples. Proportions are based on the percentage of 945 samples in which the indicated genus was not detected (white), detected by culture (light gray), detected by sequence analysis (dark gray), or detected by culture and sequence analysis (black).
The proportion of samples that was sequence positive and culture negative (false-negative cultures based on sequence analysis) for these seven genera ranged from 3.3% (Burkholderia) to 87.1% (Streptococcus), while sequence-negative and culture-positive (false-negative sequences based on culture) samples ranged from 0% (Streptococcus) to 13.7% (Staphylococcus) (Fig. 1; see also Table S1 in the supplemental material). The proportion of samples that was culture positive and sequence positive ranged from 1.6% (Streptococcus) to 72.5% (Pseudomonas).
The false-negative culture rate varied based on the relative abundance of the genus in the sample (i.e., the relative abundance of the DNA sequence reads classified as representing this genus among all sequence reads in the sample). When the relative abundance of a genus was less than 1%, the false-negative culture rate ranged between 29% (Staphylococcus) and 100% (Streptococcus) (Fig. 2; see also Table S2 in the supplemental material). When the relative abundance was ≥1%, the false-negative culture rates for Pseudomonas, Achromobacter, Burkholderia, and Staphylococcus were <10%, whereas the rates for Haemophilus and Stenotrophomonas were 79% and 26%, respectively. Of note, when the analysis of Haemophilus was limited to only the OTU representing Haemophilus influenzae, the false-negative culture rate decreased to 53% when relative abundance was ≥1% (see Table S2 footnote). With the exception of Streptococcus (see Discussion), only Haemophilus and Stenotrophomonas had false-negative culture rates of >10% when relative abundance was ≥10%.
FIG 2.
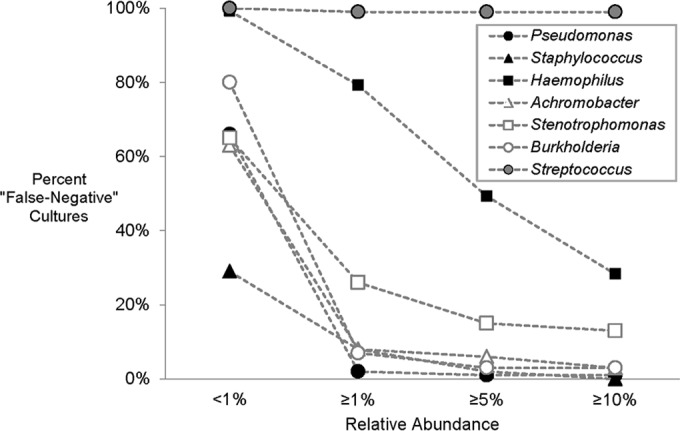
Proportion of false-negative cultures based on the relative abundance of the indicated genus in DNA sequence analysis.
Sequence-based prevalence and abundance.
Sequence analysis indicated that 31 genera were detected in at least 10% of the 945 sputum samples (see Table S3 in the supplemental material). Among these, Streptococcus was the most prevalent, as it was detected in nearly 90% of samples. The majority (74%) of the most prevalent genera detected by sequence analysis was not reported in routine culture-based results. This group included several obligate and facultative anaerobic genera, such as Prevotella (present in 82% of samples), Veillonella (79%), Rothia (67%), Granulicatella (64%), Gemella (60%), and Fusobacterium (52%).
Sequence analysis also indicated that each of 26 genera were present in samples with an average relative abundance of at least 1% when detected (see Table S4 in the supplemental material). The genera with the highest average relative abundances included the six traditional CF pathogens (Pseudomonas, Staphylococcus, Haemophilus, Achromobacter, Stenotrophomonas, and Burkholderia) as well as Streptococcus, each of which had an average relative abundance of at least 8.5% when detected in sputum samples. An OTU best classified as Polycyclovorans, a marine bacterium, was detected in six sputum samples from two patients at relatively low abundances (0.2% to 3.0%); however, this taxon was found in an additional sample from a third patient at 85% relative abundance, yielding an average relative abundance of 13%. The next most abundant genus not reported by culture was Prevotella, which had an average relative abundance of 9.6% when detected in sputum samples.
The six traditional CF pathogens varied with respect to the relationship between their prevalence and relative abundance (Fig. 3; see also Fig. S2 in the supplemental material). Pseudomonas was highly prevalent and detected in relatively high abundances. While Burkholderia was relatively uncommon, it was present in high relative abundance when detected. Achromobacter and Stenotrophomonas were also detected in a small proportion of samples, but were relatively abundant members of the bacterial community when present. In general, Streptococcus and several anaerobic genera (Prevotella, Veillonella, Rothia, Granulicatella, Gemella, and Fusobacterium) were highly prevalent but were usually present in relatively low abundances when detected.
FIG 3.

Prevalences and relative abundances of genera. The prevalences (dark bars) and mean (plus standard deviation) relative abundances (light bars) of the top 10 most abundant and/or prevalent genera detected in 945 sputum samples by sequence analysis are shown. Mean relative abundance in samples in which the specified genus was detected.
Taxa not reported by culture.
For each sputum sample, we assessed the number of taxa detected by sequence analysis relative to the number reported in culture results. The median proportion of OTUs detected by sequence but not reported in culture was 88.9% (range, 0% to 100%; median, 15 OTUs; range, 0 to 51 OTUs) (Fig. 4; see also Fig. S3A in the supplemental material). That is, among the great majority of the 945 samples, 80% to 90% of species detected by sequence analysis were not reported by culture.
FIG 4.
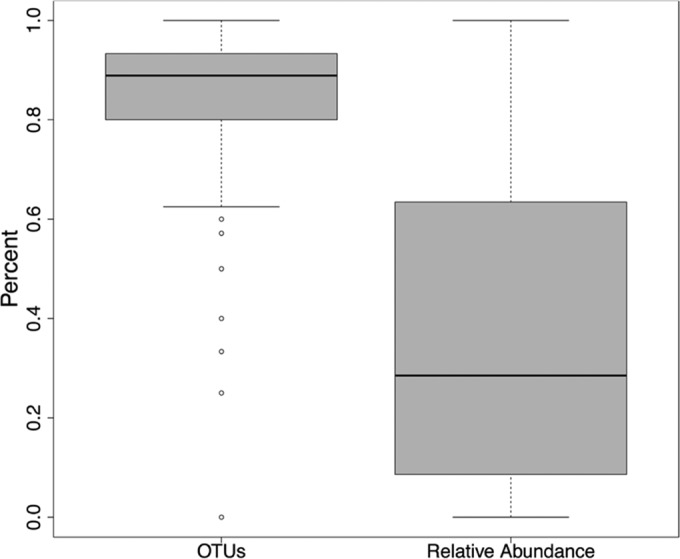
Box plot of relative abundance and proportion of OTUs not reported by culture. Boxes signify the upper and lower quartiles, black lines denote median values, whiskers show 1.5 interquartile range, and open circles designate outliers.
For each sputum sample, we also assessed the proportion of DNA sequence reads that represented taxa not reported in the culture results of that sample. The average relative abundance of genera not reported in culture was 37% (median, 28.5%; range, 0% to 100%) (Fig. 4; see also Fig. S3B in the supplemental material). In 214 (22.6%) of 945 samples, the genus with the highest relative abundance was not reported by culture. Streptococcus was the most abundant genus in 98 (10.4%) samples and was not reported by culture in 96 (98%) of these. In 310 (33%) of the 945 samples, genera not reported in culture comprised at least 50% of the total bacterial abundance; in 166 (17.6%) samples, genera not reported in culture constituted at least 75% of the bacterial load detected by sequence analysis (see Table S5 in the supplemental material). The relative abundance of taxa not reported in culture varied with the overall diversity of the bacterial community detected in each sample. The fraction of the community detected by sequence but not reported by culture was greatest when the overall Shannon diversity was highest and declined as Shannon diversity declined (Fig. 5). This trend was driven by community richness (number of OTUs detected) and by community evenness (relative abundances of OTUs) (see Fig. S4 in the supplemental material).
FIG 5.
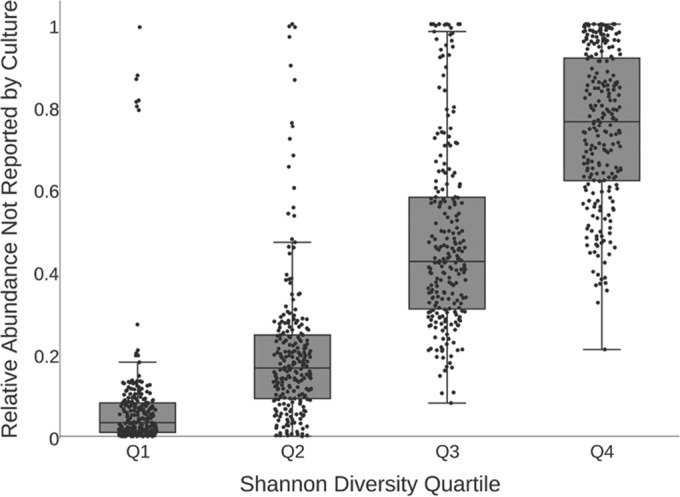
Box plot showing the relative abundances (summed) of the genera not reported by culture and grouped by Shannon diversity quartiles. The black line corresponds to the median summed relative abundance for each diversity quartile (1, low diversity; 4, high diversity). Each black dot represents one sputum sample.
DISCUSSION
Recent studies using culture-independent methods have shown that the bacterial communities inhabiting CF airways are likely more complex and dynamic than previously appreciated. These observations raise the question of whether the use of culture-independent approaches to profile airway bacterial communities may provide clinically relevant information beyond that obtained by culture of select species (13). An important consideration in addressing this question is how results from current culture-based assessment of CF respiratory specimens compare with results from culture-independent bacteriologic analysis. To address this question, we assessed a large set of CF sputum samples that had been previously analyzed by DNA sequence analysis and by selective CF culture over the course of 11 years in a single clinical microbiology laboratory. This laboratory, which serves large pediatric and adult CF care centers, is well experienced with the processing and culture of CF respiratory specimens. Our goal was to compare the results of DNA sequence analysis with the results of culture by using established guidelines for the processing and culture of CF specimens in a real-world setting. As such, we did not explore the use of extensive, nonroutine growth conditions, which in previous work have been shown to recover the majority of taxa detected by DNA sequence analysis (17).
For most of the traditional CF pathogens, the overall proportions of the 945 sputum samples that were reported to be positive by culture were comparable to the proportions that were positive by sequence analysis (Fig. 1; Table 1). Approximately 14% of samples were culture positive but sequence negative for Staphylococcus. This relatively high false-negative sequence rate most likely reflects the incomplete lysing of Staphylococcus cells by the DNA preparation method used to process the sputum samples included in this study. Modification of this method by the addition of reagents that better lyse Staphylococcus cells, such as lysozyme and lysostaphin, would be expected to increase the detection of Staphylococcus, resulting in lower discordance between these methods (18).
Over 30% of samples were sequence positive but culture negative for Haemophilus. This false-negative culture reflects, in part, the detection by sequence analysis of Haemophilus species other than Haemophilus influenzae, which is typically the only Haemophilus species sought in selective culture of CF respiratory specimens. In fact, an OTU representing Haemophilus parainfluenzae was detected in 29% of samples, while the OTU representing H. influenzae was detected in 24% of samples. Of note, whereas the average relative abundance of H. influenzae was 11%, the average relative abundance of H. parainfluenzae was 2%. When considering H. influenzae alone, a much smaller proportion of samples (19.5%) was found to be sequence positive and culture negative. That is, a considerably smaller proportion of false-negative culture results was observed for H. influenzae relative to the genus Haemophilus as a whole. We also note that in 2008 the clinical microbiology laboratory incorporated the use of chocolate agar supplemented with bacitracin in their CF culture protocol. While 219 (34%) of the 648 samples processed prior to this time were sequence positive and culture negative for Haemophilus, this result was found in 89 (30%) of the 297 samples processed after this change in protocol.
Streptococcus species, which are not generally considered among the group of traditional CF pathogens and are therefore not routinely identified in routine CF culture, were reported in culture of only a very small proportion of samples. In contrast, Streptococcus species were the most frequently detected taxa by sequence analysis (see Table S3 in the supplemental material) and, when detected, were often present in relatively high abundances (see Table S4 in the supplemental material). Many, if not most, Streptococcus species grow best under culture conditions (e.g., low oxygen concentrations) that are not typically used in the routine culture of CF specimens. Those that are recovered in culture are most often discarded or reported as oral flora. However, species of this genus, particularly members of the Streptococcus anginosus/milleri group, have been implicated in contributing to lung disease in CF (19, 20). In our analysis, an OTU representing members of the S. anginosus/milleri group was the most common Streptococcus OTU detected and was found in 802 (85%) of the 945 samples, with an average relative abundance of 7.5%. It is also noteworthy that the distinction between oral flora (i.e., bacterial inhabitants of the upper airway or species residing in the healthy oropharynx that may be considered contaminants of expectorated sputum) and species that infect the lower airways in CF has become increasingly blurred. Recent studies employing ecological models of biogeography (5, 21) and sequence-based experimental data (6) have concluded that the detection of oral microbes in CF sputum more likely represents immigration from the oropharynx to the airways rather than mere contamination of respiratory samples.
These same considerations may apply to the anaerobic species that we observed by sequence analysis to be highly prevalent in our sample set. Because CF specimens are not typically cultured under anaerobic conditions, these species are not reported in CF culture results. The high prevalence of anaerobic species that we detected is consistent with the results of several previous studies that have used culture and culture-independent methods to demonstrate the presence and diversity of anaerobic species in CF airways (17, 22–26). Further, the estimates of relative abundance provided by sequence analysis in our study demonstrate that anaerobic species often constitute a significant proportion of the total bacterial density (see Table S4 in the supplemental material). By way of example, the combined average relative abundance of the 10 most prevalent obligate and facultative anaerobic species detected in this study (Streptococcus, Prevotella, Veillonella, Rothia, Granulicatella, Gemella, Fusobacterium, Actinomyces, Oribacterium, and Porphyromonas) was 43.9%, which was comparable to the average relative abundance observed for Pseudomonas (47.3%). These results are consistent with those of Tunney et al. (23), who used culture-based analyses to show that anaerobic bacteria were most often present in equal or greater numbers than P. aeruginosa in CF sputum samples. The concern that anaerobic species merely represent contamination from the upper airway has been further assuaged by a study that identified anaerobic species in deep tissue samples of lung resected from a child with CF (27) and by other recent work that provides evidence of microbial anaerobic respiration in CF airways (28, 29).
We observed that the rate with which the traditional CF pathogens (Pseudomonas, Staphylococcus, Haemophilus, Achromobacter, Stenotrophomonas, and Burkholderia) were recovered in culture varied with respect to their relative abundance in the sample (Fig. 2; see also Table S2 in the supplemental material). These taxa were cultured from only a minority (1% to 37%) of samples wherein their relative abundances were <1%, which demonstrated that selective culture methods are not uniformly effective in recovering species of interest when present in relatively low density. While the rate of false-negative cultures decreased precipitously when relative abundance exceeded 1% for Pseudomonas, Achromobacter, and Burkholderia, this rate remained high for Haemophilus and Stenotrophomonas, even when the relative abundances of these taxa exceeded 10%. It should be noted that while culture only detects viable bacteria, sequencing-based methods may detect DNA from lysed, nonviable bacteria. However, we do not think this had a great impact on our results, given that concordance was quite high for Pseudomonas, Achromobacter, and Burkholderia above a threshold of 1% relative abundance. In addition, we saw no differences in concordance rates during antibiotic treatment (data not shown) when a preponderance of dead bacteria might be expected to create a disparity based on viable versus nonviable bacteria.
The relative abundance data provided by sequence analysis also allowed us to estimate the proportion of the total bacterial density in sputum samples that was comprised of species not reported in culture results. This generally represented a considerable proportion of the total bacterial load. In fact, in one-third of the 945 samples, species not reported in culture represented at least 50% of the bacterial load; in 18% of samples, taxa comprising 75% or more of the bacterial density were not reported in culture (see Table S5 in the supplemental material). In approximately 20% of samples, the genus with the highest relative abundance was not reported in culture. These findings are difficult to ascribe merely to oral contamination of expectorated sputum.
The role that culture-independent analyses might play in assessing the clinical microbiology of CF respiratory specimens requires further consideration (13). The utility of detecting bacteria that are not considered to be traditional CF pathogens, and that are therefore not specifically targeted in current culture-based protocols, will require a better understanding of the role that these species may play in contributing to lung disease. However, defining the relative pathogenicity of select members of a polymicrobial community—or, more specifically, the role that individual species may play in the pathogenicity of the community—will prove challenging. Further, culture-independent analyses may improve our understanding of the early natural history of infection with species already accepted to be CF pathogens. For instance, the observation that Pseudomonas infection is often intermittent for a period prior to chronic infection as determined by selective culture may be refined by the ability to more consistently detect the species when it is present in relatively low abundance in the airway community.
The ability of culture-independent analyses to provide estimates of a species relative abundance in mixed microbial communities may allow for clearer distinction between upper airway contaminants (low relative abundance) and taxa more likely to be infecting the lower airways (high relative abundance). However, such measures will need to be interpreted in the context of estimates of overall (absolute) bacterial density. Finally, as our understanding of CF airway microbial ecology continues to expand, the value of methods that enable estimation of airway community diversity will be investigated. It is conceivable, for example, that differentiating communities with or without a dominant species—or estimating overall community diversity—may impact antimicrobial treatment strategies. Although culture-independent assessment of CF respiratory specimens remains primarily in the domain of research laboratories, commercial platforms capable of providing these analyses are increasingly finding their way into the clinical microbiology laboratory (30). CF caregivers may soon expect analyses that provide detection of nontraditional pathogens, assessment of species relative abundance, and estimates of airway microbial community diversity.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge the many individuals who provided the sputum samples used in this study. We thank the UMHS clinical microbiology laboratory for generous assistance in obtaining sputum samples for this study.
The National Institutes of Health provided funding to John J. LiPuma under NHLBI grant 1RC1HL100809-01. The Cystic Fibrosis Foundation, the Charles Woodson Pediatric Research Fund, and the Nesbitt Program for Cystic Fibrosis Research also provided funding to John J. LiPuma. The National Institutes of Health provided funding to the University of Michigan under CTSA grant UL1RR024986.
The funders had no role in study design, in data collection and interpretation, or in the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JCM.02299-15.
REFERENCES
- 1.Gilligan PH. 1991. Microbiology of airway disease in patients with cystic fibrosis. Clin Microbiol Rev 4:35–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LiPuma JJ. 2010. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev 23:299–323. doi: 10.1128/CMR.00068-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang YJ, LiPuma JJ. 23 December 2015. The microbiome in cystic fibrosis. Clin Chest Med doi: 10.1016/j.ccm.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansen SK, Rau MH, Johansen HK, Ciofu O, Jelsbak L, Yang L, Folkesson A, Jarmer HO, Aanaes K, von Buchwald C, Hoiby N, Molin S. 2012. Evolution and diversification of Pseudomonas aeruginosa in the paranasal sinuses of cystic fibrosis children have implications for chronic lung infection. ISME J 6:31–45. doi: 10.1038/ismej.2011.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkataraman A, Bassis CM, Beck JM, Young VB, Curtis JL, Huffnagle GB, Schmidt TM. 2015. Application of a neutral community model to assess structuring of the human lung microbiome. mBio 6:e02284-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassis CM, Erb-Downward JR, Dickson RP, Freeman CM, Schmidt TM, Young VB, Beck JM, Curtis JL, Huffnagle GB. 2015. Analysis of the upper respiratory tract microbiotas as the source of the lung and gastric microbiotas in healthy individuals. mBio 6:e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cox MJ, Allgaier M, Taylor B, Baek MS, Huang YJ, Daly RA, Karaoz U, Andersen GL, Brown R, Fujimura KE, Wu B, Tran D, Koff J, Kleinhenz ME, Nielson D, Brodie EL, Lynch SV. 2010. Airway microbiota and pathogen abundance in age-stratified cystic fibrosis patients. PLoS One 5(6):e11044. doi: 10.1371/journal.pone.0011044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, Young VB, LiPuma JJ. 2012. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A 109:5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stokell JR, Gharaibeh RZ, Hamp TJ, Zapata MJ, Fodor AA, Steck TR. 2015. Analysis of changes in diversity and abundance of the microbial community in a cystic fibrosis patient over a multiyear period. J Clin Microbiol 53:237–247. doi: 10.1128/JCM.02555-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stressmann FA, Rogers GB, van der Gast CJ, Marsh P, Vermeer LS, Carroll MP, Hoffman L, Daniels TW, Patel N, Forbes B, Bruce KD. 2012. Long-term cultivation-independent microbial diversity analysis demonstrates that bacterial communities infecting the adult cystic fibrosis lung show stability and resilience. Thorax 67:867–873. doi: 10.1136/thoraxjnl-2011-200932. [DOI] [PubMed] [Google Scholar]
- 11.Klepac-Ceraj V, Lemon KP, Martin TR, Allgaier M, Kembel SW, Knapp AA, Lory S, Brodie EL, Lynch SV, Bohannan BJ, Green JL, Maurer BA, Kolter R. 2010. Relationship between cystic fibrosis respiratory tract bacterial communities and age, genotype, antibiotics and Pseudomonas aeruginosa. Environ Microbiol 12:1293–1303. doi: 10.1111/j.1462-2920.2010.02173.x. [DOI] [PubMed] [Google Scholar]
- 12.Carmody LA, Zhao J, Schloss PD, Petrosino JF, Murray S, Young VB, Li JZ, LiPuma JJ. 2013. Changes in cystic fibrosis airway microbiota at pulmonary exacerbation. Ann Am Thorac Soc 10:179–187. doi: 10.1513/AnnalsATS.201211-107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.LiPuma JJ. 2015. Assessing airway microbiota in cystic fibrosis: what more should be done? J Clin Microbiol 53:2006–2007. doi: 10.1128/JCM.01218-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gilligan PH, Kiska DL, Appleman MD. 2006. Cumitech 43: cystic fibrosis microbiology. ASM Press, Washington, DC. [Google Scholar]
- 15.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM. 2009. The Ribosomal Database Project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145. doi: 10.1093/nar/gkn879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sibley CD, Grinwis ME, Field TR, Eshaghurshan CS, Faria MM, Dowd SE, Parkins MD, Rabin HR, Surette MG. 2011. Culture enriched molecular profiling of the cystic fibrosis airway microbiome. PLoS One 6(7):e22702. doi: 10.1371/journal.pone.0022702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao J, Carmody LA, Kalikin LM, Li J, Petrosino JF, Schloss PD, Young VB, LiPuma JJ. 2012. Impact of enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS One 7(3):e33127. doi: 10.1371/journal.pone.0033127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Parkins MD, Sibley CD, Surette MG, Rabin HR. 2008. The Streptococcus milleri group—an unrecognized cause of disease in cystic fibrosis: a case series and literature review. Pediatr Pulmonol 43:490–497. doi: 10.1002/ppul.20809. [DOI] [PubMed] [Google Scholar]
- 20.Sibley CD, Parkins MD, Rabin HR, Duan K, Norgaard JC, Surette MG. 2008. A polymicrobial perspective of pulmonary infections exposes an enigmatic pathogen in cystic fibrosis patients. Proc Natl Acad Sci U S A 105:15070–15075. doi: 10.1073/pnas.0804326105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Whiteson KL, Bailey B, Bergkessel M, Conrad D, Delhaes L, Felts B, Harris JK, Hunter R, Lim YW, Maughan H, Quinn R, Salamon P, Sullivan J, Wagner BD, Rainey PB. 2014. The upper respiratory tract as a microbial source for pulmonary infections in cystic fibrosis. Parallels from island biogeography. Am J Respir Crit Care Med 189:1309–1315. doi: 10.1164/rccm.201312-2129PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Harris JK, De Groote MA, Sagel SD, Zemanick ET, Kapsner R, Penvari C, Kaess H, Deterding RR, Accurso FJ, Pace NR. 2007. Molecular identification of bacteria in bronchoalveolar lavage fluid from children with cystic fibrosis. Proc Natl Acad Sci U S A 104:20529–20533. doi: 10.1073/pnas.0709804104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tunney MM, Field TR, Moriarty TF, Patrick S, Doering G, Muhlebach MS, Wolfgang MC, Boucher R, Gilpin DF, McDowell A, Elborn JS. 2008. Detection of anaerobic bacteria in high numbers in sputum from patients with cystic fibrosis. Am J Respir Crit Care Med 177:995–1001. doi: 10.1164/rccm.200708-1151OC. [DOI] [PubMed] [Google Scholar]
- 24.Worlitzsch D, Rintelen C, Bohm K, Wollschlager B, Merkel N, Borneff-Lipp M, Doring G. 2009. Antibiotic-resistant obligate anaerobes during exacerbations of cystic fibrosis patients. Clin Microbiol Infect 15:454–460. doi: 10.1111/j.1469-0691.2008.02659.x. [DOI] [PubMed] [Google Scholar]
- 25.Zemanick ET, Wagner BD, Sagel SD, Stevens MJ, Accurso FJ, Harris JK. 2010. Reliability of quantitative real-time PCR for bacterial detection in cystic fibrosis airway specimens. PLoS One 5(11):e15101. doi: 10.1371/journal.pone.0015101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fodor AA, Klem ER, Gilpin DF, Elborn JS, Boucher RC, Tunney MM, Wolfgang MC. 2012. The adult cystic fibrosis airway microbiota is stable over time and infection type, and highly resilient to antibiotic treatment of exacerbations. PLoS One 7(9):e45001. doi: 10.1371/journal.pone.0045001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brown PS, Pope CE, Marsh RL, Qin X, McNamara S, Gibson R, Burns JL, Deutsch G, Hoffman LR. 2014. Directly sampling the lung of a young child with cystic fibrosis reveals diverse microbiota. Ann Am Thorac Soc 11:1049–1055. doi: 10.1513/AnnalsATS.201311-383OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quinn RA, Lim YW, Maughan H, Conrad D, Rohwer F, Whiteson KL. 2014. Biogeochemical forces shape the composition and physiology of polymicrobial communities in the cystic fibrosis lung. mBio 5:e00956-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cowley ES, Kopf SH, LaRiviere A, Ziebis W, Newman DK. 2015. Pediatric cystic fibrosis sputum can be chemically dynamic, anoxic, and extremely reduced due to hydrogen sulfide formation. mBio 6:e00767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flight WG, Smith A, Paisey C, Marchesi JR, Bull MJ, Norville PJ, Mutton KJ, Webb AK, Bright-Thomas RJ, Jones AM, Mahenthiralingam E. 2015. Rapid detection of emerging pathogens and loss of microbial diversity associated with severe lung disease in cystic fibrosis. J Clin Microbiol 53:2022–2029. doi: 10.1128/JCM.00432-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Micallef L, Rodgers P. 2014. eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS One 9(7):e101717. doi: 10.1371/journal.pone.0101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.