

Abstract
The renal handling of Na+ balance is a major determinant of the blood pressure (BP) level. The inability of the kidney to excrete the daily load of Na+ represents the primary cause of chronic hypertension. Among the different segments that constitute the nephron, those present in the distal part (i.e., the cortical thick ascending limb, the distal convoluted tubule, the connecting and collecting tubules) play a central role in the fine-tuning of renal Na+ excretion and are the target of many different regulatory processes that modulate Na+ retention more or less efficiently. G-protein coupled receptors (GPCRs) are crucially involved in this regulation and could represent efficient pharmacological targets to control BP levels. In this review, we describe both classical and novel GPCR-dependent regulatory systems that have been shown to modulate renal Na+ absorption in the distal nephron. In addition to the multiplicity of the GPCR that regulate Na+ excretion, this review also highlights the complexity of these different pathways, and the connections between them.
Keywords: Kidney, Sodium excretion, Blood pressure, G-protein coupled receptors, Peptide hormone
Core tip: The maintenance of the blood pressure depends partly on the ability of the organism to match the daily intake and excretion of Na+. The kidney, which is the main organ involved in Na+ excretion, is the target of multiple regulatory pathways that contribute to the fine-tuning of secretion/reabsorption processes occurring all along the nephron. In this review we described “classical” and “novel” G-protein coupled receptor (GPCR)-mediated pathways that impact trans-epithelial Na+ transport in the distal nephron. This detailed inventory of the GPCR-mediated pathways that affect renal Na+ handling gives a broad overview of the complexity of this integrated system.
INTRODUCTION
Sodium is the main osmotic component of extracellular compartments and its concentration (140 mmol/L) must be kept within a narrow range to maintain extracellular volume and blood pressure (BP) at a normal levels. According to Guyton’s model, dysfunctional renal sodium excretion is involved in the development of salt-sensitive hypertension[1]. Daily, Na+ consumption may varies between 50 to 150 mmol and water consumption is about 1.5 L. Nearly all the Na+ and water are transferred to the extracellular compartment, which may induce changes in the circulating volume if Na+ and water are not correctly excreted. The kidneys regulate the balance of Na+ and water balances (extra-renal losses are negligible under normal circumstances). In humans, about 25000 mmoles of Na+ in 180 L of fluid are delivered daily to the glomerular filtrate. These very large amounts of Na+ and fluid are almost entirely reabsorbed by the kidney, as urinary excretion is calibrated to match dietary intake. The bulk of the filtered load of sodium (around 60%) is reabsorbed in the proximal tubule; 25% is reabsorbed by the thick ascending limb (TAL); approximately 5% to 7% is reabsorbed along the distal convoluted tubule (DCT), and 3% to 5% along the connecting tubule (CNT) and the collecting duct (CD); the latter is composed of a cortical segment (CCD), an outer medullary part and an inner medullary part (IMCD). The distal part of the nephron as defined in this review encompasses the segments from the cortical TAL (cTAL) to the IMCD since these are all involved in the fine-tuning Na+ excretion. In these segments are found all sodium transporters in which mutations induce alterations of systemic BP. Not surprisingly, numerous hormones regulate the transport of Na+ in these segments. These regulatory systems involve the activation of various receptors among which the G-protein coupled receptors (GPCRs) represent an important component.
In this review we 1/rapidly summarize the mechanisms of sodium transport in the distal nephron and 2/describe the well-established regulatory mechanisms mediated by “classical” GPCRs as well as more recently described systems involving “novel” GPCRs.
SODIUM TRANSPORT MECHANISMS IN THE DISTAL NEPHRON
Transepithelial sodium reabsorption can take place either accoss tubular epithelial cells (transcellular route) or between cells (paracellular pathway). Transcellular Na+ reabsorption requires the successive crossing of the apical and basolateral membranes. At the basolateral membrane, the Na, K-ATPase generates the electro-chemical gradient that drives Na+ entry at the apical membrane via different channels, cotransporters or exchangers. The specificity of ion transport in different nephron segments largely stems from the nature of the Na+-coupled transport systems, that are expressed at the apical membrane (Figure 1). Transcellular transport generates a transepithelial potential difference, which, can drive the paracellular transport of solutes and/or water depending on the water and ion permeability of intercellular junctions (for review see[2,3]).
Figure 1.
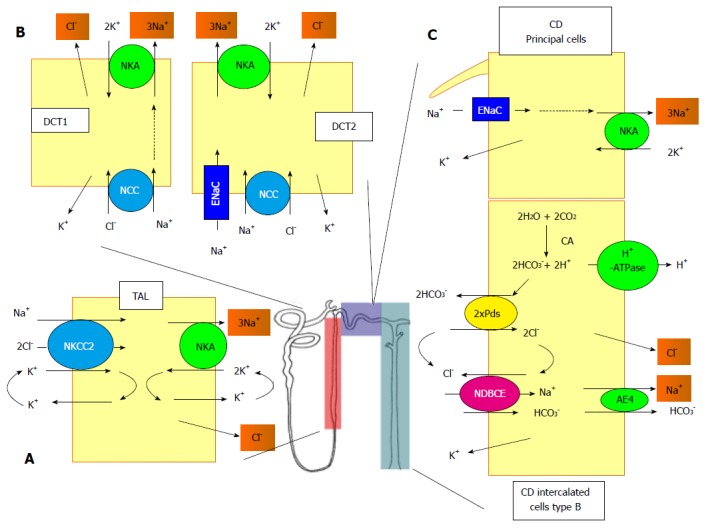
Schematic representation of the different sodium transport systems along the nephron. A: In TAL, the apical entry of Na+ is mediated by the NKCC2. At the basolateral side, the Na+ exits the cell through the NKA and the Cl- through a channel of the CLC-K family; B: DCT consists of two different sub-structures, the DCT1 and the DCT2. In DCT1, the entry of Na+ and Cl- is mediated by the NCC and in DCT2, a ENaC is also involved. In both structures, the exit of Na+ and Cl- is mediated by the NKA and a CLC-K conductance; C: In the CD, the Na+ enters the principal cells through ENaC and exits through the NKA. The system is more complex in the B-intercalated cells. The apical entry of of Na+ and Cl- is mediated by the functional association between the pendrin, bicarbonate/Cl- exchanger (Pds), and NDBCE. The system is energized by the vacuolar proton pump (H+-ATPase) and the generation of bicarbonate mediated by a CA. The exit of Na+ and Cl- is mediated by an AE4 and a Cl--conductance, respectively. TAL: Thick ascending limb; NKCC2: Na+/K+/2Cl--cotransporter; NKA: Na,K-ATPase; DCT: Distal convoluted tubule; NCC: Na+/Cl--cotransporter; ENaC: Na+ channel; CD: collecting duct; Pds: Bicarbonate/Cl- exchanger; CA: Carbonic anhydrase; NDBCE: Na+-driven bicarbonate/Cl- exchanger; AE4: Anion exchanger.
Transport mechanisms in the cTAL
The TAL is a tight epithelium that actively reabsorbs about 25% of filtered NaCl. The apical entry of Na+ is mainly mediated by the electroneutral cotransporter Na/K/2Cl (NKCC2), which is sensitive to furosemide (Figure 1). The transcellular reabsorption of Na+ in this segment is coupled to the recycling of K+ at the apical membrane through a K+ channel (ROMK) and the reabsorption of Cl- at the basolateral membrane through Cl- channels (CLCKb). Altogether, these moves generate a positive transepithelial potential difference (PDte) of about +15 mV, that allows the reabsorption of monovalent and divalent cations through the paracellular pathway. The tight junctions of TAL cells are poorly permeable to anions, which limits chloride back-flux through the paracellular route[3]. Moreover, the transport of Na+, K+ and Cl- in the TAL cells is coupled and interdependent: when one of the carriers (NKCC2, ROMK or CLCKb) is inhibited or carries a loss-of-function mutation, transport across other transcellular and paracellular pathways is impaired. In humans, these mutations are responsible for Bartter syndrome, an inherited disease characterized by excessive loss of Na+, Cl-, water and accompanied by an abnormally high urinary excretion of Mg2+ and Ca2+[4]. Moreover, certain loss-of-function mutations of NKCC2 have been shown to protect against the development of hypertension[5].
Transport mechanism in the DCT
The DCT retain 5% to 10% of the filtered NaCl, have tight waterproof junctions and reabsorb Ca2+ and Mg2+ through the transcellular pathway. In mice and rats, the DCT is divided into two parts, each of which expresses different apical Na+ transporters the DCT1, expresses only the apical NaCl cotransporter NCC. As for the DCT2, it expresses both NCC and the epithelial sodium channel ENaC. As opposed to the rodent DCT, the human DCT is uniform; it has the same expression profile as the murine DCT1[6]. Sodium reabsorption through NCC is electroneutral and sensitive to thiazide diuretics such as hydrochlorothiazide (HCTZ), whereas sodium reabsorption through ENaC is electrogenic and sensitive to amiloride.
In humans, NCC mutations inducing loss of function are responsible for Gitelman syndrome, which features hypokalemic metabolic alkalosis, hypomagnesemia and hypocalciuria[7]. In contrast, “gain-of-function” mutations of kinases regulating NCC induce an increase in NaCl reabsorption and hypertension, as encountered in Gordon’s hypertension syndrome[8].
Transport mechanisms in CNT and CD
Unlike upstream nephron segments, the epithelia of the CNT and the CD are made of two cell types, known as principal cells (PC) and intercalated cells (IC) (Figure 1) with specific and different functions. Until recently, the reabsorption of Na+ was attributed to PC whereas IC were thought to be involved in acid/base excretion. This dogma has been revisited these last few years, as discussed below.
Transport mechanisms in the PC: PC constitute about 60% of CNT/CCD cells and express ENaC at their apical membrane. Na+ reabsorption across PC is electrogenic and necessary for K+ secretion (Figure 1). The terminal part of the CNT and the CCD can reabsorb water owing to the osmotic gradient generated in part by the tubular fluid dilution that occurs in the TAL as well as Na+ reabsorption in the CNT/CCD through ENaC. The transcellular reabsorption of water is mediated by apical aquaporin 2 channels (AQP2) and the basolateral aquaporin 3 and 4 channels (AQP3 and AQP4). It is generally accepted that there is a decreasing longitudinal gradient of Na+ reabsorption and K+ secretion along the CNT and CCD. Under basal condition, in in vitro microperfusion experiments, rodent CCDs do not exhibit significant net Na+ or K+ transport[9,10]. This does not mean that there is no functional ENaC or ROMK at the apical surface of PC in the CCD. In fact, both channels have been found to be active by patch clamp experiments[11,12]. The channel activity is either not sufficient to trigger a net flux that can be measured by in vitro microperfusion experiments or is compensated by secretion processes that remain to characterized. Regarding chloride transport, it has to be noted that such a secretion pathway has been partially described; it involves the basolateral transporter NKCC1 localized in the B-type intercalated B cells of the CCD[13]. Finally, significant net fluxes of Na+, Cl- and K+ are measured in rat or mouse in vitro perfused CCD when animals are either submitted to a sodium-depleted diet or chronically treated with corticosteroids[9,10]. Therefore the CNT/CCD represents a functional “transport” reserve that can be recruited when Na+ reabsorption (hypovolemia) and/or K+ secretion (hyperkalemia) is needed[9,10].
In humans, a gain-of-function mutation of ENaC induces a constitutively active Na+ reabsorption in CNT and CD[14]. The continual passage of Na+ through the apical membrane then drives excessive K+ secretion. Thus, patients with this mutation (which causes Liddle’s syndrome) develop severe early hypertension and hypokalemia[15]. Conversely, a loss-of-function mutation of the human ENaCα-subunit results in pseudo-hypo-aldosteronism 1 and is associated with urinary Na+ wasting, low BP (at least in children) and hyperkalemia[16].
Transport mechanisms in the type B IC: Based upon immuno-histochemical studies, there are at least four types of IC: Type A, type B, type non A-non B cells and bipolar cells. Types A (A-IC) and type B (B-IC) cells are traditionally involved in the regulation of acid/base balance. However, recent results demonstrate that B-IC play a role in the reabsorption of Na+ and Cl-[10]. These cells, which are found only in the CNT and the CCD and express the H+-ATPase pumps and Cl- channels (CLCK) at the basolateral membrane and the Cl-/HCO3- exchanger pendrin (PDS) at the apical membrane. B-IC are known to secrete a high quantity of bicarbonate in response to metabolic alkalosis. Moreover, they are the only cells that reabsorb Cl- in the CNT/CCD. Even though the CNT and CCD do not express NCC, these two segments can mediate electroneutral, thiazide-sensitive sodium chloride absorption. Recently, Leviel et al[10], demonstrated a functional coupling between pendrin and a Na+-driven Cl-/HCO3- exchanger (NDCBE), using transgenic mice models and in vitro microperfusion experients. This coupling results in net and electroneutral NaCl reabsorption in the CCD (Figure 1). More recently, Chambrey et al[17] showed that this system is not energized by Na, K-ATPase as it is the case in most cells but by the H+-ATPase. Na+ enters B-ICs via NDCBE and exits through the basolateral anion exchanger AE4, which is specifically expressed in this cell type[18].
SODIUM TRANSPORT REGULATION IN THE DISTAL NEPHRON BY CLASSICAL GPCRS
In the distal nephron, the sodium transport is subject to numerous regulatory factors (endocrine, neuroendocrine and paracrine) that adjust the excretion of the cation to homeostatic needs, maintaining constant blood volume and BP. Among the different regulatory systems orchestrating the fine-tuning of the Na+ balance, those that entails the stimulation of GPCR are numerous. Some of them have been known for many years to interfere with Na+ reabsorption and their ligands are “classical” hormones of the Na+ homeostatic system. Thus, it is well known that angiotensin 2 and vasopressin have a stimulatory effect on sodium absorption all along the nephron through their respective GPCRs, whereas bradykinin, dopamine, endothelin, and PGE2 down-regulate sodium absorption (for review see[2]). More recently, novel pathways regulating Na+ absorption have been reported. They involve new types of ligands (such as proteases, lipids, and small metabolites), and receptors that have not all been fully characterized. In the following part, we will describe both “classical” and “novel” regulatory pathways of the Na+ reabsorption in the distal nephron (Table 1).
Table 1.
Recapitulation of the G-protein coupled receptors families present in the the distal nephron
| Receptors | G-proteins | Cell location | Final effector | Effect on Na+ transport |
| Vasopressin receptor (V2R) | Gs | Basolateral | Na, K-ATPase - AQP2 - NKCC2 - NCC - ENaC | Increase |
| Angiotensin II receptors (AT) | Gq/11 | Apical/basolateral | Na, K-ATPase - NKCC2 - NCC - ENaC - pendrin | Increase |
| Bradykinin receptor (B2R) | Gq | Basolateral | Na, K-ATPase - NKCC2 - NCC - ENaC (??) | Decrease |
| Endothelin receptors (ETA-ETB) | Gi, Gq, Gs, Gα12/13 | Apical/basolateral | NKCC2 - ENaC - pendrin | Decrease |
| α-adrenergic receptor (α-AR) | Gi - Gq/11 | Basolateral | Na, K-ATPase (?) - NKCC2 - ENaC | Increase (α1-AR) |
| Decrease (α2-AR) | ||||
| β-adrenergic receptor (β-AR) | Gs | Apical/basolateral | NKCC2 - NCC - ENaC - pendrin | Increase |
| Prostanoïd receptors | Gs – Gi | Apical/basolateral | NKCC2 - ENaC - pendrin | Decrease (PGE2) |
| Increase | ||||
| Dopamine receptors | Gs – Gi | Basolateral | Na, K-ATPase | Decrease |
| Proteinase-activated receptor (PAR) | Gq/11 | Basolateral | Na, K-ATPase - NKCC2 - ENaC - pendrin/NDCBE | Increase |
| Sphingosine-1 phosphate receptor | G12/13 - Gi- Gq | ? | ENaC | Decrease |
| Nucleotide-like receptors (P2X-P2Y) | Gq (P2X) Gi (P2Y) | Apical/basolateral | NKCC2 (?) - NCC - ENaC | Decrease |
| Oxaloacetate receptor (OXGR1) | Gq | Apical | Pendrin/NDCBE | Increase |
NDBCE: Na+-driven bicarbonate/Cl- exchanger; OXGR1: 2-oxoglutarate receptor 1; NKCC2: Na/K/2Cl.
“Classical” GPCRs that activates sodium absorption in the distal nephron
Vasopressin receptor V2: Vasopressin (AVP) is a peptide secreted by the hypothalamic nuclei cells and stored in the posterior pituitary. It is released into the blood stream under dehydration and cellular volume shrinkage. AVP acts on three subtypes of GPCRs: V1 (ex V1a), V2 and V3 (ex V1b). In the kidney, AVP triggers an increase in water and/or sodium reabsorption through V2 receptors. V2 receptors are located at the basolateral cell membrane of the ascending limb and DCT segments. In the CD, they are specifically present in PC[19-21]. In all these cells, the V2 receptor is coupled to the Gs protein, which rapidly activates adenylyl cyclase (AC6 and AC3 isoforms, chiefly), increases the formation of cAMP and induces the activation of PKA[22]. By this way, the activation of V2 receptor stimulates numerous Na+ transporters by phosphorylation. For instance, the Na, K-ATPase activity is enhanced by PKA activation[23]. In TAL and DCT cells, NKCC2 and NCC are also targets of PKA, which increases their membrane expression after AVP activation[24,25]. The stimulation of NKCC2 and NCC by AVP also involved intermediary kinases, such as STE20/SPS1-related proline-alanine-rich kinase (SPAK) and oxidative stress responsive kinase 1 (OSR1), and may be not solely due to a direct action of PKA on the transporters[26]. Recent studies have shown that AVP increases the abundance of phosphorylated SPAK and phosphorylated OSR1 in mTAL and DCT cells[21,27]. Thus, both SPAK and OSR1 could be important vasopressin second messengers. Interestingly, using SPAK-/- mice, Cheng et al[28] showed that SPAK was important for NKCC2 activity in both mTAL and cTAL cells but was not essential for vasopressin-induced stimulation of sodium reabsorption. In the CCD, AVP activation of V2 elicits the phosphorylation of aquaporin-2 (AQP2) leading to its translocation to the apical membrane[22]. It also increases Na+ reabsorption by activating ENaC[29,30], as PKA phosphorylation leads to the inhibition of Nedd4-2, the ubiquitin ligase that promotes the endocytosis of ENaC[31,32]. Sodium reabsorption across the CD epithelium is essential for water absorption through aquaporins[33].
Angiotensin II receptor AT1: Angiotensin II (AngII) is a hormone peptide of the renin-angiotensin-aldosterone system (RAAS) that regulates BP and other physiological processes (for review see[34]). Circulating AngII is produced by the proteolytic cleavage of angiotensinogen (which is continously produced by the liver) and involves two steps: (1) the production of angiotensin I by renin, which is released by juxtaglomerular apparatus cells; and (2) the conversion of AngI into AngII by the angiotensin converting enzyme (ACE) that is produced by the lungs. Renin is the rate-limiting factor that determines the levels of circulating AngII such that the production and release of AngII respond rapidly to hypovolemia[34]. AngII can also be produced in various tissues like the kidneys and have different effects than systemic AngII (for review see[2]).
The blood level of AngII is increased in some form of hypertension where it is involved in its development (see review[35]).
AngII binds to 2 subtypes of GPCRs called AT1 (AT1a and AT1b in rodents) and AT2. AT1 isoforms are encoded by homologous genes, have the same affinity to different ligands, trigger the same signaling pathways and are located all along the distal nephron but AT1a is the predominant renal form. AT1 receptors are expressed at both apical and basolateral membrane[36-38] and mediate the known physiological and pathological actions of AngII. These receptors are coupled to Gq/11, G12/13 and Gi/o proteins in rodents[39]. The main pathway triggered by AngII involved Gq/11-mediated inositol phosphate/Ca2+ signaling. In the cTAL, AT1a receptors activate both the PLC/PKC/ERK and protein tyrosine kinase (PTK) Src pathways thereby enhancing calcium signaling and an increase in the activity of NKCC2[40,41]. Here too, the intermediary action of SPAK has also been proposed since AngII infusion increased abundance of the phosphorylated forms of SPAK and NKCC2[42].
In the DCT, AngII increases sodium reabsorption in both the DCT1 and DCT2 by binding to its AT1 receptor. Recent experiments have shown that the AT1a receptor induces an increase in NCC phosphorylation, abundance at the apical membrane and activity through the activation of WNK4 (with no lysine kinase 4), SPAK, and OSR1[41,43].
Both the apical and basolateral addition of AngII can induce an increase in sodium reabsorption through ENaC in the rabbit CCD[44]. Using patch clamp on split open CCD, Mamenko et al[45], showed that AngII activation of AT1 receptors had a double effect on ENaC: A rapid increase in the open probability of ENaC and a slower increase in the abundance of ENaC at the apical membrane. Interestingly, AT1 receptor-induced activation of ENaC was not dependent on calcium signaling in these cells. Instead, the AT1 receptor activated a Ca2+-independent PKC and triggered the NADPH oxidase pathway[45] (see for review[35]).
In the type B IC, activation of AT1 receptors enhances Cl- uptake by pendrin[46]. Pendrin activation and the subsequent increase in Cl- reabsorption are necessary for the full pressure effects of AngII since pendrin knockout mice exhibit an attenuated BP response to this peptide hormone[46,47]. The signaling pathways between AT1 receptors and pendrin remain to be discovered, and AT1 receptors capacity to induce sodium reabsorption through NDBCE remains unknown. In summary, through the activation of AT1 receptors, AngII induces an increase in Na+ reabsorption all along the distal nephron by activating apical Na+ entry through NKCC2, NCC, ENaC and the pendrin/NDCBE functional complex[35].
Catecholamine receptors: Catecholamines [adrenaline, also called epinephrine (E), and noradrenaline, also called norepinephrine (NE)] mediate the sympathetic regulation of BP[48]. E and NE are synthetized by neurons and the adrenal gland[49]. Both cathecholamines act by binding to 9 different subtypes of adrenergic receptors (ARs) that belong to 3 different groups (α1-AR, α2-AR or β-AR). All three classes of receptors contribute to the control of BP by acting on the central nervous system (CNS), the cardio-vascular system, and the kidney. In the latter organ, all segments of the nephron as well as juxtaglomerular granular cells are in contact with sympathetic nerve terminal extremities releasing noradrenaline[50]. Renal nerve activation enhances sodium reabsorption all along the nephron and promotes renin secretion. Conversely, renal denervation inhibits sodium retention and renin secretion (for review, see[51]).
a/α1-AR and α2-AR: Three molecular species of receptors α1-AR have been cloned: α1A-AR, α1B-AR, and α1D-AR[52]. In the TAL only α1B-AR and α1D-AR are expressed, and they are coupled to Gαq/11 proteins that activate PLC-β[53,54] and trigger the IP3/DAG/PKC pathway[55,56]. In the CNT/CCD, all three α1A-AR are expressed[57] where their effects have not been determined.
The α2-AR receptors (α2A-AR, α2B-AR, and α2C-AR)[52], are coupled to Gi proteins and inhibit the formation of cAMP[58-61]. In the distal nephron, α2-AR is expressed in the TAL and CD with a higher abundance in the medulla[62]. In the rat cTAL, α2-AR activation has recently been shown to induce NO production via a PI3K/NOS pathway and to inhibit chloride absorption[63]. In the rat CCD, α2-AR activation inhibits AVP-induced cAMP production[64,65] and blunts the AVP-dependent stimulation of Na+ and water transport[66].
In the CCD, the activation of the α2A- and/or α2C-receptors by catecholamines has a dose-dependent effect on ENaC activity[67]. At low concentrations, NE triggers the inhibition of Na+ and water transport, whereas higher concentrations have the opposite effects. Since renal NE concentrations have never been measured precisely, one can only speculate that depending on the intensity of sympathetic activity, sodium transport could be either activated or inhibited along the nephron.
b/β-AR: Three subtypes of the β-AR receptors are known (β1-AR, β2-AR and β3-AR). In the distal nephron, β1-AR is expressed in the cTAL, at the apical pole of DCT cells, in all CNT cell types and preferentially in the IC of the CCD[68,69]. Using β1-AR or β2-AR knock-out mice models, Mu et al[70] demonstrated that the activation of β2-AR by isoproterenol but not that of β1-AR induces salt-sensitive hypertension. β2-AR is expressed in the DCT and in the CD where its expression increases along the cortico-medullar axis[71]. No staining of β2-AR has been found in the rat TAL[71]. β-AR receptors are coupled to GS, they stimulate cAMP production by the adenylyl cyclase and induce the activation of PKA[72]. In the cTAL, β-AR activation elicits an increase in the expression of NKCC2 at the apical membrane[73]. This increase is dependent on cAMP formation and PKA activation[73].
Using β1-AR knockout mice and β2-AR knockout mice, Mu et al[70] showed that in DCT NE induces an increase in NCC expression and phosphorylation through the activation of β2-AR. Terker et al[74], recently demonstrated that β-AR activation enhances NCC phosphorylation through the activation of oxidative stress-response kinase 1 OSR1 rather than through SPAK.
In the CCD, we found that β-AR activation leads to increased pendrin activity by triggering its translocation to the cell surface[75]. Furthermore, application of isoproterenol on in vitro microperfused mice CCD elicited rapid NaCl reabsorption. These results suggest that the CCD participates in adrenergic-dependent Na+ retention. The finding that β-AR activation enhances electroneutral Na+ reabsorption in both the DCT and CD is consistent with the results of Mu et al[70] showing that HCTZ, a specific inhibitor of NCC in the DCT and of the electroneutral pathway in the CNT/CCD, blunts the BP elevation observed under isoproterenol infusion and salt loading. Interestingly, in a more distal segment of the CD (i.e., the IMCD) β2-AR stimulation activates sodium, chloride and fluid secretion through a cAMP dependent pathway[76]. To our knowledge, the underlying mechanisms remain unknown.
Altogether, these studies show that renal sympathetic nerves can deliver NE to the basolateral membrane of distal tubule cells. Interestingly, depending on the concentration of NE and on the type of receptors (α-AR or β-AR) that are activated, the sympathetic nervous system could increase sodium absorption either in all distal tubule cells or preferentially in a few cell types. More experiments are needed to define the exact mechanisms by which of NE acts in the distal nephron. Regarding all the SNS effects on kidney, it is now evident that renal sympathetic nerves play an important role in sodium balance (for review see[77]).
Indeed, increased salt intake stimulates sympathetic activity in the kidney and in brain,. Moreover, the renal NE metabolism is modified in hypertensive rats[78] as well as in in obese patients with hypertension[79]. Renal denervation has been shown to prevent or attenuate hypertension in a wide variety of animal models of hypertension (for review see[51]). In humans, following successful clinical trials, the techniques of renal nerve ablation are now spreading in Europe[80]. However, recent clinical investigations on randomized renal denervation trials known as “symplicity HTN-3” have shown that renal denervation has limited efficacy[81,82]. These investigations, however, are subject to criticism and more studies will be needed to assess the benefits of renal denervation[83].
“Classical” GPCR that inhibits sodium absorption in the distal nephron
Bradykinin B2 receptor: Bradykinins are autocrine and paracrine hormones that are generated in different tissues by the cleavage of kininogen by kallikrein (for review see[84]). Bradykinins, via their actions on the heart, blood vessels, and kidneys, play an important role in cardiovascular homeostasis (reviewed in[85]). Along with bradykinin receptors B1 and B2 (which are two GPCRs), bradykinins and kallikrein form the kinin/kallilrein system (KKS).
In the kidney, all components of the KKS are produced in the distal nephron[86-88]. Kallikrein is a serine protease that is produced by DCT and CNT cells and secreted into the tubular lumen and extracellular space. Kininogen is produced by all distal nephron cells, which all express B2 receptors at their basolateral membrane
In vitro experiments on rodent CCDs showed that kallikrein activates ENaC by cleaving its regulatory subunit γ[89] and that it directly inhibits the H,K-ATPase type 2[90]. Therefore, kallikrein that is secreted apically could simultaneously activate ENaC and inhibit the H, K-ATPase type 2[89,90]. Kallikrein that is secreted on the basolateral side is thought to act through bradykinin formation and B2 receptors coupled to Gq proteins. In the cTAL, B2 activation in turn activates the MAPK/ERK1,2 signaling pathway and increases intracellular calcium concentrations like AT1 activation[40]. Surprisingly, NaCl reabsorption is inhibited following B2 receptor activation but enhanced following AT1 receptor activation[40]. The molecular mechanisms that allow the same transduction pathway to yield two opposite responses need to be determined in this segment. In CCDs perfused in vitro from DOCA treated rat, activation of B2 receptor sby 10-9 mol/L bradykinin inhibits the electroneutral and thiazide sensitive NaCl reabsorption[91]. Meanwhile, neither the transepithelial potential, nor the K+ secretion is inhibited by B2 receptor activation, which clearly indicates that ENaC activity is not diminished under these conditions[91]. A higher concentration of bradykinin (10-7 mol/L) has been shown to lower the open probability (Po) of ENaC via the activation of the B2/Gq/11/PLC pathway[92]. A role of B2 receptors in inhibiting salt reabsorption is confirmed by the phenotype of B2 knockout mice, which exhibit salt-sensitive hypertension[93].
KKS is a complex system having the ability to either stimulate or to inhibit Na+ reabsorption. To understand these opposite effects, it is necessary to consider the physiological context in which this system is triggered. For instance, KKS is known to be strongly upregulated by a high dietary K+ intake, a condition under which the levels of AngII and renin are low. The overall action of KKS in the distal nephron would then favor K+ secretion and limit excessive Na+ retention: Firstly, the inhibition of Na+ reabsorption in the cTAL would increase the delivery of fluid and Na+ to the distal nephron; secondly, the direct role of luminal kallikrein on ENaC and the H, K-ATPase would favor K+ secretion by stimulating its excretion (through both ROMK and MaxiK channels) and inhibiting its retention. In other situations, for instance when salt intake is increased, the stimulated KKS stimulation would inhibit the electroneutral (at low concentrations of bradykinin) or the electrogenic (at high concentration of bradykinin) Na+ reabsorption pathways. This may explain why the absence of one component of the KKS induces salt-sensitive hypertension (for review see[94]). More investigations are needed to understand how the renal effects of KKS are orchestrated to favor either K+ secretion or Na+ secretion, two apparently antagonistic situations.
Endothelin receptors ETA and ETB: Endothelin 1 (ET-1) is an endothelial cell-derived peptide that is the most potent vasoconstrictor of the body[95]. ET-1 plays important roles in the physiology and physiopathology of all organs and is an important factor in the control of sodium homeostasis and BP (reviewed by[96]). There are three different endothelins (ET-1, ET-2 and ET-3), the first of which, (ET-1) has been the most studied. The plasma levels of ET-1 are lower than its tissue levels. Moreover, ET-1 that is produced by renal tubular cells is essentially secreted at the basolateral pole. This is why ET-1 is considered to be a paracrine/autocrine hormone.
ET-1 binds to two GPCRs known as the ETA and ETB receptors[97,98]. Almost all cells express ETA and/or ETB receptors: In the nephron, ETB is the predominant endothelin receptor. These receptors are coupled to many different G proteins: Gi, Gq, Gs, and Gα12/13[99,100].
In the rat cTAL, ET-1 activates basolateral ETB receptors, which triggers the production of NO and the subsequent inhibition of NKCC2[101,102], leading to a reduction in transepithelial Cl- and Na+ fluxes. However, ET-1 does not inhibit the AVP-stimulated cAMP accumulation in the rat cTAL[103,104]. To our knowledge, the roles of ET-1 and ETB in the DCT have not been investigated. In contrast, the effects of ET-1 and ETB in the CD have been extensively studied. CD cells, particularly those in the inner medulla produce more ET-1 than any other nephron cell. ET-1 decreases Na+ reabsorption in the isolated CCD and the IMCD[105,106]. As opposed to finding in the cTAL (see above), ET-1 triggers the inhibition of the AVP-stimulated chloride transport in the CCD through PKC activation[105]. In addition to counteracting the effects of AVP, ET-1 also inhibits ENaC activity through both MAP kinase-dependent and NO-dependent pathways[107]. ETB activation has functional consequences on IC. Indeed, in IC-A, it activates H+-ATPase and in IC-B, it reduces pendrin activity (for review see[108]).
Interestingly, ET-1 production by CD cells seems to be regulated by the extracellular fluid volume (ECFV) status (for review see[96]). Mice with a specific deletion of the ET-1 gene in the CD are hypertensive under normal conditions and develop salt-sensitive-hypertension[96]. These observations could reflect an increase in sodium reabsorption in the CD but further experiments are needed to confirm this hypothesis. Several animal models of hypertension are, in fact, associated with increased vascular ET-1 synthesis (for review see[109]), which may be understood as a compensatory mechanism to try to reduce BP[96].
Prostanoid receptors: Prostaglandins are lipid mediators generated by cyclooxygenase (COX) that metabolizes arachidonic acid. The cellular concentration of COX-derived prostaglandins is modulated by several extracellular and intracellular factors. Prostaglandins have many effects in pain, inflammation, cell proliferation and body homeostasis. Signaling prostaglandins are PGE2, PGI2, PGF2, PGD2 and thromboxane A2 (TxA2). Prostaglandins are rapidly degraded which limits the scope of their action to autocrine and paracrine functions[110]. Prostaglandins act on nine specific prostaglandin receptors encoded by nine different genes. All are lipid-like GPCRs: Prostaglandin D2 receptor 1 and 2 (DP1 and DP2); Prostaglandin E receptor 1 to 4 (EP1-EP4); Prostaglandin F receptor (FP); Prostaglandin I2 receptor (IP) and thromboxane A2 receptor (TxA2R). The kidney expresses all four EP receptors, the FP receptor and TxA2R (for review see[111]).
In the cTAL, EP receptors that bind PGE2 have been reported to antagonize the action of AVP[112]. In this segment, EP3 is coupled to Gi and the presence of PGE2 was shown to inhibit AVP-induced cAMP accumulation induced by AVP and therefore to blunt the NaCl reabsorption[112]. Moreover, EP3 activates a Rho-kinase/PKC/ERK pathway that inhibits the basolateral potassium channels. Since potassium recycling at the basolateral membrane is necessary for Na,K-ATPase function, inhibition of these channels is sufficient to reduce Na+ reabsorption all along the cTAL[113]. As for the CNT/CD, these segments express all four EP receptors. In the rabbit CCD, the use of specific agonists and antagonists demonstrated that PGE2 activation of EP1 inhibits ENaC through a calcium signaling pathway[114]. In other studies, PGE2 has been shown to induce the inhibition of Na+ reabsorption by decreasing the amount of cytosolic cAMP through a Gi coupled receptor, thought to be EP3[115]. Recent studies have shown that increases in luminal flow rate activate the release by epithelial cells of PGE2 in the lumen of rabbit CCD perfused in vitro. The prostaglandin secretion is dependent on calcium and MAP-kinase signaling. The flow-dependent production of PGE2 activates luminal EP receptors and triggers the inhibition of Na+ reabsorption[116]. In the rabbit CCD, Chabardès et al[112] also reported that PGE2 antagonized the effects of AVP, as seen in the cTAL. However, this effect was not observed in the rat CCD[112].
Investigation of the regulatory system involving prostaglandin and their receptors has led to the identification of a cross-talk between the intercalated and PC of the CCD (see below)[117].
FP receptors are present in rabbit DCT cells and CCD PC where their activation was shown to inhibit sodium and water reabsorption[118]. However, nothing is known regarding the effect of FP activation in mice or rats.
Finally, the thromboxane A2 receptor is localized in the TAL segment where it is activated by a peroxidized derivative of PGF2, 8-iso-prostaglandin-F2α (8-iso-PGF2α)[119]. In the kidney, 8-iso-PGF2α induces vasoconstriction, which may impact renal blood flow[120]. In the TAL, TxA2R activation stimulates PKA and induces an increase in chloride reabsorption across NKCC2[121]. Increased concentrations of 8-iso-PGF2α and other isoprostanes have been observed in both the urine and plasma of hypertensive subjects. Enhanced NaCl retention in response to 8-iso-PGF2α may contribute to the pathogenesis of hypertension[121].
Dopamine receptors: Dopamine is a catecholaminergic neurotransmitter necessary for the function of neurons. It is involved in many different physiological functions related to CNS as well as peripheral organs or tissues[122]. In the kidney, proximal cells expressing dopamine decarboxylase produce dopamine from the precursor L-dihydroxyphenylalanine. Dopamine is then released by cells at the apical and basolateral poles possibly by multiple amino acid transporters[123]. Dopamine is subsequently eliminated either by urine excretion or methylation and deamination. Dopamine concentrations are 1000-fold higher in kidneys (nmol/L range) than in plasma (pM range)[124,125], and increase even more after sodium loading[126,127]. As described below, dopamine modulates the renal Na+ transport through an autocrine or paracrine pathway[128].
Dopamine binds to five different receptors (D1R to D5R) encoded by five different genes classified into D1-like coupled to Gs proteins (D1 and D5) and D2-like coupled to Gi/o proteins (D2, D3, and D4) subtypes[129]. Since the two receptor subtypes signal through different G proteins and are present in the kidney, one might expect dopamine to have opposite effects depending on the receptor subtypes it binds to. However, dopamine has been reported to inhibit Na+ transport at multiple sites along the renal tubule by acting on major Na+ transporters[130].
In the distal nephron, D3 is the only dopamine receptor expressed in the cTAL but its role in this segment is not known. In fact, D1-like receptors agonists have been shown to down-regulate Na, K-ATPase activity in the mTAL, but the cTAL has not been proven to be sensitive to dopamine[131]. The DCT and CNT/CCD express all 5 dopamine receptors (see[132] for review). In the CCD, D1-like receptors reduce activity of Na, K-ATPase through a cAMP/PKA pathway. Moreover, phospholipase A2 (PLA2) stimulation is necessary to inhibit the pump[133]. This inhibitory pathway has been described in the proximal tubule[134,135].
Vasopressine and aldosterone effects have been shown to be antagonized by the D4 receptor in the CD[136]. More recently, a D2-like receptor agonist was reported to induce the inhibition of basolateral potassium transporters Kir4.1 and Kir5.1. Given that these channels are essential for K+ recycling and maintaining the basolateral membrane resting potential, their inhibition induces a decrease in sodium reabsorption[137].
Recent reviews clearly delineate the antihypertensive role of dopamine and dopamine receptors[124,138]. For instance, renal dopamine production is decreased in some cases of hypertension and some human populations (African-American, Japanese) do not raise their renal production of dopamine in response to a NaCl or protein load. This may contribute to the development of salt-sensitive hypertension in these particular ethnic groups[139]. The observation that D3-/- mice have a diminished ability to excrete an acute or chronic NaCl load, leading to the expansion of ECFV support the idea that the renal dopamine pathway regulate BP (see[140] for review). However, in patients with essential hypertension or in hypertensive animal models, D1-like receptor agonists fail to induce natriuresis[141].
REGULATION OF SODIUM TRANSPORT IN THE DISTAL NEPHRON BY “NOVEL” GPCRS
“Novel” GPCR that activate sodium absorption in the distal nephron
Proteinase-activated receptors: The example of proteinase-activated receptors 2: Proteases play important roles in the regulation of sodium transport in the distal nephron and BP, by stimulating the maturation of hormones or through their direct interactions with ion transporters (see above). In addition, proteases act by activating specific receptors of the so-called protease-activated receptor family (PAR).
PARs are activated trough the proteolytic cleavage of their N-terminal domain. This cleavage unmasks a peptide sequence that binds to the inner binding site and activates the receptor. It is possible to “artificially” activate these receptors by using agonist peptides that have the same sequence as the masked ligand[142,143]. For instance, the design of human- and mouse-specific agonists of PAR2[144] has enabled investigators to elucidate the particular role of PAR2 in systems that express many PARs.
The PAR family consists of 4 members (denoted PAR1 to PAR4) that are activated by different proteases. PAR1 and PAR3 are exclusively activated by thrombin[145,146]. PAR4 can be activated by thrombin and trypsin[145,147]. PAR2 is fully insensitive to thrombin but activated by trypsin and other serine proteases[148]. It can also be activated by cysteine proteases such as Derp-1[149] and be disabled by certain serine proteases (e.g., cathepsin-G) witch cleave the N-terminal domain of the receptor beyond the peptide ligand[142]. PAR2 is expressed in all tissues tested so far. In the kidney, PAR2 is expressed in epithelial, mesangial, and endothelial cells (ECs) but also in interstitial fibroblasts and inflammatory cells that invade the kidney[150].
PAR2 is involved in inflammatory responses, and it plays a coordinating role in all inflammation steps. PAR2 activation triggers a specific response in each cell type it is expressed in: The relaxation of blood vessels, increased vascular permeability, granulocyte infiltration and leukocyte adhesion, the activation of the production of cytokines IL-6 and IL-8, and finally the transmission of pain from nerve cells[151]. In the kidney, PAR2 is involved in the renal inflammation that is observed in IgA nephropathy[152] and crescent glomerulonephritis[153]. In addition, PAR2 activates monocyte chemo-attractant protein-1[154], which stimulates the production of IL-6 and IL-8 in renal tubular cells[155]. In addition, PAR2 has been shown to modulate substrate and ion transport in many epithelia (lungs, gastro-intestinal tract, lacrimal and salivary glands)[156,157].
PAR2 is expressed along the entire nephron but at higher levels in the distal nephron[158,159]. Using in vitro microperfusion of rat and mouse renal tubules, we first reported that PAR2 may activate Na+ reabsorption in the cTAL (Figure 2). We showed that PAR2 is expressed at the basolateral pole of cTAL cells and is probably coupled to a Gq/11 protein, since it induced calcium signaling and activated a PLC/PKC/ ERK1,2 pathway. This latter pathway induces in turn the stimulation of Na, K-ATPase activity and an increase in the paracellular permeability to sodium. Interestingly, the PAR2-induced increase in sodium reabsorption was partly related to a rise in the apparent affinity of Na, K-ATPase to sodium. The latter effect was induced by another signaling pathway involving PLC and a calcium-insensitive PKC that did not activate ERK1,2 phosphorylation[158]. We next characterized the role of PAR2 in mouse and rat CCDs, where it is expressed at the basolateral membrane of ICs and PCs (Figure 3). PAR2 triggered calcium signaling in both types of cells and induced an increase in the maximum Na, K-ATPase transport rate (Vmax) through ERK phosphorylation. Under our experimental conditions, PAR2 increased sodium reabsorption through ENaC only slightly. This could be explained by the fact that the ERK pathway inhibits the apical sodium channel[160]. The increase in sodium reabsorption induced by PAR2 activation stemmed mostly from the thiazide-sensitive, electroneutral NaCl reabsorption pathway of the B-IC, which was found to be dependent on ERK phosphorylation. Using Na-depleted PAR2-/- mice, we showed that PAR2 activation was necessary to induce transport across the electroneutral NaCl reabsorption pathway of the B-IC. Indeed, compared to control mice, PAR2-/- mice exhibited excessive and rapid renal Na+ loss during sodium depletion and developed hypotension[159].
Figure 2.
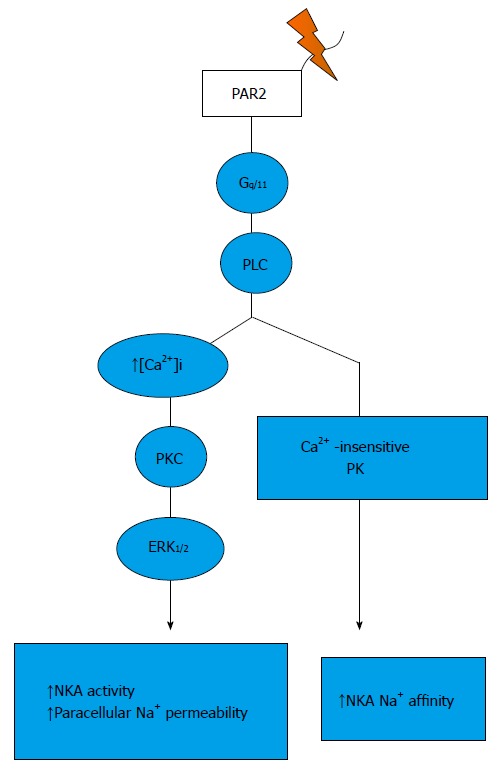
Signalling pathways trigger by protease-activated receptor 2 activation in the thick ascending limb. PAR: Protease-activated receptor.
Figure 3.
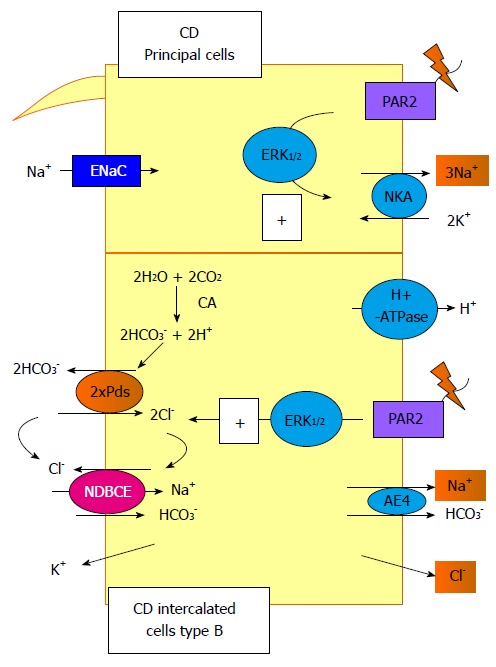
Signalling pathways trigger by protease-activated receptor 2 activation in the collecting duct. In this segment, protease-activated receptor (PAR) 2 activation increases the NKA activity in the principal cells and the Pds/Na+-driven bicarbonate/Cl- exchanger (NDBCE) system in the B-IC. Both processes involved the activation of the ERK1/2 pathway. CD: Collecting duct; PAR: Protease-activated receptor.
This hypotension cannot result from the absence of vascular PAR2 effects because in ECs, PAR2 triggers the production of NO, thereby inducing vasodilation and decreasing BP[161-163]. Proteases that cleave PAR2 at the surface of distal nephron cells in response to sodium depletion have not yet been identified. They could include serine proteases that are secreted close to PAR2. A membrane-bound serine protease such as matriptase MT-SP1 (which is encoded by the gene St14) could be a good candidate since it is one of the rare serine proteases that has been shown to activate PAR2 in vivo, at the surface of ectoderm cells during neural tube closure[164]. Moreover, matriptase is expressed in human CD cells[165]. Further investigations have to be carried out to identify the protease(s) that activate(s) renal PAR2 in vivo.
In light of its effects on Na+ reabsorption, over-activation of PAR2 in the kidney could lead to inappropriate sodium retention and hypertension. Two observations strengthen this hypothesis. First, the PAR2-encoding gene (coagulating factor II receptor like 1; F2RL1) is localized in a region of the genome associated with low renin hypertension in humans (q13 position on the fifth chromosome). Interestingly, f2rl1 is localized in a region rich in polymorphisms in the Lyon rat (q12 region on the 2nd chromosome), which is a model of low renin hypertension[166,167].
As mentioned above, PAR2 is also involved in the inflammatory response, a condition known to be related to the development of hypertension[168]. Macrophages and other inflammatory cells secrete numerous proteases that can activate or disable PAR2: Granzyme B, Elastase, tPA, plasmin, Cathepsin G and kallikrein 2, 12 and 4[169]. PAR2 could therefore play a role in inflammation-induced hypertension. Lohman et al[170] designed the first bio-compatible molecule capable of inhibiting PAR2. This molecule, called GB88, has been shown to reduce inflammation and cardiovascular damage triggered by a high fat diet in rats. Moreover, GB88 significantly diminishes BP in these animals[171]. Further investigations are needed to determine whether renal PAR2 is implicated in the BP-lowering effects of GB88.
α-Ketoglutarate receptor OXGR1: α-Ketoglutarate (αKG) is a metabolite of the Krebs cycle and may serve as a cofactor for many enzymes. Moreover, αKG is the ligand of the 2-oxoglutarate receptor 1 (OXGR1), a GPCR coupled to Gq and mainly expressed in renal distal tubules, in the pendrin-positive cells (type B IC)[172]. αKG is reabsorbed in the proximal nephron and TAL after glomerular filtration. This reabsorption is stimulated by acidosis, which, therefore, decrease αKG urinary excretion. Conversely, base loading stimulates αKG secretion by the proximal segments, leading to an increase of its urine excretion[173-175]. Since αKG is not reabsorbed after the TAL segments[173], modifications of its urine amount are proportional to variations occurring in the lumen of distal nephron. By comparing wild type mice and OXGR1-/- mice, we[176] showed that apical but not basolateral αKG acts through OXGR1 in CNT/CCD type B IC to stimulate HCO3- secretion and NaCl reabsorption. Our metabolic studies indicate that urinary αKG levels change very rapidly under acute acid-base stress. It is well known that acute acidosis activates, whereas alkalosis inhibits, NaCl reabsorption in the proximal tubule and ascending limb[177]. Since αKG concentration in the CNT/CCD decreases in acute acidosis and increases in acute alkalosis, αKG/OXGR1 could be considered as a paracrine system allowing proximal and distal parts of the nephron to communicate (Figure 4). Similarly, this communication system between proximal and distal tubule may serve to set the bicarbonate excretion by B-type IC under either acute acidosis or alkalosis. This system is sufficiently sensitive and rapid to respond to the daily variations in the dietary acid-base load as well as modifications of the metabolic production of acid and base.
Figure 4.
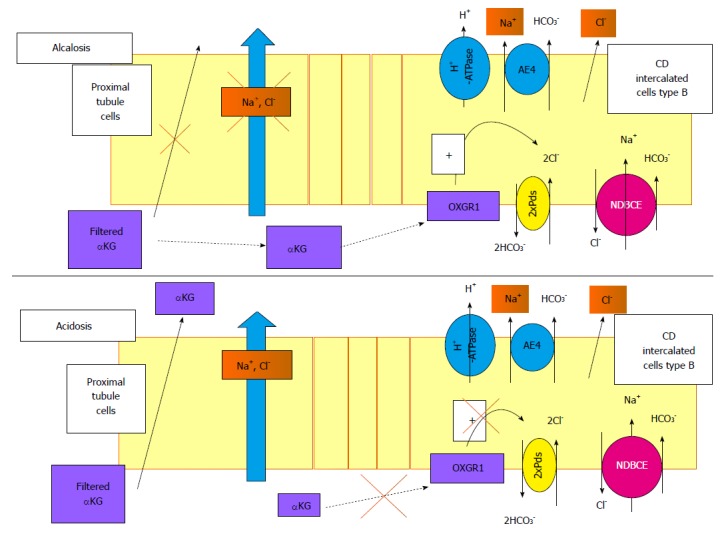
Role of α-ketoglutarate and its receptor, OXGR1, in the regulation of renal Na+ transport. α-ketoglutarate (αKG) is freely filtered by the glomerulus. During alkalosis, Na+ reabsorption in the proximal segment is reduced and the reabsorption of αKG is blunted, increasing its concentration in the lumen. This high level of αKG delivered to the distal tubule may activate its receptor in the B-intercalated cells. The signalling pathway triggered by this activation is still unknown but activates the Pds/NDBCE system, leading to an increased Na+ reabsorption. In case of acidosis, the filtered pool of αKG is reabsorbed by the proximal tubule, decreasing its luminal concentration and blunting the activation of OXGR1 in the CD. In this case, the reabsorption of Na+ in the distal nephron is not stimulated. CD: Collecting duct; NDBCE: Na+-driven bicarbonate/Cl- exchanger; OXGR1: 2-oxoglutarate receptor 1; αKG: α-ketoglutarate.
Another recent study highlights the importance of the αKG/OXGR1 paracrine system[178]. This study reports that αKG/OXGR1 mediates the adaptation of the CNT/CCD to the down-regulation of NCC in the DCT in SPAK-null mice. In these mice, Oxgr1 expression is increased but remains exclusively localized in pendrin-positive cells, the number of these cells being strikingly increased in the CNT/CCD. Interestingly, in the proximal tubule of these mice, sodium transporter expression is augmented but the expression of OAT1 (Slc22a6, that transports αKG), is down-regulated. Moreover, the expression of glutaminase and glutamate dehydrogenase, which are essential for ammoniagenesis and αKG production, is up-regulated in the proximal tubule. This should raise αKG production and secretion by proximal tubule cells and elevate luminal αKG concentration in SPAK-null mice, which is actually observed. Alkalosis is one of the consequences of NCC down-regulation. SPAK-null mice develop alkalosis, which could stimulate the αKG/OXGR1 paracrine system. The investigators postulated that the αKG/OXGR1 paracrine system may be activated by AngII, since this hormone stimulates the metabolic pathways that enhance ammoniagenesis and αKG formation in proximal tubule cells[179]. This system may be of particular importance to help restoring Na+ balance and BP in hypovolemic conditions[178].
“Novel” GPCR that inhibit sodium absorption in the distal nephron
Sphingosin-1-phosphate receptors: The first step in the synthesis of sphingosine-1-phosphate (S1P) is the hydrolysis of sphingomyelin (a component of cell membranes) into ceramide by sphingomyelinases. Ceramide is then hydrolyzed by ceraminidases to form sphingosine, which is in turn phosphorylated within the cell by sphingosine kinase SphK, an enzyme that comes in two isoforms (SphK1, SphK2), to form S1P. The intracellular concentration of S1P is maintained constant by the recycling of S1P into sphingosine by two sphingosine phosphatases and/or the degradation of S1P by a sphingosine lyase[180]. The major source of blood S1P is thought to be the red blood cells, vascular ECs, and activated platelets[181,182]. Plasma S1P concentration varies from 200 nmol/L to 900 nmol/L in humans and rodents[183]. In this compartment, the majority of S1P is bound to serum proteins such as high density lipoprotein and albumin[184]. S1P is involved in many different cellular processes from proliferation to regulation of tight junctions, etc.[185,186]. S1P may act directly upon molecular targets in the cytosol or nucleus to modulate gene expression, but S1P can also act as an extracellular signaling molecule since specific S1P membrane transporters allow it to be extruded into the extracellular compartment[187].
Sphingosin-1-phosphate is the ligand of a family of 5 GPCR, denoted S1P1-5[188]. S1P1, S1P2, and S1P3 are ubiquitously expressed, whereas S1P4 and S1P5 exhibit more restricted distribution (lung, lymphoid system and brain). S1P1 receptors are exclusively coupled to Gi/o; they can activate Akt and Rac and down-regulate PKA by inducing a decrease in cAMP cytosolic concentration. S1P2 and S1P3 receptors are most efficiently coupled to G12/13, leading to Rho activation, and to Gq, thereby activating the PLC/PKC/calcium signaling pathway[183].
Recently, the expression of SphK1 as well as S1PRs was demonstrated in whole kidney and in isolated renal cells. In the rat, S1P1-3 are more abundant in the medullary than in the cortical CD[189]. The cellular localization (i.e., apical or basolateral) remains uncertain to date. Preliminary results from in vitro microperfusion of mouse CCDs indicate that S1P administration induces a functional response, suggesting that S1P receptors are expressed at the basolateral side of the CCD cells (unpublished observation by Morla L).
Intravenous and intrarenal infusions of S1P lead to renal vasoconstriction and cause natriuresis and diuresis despite a reduction in renal blood flow. Moreover, these effects have been shown to stem from both S1P1 activation and down-regulation of the AC/PKA pathway[190]. Interestingly, the effects of S1P1 activation on sodium and water excretion are additive to those of methylisobutylamiloride, furosemide, and HCTZ -which respectively inhibit apical Na+ transporters in the proximal tubule, cTAL, and DCT/ CNT/CCD- but not to those of amiloride, a specific inhibitor of ENaC. This suggests that thenatriuretic and diuretic effects of S1P1 stem from PC. Finally it can also be noted that a S1P1 antagonist has anti-natriuretic properties[189].
The main characteristics of glomerulonephritis (proliferation of mesangial cells, stimulation of matrix production and inflammation) may involved the SphK1/S1P signaling system. Indeed, many studies have shown that S1P1, S1P2, and S1P3 are involved in inflammation and fibrosis in the glomerulus and proximal tubule[191-193]. However, so far, investigation of the specific pathophysiological contribution of each S1P receptor and SphK subtypes are not sufficiently selective and potent to yield clear-cut answers as to which of these should be targeted for treatment[194].
The recent study of Wilson et al[195] suggests that SphK1 is necessary for AngII-induced calcium signaling in isolated vascular smooth muscle cells and that store-operated calcium channels represent the exclusive intracellular target of SK1/S1P. Moreover, it shows that SphK1-/- mice do not develop hypertension following the infusion of angiotensin, suggesting its putative role in the AngII-induced hypertension[195].
Purinergic receptors (P2Y): Extracellular ATP and other nucleotides constitute local, intra-renal factors that regulate kidneys function. These nucleotides are secreted by all tubular cells on both apical and basolateral sides, either by exocytosis of nTP-filled vesicles or by specific nTP channels. ATP release occurs under mechanical stimulation. Cells express at their apical membrane a primary cilium that senses the flow rate. When luminal flow rises, the bending of the cilia is sufficient to induce ATP release at both poles of epithelial cells. ATP and other nucleotides can also be secreted under cell volume enhancement[196,197].
To date, seven P2X (P2X1-7) and eight P2Y (P2Y1,2,4,6,11-14) receptors have been identified. P2X are ligand-gated ion channels and P2Y are GPCRs. Since this review is primarily focused on GPCRs, we do not discuss here the role of P2X receptors in the distal nephron. Most P2Ys are coupled to Gq and their stimulation leads to the activation of PLCβ and intracellular calcium signaling. The P2Y12-14 receptors may form a subfamily, as they appear to be coupled to Gi/o and their stimulation triggers the inhibition of the AC/PKA pathway[198]. Many of these receptors subtypes are expressed along the nephron on both apical and basolateral cell membranes. Each nephron segment bears a specific combination of P2Y and P2X and these combinations vary from one species to another[199].
Reviews of recent studies of transgenic animals[198,200-202] indicate that P2Y2 plays an important role in sodium and water handling by the kidney.
P2Y2 receptors are expressed in cTAL[203], mDCT[204] and CD cells[205-207] and are generally found at apical and basolateral membranes, where their activation induces calcium signaling.
NKCC2 abundance and furosemide-induced natriuresis are both higher in P2Y2-/- mice, which suggests that P2Y2 receptors are an important modulator of sodium absorption in the cTAL; however their effects on sodium transport in the cTAL have not been assessed directly[199].
In the CNT/CCD of P2Y2-/- mice, the open probability (Po) of ENaC is higher but its apical expression in PC is reduced. The activation of P2Y2 by extracellular ATP should therefore inhibit ENaC activity. This effect is mediated in part by stimulation of PLC, which hydrolyzes PIP2 into IP3 and DAG. Since PIP2 binding to the β subunit of ENaC increases its open probability, reducing the amount of PIP2 in the apical membrane leads to a down-regulation of ENaC activity[201]. In addition, P2Y2 activation raises cytosolic (Ca2+) through IP3-mediated Ca2+ release from internal stores. ENaC is indirectly inhibited by intracellular Ca2+, for instance through the Ca2+-specific activation of Nedd4.2, an ubiquitin ligase that promote the endocytosis of ENaC[201]. In mDCT cells, a recent study showed that P2Y2 receptor activation and calcium signaling causes a decrease in NCC expression, owing at least partly to the destabilization of NCC mRNAs[208].
Purines and P2Y2 seem to be part of a negative feedback system that is activated to protect nephron cells from oxidative stress under high hormonal stimulation[209]. AVP, for instance, stimulates ATP release and P2Y2 activation in the cTAL and CCD[210]. Moreover, studies of P2Y2-/- mice have revealed the importance of this receptor in sodium homeostasis and BP maintenance, as these mice exhibit an increase in BP[211]. Interestingly, under aldosterone infusion and salt loading, the activity of ENaC and the salt-induced rise in BP are greater in P2Y2-/- mice than in WT mice. Thus, P2Y2 are important modulators of the action of aldosterone in the distal nephron, as they mediate the suppression of ENaC activity observed in WT mice under a high salt diet and mineralocorticoid infusion, a phenomenon known as the “aldosterone escape”[201].
Interestingly, Zhang et al[212] showed that the blunted aldosterone escape in P2Y2-/- mice is associated with an impaired increase in PGE2 and NO urine excretion under high salt loading and aldosterone infusion. These results suggest that P2Y2 are important activators of the cellular production of paracrine and autocrine messengers known to down-regulate the main sodium transporters in the nephron[213]. Gueutin et al[117] recently found that mouse CCDs are able to release PGE2 in a pathophysiological situation (Figure 5). Their study showed that ATP is released from B-type IC when H+-ATPase is down-regulated, and that luminal ATP activates P2Y2 receptors and calcium signaling in principal and IC. Interestingly, activation of the purinergic receptor induces the release of PGE2, which is known to inhibit sodium and water absorption in the distal nephron through the activation of EP1, EP3 and EP4 as described above. The effects of ATP through P2Y2 activation or those of PGE2 through PE receptors on the Na+/Cl- electroneutral transport system in IC remain unknown[117].
Figure 5.
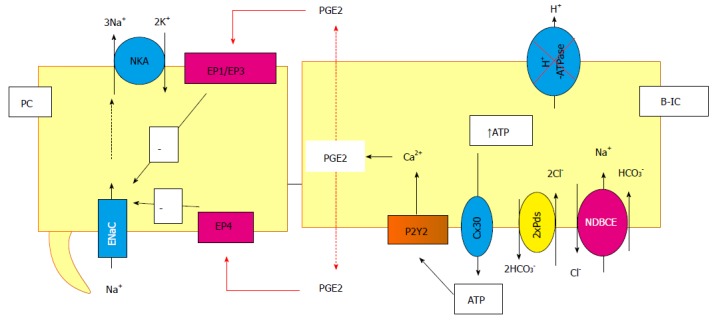
Cross-talk between principal and intercalated cells, example of the inhibition of the vacuolar proton pump. In this particular situation, the ATP is released by the B-intercalated in the lumen cell through connexion and then binds and activates the P2Y2 receptors. This activation leads to prostaglandin (PGE2) synthesis and release in both side of the cells. It binds to the EP receptors in principal cells, inducing, then, the inhibition of the Na+ reabsorption. NDBCE: Na+-driven bicarbonate/Cl- exchanger; PGE2: Prostaglandin; B-IC: Type B intercalated cells.
CONCLUSION
This review recapitulates the “classical” and “novel” signaling pathways mediated by GPCRs that regulate Na+ reabsorption in the distal nephron. The number of GPCRs that have been found to stimulate or inhibit the reabsorption of Na+ in the distal nephron is strikingly large. However, the GPCRs discussed here may only be the tip of the iceberg. Indeed, by analyzing transcriptomic data, Pradervand et al[214] have identified the expression of 78 different GPCRs in the mouse DCT/CNT and CCD, many of which have not yet been attributed a role in the kidney. Some of the most abundant GPCRs found by these investigators are known to be involved in the effects of chemokines, in the development of the CNS, in the immune system, or in the recognition of pheromones. The exploration of their role in the distal nephron and the potential discovery of their impact on Na+ transport represent a wide-open field of new regulators of extracellular volume and BP.
In our opinion, the next important challenge is not only to discover new regulatory processes but also to understand how they are linked. As shown in this review, the activation of some GPCRs induces the stimulation of Na+ reabsorption whereas others trigger the inhibition of Na+ transport. The fine-tuning of renal Na+ excretion clearly requires the simultaneous activation of different pathways (even with antagonistic effects) in the distal nephron. Moreover, depending on the physiological context (e.g., the amplitude and duration of the stimulus), these different pathways are activated or inhibited to varying extents. Two scenarios can be imagined: (1) the global response involving the activation of many GPCRs is orchestrated by an initial conductor or a few conductors, such as the RAAS or CNS; and (2) all the systems controlled by GPCRs are assembled into a complex network consisting of positive and negative feedback loops acting in series. The existence of such a connected network would explain why several models characterized by a single GPCR deficiency (a given GPCR knockout mice, for instance) exhibit strong alterations in sodium handling, since the absence of one component would perturb the entire network.
Footnotes
Conflict-of-interest statement: The authors declare having no conflict of interest.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: May 28, 2015
First decision: August 4, 2015
Article in press: November 25, 2015
P- Reviewer: Pochynyuk O, Taheri S, Thai TL S- Editor: Ji FF L- Editor: A E- Editor: Jiao XK
References
- 1.Guyton AC, Coleman TG, Young DB, Lohmeier TE, DeClue JW. Salt balance and long-term blood pressure control. Annu Rev Med. 1980;31:15–27. doi: 10.1146/annurev.me.31.020180.000311. [DOI] [PubMed] [Google Scholar]
- 2.Féraille E, Doucet A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev. 2001;81:345–418. doi: 10.1152/physrev.2001.81.1.345. [DOI] [PubMed] [Google Scholar]
- 3.Hou J. The kidney tight junction (Review) Int J Mol Med. 2014;34:1451–1457. doi: 10.3892/ijmm.2014.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greger R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol Rev. 1985;65:760–797. doi: 10.1152/physrev.1985.65.3.760. [DOI] [PubMed] [Google Scholar]
- 5.Welling PA. Rare mutations in renal sodium and potassium transporter genes exhibit impaired transport function. Curr Opin Nephrol Hypertens. 2014;23:1–8. doi: 10.1097/01.mnh.0000437204.84826.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Subramanya AR, Ellison DH. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014;9:2147–2163. doi: 10.2215/CJN.05920613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gitelman HJ, Graham JB, Welt LG. A new familial disorder characterized by hypokalemia and hypomagnesemia. Trans Assoc Am Physicians. 1966;79:221–235. [PubMed] [Google Scholar]
- 8.Wilson FH, Disse-Nicodème S, Choate KA, Ishikawa K, Nelson-Williams C, Desitter I, Gunel M, Milford DV, Lipkin GW, Achard JM, et al. Human hypertension caused by mutations in WNK kinases. Science. 2001;293:1107–1112. doi: 10.1126/science.1062844. [DOI] [PubMed] [Google Scholar]
- 9.Reif MC, Troutman SL, Schafer JA. Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest. 1986;77:1291–1298. doi: 10.1172/JCI112433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, et al. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest. 2010;120:1627–1635. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frindt G, Palmer LG. Na channels in the rat connecting tubule. Am J Physiol Renal Physiol. 2004;286:F669–F674. doi: 10.1152/ajprenal.00381.2003. [DOI] [PubMed] [Google Scholar]
- 12.Frindt G, Palmer LG. Effects of dietary K on cell-surface expression of renal ion channels and transporters. Am J Physiol Renal Physiol. 2010;299:F890–F897. doi: 10.1152/ajprenal.00323.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pech V, Thumova M, Kim YH, Agazatian D, Hummler E, Rossier BC, Weinstein AM, Nanami M, Wall SM. ENaC inhibition stimulates Cl- secretion in the mouse cortical collecting duct through an NKCC1-dependent mechanism. Am J Physiol Renal Physiol. 2012;303:F45–F55. doi: 10.1152/ajprenal.00030.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lifton RP. Molecular genetics of human blood pressure variation. Science. 1996;272:676–680. doi: 10.1126/science.272.5262.676. [DOI] [PubMed] [Google Scholar]
- 15.Pradervand S, Wang Q, Burnier M, Beermann F, Horisberger JD, Hummler E, Rossier BC. A mouse model for Liddle’s syndrome. J Am Soc Nephrol. 1999;10:2527–2533. doi: 10.1681/ASN.V10122527. [DOI] [PubMed] [Google Scholar]
- 16.Bonny O, Chraibi A, Loffing J, Jaeger NF, Gründer S, Horisberger JD, Rossier BC. Functional expression of a pseudohypoaldosteronism type I mutated epithelial Na+ channel lacking the pore-forming region of its alpha subunit. J Clin Invest. 1999;104:967–974. doi: 10.1172/JCI6821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hübner CA, et al. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA. 2013;110:7928–7933. doi: 10.1073/pnas.1221496110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hentschke M, Hentschke S, Borgmeyer U, Hübner CA, Kurth I. The murine AE4 promoter predominantly drives type B intercalated cell specific transcription. Histochem Cell Biol. 2009;132:405–412. doi: 10.1007/s00418-009-0614-0. [DOI] [PubMed] [Google Scholar]
- 19.Nonoguchi H, Owada A, Kobayashi N, Takayama M, Terada Y, Koike J, Ujiie K, Marumo F, Sakai T, Tomita K. Immunohistochemical localization of V2 vasopressin receptor along the nephron and functional role of luminal V2 receptor in terminal inner medullary collecting ducts. J Clin Invest. 1995;96:1768–1778. doi: 10.1172/JCI118222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elalouf JM, Roinel N, de Rouffignac C. Effects of antidiuretic hormone on electrolyte reabsorption and secretion in distal tubules of rat kidney. Pflugers Arch. 1984;401:167–173. doi: 10.1007/BF00583877. [DOI] [PubMed] [Google Scholar]
- 21.Pedersen NB, Hofmeister MV, Rosenbaek LL, Nielsen J, Fenton RA. Vasopressin induces phosphorylation of the thiazide-sensitive sodium chloride cotransporter in the distal convoluted tubule. Kidney Int. 2010;78:160–169. doi: 10.1038/ki.2010.130. [DOI] [PubMed] [Google Scholar]
- 22.Rieg T, Tang T, Uchida S, Hammond HK, Fenton RA, Vallon V. Adenylyl cyclase 6 enhances NKCC2 expression and mediates vasopressin-induced phosphorylation of NKCC2 and NCC. Am J Pathol. 2013;182:96–106. doi: 10.1016/j.ajpath.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morel F, Chabardès D, Imbert-Teboul M, Le Bouffant F, Hus-Citharel A, Montégut M. Multiple hormonal control of adenylate cyclase in distal segments of the rat kidney. Kidney Int Suppl. 1982;11:S55–S62. [PubMed] [Google Scholar]
- 24.Mutig K, Saritas T, Uchida S, Kahl T, Borowski T, Paliege A, Böhlick A, Bleich M, Shan Q, Bachmann S. Short-term stimulation of the thiazide-sensitive Na+-Cl- cotransporter by vasopressin involves phosphorylation and membrane translocation. Am J Physiol Renal Physiol. 2010;298:F502–F509. doi: 10.1152/ajprenal.00476.2009. [DOI] [PubMed] [Google Scholar]
- 25.Peterson LN, Wright FS. Effect of sodium intake on renal potassium excretion. Am J Physiol. 1977;233:F225–F234. doi: 10.1152/ajprenal.1977.233.3.F225. [DOI] [PubMed] [Google Scholar]
- 26.Ares GR, Caceres PS, Ortiz PA. Molecular regulation of NKCC2 in the thick ascending limb. Am J Physiol Renal Physiol. 2011;301:F1143–F1159. doi: 10.1152/ajprenal.00396.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saritas T, Borschewski A, McCormick JA, Paliege A, Dathe C, Uchida S, Terker A, Himmerkus N, Bleich M, Demaretz S, et al. SPAK differentially mediates vasopressin effects on sodium cotransporters. J Am Soc Nephrol. 2013;24:407–418. doi: 10.1681/ASN.2012040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng CJ, Yoon J, Baum M, Huang CL. STE20/SPS1-related proline/alanine-rich kinase (SPAK) is critical for sodium reabsorption in isolated, perfused thick ascending limb. Am J Physiol Renal Physiol. 2015;308:F437–F443. doi: 10.1152/ajprenal.00493.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicco C, Wittner M, DiStefano A, Jounier S, Bankir L, Bouby N. Chronic exposure to vasopressin upregulates ENaC and sodium transport in the rat renal collecting duct and lung. Hypertension. 2001;38:1143–1149. doi: 10.1161/hy1001.092641. [DOI] [PubMed] [Google Scholar]
- 30.Gaeggeler HP, Guillod Y, Loffing-Cueni D, Loffing J, Rossier BC. Vasopressin-dependent coupling between sodium transport and water flow in a mouse cortical collecting duct cell line. Kidney Int. 2011;79:843–852. doi: 10.1038/ki.2010.486. [DOI] [PubMed] [Google Scholar]
- 31.Snyder PM, Steines JC, Olson DR. Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J Biol Chem. 2004;279:5042–5046. doi: 10.1074/jbc.M312477200. [DOI] [PubMed] [Google Scholar]
- 32.Bankir L, Bichet DG, Bouby N. Vasopressin V2 receptors, ENaC, and sodium reabsorption: a risk factor for hypertension? Am J Physiol Renal Physiol. 2010;299:F917–F928. doi: 10.1152/ajprenal.00413.2010. [DOI] [PubMed] [Google Scholar]
- 33.Mironova E, Chen Y, Pao AC, Roos KP, Kohan DE, Bugaj V, Stockand JD. Activation of ENaC by AVP contributes to the urinary concentrating mechanism and dilution of plasma. Am J Physiol Renal Physiol. 2015;308:F237–F243. doi: 10.1152/ajprenal.00246.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fitzsimons JT. Angiotensin, thirst, and sodium appetite. Physiol Rev. 1998;78:583–686. doi: 10.1152/physrev.1998.78.3.583. [DOI] [PubMed] [Google Scholar]
- 35.van der Lubbe N, Zietse R, Hoorn EJ. Effects of angiotensin II on kinase-mediated sodium and potassium transport in the distal nephron. Curr Opin Nephrol Hypertens. 2013;22:120–126. doi: 10.1097/MNH.0b013e32835b6551. [DOI] [PubMed] [Google Scholar]
- 36.Harrison-Bernard LM, Navar LG, Ho MM, Vinson GP, el-Dahr SS. Immunohistochemical localization of ANG II AT1 receptor in adult rat kidney using a monoclonal antibody. Am J Physiol. 1997;273:F170–F177. doi: 10.1152/ajprenal.1997.273.1.F170. [DOI] [PubMed] [Google Scholar]
- 37.Miyata N, Park F, Li XF, Cowley AW. Distribution of angiotensin AT1 and AT2 receptor subtypes in the rat kidney. Am J Physiol. 1999;277:F437–F446. doi: 10.1152/ajprenal.1999.277.3.F437. [DOI] [PubMed] [Google Scholar]
- 38.Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30:1238–1246. doi: 10.1161/01.hyp.30.5.1238. [DOI] [PubMed] [Google Scholar]
- 39.Hunyady L, Catt KJ. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol Endocrinol. 2006;20:953–970. doi: 10.1210/me.2004-0536. [DOI] [PubMed] [Google Scholar]
- 40.Hus-Citharel A, Iturrioz X, Corvol P, Marchetti J, Llorens-Cortes C. Tyrosine kinase and mitogen-activated protein kinase/extracellularly regulated kinase differentially regulate intracellular calcium concentration responses to angiotensin II/III and bradykinin in rat cortical thick ascending limb. Endocrinology. 2006;147:451–463. doi: 10.1210/en.2005-0253. [DOI] [PubMed] [Google Scholar]
- 41.Talati G, Ohta A, Rai T, Sohara E, Naito S, Vandewalle A, Sasaki S, Uchida S. Effect of angiotensin II on the WNK-OSR1/SPAK-NCC phosphorylation cascade in cultured mpkDCT cells and in vivo mouse kidney. Biochem Biophys Res Commun. 2010;393:844–848. doi: 10.1016/j.bbrc.2010.02.096. [DOI] [PubMed] [Google Scholar]
- 42.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-γ-/- and interleukin-17A-/- mice. Hypertension. 2015;65:569–576. doi: 10.1161/HYPERTENSIONAHA.114.04975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Castañeda-Bueno M, Cervantes-Pérez LG, Vázquez N, Uribe N, Kantesaria S, Morla L, Bobadilla NA, Doucet A, Alessi DR, Gamba G. Activation of the renal Na+: Cl- cotransporter by angiotensin II is a WNK4-dependent process. Proc Natl Acad Sci USA. 2012;109:7929–7934. doi: 10.1073/pnas.1200947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peti-Peterdi J, Warnock DG, Bell PD. Angiotensin II directly stimulates ENaC activity in the cortical collecting duct via AT(1) receptors. J Am Soc Nephrol. 2002;13:1131–1135. doi: 10.1097/01.asn.0000013292.78621.fd. [DOI] [PubMed] [Google Scholar]
- 45.Mamenko M, Zaika O, Ilatovskaya DV, Staruschenko A, Pochynyuk O. Angiotensin II increases activity of the epithelial Na+ channel (ENaC) in distal nephron additively to aldosterone. J Biol Chem. 2012;287:660–671. doi: 10.1074/jbc.M111.298919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pech V, Kim YH, Weinstein AM, Everett LA, Pham TD, Wall SM. Angiotensin II increases chloride absorption in the cortical collecting duct in mice through a pendrin-dependent mechanism. Am J Physiol Renal Physiol. 2007;292:F914–F920. doi: 10.1152/ajprenal.00361.2006. [DOI] [PubMed] [Google Scholar]
- 47.Wall SM, Pech V. Pendrin and sodium channels: relevance to hypertension. J Nephrol. 2010;23 Suppl 16:S118–S123. [PubMed] [Google Scholar]
- 48.DiBona GF, Esler M. Translational medicine: the antihypertensive effect of renal denervation. Am J Physiol Regul Integr Comp Physiol. 2010;298:R245–R253. doi: 10.1152/ajpregu.00647.2009. [DOI] [PubMed] [Google Scholar]
- 49.Ehrlich ME, Evinger M, Regunathan S, Teitelman G. Mammalian adrenal chromaffin cells coexpress the epinephrine-synthesizing enzyme and neuronal properties in vivo and in vitro. Dev Biol. 1994;163:480–490. doi: 10.1006/dbio.1994.1164. [DOI] [PubMed] [Google Scholar]
- 50.Müller J, Barajas L. Electron microscopic and histochemical evidence for a tubular innervation in the renal cortex of the monkey. J Ultrastruct Res. 1972;41:533–549. doi: 10.1016/s0022-5320(72)90054-8. [DOI] [PubMed] [Google Scholar]
- 51.DiBona GF. Neural control of the kidney: functionally specific renal sympathetic nerve fibers. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1517–R1524. doi: 10.1152/ajpregu.2000.279.5.R1517. [DOI] [PubMed] [Google Scholar]
- 52.Docherty JR. Subtypes of functional alpha1- and alpha2-adrenoceptors. Eur J Pharmacol. 1998;361:1–15. doi: 10.1016/s0014-2999(98)00682-7. [DOI] [PubMed] [Google Scholar]
- 53.Slivka SR, Claeps BO. A system for simultaneous evaluation of the effect of signal transduction inhibitors on interleukin-2 synthesis and cell viability in a human T cell line. Int Arch Allergy Immunol. 1997;114:316–322. doi: 10.1159/000237688. [DOI] [PubMed] [Google Scholar]
- 54.Yu PY, Asico LD, Eisner GM, Jose PA. Differential regulation of renal phospholipase C isoforms by catecholamines. J Clin Invest. 1995;95:304–308. doi: 10.1172/JCI117656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Balboa MA, Insel PA. Stimulation of phospholipase D via alpha1-adrenergic receptors in Madin-Darby canine kidney cells is independent of PKCalpha and -epsilon activation. Mol Pharmacol. 1998;53:221–227. doi: 10.1124/mol.53.2.221. [DOI] [PubMed] [Google Scholar]
- 56.Xing M, Insel PA. Protein kinase C-dependent activation of cytosolic phospholipase A2 and mitogen-activated protein kinase by alpha 1-adrenergic receptors in Madin-Darby canine kidney cells. J Clin Invest. 1996;97:1302–1310. doi: 10.1172/JCI118546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wilborn TW, Sun D, Schafer JA. Expression of multiple alpha-adrenoceptor isoforms in rat CCD. Am J Physiol. 1998;275:F111–F118. doi: 10.1152/ajprenal.1998.275.1.F111. [DOI] [PubMed] [Google Scholar]
- 58.Lomasney JW, Cotecchia S, Lefkowitz RJ, Caron MG. Molecular biology of alpha-adrenergic receptors: implications for receptor classification and for structure-function relationships. Biochim Biophys Acta. 1991;1095:127–139. doi: 10.1016/0167-4889(91)90075-9. [DOI] [PubMed] [Google Scholar]
- 59.Woodcock EA, Johnston CI. Selective inhibition by epinephrine of parathyroid hormone-stimulated adenylate cyclase in rat renal cortex. Am J Physiol. 1982;242:F721–F726. doi: 10.1152/ajprenal.1982.242.6.F721. [DOI] [PubMed] [Google Scholar]
- 60.Woodcock EA, Johnston CI. Renal proximal tubular alpha-adrenergic receptors oppose urinary 3,5’-cyclic adenosine monophosphate response to parathyroid hormone in vivo. Endocrinology. 1985;116:1085–1089. doi: 10.1210/endo-116-3-1085. [DOI] [PubMed] [Google Scholar]
- 61.Angus JA, Cocks TM, Satoh K. The alpha adrenoceptors on endothelial cells. Fed Proc. 1986;45:2355–2359. [PubMed] [Google Scholar]
- 62.Muntz KH, Meyer L, Gadol S, Calianos TA. Alpha-2 adrenergic receptor localization in the rat heart and kidney using autoradiography and tritiated rauwolscine. J Pharmacol Exp Ther. 1986;236:542–547. [PubMed] [Google Scholar]
- 63.Plato CF, Garvin JL. Alpha(2)-adrenergic-mediated tubular NO production inhibits thick ascending limb chloride absorption. Am J Physiol Renal Physiol. 2001;281:F679–F686. doi: 10.1152/ajprenal.2001.281.4.F679. [DOI] [PubMed] [Google Scholar]
- 64.Chabardès D, Montégut M, Imbert-Teboul M, Morel F. Inhibition of alpha 2-adrenergic agonists on AVP-induced cAMP accumulation in isolated collecting tubule of the rat kidney. Mol Cell Endocrinol. 1984;37:263–275. doi: 10.1016/0303-7207(84)90096-0. [DOI] [PubMed] [Google Scholar]
- 65.Pettinger WA, Umemura S, Smyth DD, Jeffries WB. Renal alpha 2-adrenoceptors and the adenylate cyclase-cAMP system: biochemical and physiological interactions. Am J Physiol. 1987;252:F199–F208. doi: 10.1152/ajprenal.1987.252.2.F199. [DOI] [PubMed] [Google Scholar]
- 66.Chen L, Reif MC, Schafer JA. Clonidine and PGE2 have different effects on Na+ and water transport in rat and rabbit CCD. Am J Physiol. 1991;261:F126–F136. doi: 10.1152/ajprenal.1991.261.1.F126. [DOI] [PubMed] [Google Scholar]
- 67.Mansley MK, Neuhuber W, Korbmacher C, Bertog M. Norepinephrine stimulates the epithelial Na+ channel in cortical collecting duct cells via α2-adrenoceptors. Am J Physiol Renal Physiol. 2015;308:F450–F458. doi: 10.1152/ajprenal.00548.2014. [DOI] [PubMed] [Google Scholar]
- 68.Mandon B, Siga E, Champigneulle A, Imbert-Teboul M, Elalouf JM. Molecular analysis of beta-adrenergic receptor subtypes in rat collecting duct: effects on cell cAMP and Ca2+ levels. Am J Physiol. 1995;268:F1070–F1080. doi: 10.1152/ajprenal.1995.268.6.F1070. [DOI] [PubMed] [Google Scholar]
- 69.Summers RJ, Stephenson JA, Kuhar MJ. Localization of beta adrenoceptor subtypes in rat kidney by light microscopic autoradiography. J Pharmacol Exp Ther. 1985;232:561–569. [PubMed] [Google Scholar]
- 70.Mu S, Shimosawa T, Ogura S, Wang H, Uetake Y, Kawakami-Mori F, Marumo T, Yatomi Y, Geller DS, Tanaka H, et al. Epigenetic modulation of the renal β-adrenergic-WNK4 pathway in salt-sensitive hypertension. Nat Med. 2011;17:573–580. doi: 10.1038/nm.2337. [DOI] [PubMed] [Google Scholar]
- 71.Boivin V, Jahns R, Gambaryan S, Ness W, Boege F, Lohse MJ. Immunofluorescent imaging of beta 1- and beta 2-adrenergic receptors in rat kidney. Kidney Int. 2001;59:515–531. doi: 10.1046/j.1523-1755.2001.059002515.x. [DOI] [PubMed] [Google Scholar]
- 72.Kobilka BK, Kobilka TS, Daniel K, Regan JW, Caron MG, Lefkowitz RJ. Chimeric alpha 2-,beta 2-adrenergic receptors: delineation of domains involved in effector coupling and ligand binding specificity. Science. 1988;240:1310–1316. doi: 10.1126/science.2836950. [DOI] [PubMed] [Google Scholar]
- 73.Haque MZ, Caceres PS, Ortiz PA. β-Adrenergic receptor stimulation increases surface NKCC2 expression in rat thick ascending limbs in a process inhibited by phosphodiesterase 4. Am J Physiol Renal Physiol. 2012;303:F1307–F1314. doi: 10.1152/ajprenal.00019.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Terker AS, Yang CL, McCormick JA, Meermeier NP, Rogers SL, Grossmann S, Trompf K, Delpire E, Loffing J, Ellison DH. Sympathetic stimulation of thiazide-sensitive sodium chloride cotransport in the generation of salt-sensitive hypertension. Hypertension. 2014;64:178–184. doi: 10.1161/HYPERTENSIONAHA.114.03335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Azroyan A, Morla L, Crambert G, Laghmani K, Ramakrishnan S, Edwards A, Doucet A. Regulation of pendrin by cAMP: possible involvement in β-adrenergic-dependent NaCl retention. Am J Physiol Renal Physiol. 2012;302:F1180–F1187. doi: 10.1152/ajprenal.00403.2011. [DOI] [PubMed] [Google Scholar]
- 76.Wallace DP, Reif G, Hedge AM, Thrasher JB, Pietrow P. Adrenergic regulation of salt and fluid secretion in human medullary collecting duct cells. Am J Physiol Renal Physiol. 2004;287:F639–F648. doi: 10.1152/ajprenal.00448.2003. [DOI] [PubMed] [Google Scholar]
- 77.DiBona GF. Physiology in perspective: The Wisdom of the Body. Neural control of the kidney. Am J Physiol Regul Integr Comp Physiol. 2005;289:R633–R641. doi: 10.1152/ajpregu.00258.2005. [DOI] [PubMed] [Google Scholar]
- 78.Fujita T, Sato Y. Role of hypothalamic-renal noradrenergic systems in hypotensive action of potassium. Hypertension. 1992;20:466–472. doi: 10.1161/01.hyp.20.4.466. [DOI] [PubMed] [Google Scholar]
- 79.Esler MD, Hasking GJ, Willett IR, Leonard PW, Jennings GL. Noradrenaline release and sympathetic nervous system activity. J Hypertens. 1985;3:117–129. doi: 10.1097/00004872-198504000-00003. [DOI] [PubMed] [Google Scholar]
- 80.Krum H, Schlaich M, Whitbourn R, Sobotka PA, Sadowski J, Bartus K, Kapelak B, Walton A, Sievert H, Thambar S, et al. Catheter-based renal sympathetic denervation for resistant hypertension: a multicentre safety and proof-of-principle cohort study. Lancet. 2009;373:1275–1281. doi: 10.1016/S0140-6736(09)60566-3. [DOI] [PubMed] [Google Scholar]
- 81.Bhatt DL. What does the future hold for renal denervation? Eur Heart J. 2014;35:1695–1696. doi: 10.1093/eurheartj/ehu215. [DOI] [PubMed] [Google Scholar]
- 82.Kario K, Ogawa H, Okumura K, Okura T, Saito S, Ueno T, Haskin R, Negoita M, Shimada K. SYMPLICITY HTN-Japan - First Randomized Controlled Trial of Catheter-Based Renal Denervation in Asian Patients - Circ J. 2015;79:1222–1229. doi: 10.1253/circj.CJ-15-0150. [DOI] [PubMed] [Google Scholar]
- 83.Epstein M, de Marchena E. Is the failure of SYMPLICITY HTN-3 trial to meet its efficacy endpoint the “end of the road” for renal denervation? J Am Soc Hypertens. 2015;9:140–149. doi: 10.1016/j.jash.2014.12.002. [DOI] [PubMed] [Google Scholar]
- 84.Carretero OA, Scicli AG. Kinins paracrine hormone. Kidney Int Suppl. 1988;26:S52–S59. [PubMed] [Google Scholar]
- 85.Sharma JN. Role of tissue kallikrein-kininogen-kinin pathways in the cardiovascular system. Arch Med Res. 2006;37:299–306. doi: 10.1016/j.arcmed.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Figueroa CD, MacIver AG, Mackenzie JC, Bhoola KD. Localisation of immunoreactive kininogen and tissue kallikrein in the human nephron. Histochemistry. 1988;89:437–442. doi: 10.1007/BF00492599. [DOI] [PubMed] [Google Scholar]
- 87.Proud D, Vio CP. Localization of immunoreactive tissue kallikrein in human trachea. Am J Respir Cell Mol Biol. 1993;8:16–19. doi: 10.1165/ajrcmb/8.1.16. [DOI] [PubMed] [Google Scholar]
- 88.Barajas L, Powers K, Carretero O, Scicli AG, Inagami T. Immunocytochemical localization of renin and kallikrein in the rat renal cortex. Kidney Int. 1986;29:965–970. doi: 10.1038/ki.1986.94. [DOI] [PubMed] [Google Scholar]
- 89.Picard N, Eladari D, El Moghrabi S, Planès C, Bourgeois S, Houillier P, Wang Q, Burnier M, Deschenes G, Knepper MA, et al. Defective ENaC processing and function in tissue kallikrein-deficient mice. J Biol Chem. 2008;283:4602–4611. doi: 10.1074/jbc.M705664200. [DOI] [PubMed] [Google Scholar]
- 90.El Moghrabi S, Houillier P, Picard N, Sohet F, Wootla B, Bloch-Faure M, Leviel F, Cheval L, Frische S, Meneton P, et al. Tissue kallikrein permits early renal adaptation to potassium load. Proc Natl Acad Sci USA. 2010;107:13526–13531. doi: 10.1073/pnas.0913070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tomita K, Pisano JJ, Knepper MA. Control of sodium and potassium transport in the cortical collecting duct of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest. 1985;76:132–136. doi: 10.1172/JCI111935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zaika O, Mamenko M, O’Neil RG, Pochynyuk O. Bradykinin acutely inhibits activity of the epithelial Na+ channel in mammalian aldosterone-sensitive distal nephron. Am J Physiol Renal Physiol. 2011;300:F1105–F1115. doi: 10.1152/ajprenal.00606.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brochu I, Houde M, Desbiens L, Simard E, Gobeil F, Semaan W, Bkaily G, D’Orléans-Juste P. High salt-induced hypertension in B2 knockout mice is corrected by the ETA antagonist, A127722. Br J Pharmacol. 2013;170:266–277. doi: 10.1111/bph.12259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mamenko M, Zaika O, Pochynyuk O. Direct regulation of ENaC by bradykinin in the distal nephron. Implications for renal sodium handling. Curr Opin Nephrol Hypertens. 2014;23:122–129. doi: 10.1097/01.mnh.0000441053.81339.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332:411–415. doi: 10.1038/332411a0. [DOI] [PubMed] [Google Scholar]
- 96.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev. 2011;91:1–77. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hosoda K, Nakao K, Tamura N, Arai H, Ogawa Y, Suga S, Nakanishi S, Imura H. Organization, structure, chromosomal assignment, and expression of the gene encoding the human endothelin-A receptor. J Biol Chem. 1992;267:18797–18804. [PubMed] [Google Scholar]
- 98.Sakamoto A, Yanagisawa M, Sakurai T, Takuwa Y, Yanagisawa H, Masaki T. Cloning and functional expression of human cDNA for the ETB endothelin receptor. Biochem Biophys Res Commun. 1991;178:656–663. doi: 10.1016/0006-291x(91)90158-4. [DOI] [PubMed] [Google Scholar]
- 99.Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, Miller WE, Lefkowitz RJ, Olefsky JM. beta -Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem. 2001;276:43663–43667. doi: 10.1074/jbc.M105364200. [DOI] [PubMed] [Google Scholar]
- 100.Kitamura K, Shiraishi N, Singer WD, Handlogten ME, Tomita K, Miller RT. Endothelin-B receptors activate Galpha13. Am J Physiol. 1999;276:C930–C937. doi: 10.1152/ajpcell.1999.276.4.C930. [DOI] [PubMed] [Google Scholar]
- 101.Plato CF, Pollock DM, Garvin JL. Endothelin inhibits thick ascending limb chloride flux via ET(B) receptor-mediated NO release. Am J Physiol Renal Physiol. 2000;279:F326–F333. doi: 10.1152/ajprenal.2000.279.2.F326. [DOI] [PubMed] [Google Scholar]
- 102.Herrera M, Hong NJ, Ortiz PA, Garvin JL. Endothelin-1 inhibits thick ascending limb transport via Akt-stimulated nitric oxide production. J Biol Chem. 2009;284:1454–1460. doi: 10.1074/jbc.M804322200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.de Jesus Ferreira MC, Bailly C. Luminal and basolateral endothelin inhibit chloride reabsorption in the mouse thick ascending limb via a Ca(2+)-independent pathway. J Physiol. 1997;505(Pt 3):749–758. doi: 10.1111/j.1469-7793.1997.749ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tomita K, Nonoguchi H, Marumo F. Effects of endothelin on peptide-dependent cyclic adenosine monophosphate accumulation along the nephron segments of the rat. J Clin Invest. 1990;85:2014–2018. doi: 10.1172/JCI114667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tomita K, Nonoguchi H, Terada Y, Marumo F. Effects of ET-1 on water and chloride transport in cortical collecting ducts of the rat. Am J Physiol. 1993;264:F690–F696. doi: 10.1152/ajprenal.1993.264.4.F690. [DOI] [PubMed] [Google Scholar]
- 106.Zeidel ML, Brady HR, Kone BC, Gullans SR, Brenner BM. Endothelin, a peptide inhibitor of Na(+)-K(+)-ATPase in intact renaltubular epithelial cells. Am J Physiol. 1989;257:C1101–C1107. doi: 10.1152/ajpcell.1989.257.6.C1101. [DOI] [PubMed] [Google Scholar]
- 107.Bugaj V, Pochynyuk O, Mironova E, Vandewalle A, Medina JL, Stockand JD. Regulation of the epithelial Na+ channel by endothelin-1 in rat collecting duct. Am J Physiol Renal Physiol. 2008;295:F1063–F1070. doi: 10.1152/ajprenal.90321.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kohan DE. Biology of endothelin receptors in the collecting duct. Kidney Int. 2009;76:481–486. doi: 10.1038/ki.2009.203. [DOI] [PubMed] [Google Scholar]
- 109.Schiffrin EL. Role of endothelin-1 in hypertension and vascular disease. Am J Hypertens. 2001;14:83S–89S. doi: 10.1016/s0895-7061(01)02074-x. [DOI] [PubMed] [Google Scholar]
- 110.Breyer MD, Breyer RM. G protein-coupled prostanoid receptors and the kidney. Annu Rev Physiol. 2001;63:579–605. doi: 10.1146/annurev.physiol.63.1.579. [DOI] [PubMed] [Google Scholar]
- 111.Hao CM, Breyer MD. Physiological regulation of prostaglandins in the kidney. Annu Rev Physiol. 2008;70:357–377. doi: 10.1146/annurev.physiol.70.113006.100614. [DOI] [PubMed] [Google Scholar]
- 112.Chabardès D, Brick-Ghannam C, Montégut M, Siaume-Perez S. Effect of PGE2 and alpha-adrenergic agonists on AVP-dependent cAMP levels in rabbit and rat CCT. Am J Physiol. 1988;255:F43–F48. doi: 10.1152/ajprenal.1988.255.1.F43. [DOI] [PubMed] [Google Scholar]
- 113.Gu R, Jin Y, Zhai Y, Yang L, Zhang C, Li W, Wang L, Kong S, Zhang Y, Yang B, et al. PGE2 inhibits basolateral 50 pS potassium channels in the thick ascending limb of the rat kidney. Kidney Int. 2008;74:478–485. doi: 10.1038/ki.2008.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guan Y, Zhang Y, Breyer RM, Fowler B, Davis L, Hébert RL, Breyer MD. Prostaglandin E2 inhibits renal collecting duct Na+ absorption by activating the EP1 receptor. J Clin Invest. 1998;102:194–201. doi: 10.1172/JCI2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hébert RL, Jacobson HR, Breyer MD. Prostaglandin E2 inhibits sodium transport in rabbit cortical collecting duct by increasing intracellular calcium. J Clin Invest. 1991;87:1992–1998. doi: 10.1172/JCI115227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Flores D, Liu Y, Liu W, Satlin LM, Rohatgi R. Flow-induced prostaglandin E2 release regulates Na and K transport in the collecting duct. Am J Physiol Renal Physiol. 2012;303:F632–F638. doi: 10.1152/ajprenal.00169.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Gueutin V, Vallet M, Jayat M, Peti-Peterdi J, Cornière N, Leviel F, Sohet F, Wagner CA, Eladari D, Chambrey R. Renal β-intercalated cells maintain body fluid and electrolyte balance. J Clin Invest. 2013;123:4219–4231. doi: 10.1172/JCI63492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hébert RL, Carmosino M, Saito O, Yang G, Jackson CA, Qi Z, Breyer RM, Natarajan C, Hata AN, Zhang Y, et al. Characterization of a rabbit kidney prostaglandin F(2{alpha}) receptor exhibiting G(i)-restricted signaling that inhibits water absorption in the collecting duct. J Biol Chem. 2005;280:35028–35037. doi: 10.1074/jbc.M505852200. [DOI] [PubMed] [Google Scholar]
- 119.Morrow JD, Minton TA, Mukundan CR, Campbell MD, Zackert WE, Daniel VC, Badr KF, Blair IA, Roberts LJ. Free radical-induced generation of isoprostanes in vivo. Evidence for the formation of D-ring and E-ring isoprostanes. J Biol Chem. 1994;269:4317–4326. [PubMed] [Google Scholar]
- 120.Badr KF, Abi-Antoun TE. Isoprostanes and the kidney. Antioxid Redox Signal. 2005;7:236–243. doi: 10.1089/ars.2005.7.236. [DOI] [PubMed] [Google Scholar]
- 121.Cabral PD, Silva GB, Baigorria ST, Juncos LA, Juncos LI, García NH. 8-iso-prostaglandin-F2α stimulates chloride transport in thick ascending limbs: role of cAMP and protein kinase A. Am J Physiol Renal Physiol. 2010;299:F1396–F1400. doi: 10.1152/ajprenal.00225.2010. [DOI] [PubMed] [Google Scholar]
- 122.Beaulieu JM, Gainetdinov RR. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol Rev. 2011;63:182–217. doi: 10.1124/pr.110.002642. [DOI] [PubMed] [Google Scholar]
- 123.Koepsell H, Lips K, Volk C. Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res. 2007;24:1227–1251. doi: 10.1007/s11095-007-9254-z. [DOI] [PubMed] [Google Scholar]
- 124.Zeng C, Armando I, Luo Y, Eisner GM, Felder RA, Jose PA. Dysregulation of dopamine-dependent mechanisms as a determinant of hypertension: studies in dopamine receptor knockout mice. Am J Physiol Heart Circ Physiol. 2008;294:H551–H569. doi: 10.1152/ajpheart.01036.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hussain T, Lokhandwala MF. Renal dopamine receptors and hypertension. Exp Biol Med (Maywood) 2003;228:134–142. doi: 10.1177/153537020322800202. [DOI] [PubMed] [Google Scholar]
- 126.Alexander RW, Gill JR, Yamabe H, Lovenberg W, Keiser HR. Effects of dietary sodium and of acute saline infusion on the interrelationship between dopamine excretion and adrenergic activity in man. J Clin Invest. 1974;54:194–200. doi: 10.1172/JCI107743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carey RM, Van Loon GR, Baines AD, Ortt EM. Decreased plasma and urinary dopamine during dietary sodium depletion in man. J Clin Endocrinol Metab. 1981;52:903–909. doi: 10.1210/jcem-52-5-903. [DOI] [PubMed] [Google Scholar]
- 128.Siragy HM, Felder RA, Howell NL, Chevalier RL, Peach MJ, Carey RM. Evidence that intrarenal dopamine acts as a paracrine substance at the renal tubule. Am J Physiol. 1989;257:F469–F477. doi: 10.1152/ajprenal.1989.257.3.F469. [DOI] [PubMed] [Google Scholar]
- 129.Holmes A, Lachowicz JE, Sibley DR. Phenotypic analysis of dopamine receptor knockout mice; recent insights into the functional specificity of dopamine receptor subtypes. Neuropharmacology. 2004;47:1117–1134. doi: 10.1016/j.neuropharm.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 130.Jose PA, Soares-da-Silva P, Eisner GM, Felder RA. Dopamine and G protein-coupled receptor kinase 4 in the kidney: role in blood pressure regulation. Biochim Biophys Acta. 2010;1802:1259–1267. doi: 10.1016/j.bbadis.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nishi A, Eklöf AC, Bertorello AM, Aperia A. Dopamine regulation of renal Na+,K(+)-ATPase activity is lacking in Dahl salt-sensitive rats. Hypertension. 1993;21:767–771. doi: 10.1161/01.hyp.21.6.767. [DOI] [PubMed] [Google Scholar]
- 132.Zhang MZ, Harris RC. Antihypertensive mechanisms of intra-renal dopamine. Curr Opin Nephrol Hypertens. 2015;24:117–122. doi: 10.1097/MNH.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Satoh T, Cohen HT, Katz AI. Regulation of renal Na-K-ATPase by eicosanoids: central role of the cytochrome P450-monooxygenase pathway. Trans Assoc Am Physicians. 1992;105:86–92. [PubMed] [Google Scholar]
- 134.Aperia A, Bertorello A, Seri I. Dopamine causes inhibition of Na+-K+-ATPase activity in rat proximal convoluted tubule segments. Am J Physiol. 1987;252:F39–F45. doi: 10.1152/ajprenal.1987.252.1.F39. [DOI] [PubMed] [Google Scholar]
- 135.Bertorello A, Aperia A. Inhibition of proximal tubule Na(+)-K(+)-ATPase activity requires simultaneous activation of DA1 and DA2 receptors. Am J Physiol. 1990;259:F924–F928. doi: 10.1152/ajprenal.1990.259.6.F924. [DOI] [PubMed] [Google Scholar]
- 136.Li L, Schafer JA. Dopamine inhibits vasopressin-dependent cAMP production in the rat cortical collecting duct. Am J Physiol. 1998;275:F62–F67. doi: 10.1152/ajprenal.1998.275.1.F62. [DOI] [PubMed] [Google Scholar]
- 137.Zaika OL, Mamenko M, Palygin O, Boukelmoune N, Staruschenko A, Pochynyuk O. Direct inhibition of basolateral Kir4.1/5.1 and Kir4.1 channels in the cortical collecting duct by dopamine. Am J Physiol Renal Physiol. 2013;305:F1277–F1287. doi: 10.1152/ajprenal.00363.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Choi MR, Kouyoumdzian NM, Rukavina Mikusic NL, Kravetz MC, Rosón MI, Rodríguez Fermepin M, Fernández BE. Renal dopaminergic system: Pathophysiological implications and clinical perspectives. World J Nephrol. 2015;4:196–212. doi: 10.5527/wjn.v4.i2.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sanada H, Jones JE, Jose PA. Genetics of salt-sensitive hypertension. Curr Hypertens Rep. 2011;13:55–66. doi: 10.1007/s11906-010-0167-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zeng C, Jose PA. Dopamine receptors: important antihypertensive counterbalance against hypertensive factors. Hypertension. 2011;57:11–17. doi: 10.1161/HYPERTENSIONAHA.110.157727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Asico LD, Ladines C, Fuchs S, Accili D, Carey RM, Semeraro C, Pocchiari F, Felder RA, Eisner GM, Jose PA. Disruption of the dopamine D3 receptor gene produces renin-dependent hypertension. J Clin Invest. 1998;102:493–498. doi: 10.1172/JCI3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ossovskaya VS, Bunnett NW. Protease-activated receptors: contribution to physiology and disease. Physiol Rev. 2004;84:579–621. doi: 10.1152/physrev.00028.2003. [DOI] [PubMed] [Google Scholar]
- 143.Hollenberg MD, Compton SJ. International Union of Pharmacology. XXVIII. Proteinase-activated receptors. Pharmacol Rev. 2002;54:203–217. doi: 10.1124/pr.54.2.203. [DOI] [PubMed] [Google Scholar]
- 144.Nystedt S, Emilsson K, Larsson AK, Strömbeck B, Sundelin J. Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur J Biochem. 1995;232:84–89. doi: 10.1111/j.1432-1033.1995.tb20784.x. [DOI] [PubMed] [Google Scholar]
- 145.Ishihara H, Connolly AJ, Zeng D, Kahn ML, Zheng YW, Timmons C, Tram T, Coughlin SR. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature. 1997;386:502–506. doi: 10.1038/386502a0. [DOI] [PubMed] [Google Scholar]
- 146.Vu TK, Hung DT, Wheaton VI, Coughlin SR. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 147.Kahn ML, Nakanishi-Matsui M, Shapiro MJ, Ishihara H, Coughlin SR. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J Clin Invest. 1999;103:879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nystedt S, Emilsson K, Wahlestedt C, Sundelin J. Molecular cloning of a potential proteinase activated receptor. Proc Natl Acad Sci USA. 1994;91:9208–9212. doi: 10.1073/pnas.91.20.9208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Asokananthan N, Graham PT, Stewart DJ, Bakker AJ, Eidne KA, Thompson PJ, Stewart GA. House dust mite allergens induce proinflammatory cytokines from respiratory epithelial cells: the cysteine protease allergen, Der p 1, activates protease-activated receptor (PAR)-2 and inactivates PAR-1. J Immunol. 2002;169:4572–4578. doi: 10.4049/jimmunol.169.8.4572. [DOI] [PubMed] [Google Scholar]
- 150.Hollenberg MD. Proteinase-mediated signaling: proteinase-activated receptors (PARs) and much more. Life Sci. 2003;74:237–246. doi: 10.1016/j.lfs.2003.09.010. [DOI] [PubMed] [Google Scholar]
- 151.Vergnolle N. The enteric nervous system in inflammation and pain: the role of proteinase-activated receptors. Can J Gastroenterol. 2003;17:589–592. doi: 10.1155/2003/683731. [DOI] [PubMed] [Google Scholar]
- 152.Grandaliano G, Pontrelli P, Cerullo G, Monno R, Ranieri E, Ursi M, Loverre A, Gesualdo L, Schena FP. Protease-activated receptor-2 expression in IgA nephropathy: a potential role in the pathogenesis of interstitial fibrosis. J Am Soc Nephrol. 2003;14:2072–2083. doi: 10.1097/01.asn.0000080315.37254.a1. [DOI] [PubMed] [Google Scholar]
- 153.Moussa L, Apostolopoulos J, Davenport P, Tchongue J, Tipping PG. Protease-activated receptor-2 augments experimental crescentic glomerulonephritis. Am J Pathol. 2007;171:800–808. doi: 10.2353/ajpath.2007.061155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Viedt C, Dechend R, Fei J, Hänsch GM, Kreuzer J, Orth SR. MCP-1 induces inflammatory activation of human tubular epithelial cells: involvement of the transcription factors, nuclear factor-kappaB and activating protein-1. J Am Soc Nephrol. 2002;13:1534–1547. doi: 10.1097/01.asn.0000015609.31253.7f. [DOI] [PubMed] [Google Scholar]
- 155.Vesey DA, Kruger WA, Poronnik P, Gobé GC, Johnson DW. Proinflammatory and proliferative responses of human proximal tubule cells to PAR-2 activation. Am J Physiol Renal Physiol. 2007;293:F1441–F1449. doi: 10.1152/ajprenal.00088.2007. [DOI] [PubMed] [Google Scholar]
- 156.Böhm SK, Khitin LM, Grady EF, Aponte G, Payan DG, Bunnett NW. Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J Biol Chem. 1996;271:22003–22016. doi: 10.1074/jbc.271.36.22003. [DOI] [PubMed] [Google Scholar]
- 157.Vergnolle N, Macnaughton WK, Al-Ani B, Saifeddine M, Wallace JL, Hollenberg MD. Proteinase-activated receptor 2 (PAR2)-activating peptides: identification of a receptor distinct from PAR2 that regulates intestinal transport. Proc Natl Acad Sci USA. 1998;95:7766–7771. doi: 10.1073/pnas.95.13.7766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Morla L, Crambert G, Mordasini D, Favre G, Doucet A, Imbert-Teboul M. Proteinase-activated receptor 2 stimulates Na,K-ATPase and sodium reabsorption in native kidney epithelium. J Biol Chem. 2008;283:28020–28028. doi: 10.1074/jbc.M804399200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Morla L, Brideau G, Fila M, Crambert G, Cheval L, Houillier P, Ramakrishnan S, Imbert-Teboul M, Doucet A. Renal proteinase-activated receptor 2, a new actor in the control of blood pressure and plasma potassium level. J Biol Chem. 2013;288:10124–10131. doi: 10.1074/jbc.M112.446393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Shi H, Asher C, Chigaev A, Yung Y, Reuveny E, Seger R, Garty H. Interactions of beta and gamma ENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J Biol Chem. 2002;277:13539–13547. doi: 10.1074/jbc.M111717200. [DOI] [PubMed] [Google Scholar]
- 161.Danahay H, Withey L, Poll CT, van de Graaf SF, Bridges RJ. Protease-activated receptor-2-mediated inhibition of ion transport in human bronchial epithelial cells. Am J Physiol Cell Physiol. 2001;280:C1455–C1464. doi: 10.1152/ajpcell.2001.280.6.C1455. [DOI] [PubMed] [Google Scholar]
- 162.Gui Y, Loutzenhiser R, Hollenberg MD. Bidirectional regulation of renal hemodynamics by activation of PAR1 and PAR2 in isolated perfused rat kidney. Am J Physiol Renal Physiol. 2003;285:F95–104. doi: 10.1152/ajprenal.00396.2002. [DOI] [PubMed] [Google Scholar]
- 163.Kunzelmann K, Sun J, Markovich D, König J, Mürle B, Mall M, Schreiber R. Control of ion transport in mammalian airways by protease activated receptors type 2 (PAR-2) FASEB J. 2005;19:969–970. doi: 10.1096/fj.04-2469fje. [DOI] [PubMed] [Google Scholar]
- 164.Camerer E, Barker A, Duong DN, Ganesan R, Kataoka H, Cornelissen I, Darragh MR, Hussain A, Zheng YW, Srinivasan Y, et al. Local protease signaling contributes to neural tube closure in the mouse embryo. Dev Cell. 2010;18:25–38. doi: 10.1016/j.devcel.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wang JK, Lee MS, Tseng IC, Chou FP, Chen YW, Fulton A, Lee HS, Chen CJ, Johnson MD, Lin CY. Polarized epithelial cells secrete matriptase as a consequence of zymogen activation and HAI-1-mediated inhibition. Am J Physiol Cell Physiol. 2009;297:C459–C470. doi: 10.1152/ajpcell.00201.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Samani NJ, Gauguier D, Vincent M, Kaiser MA, Bihoreau MT, Lodwick D, Wallis R, Parent V, Kimber P, Rattray F, et al. Analysis of quantitative trait loci for blood pressure on rat chromosomes 2 and 13. Age-related differences in effect. Hypertension. 1996;28:1118–1122. doi: 10.1161/01.hyp.28.6.1118. [DOI] [PubMed] [Google Scholar]
- 167.Sassard J, Lo M, Liu KL. Lyon genetically hypertensive rats: an animal model of “low renin hypertension”. Acta Pharmacol Sin. 2003;24:1–6. [PubMed] [Google Scholar]
- 168.Harrison DG, Marvar PJ, Titze JM. Vascular inflammatory cells in hypertension. Front Physiol. 2012;3:128. doi: 10.3389/fphys.2012.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mason SD, Joyce JA. Proteolytic networks in cancer. Trends Cell Biol. 2011;21:228–237. doi: 10.1016/j.tcb.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Lohman RJ, Cotterell AJ, Barry GD, Liu L, Suen JY, Vesey DA, Fairlie DP. An antagonist of human protease activated receptor-2 attenuates PAR2 signaling, macrophage activation, mast cell degranulation, and collagen-induced arthritis in rats. FASEB J. 2012;26:2877–2887. doi: 10.1096/fj.11-201004. [DOI] [PubMed] [Google Scholar]
- 171.Lim J, Iyer A, Liu L, Suen JY, Lohman RJ, Seow V, Yau MK, Brown L, Fairlie DP. Diet-induced obesity, adipose inflammation, and metabolic dysfunction correlating with PAR2 expression are attenuated by PAR2 antagonism. FASEB J. 2013;27:4757–4767. doi: 10.1096/fj.13-232702. [DOI] [PubMed] [Google Scholar]
- 172.He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature. 2004;429:188–193. doi: 10.1038/nature02488. [DOI] [PubMed] [Google Scholar]
- 173.Ferrier B, Martin M, Baverel G. Reabsorption and secretion of alpha-ketoglutarate along the rat nephron: a micropuncture study. Am J Physiol. 1985;248:F404–F412. doi: 10.1152/ajprenal.1985.248.3.F404. [DOI] [PubMed] [Google Scholar]
- 174.Martin M, Ferrier B, Baverel G. Transport and utilization of alpha-ketoglutarate by the rat kidney in vivo. Pflugers Arch. 1989;413:217–224. doi: 10.1007/BF00583533. [DOI] [PubMed] [Google Scholar]
- 175.Cheema-Dhadli S, Halperin ML. Influence of hypernatraemia and urea excretion on the ability to excrete a maximally hypertonic urine in the rat. J Physiol. 2002;541:929–936. doi: 10.1113/jphysiol.2002.019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tokonami N, Morla L, Centeno G, Mordasini D, Ramakrishnan SK, Nikolaeva S, Wagner CA, Bonny O, Houillier P, Doucet A, et al. α-Ketoglutarate regulates acid-base balance through an intrarenal paracrine mechanism. J Clin Invest. 2013;123:3166–3171. doi: 10.1172/JCI67562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bobulescu IA, Moe OW. Luminal Na(+)/H (+) exchange in the proximal tubule. Pflugers Arch. 2009;458:5–21. doi: 10.1007/s00424-008-0595-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Grimm PR, Lazo-Fernandez Y, Delpire E, Wall SM, Dorsey SG, Weinman EJ, Coleman R, Wade JB, Welling PA. Integrated compensatory network is activated in the absence of NCC phosphorylation. J Clin Invest. 2015;125:2136–2150. doi: 10.1172/JCI78558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Nagami GT, Plumer AK, Beyda RM, Schachter O. Effects of acid challenges on type 2 angiotensin II receptor-sensitive ammonia production by the proximal tubule. Am J Physiol Renal Physiol. 2014;307:F53–F57. doi: 10.1152/ajprenal.00466.2013. [DOI] [PubMed] [Google Scholar]
- 180.Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6:489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Yatomi Y, Ozaki Y, Ohmori T, Igarashi Y. Sphingosine 1-phosphate: synthesis and release. Prostaglandins. 2001;64:107–122. doi: 10.1016/s0090-6980(01)00103-4. [DOI] [PubMed] [Google Scholar]
- 182.Venkataraman K, Thangada S, Michaud J, Oo ML, Ai Y, Lee YM, Wu M, Parikh NS, Khan F, Proia RL, et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem J. 2006;397:461–471. doi: 10.1042/BJ20060251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 184.Kihara A, Mitsutake S, Mizutani Y, Igarashi Y. Metabolism and biological functions of two phosphorylated sphingolipids, sphingosine 1-phosphate and ceramide 1-phosphate. Prog Lipid Res. 2007;46:126–144. doi: 10.1016/j.plipres.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 185.Hla T, Brinkmann V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology. 2011;76:S3–S8. doi: 10.1212/WNL.0b013e31820d5ec1. [DOI] [PubMed] [Google Scholar]
- 186.Maceyka M, Harikumar KB, Milstien S, Spiegel S. Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 2012;22:50–60. doi: 10.1016/j.tcb.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fukuhara S, Simmons S, Kawamura S, Inoue A, Orba Y, Tokudome T, Sunden Y, Arai Y, Moriwaki K, Ishida J, et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122:1416–1426. doi: 10.1172/JCI60746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Chun J, Goetzl EJ, Hla T, Igarashi Y, Lynch KR, Moolenaar W, Pyne S, Tigyi G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol Rev. 2002;54:265–269. doi: 10.1124/pr.54.2.265. [DOI] [PubMed] [Google Scholar]
- 189.Zhu Q, Xia M, Wang Z, Li PL, Li N. A novel lipid natriuretic factor in the renal medulla: sphingosine-1-phosphate. Am J Physiol Renal Physiol. 2011;301:F35–F41. doi: 10.1152/ajprenal.00014.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Bischoff A, Meyer Zu Heringdorf D, Jakobs KH, Michel MC. Lysosphingolipid receptor-mediated diuresis and natriuresis in anaesthetized rats. Br J Pharmacol. 2001;132:1925–1933. doi: 10.1038/sj.bjp.0703969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Park SW, Kim M, Brown KM, D’Agati VD, Lee HT. Inhibition of sphingosine 1-phosphate receptor 2 protects against renal ischemia-reperfusion injury. J Am Soc Nephrol. 2012;23:266–280. doi: 10.1681/ASN.2011050503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Shiohira S, Yoshida T, Sugiura H, Nishida M, Nitta K, Tsuchiya K. Sphingosine-1-phosphate acts as a key molecule in the direct mediation of renal fibrosis. Physiol Rep. 2013;1:e00172. doi: 10.1002/phy2.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Koch A, Völzke A, Puff B, Blankenbach K, Meyer Zu Heringdorf D, Huwiler A, Pfeilschifter J. PPARγ agonists upregulate sphingosine 1-phosphate (S1P) receptor 1 expression, which in turn reduces S1P-induced [Ca(2+)]i increases in renal mesangial cells. Biochim Biophys Acta. 2013;1831:1634–1643. doi: 10.1016/j.bbalip.2013.07.011. [DOI] [PubMed] [Google Scholar]
- 194.Schwalm S, Pfeilschifter J, Huwiler A. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway to treat chronic inflammatory kidney diseases. Basic Clin Pharmacol Toxicol. 2014;114:44–49. doi: 10.1111/bcpt.12103. [DOI] [PubMed] [Google Scholar]
- 195.Wilson PC, Fitzgibbon WR, Garrett SM, Jaffa AA, Luttrell LM, Brands MW, El-Shewy HM. Inhibition of Sphingosine Kinase 1 Ameliorates Angiotensin II-Induced Hypertension and Inhibits Transmembrane Calcium Entry via Store-Operated Calcium Channel. Mol Endocrinol. 2015;29:896–908. doi: 10.1210/me.2014-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Jensen ME, Odgaard E, Christensen MH, Praetorius HA, Leipziger J. Flow-induced [Ca2+]i increase depends on nucleotide release and subsequent purinergic signaling in the intact nephron. J Am Soc Nephrol. 2007;18:2062–2070. doi: 10.1681/ASN.2006070700. [DOI] [PubMed] [Google Scholar]
- 197.Praetorius HA, Leipziger J. Fluid flow sensing and triggered nucleotide release in epithelia. J Physiol. 2008;586:2669. doi: 10.1113/jphysiol.2008.155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Guan Z, Fellner RC, Van Beusecum J, Inscho EW. P2 receptors in renal autoregulation. Curr Vasc Pharmacol. 2014;12:818–828. doi: 10.2174/15701611113116660152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Van Beusecum J, Inscho EW. Regulation of renal function and blood pressure control by P2 purinoceptors in the kidney. Curr Opin Pharmacol. 2015;21:82–88. doi: 10.1016/j.coph.2015.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Vallon V, Rieg T. Regulation of renal NaCl and water transport by the ATP/UTP/P2Y2 receptor system. Am J Physiol Renal Physiol. 2011;301:F463–F475. doi: 10.1152/ajprenal.00236.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Vallon V, Stockand J, Rieg T. P2Y receptors and kidney function. Wiley Interdiscip Rev Membr Transp Signal. 2012;1:731–742. doi: 10.1002/wmts.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Toney GM, Vallon V, Stockand JD. Intrinsic control of sodium excretion in the distal nephron by inhibitory purinergic regulation of the epithelial Na(+) channel. Curr Opin Nephrol Hypertens. 2012;21:52–60. doi: 10.1097/MNH.0b013e32834db4a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Paulais M, Baudouin-Legros M, Teulon J. Extracellular ATP and UTP trigger calcium entry in mouse cortical thick ascending limbs. Am J Physiol. 1995;268:F496–F502. doi: 10.1152/ajprenal.1995.268.3.F496. [DOI] [PubMed] [Google Scholar]
- 204.Bidet M, De Renzis G, Martial S, Rubera I, Tauc M, Poujeol P. Extracellular ATP increases [CA(2+)](i) in distal tubule cells. I. Evidence for a P2Y2 purinoceptor. Am J Physiol Renal Physiol. 2000;279:F92–F101. doi: 10.1152/ajprenal.2000.279.1.F92. [DOI] [PubMed] [Google Scholar]
- 205.Rouse D, Leite M, Suki WN. ATP inhibits the hydrosmotic effect of AVP in rabbit CCT: evidence for a nucleotide P2u receptor. Am J Physiol. 1994;267:F289–F295. doi: 10.1152/ajprenal.1994.267.2.F289. [DOI] [PubMed] [Google Scholar]
- 206.Woda CB, Leite M, Rohatgi R, Satlin LM. Effects of luminal flow and nucleotides on [Ca(2+)](i) in rabbit cortical collecting duct. Am J Physiol Renal Physiol. 2002;283:F437–F446. doi: 10.1152/ajprenal.00316.2001. [DOI] [PubMed] [Google Scholar]
- 207.Deetjen P, Thomas J, Lehrmann H, Kim SJ, Leipziger J. The luminal P2Y receptor in the isolated perfused mouse cortical collecting duct. J Am Soc Nephrol. 2000;11:1798–1806. doi: 10.1681/ASN.V11101798. [DOI] [PubMed] [Google Scholar]
- 208.Gailly P, Szutkowska M, Olinger E, Debaix H, Seghers F, Janas S, Vallon V, Devuyst O. P2Y2 receptor activation inhibits the expression of the sodium-chloride cotransporter NCC in distal convoluted tubule cells. Pflugers Arch. 2014;466:2035–2047. doi: 10.1007/s00424-013-1438-2. [DOI] [PubMed] [Google Scholar]
- 209.Briet M, Schiffrin EL. Aldosterone: effects on the kidney and cardiovascular system. Nat Rev Nephrol. 2010;6:261–273. doi: 10.1038/nrneph.2010.30. [DOI] [PubMed] [Google Scholar]
- 210.Odgaard E, Praetorius HA, Leipziger J. AVP-stimulated nucleotide secretion in perfused mouse medullary thick ascending limb and cortical collecting duct. Am J Physiol Renal Physiol. 2009;297:F341–F349. doi: 10.1152/ajprenal.00190.2009. [DOI] [PubMed] [Google Scholar]
- 211.Rieg T, Bundey RA, Chen Y, Deschenes G, Junger W, Insel PA, Vallon V. Mice lacking P2Y2 receptors have salt-resistant hypertension and facilitated renal Na+ and water reabsorption. FASEB J. 2007;21:3717–3726. doi: 10.1096/fj.07-8807com. [DOI] [PubMed] [Google Scholar]
- 212.Zhang Y, Listhrop R, Ecelbarger CM, Kishore BK. Renal sodium transporter/channel expression and sodium excretion in P2Y2 receptor knockout mice fed a high-NaCl diet with/without aldosterone infusion. Am J Physiol Renal Physiol. 2011;300:F657–F668. doi: 10.1152/ajprenal.00549.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Harris RC. Cyclooxygenase-2 and the kidney: functional and pathophysiological implications. J Hypertens Suppl. 2002;20:S3–S9. [PubMed] [Google Scholar]
- 214.Pradervand S, Zuber Mercier A, Centeno G, Bonny O, Firsov D. A comprehensive analysis of gene expression profiles in distal parts of the mouse renal tubule. Pflugers Arch. 2010;460:925–952. doi: 10.1007/s00424-010-0863-8. [DOI] [PubMed] [Google Scholar]