

Abstract
Lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) is the most common extranodal B cell tumor and accounts for 8% of non-Hodgkin’s lymphomas. Gastric MALT lymphoma is the best-studied example and is a prototypical neoplasm that occurs in the setting of chronic inflammation brought on by persistent infection or autoimmune disease. Cytogenetic abnormalities are commonly acquired during the course of disease and the most common is chromosomal translocation t(11;18)(q21;q21), which creates the API2-MALT1 fusion oncoprotein. t(11;18)-positive lymphomas can be clinically aggressive and have a higher rate of dissemination than t(11;18)-negative tumors. Many cancers, including MALT lymphomas, characteristically exhibit deregulated over-activation of cellular survival pathways, such as the nuclear factor-κB (NF-κB) pathway. Molecular characterization of API2-MALT1 has revealed it to be a potent activator of NF-κB, which is required for API2-MALT1-induced cellular transformation, however the mechanisms by which API2-MALT1 exerts these effects are only recently becoming apparent. The API2 moiety of the fusion binds tumor necrosis factor (TNF) receptor associated factor (TRAF) 2 and receptor interacting protein 1 (RIP1), two proteins essential for TNF receptor-induced NF-κB activation. By effectively mimicking ligand-bound TNF receptor, API2-MALT1 promotes TRAF2-dependent ubiquitination of RIP1, which then acts as a scaffold for nucleating and activating the canonical NF-κB machinery. Activation occurs, in part, through MALT1 moiety-dependent recruitment of TRAF6, which can directly modify NF-κB essential modulator, the principal downstream regulator of NF-κB. While the intrinsic MALT1 protease catalytic activity is dispensable for this canonical NF-κB signaling, it is critical for non-canonical NF-κB activation. In this regard, API2-MALT1 recognizes NF-κB inducing kinase (NIK), the essential upstream regulator of non-canonical NF-κB, and cleaves it to generate a stable, constitutively active fragment. Thus, API2-MALT1 harnesses multiple unique pathways to achieve deregulated NF-κB activation. Emerging data from our group and others have also detailed additional gain-of-function activities of API2-MALT1 that extend beyond NF-κB activation. Specifically, API2-MALT1 recruits and subverts multiple other signaling factors, including LIM domain and actin-binding protein 1 (LIMA1) and Smac/DIABLO. Like NIK, LIMA1 represents a unique substrate for API2-MALT1 protease activity, but unlike NIK, its cleavage sets in motion a major NF-κB-independent pathway for promoting oncogenesis. In this review, we highlight the most recent results characterizing these unique and diverse gain-of-function activities of API2-MALT1 and how they contribute to lymphomagenesis.
Keywords: Oncogene, Fusion oncoprotein, Lymphoma, Chromosomal translocation, Ubiquitination, Apoptosis, Nuclear factor-κB, Caspases
Core tip: We summarize the identification of novel API2-mucosa-associated lymphoid tissue (MALT) 1-interacting proteins that uniquely mediate the cellular effects of the fusion oncoprotein but not wild-type API2 or MALT1. API2-MALT1 recruits receptor interacting protein 1 and tumor necrosis factor (TNF) receptor associated factor 2, which normally function downstream of the TNF receptor, and utilizes these proteins to communicate unregulated canonical nuclear factor-κB (NF-κB) in a manner that does not depend on the protease activity of MALT1. Simultaneously, NF-κB inducing kinase is recruited to API2-MALT1 and is proteolytically cleaved by the MALT1 protease domain to generate a stable, non-canonical NF-κB-activating fragment. Finally, LIM domain and actin-binding protein 1 is similarly recruited and cleaved as an API2-MALT1 specific target and its cleavage mediates an NF-κB-independent mechanism of oncogenesis. Additional factors, including SMAC and BCL10, may also play key roles as API2-MALT1 binding partners and downstream signaling factors. Thus, the API2-MALT1 fusion utilizes a distinct set of protein-protein interactions to leverage multiple, divergent mechanisms and achieve potent oncogenic reprogramming of affected B cells.
INTRODUCTION
Extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue (MALT lymphoma) is a prototypical cancer that occurs at sites affected by chronic inflammation due to persistent pathogenic infection or autoimmune disease[1]. The best-studied example of MALT lymphoma affects the gastric mucosa and occurs in the setting of Helicobacter pylori infection. Lymphomas of this type are commonly treated with antibiotics to eradicate the causative microorganism, which prevents further antigenic stimulation of the oncogenic B cells. However, in a subset of MALT lymphomas, acquired genetic abnormalities relieve the neoplastic cells of their dependence on the inflammatory milieu and induce a state of stimulus-independent growth[2].
Interestingly, some MALT lymphomas arise in an atypical fashion and occur in the absence of underlying chronic inflammation or autoimmune disorders. These lymphomas typically harbor the most common and first-described genetic abnormality in MALT lymphoma, namely the chromosomal translocation t(11;18)(q21;q21), which fuses a portion of the inhibitor of apoptosis 2 (API2; also known as cIAP2) and the MALT lymphoma translocation gene 1 (MALT1) genes creating an expressed fusion protein known as API2-MALT1[3-5]. This chromosomal translocation is specific to MALT lymphoma, and when it occurs, it is generally seen as the sole cytogenetic abnormality[6,7]. The fact that API2-MALT1-positive lymphomas arise in the absence of inflammatory conditions that typically predispose to the development of MALT lymphoma suggest that the fusion itself is the oncogenic driver. In addition, t(11;18)-positive lymphomas are associated with treatment resistance and a higher tendency to disseminate[8-11].
While many chromosomal breakpoints have been identified, the resultant API2-MALT1 chimeric proteins invariably contain the amino terminus of API2, which includes three intact baculovirus IAP repeat (BIR) domains, and the carboxy terminus of MALT1, which contains immunoglobulin (Ig)-like domains that also mediate protein-protein interactions (Figure 1)[12]. Also invariably included in the fusion is the enzymatically active “caspase-like” protease domain of MALT1. The proteolytic activity of MALT1 has been well-documented, but only a few natural substrates are known and our understanding of the overall impact of MALT1 proteolytic activity to cell signaling and physiology is still unfolding[13].
Figure 1.
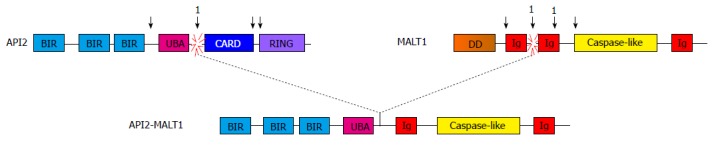
Schematic illustration of the API2-mucosa-associated lymphoid tissue 1 fusion oncoprotein. The most commonly occurring API2-MALT1 fusion is depicted. Recurrent breakpoint positions for both MALT1 and API2 are indicated by arrowheads. The most common breakpoint for API2, by far, is indicated by 1 (approximately 93%). There are two relatively common breakpoints for MALT1, both indicated by asterisks (approximately 42% and 32%, respectively). CARD: Caspase recruitment domain; DD: Death domain; Ig: Immunoglobulin-like; BIR: Baculovirus inhibitor of apoptosis repeat; UBA: Ubiquitin-associated domain; RING: Really interesting new gene; MALT: Mucosa-associated lymphoid tissue.
Wild-type API2 is a member of the IAP family of anti-apoptotic proteins and also contains a C-terminal caspase recruitment domain (CARD) that is not present in the commonest API2-MALT1 chimeras[14]. More importantly, API2 possesses a really interesting new gene (RING) domain that is never included in API2-MALT1 chimeras; the RING domain of API2 possesses E3 ubiquitin ligase activity and mediates covalent conjugation of ubiquitin to interacting proteins, which either targets the substrate for degradation via the proteasome or modulates the function of the substrate protein[15]. Wild-type MALT1 contains an amino-terminal death domain (DD) and an additional Ig-like domain; the DD is never present in API2-MALT1 fusion proteins and this N-terminal Ig-like domain is only rarely included[13]. MALT1 is a crucial mediator of antigen receptor-induced activation and proliferation in lymphocytes and also mediates cellular signaling in other immune cells downstream of immunoreceptor tyrosine-based activation motif -containing receptors. Additionally, MALT1 has been found to mediate inflammatory responses to agonistic activation of various G protein-coupled receptors in non-immune cells[12].
MALT1 couples to these various receptors via interaction with B-cell CLL/lymphoma 10 (BCL10), an adaptor protein, and members of the CARD and membrane-associated guanylate kinase-like (MAGUK) domain-containing protein (CARMA) family of proteins (Figure 2). Formation of a receptor-induced complex consisting of the CARMA, Bcl10, and MALT1 proteins, known as the CBM signalosome, results in oligomerization of MALT1 and its coupling to downstream signaling mediators to effect a response[12]. The best-studied signaling pathway that is activated by the CBM signalosome is that which controls nuclear factor-κB (NF-κB), a family of transcription factors that mediates the response to a growing cadre of diverse extracellular substances including growth factors, antigenic stimuli, peptide hormones, and chemotactic agents. There are actually two principal pathways for activating NF-κB subunits: The canonical (or classical) and non-canonical (or alternative) pathways (Figure 2)[16]. Both depend on stimulus-specific activation of kinase cascades that trigger degradation of NF-κB-inhibitory proteins and free the active transcription factor dimers; canonical dimers typically comprise RelA (p65) and p50 whereas non-canonical dimers comprise RelB and p52 (Figure 2). Active NF-κB dimers translocate to the nucleus and bind consensus sequences in the promoters of many genes to rapidly induce expression of proteins that drive proliferation, survival, and differentiation of cells of the immune system, all of which must be properly and appropriately controlled to minimize the risk of tissue damage and disease. Indeed, genetic defects that impair proper activation of NF-κB in lymphocytes can result in immunodeficiency, while aberrant hyper-activation of NF-κB is associated with autoimmune disease and various cancers. The precise mechanisms governing NF-κB activation are described in greater detail in the legend to Figure 2. In addition, many comprehensive reviews of NF-κB signaling have been published[16-19], including reviews focused on how the CBM signalosome interacts with the NF-κB machinery[20,21], and the reader is also referred to these for a more detailed treatment of the subject.
Figure 2.
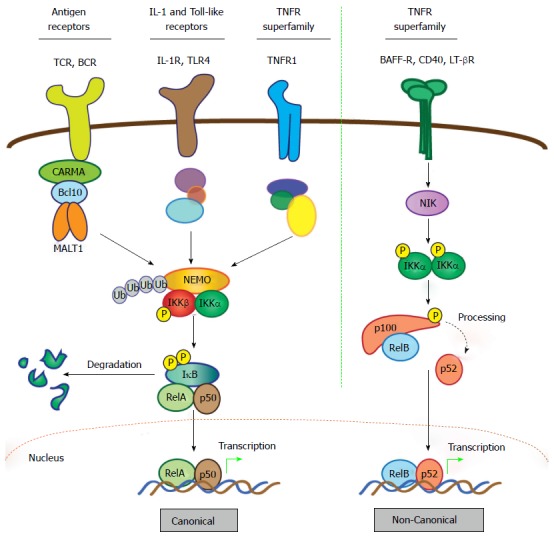
Activation of nuclear factor-κB: Canonical and non-canonical pathways. The NF-κB family comprises five members characterized by the presence of a Rel homology domain, which mediates DNA binding and dimerization. To allow prompt and efficient activation in response to stimuli, NF-κB dimers are sequestered in the cytoplasm in an inactive form via interaction with IκB proteins. Activation of NF-κB quickly follows ligand-induced stimulation of a variety of receptors, including TNF, IL-1, Toll-like, and BCR and TCR. The two principal signaling mechanisms that lead to activation of specific NF-κB heterodimers are the canonical and non-canonical pathways. In the case of canonical signaling, receptor-mediated signaling is transmitted via recruitment and/or activation of various upstream signaling components that promote activation of the IKK complex, which consists of catalytic kinase subunits IKKα and IKKβ and a regulatory scaffold protein called NF-κB essential modulator (NEMO; also called IKKγ). Once the IKK complex is activated, the steps directing canonical NF-κB stimulation are generally shared, regardless of the upstream stimulus. Selectivity is thought to reside mostly in the unique, “private” pathways utilized by distinct receptors for communication with the IKK complex. In the case of antigen receptors, assembly of the CBM signalosome is critical for this to occur. Upon activation of the IKK complex, the IKKβ kinase subunit phosphorylates IκBα, marking it for lysine (K)48-linked ubiquitination and subsequent degradation via the proteasome, which represents the commitment step for canonical NF-κB activation. Newly freed NF-κB heterodimers, most commonly RelA (p65) and p50, translocate to the nucleus where they bind target sites within the promoters of a vast array of responsive genes to induce expression. Notably, the gene encoding IκBα is directly regulated by NF-κB and its synthesis serves as a negative feedback loop, thus attenuating and resetting the canonical NF-κB machinery. In contrast to the multitude of stimuli and signaling intermediates that activate canonical NF-κB, a relatively small number of cell-surface receptors promote non-canonical NF-κB activation. These include the BAFF, LT-β, and CD40 TNF superfamily receptors. Stimulation of non-canonical NF-κB centers on stabilization and activation of the critical upstream regulator, NIK. In resting cells, NIK is targeted for K48-linked ubiquitination and proteasomal degradation by a complex of proteins that includes cIAP1, cIAP2, TRAF2 and TRAF3, which together function as a “destruction complex” and constitutively represses NIK activity. Ligand binding to selective TNF superfamily receptors promotes degradation of TRAF3, the protein which links NIK to cIAP1/2 and TRAF2, thereby freeing NIK from the destruction complex and allowing its stabilization and activation. Then, NIK phosphorylates and activates the IKKα kinase subunit, which in turn phosphorylates the NF-κB2 precursor p100, targeting it for partial proteasomal degradation to p52. Like IκBα, p100 contains C-terminal ankyrin repeats that are responsible for cytoplasmic retention of RelB/p100 complexes in unstimulated cells. Freed RelB/p52 dimers also undergo nuclear translocation and drive transcription of appropriate genes in a manner analogous to the canonical RelA/p50 heterodimer complexes. Unlike canonical NF-κB signal termination, negative regulation of non-canonical NF-κB is less well-characterized. It appears to depend on reciprocal IKKα-dependent phosphorylation of NIK on C-terminal serine residues, which leads to TRAF/cIAP complex-independent NIK destabilization. However, more work is needed to fully define the mechanism involved in attenuating the non-canonical NF-κB signaling pathway. CARMA: CARD and membrane-associated guanylate kinase-like domain-containing protein; NF-κB: Nuclear factor-κB; MALT: Mucosa-associated lymphoid tissue; IκB: Inhibitor of NF-κB; IL: Interleukin; IKK: IκB kinase; BAFF: B-cell activating factor belonging to the TNF family; LT: Lymphotoxin; NIK: NF-κB-inducing kinase; BCR: B-cell receptor; TCR: T-cell antigen receptor.
In this review, we will summarize the unique, gain-of-function properties that have been described recently for the API2-MALT1 fusion oncoprotein. In particular, we will highlight the contributions of the API2 moiety which, through its BIR protein interaction domains, creates a special platform for downstream signaling and works together in surprising fashion with the protease domain of the MALT1 moiety.
UNIQUE PROPERTIES OF API2-MALT1: AUTO-OLIGOMERIZATION
Like wild-type MALT1, the API2-MALT1 fusion protein is able to promote activation of NF-κB[22-24]. In both cases, it is thought that oligomerization of the MALT1 C-terminus is critical for downstream communication with the NF-κB machinery. However, while wild-type MALT1 must rely on its association with the CARMA and BCL10 components of the CBM signalosome for stimulus-induced oligomerization, API2-MALT1 is able to auto-oligomerize via heterotypic association between the BIR1 domain of the API2 moiety and the C-terminal caspase-like domain of MALT1[25,26]. Juxtaposing these two protein components imparts oncogenic potential to API2-MALT1, per se, that the wild-type counterparts are unable to achieve and depends on the presence of an intact BIR1 domain. Because the homo-oligomerization of API2-MALT1 occurs constitutively, and is not influenced by any known regulatory processes such as those which govern assembly and disassembly of the CBM signalosome, the presence of API2-MALT1 in a cell induces exceptionally potent and sustained NF-κB activation. This high level of NF-κB activity is associated with protection against apoptotic stimuli, and importantly, confers anchorage-independent growth to NIH-3T3 fibroblasts, underscoring the transformative potential of API2-MALT1 and marking it as a bone fide oncogene. It is important to note that these effects are dependent on the fusion of these two protein moieties and result from the invariant inclusion of the BIR domains from wild-type API2 and the protease and Ig-like domains of wild-type MALT1. These seminal findings provided an excellent framework on which to interrogate in greater detail how the API2 and MALT1 fusion partners contribute to the overall potential of API2-MALT1 and offer explanations for the advanced clinical nature of t(11;18)-positive lymphomas. In particular, these observations prompted exploration of whether heterotypic protein interactions, in addition to this homotypic interaction, occur via either the API2 or MALT1 domains to affect function of the overall fusion protein.
UNIQUE PROPERTIES OF API2-MALT1: ASSEMBLY OF NOVEL DOWNSTREAM EFFECTOR COMBINATIONS
Smac/DIABLO and BCL10
In addition to auto-oligomerization, the API2 moiety of API2-MALT1 also mediates other critical protein-protein interactions. The API2 BIR domains interact with multiple signaling proteins that play important roles as regulators of cellular apoptosis and NF-κB activity, and these interactions likely contribute to API2-MALT1-dependent promotion of cell survival. For example, second mitochondrial activator of caspases (Smac; also known as DIABLO) is a mitochondrial protein that is released into the cytoplasm in response to pro-apoptotic stimuli and binds to the API2 moiety of API2-MALT1[27]. Smac can promote apoptosis by binding to X-linked inhibitor apoptosis, which then liberates caspases from inhibition and allows apoptosis to proceed[28]. It has been proposed that the binding of Smac to the API2 moiety of API2-MALT1 sequesters it and prevents it from performing this function, which is critical to API2-MALT1’s ability to suppress Smac-dependent apoptosis[27]. However, the precise molecular mechanism by which API2-MALT1 interferes with Smac-dependent apoptosis is not yet fully understood, nor is it known how the MALT1 moiety of the fusion contributes to this process.
One study demonstrated that like Smac, BCL10 also binds to the API2 moiety of the API2-MALT1 fusion[29]. The authors first found that wild-type API2 interacts with BCL10 through its BIR region and ubiquitinates BCL10 via its RING E3 ubiquitin ligase domain. They then showed that API2-mediated ubiquitination targets BCL10 for proteasomal degradation. In contrast to wild-type API2, the API2-MALT1 fusion, regardless of specific translocation breakpoint, always lacks the API2 RING domain and therefore does not posses E3 ubiquitin ligase activity. The authors thus demonstrated that the API2-MALT1 fusion retains the ability to bind BCL10 via the BIR region but fails to ubiquitinate BCL10. This leads to stabilization and upregulation of BCL10 in t(11;18)/API2-MALT1 positive lymphomas and could theoretically sensitize cells to prosurvival signaling through the B cell receptor. The role of the MALT1 moiety in API2-MALT1-dependent stabilization/upregulation of BCL10, and the contribution of BCL10 upregulation to the oncogenic potential of API2-MALT1, has yet to be determined.
Receptor interacting protein 1, TNF receptor associated factor 2 and TNF receptor associated factor 6
The BIR domains of wild-type IAP proteins, including API2, mediate unique interactions with members of the TNF receptor associated factor (TRAF) family of signaling proteins to modulate NF-κB activation in the setting of tumor necrosis factor (TNF) receptor stimulation[30-33]. Here, the activated TNF receptor recruits the adaptor TNF receptor associated DD protein (TRADD), which in turn binds TRAF2 and receptor interacting protein 1 (RIP1)[34,35]. TRAF2 also recruits wild-type API2 (cIAP2) and cIAP1 proteins, which together mediate the non-degradative (K63-linked) poly-ubiquitination of RIP1 (Figure 3)[15,31,33,36]. Since TRAF2 and the IAP proteins each contain similar E3 ubiquitin ligase (RING) domains, it remains somewhat unclear which protein is the critical ligase for RIP1 ubiquitination, or if all three work in concert. However, in isolation, each is capable of catalyzing the ubiquitination given the right conditions so it is thought that each may play an active role[37]. Next, the poly-ubiquitin chains on RIP1 create a docking site for the NF-κB essential modulator (NEMO) subunit of the IKK complex, promoting canonical NF-κB activation[37-41].
Figure 3.
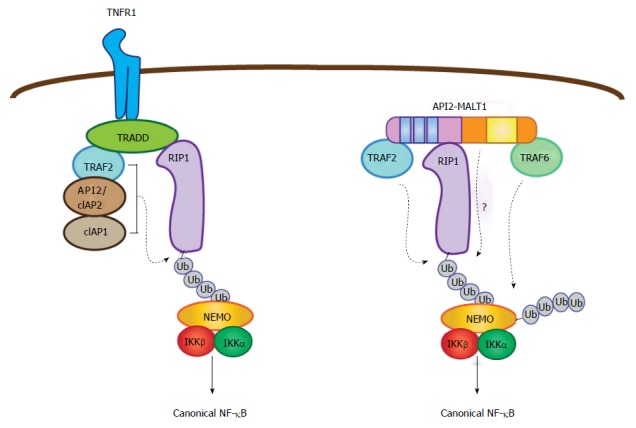
Parallel pathways for canonical nuclear factor-κB activation mediated by tumor necrosis factor receptor and API2-mucosa-associated lymphoid tissue 1. In the context of TNF receptor activation, TNF receptor associated death domain protein brokers the assembly of a complex containing three E3 ubiquitin ligases (API2/cIAP2, cIAP1, and TRAF2), which together promote RIP1 ubiquitination. This then serves as a platform for recruitment of NEMO. In parallel fashion, the API2 moiety of API2-MALT1 binds TRAF2 and RIP1 to promote RIP1 ubiquitination, but in this case the ubiquitination event seems to be mediated only by TRAF2. Importantly, the MALT1 moiety facilitates the ubiquitination of RIP1 through an as of yet undefined mechanism. Recruitment of NEMO via the RIP1 ubiquitin scaffold allows MALT1 moiety-associated TRAF6 to promote NEMO ubiquitination, IKK complex activation and subsequent canonical NF-κB activation. TRAF: TNF receptor associated factor; NEMO: NF-κB essential modulator; RIP1: Receptor interacting protein 1; TRADD: TNF receptor associated death domain protein; IKK: IκB kinase; MALT: Mucosa-associated lymphoid tissue; TNF: Tumor necrosis factor; NF-κB: Nuclear factor-κB.
Since the API2 moiety of API2-MALT1 retains the BIR domains, the possibility existed that this portion of the fusion protein recruits TRAF2, and although the API2 RING domain is missing from the API2-MALT1 fusion, the recruited wild-type TRAF2 might mediate RIP1 ubiquitination (Figure 3). Indeed, TRAF2 binding occurs through the BIR1 domain of the API2 moiety and deletion of this motif from the oncoprotein compromises its ability to activate NF-κB[26]. Further, deletion of the RING domain of TRAF2 inhibits API2-MALT1-induced NF-κB activation[26]. To further characterize the essential nature of TRAF2 and the manner in which it contributes to API2-MALT1-induced effects, we turned again to the TNF receptor signaling complex as a potential analogous model. We determined that, somewhat akin to TRADD, API2-MALT1 can recruit RIP1 through an interaction that is independent of both the BIR1 domain and TRAF2, and that this is required for API2-MALT1-induced canonical NF-κB activation (Figure 3)[42]. API2-MALT1 expression also promotes ubiquitination of RIP1 and recruitment of NEMO, but only in the presence of wild-type TRAF2 and its intact RING domain. Mutants of API2-MALT1 that fail to recruit TRAF2 cannot induce RIP1 ubiquitination, do not recruit NEMO or activate NF-κB, and cannot promote transformation of NIH-3T3 cells[42].
Although the API2 moiety binds both RIP1 and TRAF2, expression of the API2 moiety alone is insufficient to promote either RIP1 ubiquitination or NF-κB activation[42]. Therefore, the MALT1 moiety must provide additional necessary functionality. However, MALT1 protease activity, the principal activity ascribed to the MALT1 C-terminus, is not required since disabling the catalytic domain by site-directed mutagenesis has no effect on RIP1 ubiquitination. Since the C-terminus of MALT1 is also known to bind TRAF6, another RING-containing E3 ligase, we considered the possibility that TRAF6 recruitment to the API2-MALT1 fusion might represent the critical event and that TRAF6 might work together with TRAF2 to mediate the RIP1 ubiquitination; but this is not the case either since site-directed mutagenesis of the TRAF6 binding sites within API2-MALT1 has little effect on RIP1 ubiquitination[42]. Taken together, these results suggest that there is some, still-undetermined functionality of the MALT1 moeity within the API2-MALT1 fusion protein that allows the API2 moiety to direct efficient TRAF2-dependent RIP1 ubiquitination. Further work will be required to reveal the nature of this enabling activity.
Nevertheless, it is important to underscore that maximal NF-κB induction does appear to depend on TRAF6 recruitment to API2-MALT1, since NF-κB activity is significantly dampened upon mutation or deletion of the C-terminal TRAF6 binding sites[42,43]. This is because TRAF6 appears to direct a pathway of K63-linked NEMO ubiquitination that is parallel to the TRAF2-dependent pathway of RIP1 ubiquitination. Thus, we speculate that API2-MALT1 coordinates a sequential process whereby RIP1 recruitment and TRAF2-dependent ubiquitination occurs first and provides a platform for subsequent NEMO recruitment and TRAF6-dependent ubiquitination (Figure 3). By working together in this fashion, the tethered API2 and MALT1 moieties appear to provide an especially potent activating signal for the IKK complex.
To fully understand this complex process of API2-MALT1 driven substrate ubiquitination, it is also important to identify the essential E2 ubiquitin conjugating enzymes that are involved. Originally, API2-MALT1 expression was shown to promote deregulated ubiquitination of NEMO and NF-κB activation in a manner that depended on the E2 enzyme, ubiquitin conjugating enzyme 13 (Ubc13)[25]. This is analogous to the situation for wild-type MALT1, whereby CBM-dependent NF-κB activation in response to antigen receptor engagement requires both Ubc13 and TRAF6-mediated ubiquitination of NEMO[44]. Thus, it appears that Ubc13 plays a major role in API2-MALT1-mediated canonical NF-κB activation, but additional investigation is necessary to definitively conclude this supposition and determine if additional E2 conjugating enzymes are involved. In conclusion, considerable progress has been made in understanding the elaborate mechanisms by which API2-MALT1 brokers IKK complex activation. In addition to utilizing a MALT1 centric mechanism for ubiquitinating NEMO, the fusion protein also appears to adopt and exploit a mechanism that is ordinarily used by the TNF receptor for ubiquitination of RIP1.
UNIQUE PROPERTIES OF API2-MALT1: RECRUITMENT AND CLEAVAGE OF UNIQUE PROTEOLYTIC SUBSTRATES
Recent studies have highlighted a particularly intriguing aspect of API2-MALT1 function, namely its ability to recruit and proteolytically cleave substrates that are normally not recognized as substrates by the protease domain of wild-type MALT1. Evidence suggests that this occurs because the API2 moiety provides substrate binding functionality, bringing these substrates into close proximity of the MALT1 proteolytic active site. To date, two such substrates have been identified, but we speculate that more may exist and could be revealed through detailed proteomic screens designed to capture unique API2-MALT1 binding partners.
NIK
Early studies investigating the protease dependent-function of API2-MALT1 established that optimal activation of NF-κB required the intact MALT1 protease domain of the fusion oncoprotein[23,24]. Because both canonical and non-canonical NF-κB dimers bind overlapping DNA consensus sequences[45], we reasoned that perhaps the protease activity of API2-MALT1 was preferentially more important for one or the other of these pathways. By assessing the subcellular distribution of NF-κB subunits in response to either wild-type or protease-dead API2-MALT1 expression, we found no difference in the nuclear translocation of the canonical NF-κB subunit RelA (p65); that is, protease-dead API2-MALT1 was just as capable as wild-type API2-MALT1 at promoting RelA translocation[46]. However, translocation of the non-canonical NF-κB subunit p52 was much less effective in response to expression of protease-dead API2-MALT1, and this mutant was unable to promote processing of p100 to p52. This suggested to us the possibility that API2-MALT1 protease activity may influence the initial steps of non-canonical NF-κB activation. Because the fusion oncoprotein is able to interact with components of the NF-κB inducing kinase (NIK) destruction complex, including TRAF2, we hypothesized that perhaps API2-MALT1 was sequestering and/or triggering degradation of one or more of the components in a protease-dependent manner, leading to NIK stabilization. Instead, we found that API2-MALT1 was able to directly associate with NIK via the API2 moiety, bringing it into close proximity to the MALT1 protease domain and thereby making it an unexpected cleavage target[46]. We then determined that MALT1 triggers cleavage of NIK at Arg325, creating a stable, catalytically-active C-terminal NIK fragment (Figure 4). Furthermore, we found that suppressing NIK levels in API2-MALT1-expressing B cells impaired non-canonical NF-κB activation and blocked expression of downstream targets, specifically those involved in apoptosis resistance and B-cell adhesion, sensitizing API2-MALT1-expressing cells to dexamethasone-induced cell death and blocking vascular cell adhesion molecule -1-dependent adhesion. Importantly, interrogation of primary MALT lymphoma patient samples revealed the presence of cleaved NIK and enhanced p100 processing only in API2-MALT1 translocation-positive specimens. Furthermore, gene expression analyses of two independent MALT lymphoma tumor collections established that API2-MALT1-positive tumors are enriched in expression of non-canonical NF-κB target genes[46]. NIK is thus far known as a selective cleavage substrate for API2-MALT1; wild-type MALT1 appears unable to cleave NIK, likely because MALT1 has no mechanism for recruiting NIK and therefore has limited opportunity to position it in proximity to its proteolytic active site. These results uncovered the first substrate of the MALT1 protease that is specifically targeted by the API2-MALT1 fusion oncoprotein. In addition, the results underscored how the two moieties of the fusion oncoprotein work together in gain-of-function fashion, with the API2 moiety recruiting an unnatural substrate, and the MALT1 moiety mediating enzymatic cleavage of that substrate.
Figure 4.
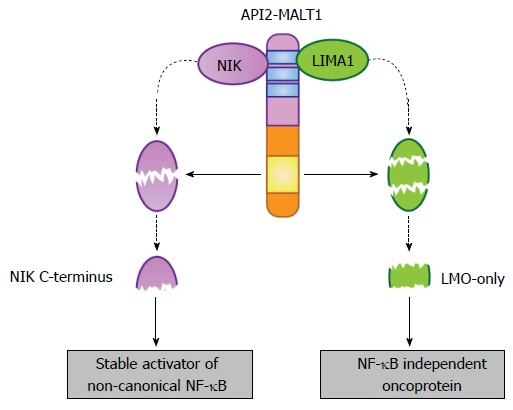
Selective substrates of the API2-mucosa-associated lymphoid tissue 1 fusion oncoprotein. The API2 moiety recruits at least two proteins (NIK and LIMA1), making them available as targets for the MALT1 protease domain (yellow box) preserved in the MALT1 moiety. In the case of NIK, site-specific cleavage frees a stable, catalytically active fragment that promotes deregulated non-canonical NF-κB activation. This fragment is stable because it lacks the C-terminal domains necessary for its recruitment to the NIK “destruction complex”. In the case of LIMA1, site-specific cleavage at two sites not only destroys the tumor suppressor function of the full-length LIMA1 protein but also liberates a central, LMO-domain only fragment that displays gain-of-function oncogenic activity. MALT: Mucosa-associated lymphoid tissue; NF-κB: Nuclear factor-κB; NIK: NF-κB inducing kinase; LIMA: LIM domain and actin-binding protein 1.
LIM domain and actin-binding protein 1
The majority of the literature on API2-MALT1 has focused on the mechanisms by which API2-MALT1 subverts cellular signaling proteins to manifest robust and sustained NF-κB activation, thereby promoting MALT lymphomagenesis. Recently, however, a novel NF-κB-independent, but MALT1 protease domain-dependent mechanism of API2-MALT1-mediated oncogenesis was revealed[47]. Mass spectrometric analysis of immunoprecipitated complexes from cells expressing either wild-type or catalytically inactive API2-MALT1 identified a novel association with the tumor suppressor, LIM domain and actin-binding protein 1 (LIMA1), also known as epithelial protein lost in neoplasm. As for NIK, the LIMA1 interaction was mapped to the API2 moiety of API2-MALT1. Also similar to the situation for NIK, the recruitment allows MALT1 protease-dependent cleavage of LIMA1, but in this case cleavage occurs in two foci, thereby releasing a central LIM-domain only (LMO) fragment (Figure 4).
Since LIMA1 acts as a tumor suppressor, its expression in B lymphocytes serves to reduce growth, proliferation, colony formation, and adhesion to extracellular matrix. Experimentally silencing LIMA1 was sufficient to reverse these effects and cause enhanced cell proliferation, colony formation, and B cell adhesion, effects that mimic API2-MALT1 over-expression[47]. Interestingly, API2-MALT1 expression was even more potent at promoting cell proliferation than was LIMA1 silencing, which led to the hypothesis that generation of the LMO cleavage fragment might unmask a pro-tumorigenic property of the LMO fragment. Indeed, engineered overexpression of the LMO cleavage fragment alone led to increased cell proliferation, colony formation, and fibronectin-mediated adhesion[47]. Thus, it appears that API2-MALT1 dependent LIMA1 cleavage serves the dual purpose of depleting a tumor suppressor from the cell while simultaneously generating an oncogenic LMO cleavage fragment.
In contrast, enforced expression of a cleavage resistant LIMA1 was sufficient to suppress API2-MALT1-dependent growth, proliferation, and colony formation. Further, cleavage resistant LIMA1 was capable of blocking API2-MALT1-dependent tumorigenesis in a murine xenograft model[47]. Importantly, the cleavage of LIMA1 was detected in API2-MALT1-positive MALT lymphoma patient samples, providing strong clinical evidence that, like NIK cleavage, this occurs in human tumors. Taken together, these studies revealed that API2-MALT1-dependent cleavage of LIMA1 contributes to the overall mechanism of action of API2-MALT1-induced lymphomagenesis, and broadens our appreciation of API2-MALT1 as a fusion oncoprotein that affects cell biology through mechanisms that go beyond NF-κB activation. Further investigation is required to determine if LIMA1 is ever targeted for cleavage by wild-type MALT1, but from what we have learned regarding NIK cleavage, we suspect that LIMA1 is only targeted via the combined actions of the API2 and MALT1 moieties. Supporting this notion, overexpression of wild-type MALT1 is insufficient to reverse the tumor suppressive effects of LIMA1.
CONCLUSION
The t(11;18) translocation, identified only in a subset of MALT lymphomas, creates the API2-MALT1 fusion oncoprotein and is associated with clinically aggressive disease. Molecular characterization of the mechanisms by which API2-MALT1 exerts its effects has revealed two general strategies of cellular protein subversion: (1) recruitment and exploitation of unique combinations of signaling proteins as a mechanism for unregulated downstream NF-κB activation; and (2) recruitment and cleavage of unnatural substrates, generating substrate fragments that possess gain-of-function oncogenic activity. Like other fusion oncoproteins (e.g., BCR-ABL, NPM-ALK) each of these mechanistic strategies requires the presence and concerted activities of both fusion partners. Fully understanding the breadth and scope of API2-MALT1-dependent interactions may inform design of targeted therapies to block associations that arm API2-MALT1 with the means to perturb cellular signaling pathways and promote lymphomagenesis.
Footnotes
Conflict-of-interest statement: The authors declare no conflicts of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: July 14, 2015
First decision: October 13, 2015
Article in press: December 8, 2015
P- Reviewer: Chi SG, Hu XT, Voulgarelis M S- Editor: Ji FF L- Editor: A E- Editor: Jiao XK
References
- 1.Sagaert X, Van Cutsem E, De Hertogh G, Geboes K, Tousseyn T. Gastric MALT lymphoma: a model of chronic inflammation-induced tumor development. Nat Rev Gastroenterol Hepatol. 2010;7:336–346. doi: 10.1038/nrgastro.2010.58. [DOI] [PubMed] [Google Scholar]
- 2.Isaacson PG, Du MQ. MALT lymphoma: from morphology to molecules. Nat Rev Cancer. 2004;4:644–653. doi: 10.1038/nrc1409. [DOI] [PubMed] [Google Scholar]
- 3.Dierlamm J, Baens M, Wlodarska I, Stefanova-Ouzounova M, Hernandez JM, Hossfeld DK, De Wolf-Peeters C, Hagemeijer A, Van den Berghe H, Marynen P. The apoptosis inhibitor gene API2 and a novel 18q gene, MLT, are recurrently rearranged in the t(11; 18)(q21; q21) associated with mucosa-associated lymphoid tissue lymphomas. Blood. 1999;93:3601–3609. [PubMed] [Google Scholar]
- 4.Akagi T, Motegi M, Tamura A, Suzuki R, Hosokawa Y, Suzuki H, Ota H, Nakamura S, Morishima Y, Taniwaki M, et al. A novel gene, MALT1 at 18q21, is involved in t(11; 18) (q21; q21) found in low-grade B-cell lymphoma of mucosa-associated lymphoid tissue. Oncogene. 1999;18:5785–5794. doi: 10.1038/sj.onc.1203018. [DOI] [PubMed] [Google Scholar]
- 5.Morgan JA, Yin Y, Borowsky AD, Kuo F, Nourmand N, Koontz JI, Reynolds C, Soreng L, Griffin CA, Graeme-Cook F, et al. Breakpoints of the t(11; 18)(q21; q21) in mucosa-associated lymphoid tissue (MALT) lymphoma lie within or near the previously undescribed gene MALT1 in chromosome 18. Cancer Res. 1999;59:6205–6213. [PubMed] [Google Scholar]
- 6.Ott G, Katzenberger T, Greiner A, Kalla J, Rosenwald A, Heinrich U, Ott MM, Müller-Hermelink HK. The t(11; 18)(q21; q21) chromosome translocation is a frequent and specific aberration in low-grade but not high-grade malignant non-Hodgkin’s lymphomas of the mucosa-associated lymphoid tissue (MALT-) type. Cancer Res. 1997;57:3944–3948. [PubMed] [Google Scholar]
- 7.Auer IA, Gascoyne RD, Connors JM, Cotter FE, Greiner TC, Sanger WG, Horsman DE. t(11; 18)(q21; q21) is the most common translocation in MALT lymphomas. Ann Oncol. 1997;8:979–985. doi: 10.1023/a:1008202303666. [DOI] [PubMed] [Google Scholar]
- 8.Liu H, Ye H, Dogan A, Ranaldi R, Hamoudi RA, Bearzi I, Isaacson PG, Du MQ. T(11; 18)(q21; q21) is associated with advanced mucosa-associated lymphoid tissue lymphoma that expresses nuclear BCL10. Blood. 2001;98:1182–1187. doi: 10.1182/blood.v98.4.1182. [DOI] [PubMed] [Google Scholar]
- 9.Liu H, Ruskon-Fourmestraux A, Lavergne-Slove A, Ye H, Molina T, Bouhnik Y, Hamoudi RA, Diss TC, Dogan A, Megraud F, et al. Resistance of t(11; 18) positive gastric mucosa-associated lymphoid tissue lymphoma to Helicobacter pylori eradication therapy. Lancet. 2001;357:39–40. doi: 10.1016/S0140-6736(00)03571-6. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Ye H, Ruskone-Fourmestraux A, De Jong D, Pileri S, Thiede C, Lavergne A, Boot H, Caletti G, Wündisch T, et al. T(11; 18) is a marker for all stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology. 2002;122:1286–1294. doi: 10.1053/gast.2002.33047. [DOI] [PubMed] [Google Scholar]
- 11.Thieblemont C, Berger F, Dumontet C, Moullet I, Bouafia F, Felman P, Salles G, Coiffier B. Mucosa-associated lymphoid tissue lymphoma is a disseminated disease in one third of 158 patients analyzed. Blood. 2000;95:802–806. [PubMed] [Google Scholar]
- 12.Rosebeck S, Rehman AO, Lucas PC, McAllister-Lucas LM. From MALT lymphoma to the CBM signalosome: three decades of discovery. Cell Cycle. 2011;10:2485–2496. doi: 10.4161/cc.10.15.16923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Afonina IS, Elton L, Carpentier I, Beyaert R. MALT1--a universal soldier: multiple strategies to ensure NF-κB activation and target gene expression. FEBS J. 2015;282:3286–3297. doi: 10.1111/febs.13325. [DOI] [PubMed] [Google Scholar]
- 14.Deveraux QL, Reed JC. IAP family proteins--suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 15.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, Durkin J, Gillard JW, Jaquith JB, Morris SJ, Barker PA. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 16.Oeckinghaus A, Ghosh S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1:a000034. doi: 10.1101/cshperspect.a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oeckinghaus A, Hayden MS, Ghosh S. Crosstalk in NF-κB signaling pathways. Nat Immunol. 2011;12:695–708. doi: 10.1038/ni.2065. [DOI] [PubMed] [Google Scholar]
- 18.Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 19.Wertz IE, Dixit VM. Signaling to NF-kappaB: regulation by ubiquitination. Cold Spring Harb Perspect Biol. 2010;2:a003350. doi: 10.1101/cshperspect.a003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thome M. Multifunctional roles for MALT1 in T-cell activation. Nat Rev Immunol. 2008;8:495–500. doi: 10.1038/nri2338. [DOI] [PubMed] [Google Scholar]
- 21.Thome M. CARMA1, BCL-10 and MALT1 in lymphocyte development and activation. Nat Rev Immunol. 2004;4:348–359. doi: 10.1038/nri1352. [DOI] [PubMed] [Google Scholar]
- 22.Stoffel A, Chaurushiya M, Singh B, Levine AJ. Activation of NF-kappaB and inhibition of p53-mediated apoptosis by API2/mucosa-associated lymphoid tissue 1 fusions promote oncogenesis. Proc Natl Acad Sci USA. 2004;101:9079–9084. doi: 10.1073/pnas.0402415101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uren AG, O’Rourke K, Aravind LA, Pisabarro MT, Seshagiri S, Koonin EV, Dixit VM. Identification of paracaspases and metacaspases: two ancient families of caspase-like proteins, one of which plays a key role in MALT lymphoma. Mol Cell. 2000;6:961–967. doi: 10.1016/s1097-2765(00)00094-0. [DOI] [PubMed] [Google Scholar]
- 24.Lucas PC, Yonezumi M, Inohara N, McAllister-Lucas LM, Abazeed ME, Chen FF, Yamaoka S, Seto M, Nunez G. Bcl10 and MALT1, independent targets of chromosomal translocation in malt lymphoma, cooperate in a novel NF-kappa B signaling pathway. J Biol Chem. 2001;276:19012–19019. doi: 10.1074/jbc.M009984200. [DOI] [PubMed] [Google Scholar]
- 25.Zhou H, Du MQ, Dixit VM. Constitutive NF-kappaB activation by the t(11; 18)(q21; q21) product in MALT lymphoma is linked to deregulated ubiquitin ligase activity. Cancer Cell. 2005;7:425–431. doi: 10.1016/j.ccr.2005.04.012. [DOI] [PubMed] [Google Scholar]
- 26.Lucas PC, Kuffa P, Gu S, Kohrt D, Kim DS, Siu K, Jin X, Swenson J, McAllister-Lucas LM. A dual role for the API2 moiety in API2-MALT1-dependent NF-kappaB activation: heterotypic oligomerization and TRAF2 recruitment. Oncogene. 2007;26:5643–5654. doi: 10.1038/sj.onc.1210342. [DOI] [PubMed] [Google Scholar]
- 27.Hosokawa Y, Suzuki H, Suzuki Y, Takahashi R, Seto M. Antiapoptotic function of apoptosis inhibitor 2-MALT1 fusion protein involved in t(11; 18)(q21; q21) mucosa-associated lymphoid tissue lymphoma. Cancer Res. 2004;64:3452–3457. doi: 10.1158/0008-5472.CAN-03-3677. [DOI] [PubMed] [Google Scholar]
- 28.Silke J, Vucic D. IAP family of cell death and signaling regulators. Methods Enzymol. 2014;545:35–65. doi: 10.1016/B978-0-12-801430-1.00002-0. [DOI] [PubMed] [Google Scholar]
- 29.Hu S, Du MQ, Park SM, Alcivar A, Qu L, Gupta S, Tang J, Baens M, Ye H, Lee TH, et al. cIAP2 is a ubiquitin protein ligase for BCL10 and is dysregulated in mucosa-associated lymphoid tissue lymphomas. J Clin Invest. 2006;116:174–181. doi: 10.1172/JCI25641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Samuel T, Welsh K, Lober T, Togo SH, Zapata JM, Reed JC. Distinct BIR domains of cIAP1 mediate binding to and ubiquitination of tumor necrosis factor receptor-associated factor 2 and second mitochondrial activator of caspases. J Biol Chem. 2006;281:1080–1090. doi: 10.1074/jbc.M509381200. [DOI] [PubMed] [Google Scholar]
- 31.Vince JE, Pantaki D, Feltham R, Mace PD, Cordier SM, Schmukle AC, Davidson AJ, Callus BA, Wong WW, Gentle IE, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284:35906–35915. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mahoney DJ, Cheung HH, Mrad RL, Plenchette S, Simard C, Enwere E, Arora V, Mak TW, Lacasse EC, Waring J, et al. Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc Natl Acad Sci USA. 2008;105:11778–11783. doi: 10.1073/pnas.0711122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varfolomeev E, Goncharov T, Fedorova AV, Dynek JN, Zobel K, Deshayes K, Fairbrother WJ, Vucic D. c-IAP1 and c-IAP2 are critical mediators of tumor necrosis factor alpha (TNFalpha)-induced NF-kappaB activation. J Biol Chem. 2008;283:24295–24299. doi: 10.1074/jbc.C800128200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 35.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 36.Alvarez SE, Harikumar KB, Hait NC, Allegood J, Strub GM, Kim EY, Maceyka M, Jiang H, Luo C, Kordula T, et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature. 2010;465:1084–1088. doi: 10.1038/nature09128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Justus SJ, Ting AT. Cloaked in ubiquitin, a killer hides in plain sight: the molecular regulation of RIPK1. Immunol Rev. 2015;266:145–160. doi: 10.1111/imr.12304. [DOI] [PubMed] [Google Scholar]
- 38.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 39.Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281:13636–13643. doi: 10.1074/jbc.M600620200. [DOI] [PubMed] [Google Scholar]
- 40.Wu CJ, Conze DB, Li T, Srinivasula SM, Ashwell JD. Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected] Nat Cell Biol. 2006;8:398–406. doi: 10.1038/ncb1384. [DOI] [PubMed] [Google Scholar]
- 41.O’Donnell MA, Ting AT. NFκB and ubiquitination: partners in disarming RIPK1-mediated cell death. Immunol Res. 2012;54:214–226. doi: 10.1007/s12026-012-8321-7. [DOI] [PubMed] [Google Scholar]
- 42.Rosebeck S, Rehman AO, Apel IJ, Kohrt D, Appert A, O’Donnell MA, Ting AT, Du MQ, Baens M, Lucas PC, et al. The API2-MALT1 fusion exploits TNFR pathway-associated RIP1 ubiquitination to promote oncogenic NF-κB signaling. Oncogene. 2014;33:2520–2530. doi: 10.1038/onc.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Noels H, van Loo G, Hagens S, Broeckx V, Beyaert R, Marynen P, Baens M. A Novel TRAF6 binding site in MALT1 defines distinct mechanisms of NF-kappaB activation by API2middle dotMALT1 fusions. J Biol Chem. 2007;282:10180–10189. doi: 10.1074/jbc.M611038200. [DOI] [PubMed] [Google Scholar]
- 44.Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ. The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell. 2004;14:289–301. doi: 10.1016/s1097-2765(04)00236-9. [DOI] [PubMed] [Google Scholar]
- 45.Siggers T, Chang AB, Teixeira A, Wong D, Williams KJ, Ahmed B, Ragoussis J, Udalova IA, Smale ST, Bulyk ML. Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat Immunol. 2012;13:95–102. doi: 10.1038/ni.2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rosebeck S, Madden L, Jin X, Gu S, Apel IJ, Appert A, Hamoudi RA, Noels H, Sagaert X, Van Loo P, et al. Cleavage of NIK by the API2-MALT1 fusion oncoprotein leads to noncanonical NF-kappaB activation. Science. 2011;331:468–472. doi: 10.1126/science.1198946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nie Z, Du MQ, McAllister-Lucas LM, Lucas PC, Bailey NG, Hogaboam CM, Lim MS, Elenitoba-Johnson KS. Conversion of the LIMA1 tumour suppressor into an oncogenic LMO-like protein by API2-MALT1 in MALT lymphoma. Nat Commun. 2015;6:5908. doi: 10.1038/ncomms6908. [DOI] [PubMed] [Google Scholar]