

Abstract
Hearing impairment is the most frequent sensory deficit in humans. Deafness genes, which harbor pathogenic mutations that have been identified in families with hereditary hearing loss, are commonly expressed in the auditory end organ or the cochlea and may contribute to normal hearing function, yet some of the mouse models carrying these mutations fail to recapitulate the hearing loss phenotype. In this study, we find that distinct expression patterns of those deafness genes in the cochlea of a non-human primate, the common marmoset (Callithrix jacchus). We examined 20 genes whose expression in the cochlea has already been reported. The deafness genes GJB3, CRYM, GRHL2, DFNA5, and ATP6B1 were expressed in marmoset cochleae in patterns different from those in mouse cochleae. Of note, all those genes are causative for progressive hearing loss in humans, but not in mice. The other tested genes, including the deafness gene COCH, in which mutation recapitulates deafness in mice, were expressed in a similar manner in both species. The result suggests that the discrepancy in the expression between rodents and primates may account for the phenotypic difference. This limitation of the rodent models can be bypassed by using non-human primate models such as the marmoset.
Deafness is one of the most common types of congenital impairment, and at least half of which are caused by hereditary mutations. In some patients, hearing loss is progressive, with hearing loss developing gradually during childhood or youth after the acquisition of speech abilities. In these post-lingual hearing loss cases, a hearing aid or cochlea implant is effective because the auditory cortex has already developed1. Therefore, the prevention of progressive hearing loss would be one of the most promising therapeutics for long-term hearing ability.
As of 2015, the genes responsible for hearing loss have been identified in 99 autosomal recessive2, 67 autosomal dominant3, and six X-chromosomal recessive loci4. While mouse models for the vast majority of deafness genes recapitulate the human phenotype, in a few cases mouse models for human deafness did not result in hearing loss5,6,7. Especiallycin such cases, detailed mechanisms by which they cause hearing loss are not fully understood because of the difficulty in obtaining human cochleae suitable for pathological analyses. First, the number of human subjects during the development in hearing impairment is limited because it is non-lethal symptom. Second, there are technical difficulties of fixation and decalcification with the immunoreactivity retained. Third, the cochlea atrophies during normal aging8 and age-matching healthy controls can be difficult. Finally, there are differences in the physiology of individual cell types and/or proteins in the cochlea between humans and rodents (including mice, rats, guinea pigs, and gerbils), which are frequently used as animal models for examining gene expression patterns and determining the pathophysiology. Indeed, frequently such models recapitulate the human pathophysiology. However, some rodent models for hereditary hearing loss fail to recapitulate the phenotypes of human patients5,7. Such discrepancies can be accounted by a biological difference between species, but the details remain unknown.
Therefore, we wanted to use non-human primates to investigate the anatomy of hearing organs and the pathophysiology of hearing loss. In particular, we were interested in a small New World monkey species, the common marmoset (Callithrix jacchus), which exhibits more human-associated traits than Old World monkeys, including a richer variety of vocalizations and verbal communication9. Furthermore, the marmoset’s hearing range overlaps with that of humans10, suggesting the significance of the marmoset as a model for hearing research. Because genetic modification is now possible in the common marmoset11, we can use this species as a non-human primate model for the investigation of the detailed pathogenic mechanisms of hearing loss.
In this study, we established a successful immunohistochemical approach for the common marmoset and examined the expression patterns of 20 genes in the inner ear, genes whose expression has already been investigated in rodent or human cochleae (Fig. 1a). The genes tested include well-known genes causal for hereditary progressive hearing loss such as GJB3 (CX31, DFNA2B, DFNB91), CRYM, GRHL2 (DFNA28), DFNA5, and ATP6B1 (distal renal tubular acidosis (dRTA) with hearing loss), all of whose pathophysiology remains unknown because their rodent models did not recapitulate the deafness phenotype. We found that these genes had distinct expression patterns, in which the proteins were distributed in the cochlea differently in primates and rodents. We also examined the expression profiles of COCH (DFNA9) and CONNEXIN26 (CX26, DFNA3A, DFNB1A), which have mouse models that recapitulate the deafness phenotype. We found that those proteins were expressed in a pattern similar to that reported for rodents.
Figure 1.
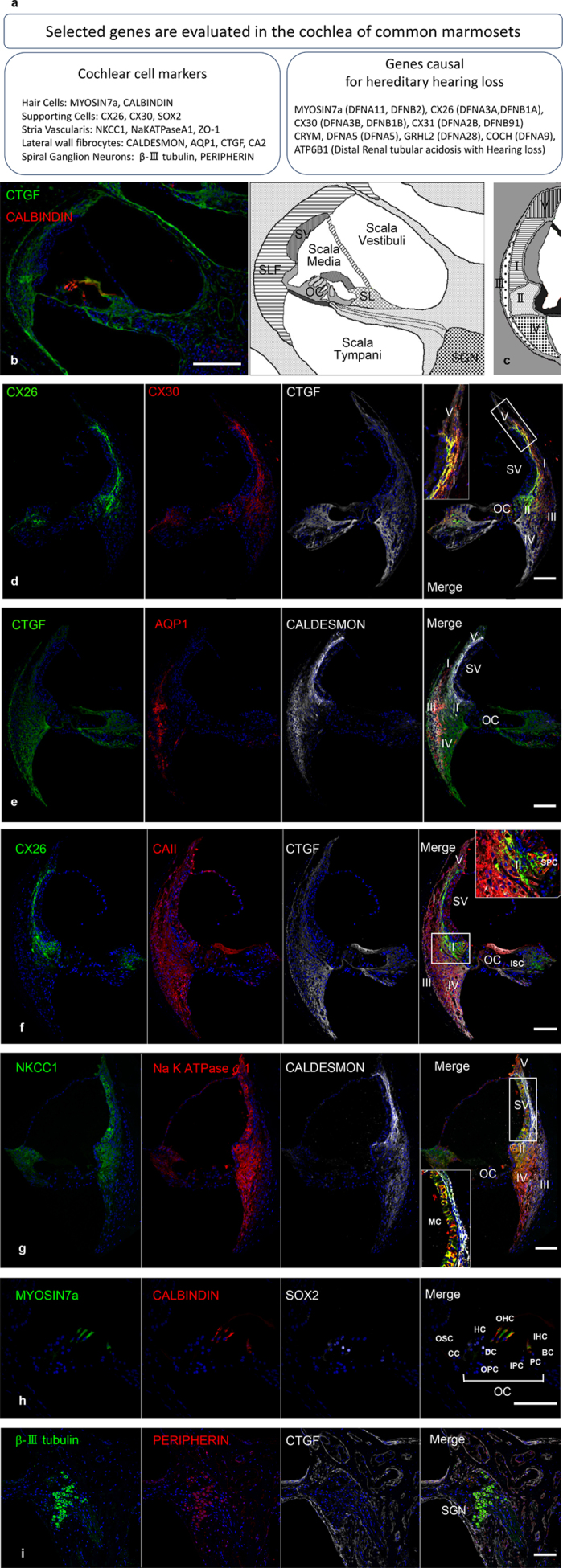
Expression of conventional markers in the cochlea of the common marmoset (a) Selected genes are evaluated in the cochlea of common marmosets. We selected 14 cochlear cell markers and nine genes causal for hereditary hearing loss. Antibodies specific to them were used in this study. (b) A cross-sectional view of the common marmoset cochlea duct under low magnification (left). The corresponding schema with anatomical landmarks is shown (right). (c) Schema for the subtypes of lateral wall fibrocytes. (d–g) The expression patterns of conventional markers in the lateral wall. CX26, CX30, CTGF, AQP1, CALDESMON, NKCC1, and Na+/K+ ATPase α1 expression were all observed in the cochleae, as well as previously in rodent. High magnification images were shown in the most right panels of (f,g). (h) Expression patterns of conventional markers in OC. MYOSIN 7a and CALBINDIN expression was observed in three rows of outer hair cells and one row of inner hair cells. SOX2 expression was observed in inner pillar cells, outer pillar cells, Deiters’ cells, and Hensen’s cells. (i) Expression pattern of conventional markers in SGNs. Expression of βIII TUBULIN and PERIPHERIN was observed in marmoset SGNs. The nuclei were counterstained with Hoechst (blue). Scale bar: 200 μm in b, 100 μm in (c–i). Basal turns in (d–g,i), apical turn in h. SV: stria vascularis, SLF: spiral ligament fibrocytes (I-V: Type I-V), OC: organ of Corti, SL: spiral limbus, SGN: spiral ganglion neurons, MC: Marginal cells, SPC: Spiral prominence cells, ISC: Inner sulcus cells, OSC: Outer sulcus cells, CC: Claudius cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells, OPC: outer pillar cells, PC: inner phalangeal cells, BC: border cells, OHC: Outer hair cells, IHC: Inner hair cells.
Taken together, our results suggest that the discrepancy of the expression between rodents and primates accounts for the phenotypic difference between them, and that the limitations of rodent models in researching human hereditary hearing loss could be bypassed by using non-human primate models such as the common marmoset.
Results
Immunohistochemical analysis of marmoset cochleae
Few studies12,13 have been conducted of the morphology of the temporal bone in the common marmoset, which have the cochlea with 2 and 3/4 turns (Supplementary Fig. S1), and no experiments using immunohistochemistry have been reported. To fully optimize the final protocol for histochemical analyses, we tested several methods including modifications of local perfusion methods for fixations, decalcification periods, thickness of frozen sections and antigen retrievals. The smaller size of the temporal bone in marmosets enabled us to investigate the histopathology using an easier and shorter decalcification process without excess fixation, resulting in the preservation of structures and immunoreactivity. Whereas it takes 3–5 months on average to decalcify human temporal bones14,15, it took 3–4 weeks to sufficiently decalcify the marmoset bones. As shown in Fig. 1b, we successfully immunostained samples using the optimized frozen section methods. Notably, the temporal bone sections showed that the general morphology of the marmosets’ cochleae is similar to that of humans.
Expression of conventional markers in the cochlea
We next collected basic information on the marmoset cochlea using immunostaining. First, we examined several lateral wall cell markers, CONNEXIN 26 (CX26), CX30, CTGF, AQUAPORIN 1 (AQP1), CALDESMON, CARBONIC ANHYDRASE II (CAII), Na-K-Cl COTRANSPORTER 1 (NKCC1), Na+/K+ ATPase α1, and CTGF. The lateral wall of the cochlea is composed of type I–V lateral wall fibrocytes and three layers of cells in the stria vascularis (marginal cells, intermediate cells, and basal cells). The cell types of the lateral wall fibrocytes are defined by their spatial distribution and structure16,17 (Fig. 1c). Immunoreactivity for CX26 was observed in type I and II fibrocytes, the basal cells of the stria vascularis, spiral prominence cells, outer sulcus cells, supporting cells, and spiral limbus cells. CX30 immunoreactivity was observed in type I, II, III, and V fibrocytes, spiral prominence cells, outer sulcus cells, supporting cells, and spiral limbus cells. AQP1 immunoreactivity was observed specifically in type III fibrocytes. CALDESMON immunoreactivity was observed in type I and III fibrocytes. CAII immunoreactivity was observed in type I, III, and IV fibrocytes and the intermediate cells of the stria vascularis. NKCC1 immunoreactivity was observed in type II and IV fibrocytes, marginal cells, and spiral limbus cells. Na+/K+ ATPase α1 immunoreactivity was observed in type II, IV, and V fibrocytes and marginal cells. CTGF immunoreactivity was observed in type I, II, III, IV, and V fibrocytes, the basilar membrane, and the spiral limbus (Fig. 1d–g).
In the organ of Corti, immunoreactivity for MYOSIN 7a (MYO7a) and CALBINDIN was observed in three rows of outer hair cells and one row of inner hair cells. SOX2, a well-established supporting cell marker, was observed in some supporting cells: Hensen’s cells, Deiters’ cells, inner pillar cells, outer pillar cells, inner phalangeal cells, and border cells (Fig. 1h). In spiral ganglion neurons (SGNs), immunoreactivity for β-III TUBULIN and PERIPHERIN was observed.
The expression of CX26, CX30, CTGF, AQP1, CALDESMON, CAII, NKCC1, Na+/K+ ATPase α1, MYO7a, SOX2, and β-III TUBULIN in the cochlea of common marmosets was similar to that previously reported in mouse and other rodents. On the other hand, two proteins, CTGF and PERIPHERIN, were expressed differently between marmosets and rodents: CTGF expression was observed in all types of fibrocytes in the common marmoset, whereas its expression in mice is limited to type IV fibrocytes18. PERIPHERIN expression was observed in most SGNs, and its expression pattern was different from that of rodents, but quite similar to that of humans19. The gross morphology of the cross section and AQP1 expression suggest that, in the common marmoset, several rows of type III fibrocytes are distributed in the outer part of the lateral wall, but not in the bone capsule, whereas in the rodent, the cells are distributed in a single border layer next to the bone20. This distribution pattern of type I fibrocytes is similar to that of human temporal bones21.
Expression pattern of the deafness gene GJB3 (CX31, DFNA2B/DFNB91)
CX31, also known as gap junction beta-3 protein (GJB3) or DFNA2B/DFNB91, is a gap junction protein, and in humans it is encoded by the GJB3 gene22. Its expression in mouse cochlea is limited to the lateral wall and spiral limbus23. In marmoset cochlea, CX31 immunoreactivity was observed in the type I fibrocytes of the spiral ligaments, the basal cells of the stria vascularis, Reissner’s membrane, and supporting cells (inner sulcus cells, Hensen’s cells, Claudius cells, and outer sulcus cells) (Fig. 2a,b). Immunoreactivity for CX31 in other supporting cells was not observed, specifically in Deiters’ cells, outer pillar cells, inner pillar cells, and hair cells. Immunoreactivity for CX31 in type I fibrocytes was partially co-localized with that of CX26. In the stria vascularis, immunoreactivity for CX31 was observed in basal cells and co-localized with ZO-1 (Fig. 2c).
Figure 2.

CONNEXIN 31 expression in the cochlea of the common marmoset (a) CX31 expression was observed in the type I fibrocytes of the spiral ligaments, the basal cells of the stria vascularis, Reissner’s membrane, and supporting cells. (b) CX31 expression was observed in inner sulcus cells, Hensen’s cells, Claudius cells, and outer sulcus cells. (c) Cx31 expression was observed in the stria vascularis and type I fibrocytes. CX31 expression was observed in the ZO-1-positive basal cells of the stria vascularis and type I fibrocytes. In type I fibrocytes, CX31 expression partially overlapped with CX26 expression. The nuclei were counterstained with Hoechst (blue). Scale bar: 100 μm in (a,b), 50 μm in (c). Basal turns in (a,c), middle turn in b. OC: organ of Corti, ISC: Inner sulcus cells, OSC: Outer sulcus cells, CC: Claudius cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells, I: spiral ligament fibrocytes Type I, MC: Marginal cells, IC: Intermediate cells, BC: Basal cells.
Expression pattern of the deafness gene CRYM
CRYM (mu-crystallin homolog), also known as NADP-regulated thyroid-hormone-binding protein (THBP), is a crystallin structural protein encoded by the CRYM gene. In humans, CRYM is expressed exclusively in the inner ear, as shown by a cDNA microarray analysis of gene expression24. In mice it exhibits limited expression in the lateral wall and the spiral limbus24,25. We found that the marmoset had broader expression, not only in the lateral wall spiral ligament and the spiral limbus, but also in both inner and outer hair cells, supporting cells, (Fig. 3a). Expression was found in all types of the supporting cells between the inner sulcus and outer sulcus cell (Fig. 3b).
Figure 3.

CRYM expression in the cochlea of the common marmoset (a) CRYM expression was observed in the lateral wall spiral ligament, both inner and outer hair cells, supporting cells, and the spiral limbus. (b) CRYM expression in the supporting cells was broadly observed between the inner sulcus and outer sulcus cells. CRYM expression was also observed in inner and outer hair cells. The nuclei were counterstained with Hoechst (blue). Scale bar: 100 μm. Middle turns in (a,b). SV: stria vascularis, OC: organ of Corti, ISC: Inner sulcus cells, OSC: Outer sulcus cells, CC: Claudius cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells.
Expression pattern of the deafness gene GRHL2 (DFNA28)
GRHL2, also known as TFCP2L3, is a transcriptional member of the Grh/CP2 family and is a mammalian homolog of the Drosophila gene grainyhead. In the common marmoset, GRHL2 expression was observed in the lateral wall spiral ligament, hair cells, supporting cells, and SGNs (Fig. 4a,b). No expression was observed in the stria vascularis, whereas it is expressed in the mouse stria vascularis. Supporting cell expression was broad, with both inner sulcus cells and outer sulcus cells expressing it, as well as inner and outer hair cells (Fig. 4c). In the spiral ligament, GRHL2 expression was observed in type I and III fibrocytes, which were co-labeled with CALDESMON (Fig. 4d,e). Notably, as shown in a previous report about human keratinocytes26, cytoplasmic expressions of GRHL2 were observed and we confirmed that this expression is not nonspecific signal using two different anti-GRHL2 antibodies (Supplementary Fig. 2).
Figure 4.

GRHL2 expression in the cochlea of the common marmoset (a, b) GRHL2 expression was observed in the lateral wall spiral ligament, hair cells, supporting cells, and spiral ganglion neurons. No expression was observed in the stria vascularis. (c) Supporting cell expression was broadly observed between the inner sulcus and outer sulcus cells, and GRHL2 expression was observed in the inner and outer hair cells. (d,e) In the spiral ligament, GRHL2 expression was observed in type I and III fibrocytes and co-localized with CALDESMON. The nuclei were counterstained with Hoechst (blue). Scale bar: 200 μm in a, 100 μm in (b–e). Middle turns in (a–e). SV: stria vascularis, OC: organ of Corti, SGN: spiral ganglion neurons, ISC: Inner sulcus cells, OSC: Outer sulcus cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells, OHC: Outer hair cells, IHC: Inner hair cells, SLF: spiral ligament fibrocytes (I-V: Type I-V).
Expression pattern of the deafness gene DFNA5
DFNA5 is the fifth mapped autosomal dominant locus for hereditary hearing impairment in humans27, yet its physiological function is still unknown. Its expression in mouse cochlea is thought to be in the stria vascularis28, but no detailed evaluation using immunostaining has ever been reported. In the common marmoset, we observed it in the stria vascularis, spiral ligament fibrocytes, both inner and outer hair cells, supporting cells, and SGNs (Fig. 5a). Expression was found in all types of the supporting cells between the inner sulcus and outer sulcus cell (Fig. 5b). In spiral ligaments, DFNA5 expression was limited to type II fibrocytes (Fig. 5c). In the marmoset stria vascularis, DFNA5 expression was limited to basal cells (Fig. 5d).
Figure 5.

DFNA5 expression in the cochlea of the common marmoset (a) DFNA5 expression was observed in the spiral ligament fibrocytes, supporting cells, inner and outer hair cells, and spiral ganglion neurons. (b) Expression in the supporting cells was broadly observed between the inner sulcus and outer sulcus cells. (c) DFNA5 expression was observed in type II fibrocytes. (d) DFNA5 expression was observed in the ZO-1- and CX26-positive basal cells of the stria vascularis. The nuclei were counterstained with Hoechst (blue). Scale bar: 200 μm in (a), 100 μm in (c), 50 μm in (b,d). Middle turns in (a,c), apical turn in (b), and basal turn in d. SV: stria vascularis, OC: organ of Corti, SGN: spiral ganglion neurons, ISC: Inner sulcus cells, OSC: Outer sulcus cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells, MC: Marginal cells, IC: Intermediate cells BC: basal cells, I-V: spiral ligament fibrocytes Type I-V.
Expression pattern of the deafness gene ATP6B1 (dRTA with Hearing loss)
ATP6B1 is one of the components of the vacuolar ATPase (V-ATPase), a multi-subunit enzyme that mediates acidification of eukaryotic intracellular organelles. In contrast to the limited expression in the spiral limbus in mouse29, expression in the marmoset was broadly observed in the lateral wall spiral ligament, in both inner and outer hair cells, in supporting cells, in the spiral limbus, and in SGNs (Fig. 6a). ATP6B1 expression in supporting cells was detected between the inner sulcus and outer sulcus cells (Fig. 6b).
Figure 6.

ATP6B1 expression in the cochlea of the common marmoset (a) ATP6B1 expression was observed in all types of spiral ligament fibrocytes, inner and outer hair cells, supporting cells, and the spiral limbus. (b) In the organ of Corti, ATP6B1 expression was observed in all hair cells and supporting cells between the inner sulcus and outer sulcus cells. The nuclei were counterstained with Hoechst (blue). Scale bar: 100 μm. Middle turn in (a) basal turn in (b). SV: stria vascularis, OC: organ of Corti, ISC: Inner sulcus cells, OSC: Outer sulcus cells, CC: Claudius cells, HC: Hensen’s cells, DC: Deiters’ cells, IPC: inner pillar cells, OHC: Outer hair cells, IHC: Inner hair cells.
Expression pattern of the deafness gene COCH (DFNA9)
COCH (cochlin, coagulation factor C homology) is coded by the COCH gene. In humans, the expression of COCH is restricted to the inner ear24, and it is clinically used as a perilymph-specific protein30. Among the deafness genes tested in this experiment, this is the only gene for which a mouse model recapitulates the human symptoms31. In our study, the expression of COCH was observed in the spiral ligaments and spiral limbus (Fig. 7a,b). No expression was observed in epithelial cells. There was no difference between our results in the common marmoset and those reported for humans and mice32. The expression pattern of COCH appears to be conserved between rodents and non-human primates.
Figure 7.

COCH expression in the cochlea of the common marmoset (a,b) COCH expression was observed in the spiral ligaments and spiral limbus. No expression was observed in epithelial cells including supporting cells and hair cells. The nuclei were counterstained with Hoechst (blue). Scale bar: 200 μm in (a) 100 μm in (b). Basal turn in (a) middle turn in (b). SV: stria vascularis, OC: organ of Corti, SGN: spiral ganglion neurons, I-V: spiral ligament fibrocytes Type I-V.
Validations of antibodies used in this study
To validate specificities of the antibodies used in this study, we performed immunostaining of the skin, the small intestine or the kidney of common marmoset (Supplementary Fig. S3). Antibodies for CX31, CRYM and GRHL2 used in this study showed immunoreactivities in epidermis or hair follicles in the skin samples and no expression was detected in connective tissues as reported previously33,34,35,36. In small intestine samples, antibodies for DFNA5 used in this study showed immunoreactivities in villus and no immunoreactivities was detected in crypt as previously reported37.In kidney samples, antibodies for ATP6B1 used in this study showed immunoreactivities in collecting ducts and tubules and no immunoreactivities was detected in glomeruli as previously reported38. Additionally we performed antibody absorption tests with the antibodies which antigens used for producing them were commercially available. Immunoreactivities were abolished by incubation with antigen peptides (Supplementary Fig. S4). We also confirmed specificities of secondary antibodies used in this study (Supplementary Fig. S5).
Discussion
Hearing loss is the most frequent congenital sensory impairment. Approximately 1 out of every 1000 newborns suffers from deafness39, and in about half the cases, the cause is monogenic40. Slowly progressive hereditary hearing loss may be accounted by the gradual loss of auditory cells thus treatments for preventing damage to the cells would be an effective therapeutic strategy. Here we examined the expression of 20 genes in the cochlea of the common marmoset and compared their patterns in between rodent and non-human primate cochleae by immunohistochemistry.
We chose six genes that cause congenital hereditary progressive hearing loss and we characterized their differential expression pattern between the common marmoset and rodents.
Mutations in the GJB3 gene, which encodes CX31, cause DFNA2B/DFNB91. Hearing loss in DFNA2B has a progressive and down-sloping pattern. Remarkably, CX31 knockout mice do not suffer from hearing loss5; therefore, the detailed pathophysiology still remains to be elucidated, although disruptions in potassium recycling are suspected to be involved due to the function of CX3141. We found species-specific expression pattern of CX31 in cochleae. In mice, it is expressed not in the stria vascularis or the organ of Corti23. In the common marmoset, it is expressed in the basal cells of the stria vascularis and broadly in the supporting cells of the organ of Corti. These results suggest that the expression of CX31 in the organ of Corti plays a more essential role in the hearing of primates. The divergence of the connexin families during evolution, in their expression and functions, has been reported in the skin33,42 and heart43. Our findings suggest that the species-difference in the recycling of ions, due to the differential expression of these gap junction genes in the cochlea, accounts for the phenotypic difference between attempted mouse models and human cases – a conclusion that may bring us closer to new therapeutic strategies for preventing hearing loss.
Mutations in the CRYM gene cause progressive and down-sloping hearing loss24. However, knockout mouse models do not suffer from hearing loss7. In rodents, it is not expressed in the organ of Corti24,25. However, the common marmoset expressed it in the organ of Corti. A previous functional study found that CRYM binds thyroid hormone (T3), and suggested that CRYM mutations cause hearing loss through disturbing the thyroid hormone-binding properties of the cochlea44. Our results suggest that, in primates, the expression of CRYM and the subsequent interaction with thyroid hormone in the organ of Corti is essential for maintaining cochlear cells and preserving normal hearing.
Mutations in the GRHL2 gene cause DFNA28, resulting in progressive, down-sloping hearing loss45. Knockout mouse model of GRHL2 is embryonic lethal, and no established hearing loss model is available46. GRHL2 is a transcription factor that binds to and regulates the activity of the human telomerase reverse transcriptase gene promoter47 and age-related hearing loss genes48. The progressive hearing loss of DFNA28 and age-related hearing loss suggest that GRHL2 is essential for maintaining epithelial cells49. In mice, it is expressed in all epithelial cells of the cochlea45. In the common marmoset, GRHL2 was not expressed as broadly, and no expression was detected in the stria vascularis. Instead, it was expressed in the spiral ligament and limbus. Our detailed investigation into the expression patterns of GRHL2 will be useful for researching the disease mechanisms of DFNA28 and of age-related hearing loss, with our primate model offering more reliable information than rodents.
Mutations in DFNA5 cause progressive, down-sloping hearing impairment28. In vitro studies have shown that the transfection of mutant DFNA5 causes cell death, whereas transfection of wild-type DFNA5 does not50. In addition, mice lacking DFNA5 do not suffer from deafness6. Therefore, the mutations leading to DFNA5-induced hearing loss are thought to be gain-of-function mutations. In mice, it is expressed (at postnatal day 3) in the stria vascularis and the greater epithelial ridge, as determined by RT-PCR27, but the exact spatial expression pattern has not been characterized. We found that its expression in the stria vascularis was limited to the basal cells and it was also expressed in the SGNs. Considering the reported functional assay, knockout mouse model is not suitable and a gain-of-function mouse model with missense mutations will be awaited. Our detailed characterization of the expression pattern of DFNA5 will be useful for researching its role in disease.
Mutation of the ATP6B1 gene causes dRTA with hearing loss. In autosomal recessive dRTA, a substantial fraction of the patients have sensorineural hearing loss with a progressive, down-sloping pattern29. Knockout mice model do not suffer from hearing loss51. In mice, it is expressed only in the spiral limbus29. In the common marmoset, it was detected not only in the spiral limbus, but also broadly across supporting cells.
Mutations in the COCH gene cause DFNA9, which is associated with progressive hearing loss52. Whereas knockout mice do not suffer from hearing loss53, mice carrying the same missense mutation as human patients show late onset hearing impairment54. We found no difference in the expression of the COCH gene between mouse, human32, and marmoset. This observation may account for rodent models being able to recapitulate human deafness; in other words, conservation of the expression pattern between primates and rodents should be a prerequisite for studying mouse models.
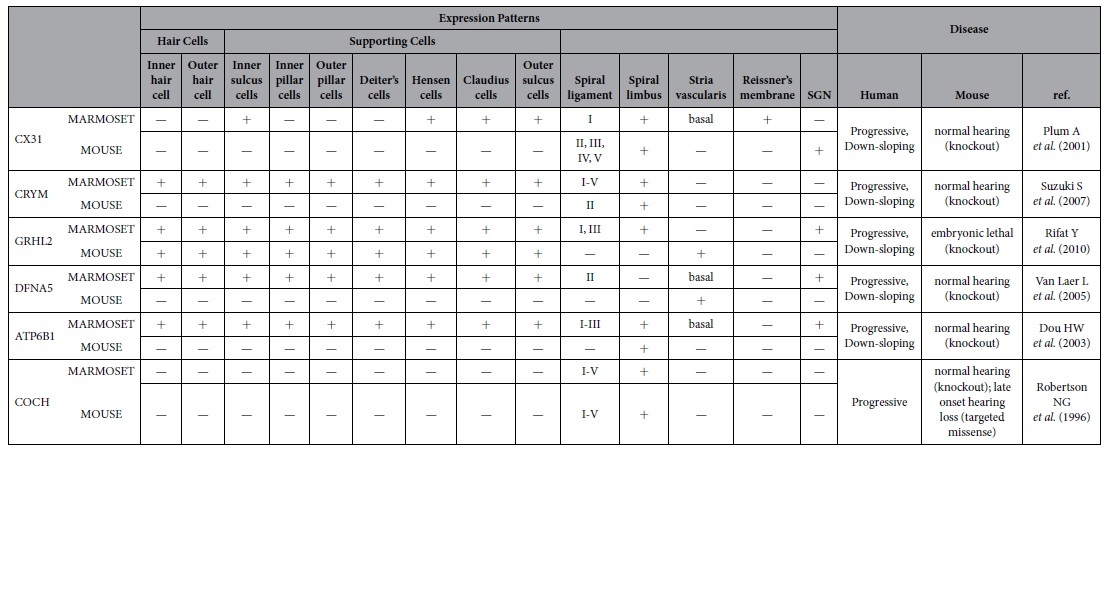
In this study, we analyzed six proteins causing progressive hereditary hearing loss, five of which (CX31, CRYM, GRHL2, DFNA5, and ATP6B1) do not recapitulate human symptoms in the mouse model (Table 1). Their amino acid sequences are much more similar to common marmoset than that of mouse (Supplementary Fig. S6) and immunostaining showed that the five genes had different expression patterns between the marmoset and mouse. On the other hand, the expression pattern of COCH, which has a mouse model that recapitulates the deafness phenotype of patients, was conserved between mouse, human, and marmoset (Supplementary Fig. S7).
Table 1. Differential expression of deafness genes in marmosets and rodents.
Mouse mutants are a useful and powerful tool for examining the pathophysiologies of hereditary diseases, including deafness, but they are not always applicable. For missense mutations, a gain-of-function (including a dominant negative function) model may account for the symptoms, and, in these instances, a transgenic or knock-in approach is effective. For example, transgenic mice expressing a deafness-causing mutant of connexin 26, a mutant carrying the R75W mutation, have hearing impairments similar to those observed in human patients55. For nonsense or truncating mutations, a loss-of-function (i.e., knockout) model may sometimes explain the symptoms. For example, deficiency of Connexin 30 in mice causes severe hearing impairment as observed in human patients of GJB6, which is caused by mutations in CX3056. Choosing appropriate approaches regarding type of mutations in the human cases may offer rodent disease models.
However, these models are based on the assumption that the distributions of targeted gene products are same in humans and rodents, and we have found examples showing that this assumption is not always true by examining non-human primate cochleae. Considering the correlation of phenotypical difference and differential expression patterns, it may be helpful to examine the expression profiles of target genes in primates and rodents before creating a mouse mutant. The common marmoset model offers a useful platform to evaluate expression patterns in the primate because it avoids the difficulties of immunostaining human temporal bone, which is time-consuming and technically difficult.
Also, the difference in the life span of human and mouse may result in the failure of recapitulating human disease phenotypes in mice, especially for the bilateral progressive hearing loss or adult-onset hearing loss. In those diseases, the life-span of mouse may not be long enough considering the onset of the phenotypes. For these diseases, non-human primates, which have longer life span than mice, would be more appropriate. Another possibility that should be raised in the discussion of the discrepancies in phenotypes of human cases and mouse models is that of a human deafness gene being erroneously assigned as a deafness gene. Whether a mutation found in patients is pathogenic is essentially defined through an inductive process; thus, supporting experiments in disease models evaluating such mutation are of great importance. In this process, confirmations using human disease cells or non-human primate in vivo model will be helpful in the future. Knowledge of the inner ear has so far been largely dependent on experiments performed in rodent models (including mouse, rat, guinea pig, and gerbil), primarily because of well-established techniques in the biotechnology: The reagents are optimized to those species and the genetic modification is enabled. However, our results indicate that the difference in the expression patterns may lead to the phenotypical discrepancy between rodent models and human patients. We propose the common marmoset, a non-human primate that is easy to handle, as a promising option for bridging the species gap. Studies using genetically modified marmosets57,58 would be a fascinating and feasible strategy for elucidating the mechanisms of hereditary hearing loss that cannot be determined by using rodent models.
Methods
Specimens of the common marmoset
Fixed and decapitated cadaverous heads of the 3–6 years old common marmosets were kindly provided from Reona Kobayashi, Takahiro Kondo, Kimika Yoshino-Saito, and Seiji Shiozawa. Fixed small intestine and kidney samples were also kindly provided from them. The animal experiments were approved by the ethics committee of Keio University (number: 11006) and were in accordance with the guidelines of the National Institutes of Health, and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Tissue preparation
Temporal bones from young adult marmosets were dissected, fixed, decalcified with Decalcifying Solution B (Wako, Saitama, Japan) for 3–4 weeks, and embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Tokyo, Japan) for cryosection. The 7 μm sections were used for immunohistochemistry.
Small intestines, kidneys and facial skins from young adult marmosets were used for validations of antibodies. Those tissues were dissected and fixed with 4% PFA, and the specimens were embedded in Tissue-Tek O.C.T. compound for cryosection. The 7 μm sections were used for immunohistochemistry.
Temporal bones from 3 weeks old mice (C57BL/6) were dissected, fixed, decalcified with Decalcifying Solution B for 3 days, and embedded in Tissue-Tek O.C.T. compound for cryosection. The 7 μm sections were used for immunohistochemistry. This animal experiments using mice were approved by the ethics committee of Keio University (number: 08020) and were in accordance with the guidelines of the National Institutes of Health, and the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Immunohistochemistry
After a brief wash with PBS, sections were heated (80 °C) in 10 mM citrate buffer (pH 6) for 1 h. After a brief wash, the sections were preblocked for 1 h at room temperature with 10% normal serum in PBS, incubated with primary antibodies at 4 °C overnight, and incubated with Alexa Fluor-conjugated secondary antibodies (Alexa488, Alexa555 and Alexa647) for 60 min at room temperature. The nuclei were counterstained with Hoechst 33342. For validations of secondary antibodies immunohistochemistry was performed without primary antibody in an overnight incubation step.
Antibody absorption test
Primary antibodies were incubated with or without (control) peptide used for generating antigens provided from antibody suppliers. After overnight incubations following immunohistochemical procedures were performed with using these antibody as primary antibody.
Antibodies
The primary antibodies used in this study are as follows: anti-SOX2 (goat IgG, Santa Cruz Biotechnology, Dallas, TX, USA, sc17320, 1:100), anti-CX26 (mouse IgG, Zymed, San Francisco, CA, USA, 13–8100, 1:300), anti-CX30 (rabbit IgG, Sigma-Aldrich, St. Louis, MO, USA, HPA014846, 1:200), anti-CX31 (rabbit IgG, Proteintech, Manchester, UK, 12880–1-AP, 1:100), anti-CTGF (goat IgG, Santa Cruz Biotechnology, sc14939, 1:100), anti-AQP1 (rabbit IgG, Millipore, Billerica, MA, USA, AB3272, 1:300), anti-CALDESMON (Sigma-Aldrich, C0297, 1:100), anti-CA II (rabbit IgG, Santa Cruz Biotechnology, sc25596, 1:100), anti-NKCC1 (goat IgG, Santa Cruz Biotechnology, sc21545, 1:300), anti-Na+/K+-ATPase α1 (rabbit IgG, Novus Biologicals, Littleton, CO, USA, EP1845Y, 1:500), anti-CALBINDIN (rabbit IgG, Abcam, Cambridge, UK, ab11426, 1:1000), anti-ZO-1 (goat IgG, Abcam, ab190085, 1:100), anti-CRYM (mouse IgG, GeneTex, Irvine, CA, USA, GTX84654, 1:100), anti-MYOSIN 7a (mouse IgG, DSHB, Iowa City, IA,USA, 138–1-s, 1:30; rabbit IgG, Proteus, 25–6790), anti-GRHL2 (rabbit IgG, Sigma-Aldrich, HPA004820, 1:200, rabbit IgG, LSBio, LS-B3983, 1:150), anti-DFNA5 (rabbit IgG, Sigma-Aldrich, HPA011326, 1:400), anti-COCH (rabbit IgG, Sigma-Aldrich, HPA050122, 1:100), anti-β-TUBULIN III (mouse IgG, Sigma-Aldrich, T8660, 1:250), anti-ATP6B1 (goat IgG, Santa Cruz Biotechnology, sc21206, 1:50), and anti-PERIPHERIN (rabbit IgG, Millipore, AB1530, 1:100).
Antigen peptides
For GRHL2, PrEST Antigen GRHL2 (Sigma-Aldrich, APREST86696) and for ATP6B1 (Santa Cruz Biotechnology, sc21206P).
Additional Information
How to cite this article: Hosoya, M. et al. Distinct Expression Patterns Of Causative Genes Responsible For Hereditary Progressive Hearing Loss In Non-Human Primate Cochlea. Sci. Rep. 6, 22250; doi: 10.1038/srep22250 (2016).
Supplementary Material
Acknowledgments
We thank Kotaro Watanabe and Ayano Mitsui for their technical support, and Reona Kobayashi, Takahiro Kondo, Kimika Yoshino-Saito, and Seiji Shiozawa for materials. Research was supported by JSPS research fellowships for Young Scientists (DC) to M.H. by MHLW, Research on Sensory and Communicative Disorders, MEXT, Grant-in-Aid for Scientific Research (C) and (B) (24592560, 15H04991), and Takeda Science Foundation to M.F.
Footnotes
H.O. is a founding scientist and a paid Scientific Advisory Board of San Bio Co. Ltd.
Author Contributions M.H. and M.F. designed the study, performed the research, and analyzed the data; M.F., M.H., K.O. and H.O. wrote the paper.
References
- Kral A., Hartmann R., Tillein J., Heid S. & Klinke R. Delayed maturation and sensitive periods in the auditory cortex. Audiol Neurootol 6, 346–362, doi: 46845 (2001). [DOI] [PubMed] [Google Scholar]
- Li J. et al. Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum Mutat 36, 98–105, doi: 10.1002/humu.22712 (2015). [DOI] [PubMed] [Google Scholar]
- Thoenes M. et al. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J Rare Dis 10, 15, doi: 10.1186/s13023-015-0238-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rost S. et al. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur J Hum Genet 22, 208–215, doi: 10.1038/ejhg.2013.108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plum A. et al. Connexin31-deficiency in mice causes transient placental dysmorphogenesis but does not impair hearing and skin differentiation. Dev Biol 231, 334–347, doi: 10.1006/dbio.2000.0148 (2001). [DOI] [PubMed] [Google Scholar]
- Van Laer L. et al. Mice lacking Dfna5 show a diverging number of cochlear fourth row outer hair cells. Neurobiol Dis 19, 386–399, doi: 10.1016/j.nbd.2005.01.019 (2005). [DOI] [PubMed] [Google Scholar]
- Suzuki S. et al. micro-Crystallin as an intracellular 3,5,3′-triiodothyronine holder in vivo. Mol Endocrinol 21, 885–894, doi: 10.1210/me.2006-0403 (2007). [DOI] [PubMed] [Google Scholar]
- Schuknecht H. F. Further Observations on the Pathology of Presbycusis. Archiv Otolaryngol 80, 369–382 (1964). [DOI] [PubMed] [Google Scholar]
- Yamazaki Y. & Watanabe S. Marmosets as a next-generation model of comparative cognition. Japanese Psychological Research 51, 182–196, doi: 10.1111/j.1468-5884.2009.00398.x (2009). [DOI] [Google Scholar]
- Osmanski M. S. & Wang X. Measurement of absolute auditory thresholds in the common marmoset (Callithrix jacchus). Hear Res 277, 127–133, doi: 10.1016/j.heares.2011.02.001 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki E. et al. Generation of transgenic non-human primates with germline transmission. Nature 459, 523–527, doi: 10.1038/nature08090 (2009). [DOI] [PubMed] [Google Scholar]
- Johnson L. A., Della Santina C. C. & Wang X. Temporal bone characterization and cochlear implant feasibility in the common marmoset (Callithrix jacchus). Hear Res 290, 37–44, doi: 10.1016/j.heares.2012.05.002 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borin A. et al. Anatomical study of a temporal bone from a non-human primate (Callithrix sp). Braz J Otorhinolaryngol 74, 370–373 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold W. Immunohistochemical investigation of the human inner ear. Limitations and prospects. Acta Otolaryngol 105, 392–397 (1988). [DOI] [PubMed] [Google Scholar]
- Bianchi L. M. & Gale N. W. Distribution of Eph-related molecules in the developing and mature cochlea. Hear Res 117, 161–172 (1998). [DOI] [PubMed] [Google Scholar]
- Hirose K. & Liberman M. C. Lateral wall histopathology and endocochlear potential in the noise-damaged mouse cochlea. J Assoc Res Otolaryngol 4, 339–352, doi: 10.1007/s10162-002-3036-4 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer S. S. & Schulte B. A. The fine structure of spiral ligament cells relates to ion return to the stria and varies with place-frequency. Hear Res 100, 80–100 (1996). [DOI] [PubMed] [Google Scholar]
- Adams J. C. Immunocytochemical traits of type IV fibrocytes and their possible relations to cochlear function and pathology. J Assoc Res Otolaryngol 10, 369–382, doi: 10.1007/s10162-009-0165-z (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Kinnefors A., Bostrom M. & Rask-Andersen H. Expression of peripherin in human cochlea. Cell Tissue Res 342, 345–351, doi: 10.1007/s00441-010-1081-6 (2010). [DOI] [PubMed] [Google Scholar]
- Stankovic K. M., Adams J. C. & Brown D. Immunolocalization of aquaporin CHIP in the guinea pig inner ear. Am J Physiol 269, C1450–1456 (1995). [DOI] [PubMed] [Google Scholar]
- Lopez I. A., Ishiyama G., Lee M., Baloh R. W. & Ishiyama A. Immunohistochemical localization of aquaporins in the human inner ear. Cell Tissue Res 328, 453–460, doi: 10.1007/s00441-007-0380-z (2007). [DOI] [PubMed] [Google Scholar]
- Xia J. H. et al. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat Genet 20, 370–373, doi: 10.1038/3845 (1998). [DOI] [PubMed] [Google Scholar]
- Xia A. P. et al. Expression of connexin 31 in the developing mouse cochlea. Neuroreport 11, 2449–2453 (2000). [DOI] [PubMed] [Google Scholar]
- Abe S. et al. Identification of CRYM as a Candidate Responsible for Nonsyndromic Deafness, through cDNA Microarray Analysis of Human Cochlear and Vestibular Tissues* *Nucleotide sequence data reported herein are available in the DDBJ/EMBL/GenBank databases; for details, see the Electronic-Database Information section of this article. The American Journal of Human Genetics 72, 73–82, doi: 10.1086/345398 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usami S. et al. The localization of proteins encoded by CRYM, KIAA1199, UBA52, COL9A3 and COL9A1, genes highly expressed in the cochlea. Neuroscience 154, 22–28, doi: 10.1016/j.neuroscience.2008.03.018 (2008). [DOI] [PubMed] [Google Scholar]
- Petrof G. et al. Mutations in GRHL2 result in an autosomal-recessive ectodermal Dysplasia syndrome. Am J Hum Genet 95, 308–314, doi: 10.1016/j.ajhg.2014.08.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer L. et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nature Genetics 20, 194–197 (1998). [DOI] [PubMed] [Google Scholar]
- Van Laer L. et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet 20, 194–197, doi: 10.1038/2503 (1998). [DOI] [PubMed] [Google Scholar]
- Karet F. E. et al. Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat Genet 21, 84–90, doi: 10.1038/5022 (1999). [DOI] [PubMed] [Google Scholar]
- Ikezono T. et al. Cochlin-tomoprotein: a novel perilymph-specific protein and a potential marker for the diagnosis of perilymphatic fistula. Audiol Neurootol 14, 338–344, doi: 10.1159/000212113 (2009). [DOI] [PubMed] [Google Scholar]
- Jones S. M. et al. Hearing and vestibular deficits in the Coch(−/−) null mouse model: comparison to the Coch(G88E/G88E) mouse and to DFNA9 hearing and balance disorder. Hear Res 272, 42–48, doi: 10.1016/j.heares.2010.11.002 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson N. G. et al. Inner ear localization of mRNA and protein products of COCH, mutated in the sensorineural deafness and vestibular disorder, DFNA9. Hum Mol Genet 10, 2493–2500 (2001). [DOI] [PubMed] [Google Scholar]
- Di W. L., Rugg E. L., Leigh I. M. & Kelsell D. P. Multiple epidermal connexins are expressed in different keratinocyte subpopulations including connexin 31. J Invest Dermatol 117, 958–964, doi: 10.1046/j.0022-202x.2001.01468.x (2001). [DOI] [PubMed] [Google Scholar]
- Kurek D., Garinis G. A., van Doorninck J. H., van der Wees J. & Grosveld F. G. Transcriptome and phenotypic analysis reveals Gata3-dependent signalling pathways in murine hair follicles. Development 134, 261–272, doi: 10.1242/dev.02721 (2007). [DOI] [PubMed] [Google Scholar]
- Chen W. et al. Grainyhead-like 2 (GRHL2) inhibits keratinocyte differentiation through epigenetic mechanism. Cell Death Dis 3, e450, doi: 10.1038/cddis.2012.190 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto Y. et al. Subgroups of enlarged vestibular aqueduct in relation to SLC26A4 mutations and hearing loss. Laryngoscope 124, E134–140, doi: 10.1002/lary.24368 (2014). [DOI] [PubMed] [Google Scholar]
- Gassler N. et al. Molecular characterisation of non-absorptive and absorptive enterocytes in human small intestine. Gut 55, 1084–1089, doi: 10.1136/gut.2005.073262 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidarsson H. et al. The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS One 4, e4471, doi: 10.1371/journal.pone.0004471 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgert N., Smith R. J. & Van Camp G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 681, 189–196, doi: 10.1016/j.mrrev.2008.08.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton C. C. & Nance W. E. Newborn hearing screening–a silent revolution. New Engl J Med 354, 2151–2164, doi: 10.1056/NEJMra050700 (2006). [DOI] [PubMed] [Google Scholar]
- Steel K. P. & Kros C. J. A genetic approach to understanding auditory function. Nat Genet 27, 143–149, doi: 10.1038/84758 (2001). [DOI] [PubMed] [Google Scholar]
- Richard G. Connexins: a connection with the skin. Exp Dermatol 9, 77–96 (2000). [DOI] [PubMed] [Google Scholar]
- van Kempen M. J. et al. Differential connexin distribution accommodates cardiac function in different species. Microsc Res Tech 31, 420–436, doi: 10.1002/jemt.1070310511 (1995). [DOI] [PubMed] [Google Scholar]
- Oshima A. et al. CRYM mutations cause deafness through thyroid hormone binding properties in the fibrocytes of the cochlea. J Med Genet 43, e25, doi: 10.1136/jmg.2005.034397 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters L. M. et al. Mutation of a transcription factor, TFCP2L3, causes progressive autosomal dominant hearing loss, DFNA28. Hum Mol Genet 11, 2877–2885, doi: 10.1093/hmg/11.23.2877 (2002). [DOI] [PubMed] [Google Scholar]
- Rifat Y. et al. Regional neural tube closure defined by the Grainy head-like transcription factors. Dev Biol 345, 237–245, doi: 10.1016/j.ydbio.2010.07.017 (2010). [DOI] [PubMed] [Google Scholar]
- Kang X., Chen W., Kim R. H., Kang M. K. & Park N. H. Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D and GRHL2 in human oral squamous cell carcinoma cells. Oncogene 28, 565–574, doi: 10.1038/onc.2008.404 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer L. et al. The grainyhead like 2 gene (GRHL2), alias TFCP2L3, is associated with age-related hearing impairment. Hum Mol Genet 17, 159–169, doi: 10.1093/hmg/ddm292 (2008). [DOI] [PubMed] [Google Scholar]
- Hilgert N., Smith R. J. & Van Camp G. Function and expression pattern of nonsyndromic deafness genes. Curr Mol Med 9, 546–564 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Laer L. et al. DFNA5: hearing impairment exon instead of hearing impairment gene? J Med Genet 41, 401–406, doi: 10.1136/jmg.2003.015073 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H. W., Finberg K., Cardell E. L., Lifton R. & Choo D. Mice lacking the B1 subunit of H+-ATPase have normal hearing. Hearing Research 180, 76–84, doi: 10.1016/S0378-5955(03)00108-4 (2003). [DOI] [PubMed] [Google Scholar]
- Robertson N. G. et al. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat Genet 20, 299–303, doi: 10.1038/3118 (1998). [DOI] [PubMed] [Google Scholar]
- Makishima T. et al. Targeted disruption of mouse Coch provides functional evidence that DFNA9 hearing loss is not a COCH haploinsufficiency disorder. Hum Genet 118, 29–34, doi: 10.1007/s00439-005-0001-4 (2005). [DOI] [PubMed] [Google Scholar]
- Robertson N. G. et al. A targeted Coch missense mutation: a knock-in mouse model for DFNA9 late-onset hearing loss and vestibular dysfunction. Hum Mol Genet 17, 3426–3434, doi: 10.1093/hmg/ddn236 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo T. et al. Transgenic expression of a dominant-negative connexin26 causes degeneration of the organ of Corti and non-syndromic deafness. Hum Mol Genet 12, 995–1004, doi: 10.1093/hmg/ddg116 (2003). [DOI] [PubMed] [Google Scholar]
- Teubner B. et al. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet 12, 13–21, doi: 10.1093/hmg/ddg001 (2003). [DOI] [PubMed] [Google Scholar]
- Kishi N., Sato K., Sasaki E. & Okano H. Common marmoset as a new model animal for neuroscience research and genome editing technology. Dev Growth Differ 56, 53–62, doi: 10.1111/dgd.12109 (2014). [DOI] [PubMed] [Google Scholar]
- Izpisua Belmonte J. C. et al. Brains, genes and primates. Neuron 86, 617–631, doi: 10.1016/j.neuron.2015.03.021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.