

Abstract
AIM: To determine whether Helicobacter pylori (H. pylori) infection confers a higher risk of Nonalcoholic fatty liver disease (NAFLD).
METHODS: Healthy people who underwent health screening were analyzed retrospectively. Inclusion criteria were age ≥ 20 years, history of H. pylori infection, and recorded insulin level. Participants were classified as H. pylori positive or negative according to 13C urea breath tests. NAFLD was defined using the hepatic steatosis index (HSI) and NAFLD liver fat score (NAFLD-LFS). Those with an HSI > 36 or NAFLD-LFS > -0.640 were considered to have NAFLD. Multivariable logistic regression was performed to identify risk factors for NAFLD.
RESULTS: Three thousand six hundred and sixty-three people were analyzed and 1636 (44.7%) were H. pylori positive. H. pylori infection was associated with older age, male gender, hypertension, higher body mass index, and a dyslipidemic profile. HSI differed significantly between H. pylori positive and negative subjects (median 33.2, interquartile range (IQR) 30.0-36.2 for H. pylori-positive vs median 32.6, IQR 29.8-36.0 for negative participants, P = 0.005), but NAFLD-LSF did not [median -1.7, IQR -2.4 - -0.7 vs median -1.8, IQR -2.4-(-0.7), respectively, P = 0.122]. The percentage of people with NAFLD did not differ between infected and uninfected groups: HIS, 26.9% vs 27.1%, P = 0.173; NAFLD-LFS, 23.5% vs 23.1%, P = 0.778. H. pylori infection was not a risk factor, but C-reactive protein concentration and smoking were significant risk factors for NAFLD.
CONCLUSION: H. pylori infection is not a risk factor for NAFLD as indicated by HSI or NAFLD-LFS. Prospective, large-scale studies involving liver biopsies should be considered.
Keywords: Helicobacter pylori, Nonalcoholic fatty liver disease, Hepatic steatosis index, Nonalcoholic fatty liver disease liver fat score, Urea breath test
Core tip: Nonalcoholic fatty liver disease (NAFLD) is a common disorder which affects 20%-45% of the general population. Helicobacter pylori (H. pylori) infection has been suggested as a contributing to NAFLD. We investigated the association between H. pylori infection and NAFLD by using two non-invasive scoring formula, NAFLD-fat score and hepatic steatosis index. Our study showed that H. pylori infection was not a risk factor for NAFLD by either formula. However, c-reactive protein and smoking were significant risk factors for NAFLD. Prospective studies involving liver biopsies should be carried out to further investigate the association between H. pylori infection and NAFLD.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a common disorder that is reported to affect 20%-45% of the general population and 60%-75% of obese people[1,2]. NAFLD is believed to be the hepatic expression of the metabolic syndrome and is closely associated with visceral obesity, dyslipidemia, insulin resistance, and type 2 diabetes[3]. NAFLD is clinically significant because it confers higher all-cause mortality and increases the risk of cardiovascular diseases and liver-related death[2,4].
NAFLD is a complex disorder that is influenced by diverse mechanisms, including genetic, environmental, and metabolic factors[2]. Recent studies have focused on the microbiota of the gastrointestinal tract as a cause of NAFLD[5,6]. These may act through a variety of mechanisms, such as by increasing gut permeability through small intestinal overgrowth; influencing the innate immune system; fermenting indigestible carbohydrates, which increases nutrient absorption; decreasing glucagon-like-peptide-1 expression; modifying conjugated bile acid patterns; and producing endogenous ethanol[7-12]. However, NAFLD treatment by targeting the gut microbiota is limited because the relevant bacterial strains and treatment modalities are under investigation[8].
In this respect, Helicobacter pylori (H. pylori) infection is appealing as its diagnostic and eradication methods are easy and inexpensive[13]. H. pylori is a gram-negative, microaerophilic bacteria that colonizes the stomach[14]. Although H. pylori is a cause of gastrointestinal disease[14], recent attention has focused on whether H. pylori contributes to metabolic disorders including NAFLD[15-17]. H. pylori is thought to contribute to the pathogenesis of NAFLD by increasing insulin resistance, stimulating the release of proinflammatory cytokines, and increasing intestinal permeability[16,18]. However, clinical data linking H. pylori with NAFLD are limited because the studies included small sample sizes or relied on serological tests to identify H. pylori infection[19-22]. Therefore, we performed a large cross-sectional screening study of asymptomatic healthy people to investigate whether H. pylori infection is associated with NAFLD.
MATERIALS AND METHODS
Study population
We conducted a cross-sectional study of people who underwent routine health screening examinations at the Center for Health Promotion of Seoul St. Mary’s Hospital (Seoul, South Korea) between January 2010 and December 2011. The inclusion criteria were asymptomatic people who (1) had undergone tests to identify the presence of H. pylori; (2) were tested to obtain the serum insulin concentration; and (3) were aged ≥ 20 years.
We excluded subjects who (1) were heavy drinkers (> 20.0 g alcohol/d for women and > 30.0 g alcohol/d for men); (2) were seropositive for either hepatitis B virus surface antigen or anti-hepatitis C virus antibody; (3) had been diagnosed with liver cirrhosis; (4) had a history of malignancy; or (5) had missing records. This study was reviewed and approved by the institutional review board of the Seoul St. Mary’s Hospital (IRB No. KC12RISI0317), which waived the need for consent forms because this was a retrospective study with blinded records.
Data collection
All participants completed a standardized, self-validated questionnaire during the routine health screening program. The questionnaire asked about smoking habits, alcohol consumption, and medical history, including prior malignancy, surgery, diabetes, hypertension, dyslipidemia, and liver cirrhosis. Medication history included the regular use of aspirin, nonsteroidal anti-inflammatory drugs, antidiabetic medication, antihypertensive medication, or medication for dyslipidemia. Anthropometric measures were obtained by trained medical personnel. Waist circumference was measured in the horizontal plane at the midpoint of the distance between the lowest rib and the iliac crest. Hip circumference was measured as the greatest circumference of the buttocks. Blood pressure was measured in the right arm using a mercury sphygmomanometer with an adequate cuff size with the participant seated and after at least 10 min of rest. Body mass index (BMI) was calculated as weight divided by the square of height (kg/m2).
H. pylori status was determined using the 13C urea breath test (Helifinder™; Medichems, Seoul, South Korea). Venous blood samples were taken in the morning after an overnight fast of at least 12 h. Fasting serum insulin level was measured using a radioimmunoassay kit (Insulin RIA beads; TFB-Japan Co. Ltd., Tokyo, Japan). Fasting plasma glucose, glycated hemoglobin, total cholesterol, triglyceride, high-density lipoprotein cholesterol (HDL-C), and low-density lipoprotein cholesterol (LDL-C) levels were measured on the Hitachi 7150 Autoanalyzer (Hitachi Ltd., Tokyo, Japan). Alanine aminotransferase (ALT), aspartate aminotransferase (AST), and gamma-glutamyltransferase concentrations were measured on the Hitachi 7600 Autoanalyzer (Hitachi Ltd., Tokyo, Japan). Complete blood cell counts were performed using a Sysmex XE-2100 analyzer (Sysmex, Kobe, Japan).
Definitions
The participants were classified as either positive or negative for H. pylori according to the results of their H. pylori test. Determination of NAFLD was based on a previously published noninvasive steatosis formula[23,24]. The hepatic steatosis index (HSI) and NAFLD liver fat score (NAFLD-LFS) were used to identify the presence of NAFLD. Participants with an HSI > 36 or NAFLD-LFS > -0.640 were classified as having NAFLD[23,24]. The following equations were used to calculate the HSI and NAFLD-LFS.
HSI = 8 × ALT/AST + BMI (if diabetes mellitus is present, +2; if the participant is female, +2)[23].
NAFLD-LFS = -2.89 + 1.18 × metabolic syndrome (yes = 1, no = 0) + 0.45 × type 2 diabetes (yes = 2, no = 0) + 0.15 × insulin (mU/L) + 0.04 × AST (U/L) - 0.94 × AST/ALT[24].
Obesity was defined according to the World Health Organization Regional Office for the Western Pacific Region criteria (BMI > 25 kg/m2)[25]. Type 2 diabetes was defined as a hemoglobin A1c level ≥ 6.5%, previous diagnosis of type 2 diabetes, or current use of antidiabetic medication. The metabolic syndrome was defined according to the definitions of the American Heart Association and the National Heart, Lung, and Blood Institute, and the International Diabetes Federation as ≥ 3 of the following: (1) waist circumference ≥ 90 cm in men and ≥ 80 cm in women, which are the modified criteria for the Asian population; (2) triglyceride concentration ≥ 150 mg/dL or use of triglyceride-lowering medication; (3) low HDL-C concentration (< 40 mg/dL in men and < 50 mg/dL in women); (4) systolic blood pressure ≥ 130 mmHg, diastolic blood pressure ≥ 85 mmHg, or use of antihypertensive medication; or (5) fasting glucose level ≥ 100 mg/dL or use of antidiabetic medication or previously diagnosed type 2 diabetes[26]. The participants were categorized based on their alcohol consumption behavior as either nondrinkers or mild-to-moderate drinkers (1.0-30.0 g alcohol/d in men and 1.0-20.0 g alcohol/d in women). Smoking was defined as either ‘‘yes’’ (participants who had smoked ≥ 100 cigarettes over their lifetime) or ‘‘no’’ (< 100 cigarettes).
Statistical analysis
Categorical variables were examined by Pearson’s χ2 test, and differences in continuous variables were identified using the Mann-Whitney U test. Results are presented as numbers (%) and medians [interquartile range (IQR)]. Agreement between the HSI and NAFLD-LFS scores was determined by overall agreement, Goodman and Kruskal’s gamma, and Cohen’s kappa. Multivariable analysis of the risk factors for both NAFLD scores was performed using logistic regression by excluding variables included in the scoring formula. Odds ratios (ORs) and 95%CIs were calculated for each variable in the multivariable analysis. All tests were 2-sided and were performed at the 5% level of significance using SAS software (SAS; SAS Institute, Cary, NC, United States). Statistical review of the study was performed by a biomedical statistician from the Catholic University of Korea College of Medicine. All authors had access to the study data and reviewed and approved the final manuscript.
RESULTS
Routine health screening was completed by 18216 asymptomatic people from January 2010 to December 2011. Of these people, 4030 had results for both H. pylori infection and insulin concentration, and were included in this study. Three-hundred sixty-seven were excluded for the following reasons: (1) 121 were heavy drinkers; (2) 181 were seropositive for either hepatitis B virus surface antigen or hepatitis C virus antibody; (3) 18 had been diagnosed with liver cirrhosis; (4) 15 had a history of malignancy; and (5) 32 had missing records. Of the remaining 3663 people, 1636 (44.7%) were H. pylori positive and 2027 H. pylori negative (Figure 1).
Figure 1.
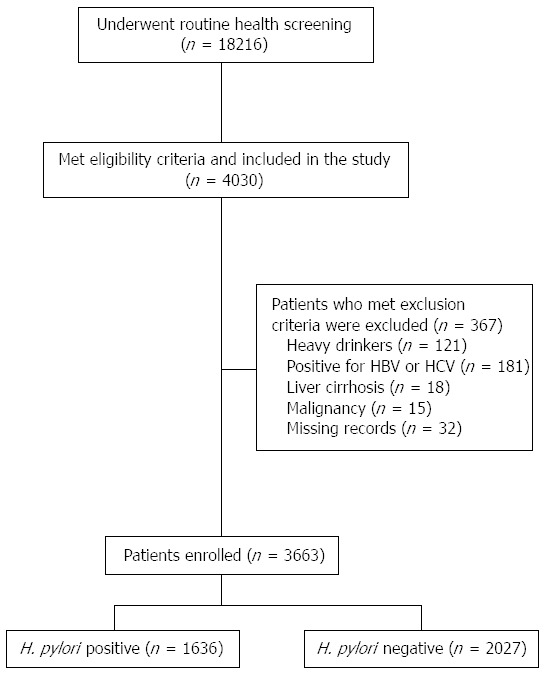
Flow chart of the study design.
A comparison between the participants according to H. pylori infection status showed a significant difference in HSI (median 33.2, IQR 30.0-36.2 for H. pylori-positive vs median 32.6, IQR 29.8-36.0 for negative participants, P = 0.005) but none for NAFLD-LFS [median -1.7, IQR -2.4-(-0.7) vs median -1.8, IQR - 2.4-(-0.7), respectively, P = 0.122] scores. There were no differences between groups in the percentages of participants classified as having NAFLD according to the HSI (26.9% vs 27.1%, P = 0.173) or NAFLD-LFS (23.5% vs 23.1%, P = 0.778).
The H. pylori-positive group was significantly older (median 54, IQR 46-61 vs median 53, IQR 43-60, P < 0.001) and included more males (60.4% vs 56.9%, P = 0.032). A higher percentage of the positive group had hypertension (27.7% vs 23.8%, P = 0.008). The positive group had a higher BMI (median 23.8, IQR 21.8-25.7 vs median 23.5, IQR 21.4-25.5 kg/m2, P = 0.002), total cholesterol concentration (median 201, IQR 180-225 vs median 195, IQR 173-220 mg/dL, P < 0.001), and LDL-C concentration (median 126, IQR 104-147.8 vs median 120, IQR 98-141 mg/dL, P < 0.001), and a lower HDL-C concentration (median 50, IQR 43-59 vs median 51, IQR 43-60 mg/dL, P = 0.007) (Table 1).
Table 1.
Clinico-demographic characteristics of the study population n (%)
| H. pylori (+)n = 1636 | H. pylori (-)n = 2027 | P value | |
| Age (yr) | 54 (46-61) | 53.0 (43-60) | < 0.001 |
| Male | 988 (60.4) | 1153 (56.9) | 0.032 |
| Body mass index (kg/m2) | 23.8 (21.8-25.7) | 23.5 (21.4-25.5) | 0.002 |
| Diabetes | 215 (13.1) | 225 (11.1) | 0.059 |
| Hypertension | 453 (27.7) | 483 (23.8) | 0.008 |
| Metabolic syndrome | 334 (20.4) | 385 (19.0) | 0.281 |
| Smoking | 699 (42.7) | 894 (44.1) | 0.403 |
| Alcohol | 893 (54.6) | 1135 (56.0) | 0.394 |
| Fasting glucose (mg/dL) | 93 (86-103) | 93 (86-102) | 0.611 |
| Insulin (mIU/mL) | 5.9 (3.9-8.7) | 5.8 (3.9-8.5) | 0.732 |
| HOMA-IR1 | 1.4 (0.9-2.2) | 1.3 (0.9-2.1) | 0.582 |
| Hemoglobin A1c (%) | 5.5 (5.2-5.7) | 5.5 (5.2-5.8) | 0.763 |
| Total cholesterol (mg/dL) | 201 (180-225) | 195 (173-220) | < 0.001 |
| Triglyceride (mg/dL) | 89 (58-137) | 86 (57-131) | 0.202 |
| HDL-C (mg/dL) | 50 (43-59) | 51 (43-60) | 0.020 |
| LDL-C (mg/dL) | 126 (104.0-147.8) | 120 (98-141) | < 0.001 |
| AST (IU/L) | 24 (20-30) | 24 (20-29) | 0.596 |
| ALT (IU/L) | 24 (18-34) | 24 (18-34) | 0.249 |
| AST/ALT | 1.0 (0.8-1.2) | 1.0 (0.8-1.2) | 0.198 |
| GGT (IU/L) | 27 (18-43) | 25 (17-43) | 0.043 |
| C-reactive protein (mg/dL) | 0.06 (0.03-0.13) | 0.06 (0.03-0.13) | 0.905 |
| HSI2 | 33.2 (30.3-36.2) | 32.6 (29.8-36.0) | 0.005 |
| HSI > 36 | 505 (30.9) | 440 (21.7) | 0.173 |
| NAFLD-LFS3 | -1.7 (-2.4 - -0.7) | -1.8 (-2.4 - -0.7) | 0.122 |
| NAFLD > -0.640 | 469 (28.7) | 385 (19.0) | 0.778 |
1HOMA-IR = (fasting glucose × fasting insulin)/405;
HSI = 8 × ALT/AST + body mass index (if diabetes mellitus, +2; if female, +2); 3NAFLD-LFS = -2.89 + 1.18 × metabolic syndrome (yes = 1, no = 0) + 0.45 × type 2 diabetes (yes = 2, no = 0) + 0.15 × insulin (mU/L) + 0.04 × AST (U/L) - 0.94 × AST/ALT. HOMA-IR: Homeostatic model assessment of insulin resistance; HDL-C: High-density lipoprotein cholesterol; LDL-C: Low-density lipoprotein cholesterol; AST: Aspartate aminotransferase; ALT: Alanine aminotransferase; GGT: Gamma-glutamyltransferase; HSI: Hepatic steatosis index; NAFLD: Nonalcoholic fatty liver disease; NAFLD-LFS: Nonalcoholic fatty liver disease liver fat score.
There was significant agreement in NAFLD diagnosis between the 2 scoring formulas. Overall agreement was 81.5%, Goodman and Kruskal’s gamma was 0.846 (P < 0.001), and Cohen’s kappa was 0.500 (P < 0.001).
Univariable analysis identified the risk factors for NAFLD defined by the HSI as the metabolic syndrome, hypertension, C-reactive protein (CRP) concentration, and smoking. Protective factors were age and HDL-C concentration. Univariable analysis of the risk factors for NAFLD defined by the NAFLD-LFS showed that the risk factors for NAFLD were age, male gender, BMI, smoking, and CRP concentration (Table 2). In both cases, the presence of H. pylori was not significant. Multivariable analysis was performed for NAFLD as defined according to each scoring formula after exclusion of the risk factors included in the scoring formula, such as gender, BMI, insulin resistance, liver enzyme levels, and metabolic syndrome components. The presence of H. pylori was not significant for either the HSI or NAFLD-LFS. In both formulas, CRP concentration and smoking were significant risk factors for NAFLD (Table 3).
Table 2.
Univariable analysis of nonalcoholic fatty liver disease according to hepatic steatosis index and nonalcoholic fatty liver disease liver fat score scores
|
HSI1 |
NAFLD-LFS2 |
|||||
| OR | 95%CI | P value | OR | 95%CI | P value | |
| Age | 0.993 | 0.986-0.999 | 0.034 | 1.017 | 1.010-1.024 | < 0.001 |
| Male | 2.043 | 1.731-2.410 | < 0.001 | |||
| Body mass index | 1.460 | 1.413-1.509 | < 0.001 | |||
| Hypertension | 2.001 | 1.703-2.350 | < 0.001 | |||
| Metabolic syndrome | 4.757 | 4.002-5.654 | < 0.001 | |||
| Smoking | 1.327 | 1.144-1.539 | < 0.001 | 1.301 | 1.115-1.517 | 0.001 |
| Alcohol | 1.022 | 0.881-1.186 | 0.772 | 0.902 | 0.773-1.051 | 0.186 |
| Presence of H. pylori | 1.109 | 0.956-1.286 | 0.173 | 1.022 | 0.876-1.193 | 0.778 |
| Fasting glucose | 1.024 | 1.020-1.027 | < 0.001 | |||
| Insulin | 1.244 | 1.218-1.270 | < 0.001 | |||
| HOMA-IR | 2.240 | 2.076-2.417 | < 0.001 | |||
| Total cholesterol | 1.005 | 1.003-1.007 | < 0.001 | 1.004 | 1.001-1.006 | 0.001 |
| Triglyceride | 1.009 | 1.007-1.010 | < 0.001 | |||
| HDL-C | 0.932 | 0.925-0.940 | < 0.001 | |||
| LDL-C | 1.008 | 1.006-1.010 | < 0.001 | 1.003 | 1.000-1.005 | 0.021 |
| C-reactive protein | 1.410 | 1.163-1.710 | < 0.001 | 1.602 | 1.298-1.978 | < 0.001 |
Factors included in each scoring formula, such as sex, body mass index, diabetes, insulin, and metabolic syndrome components, were excluded from the analysis.
HSI: 8 × ALT/AST + body mass index (if diabetes mellitus, +2; if female, +2);
NAFLD-LFS: -2.89 + 1.18 × metabolic syndrome (yes = 1, no = 0) + 0.45 × type 2 diabetes (yes = 2, no = 0) + 0.15 × insulin (mU/L) + 0.04 × AST (U/L) - 0.94 × AST/ALT. NAFLD: Nonalcoholic fatty liver disease; HSI: Hepatic steatosis index; NAFLD-LFS: Nonalcoholic fatty liver disease liver fat score; HOMA-IR: Homeostatic model assessment of insulin resistance; HDL-C: High-density lipoprotein cholesterol; LDL-C: Low-density lipoprotein cholesterol.
Table 3.
Multivariable analysis of nonalcoholic fatty liver disease according to hepatic steatosis index and nonalcoholic fatty liver disease-liver fat score scores
|
HSI1 |
NAFLD-LFS2 |
|||||
| OR | 95%CI | P value | OR | 95%CI | P value | |
| Age | 0.993 | 0.986-1.000 | 0.038 | 1.018 | 1.010-1.025 | < 0.001 |
| Smoking | 1.300 | 1.119-1.511 | 0.001 | 1.342 | 1.148-1.568 | < 0.001 |
| Presence of H. pylori | 1.129 | 0.972-1.311 | 0.113 | 1.007 | 0.862-1.176 | 0.935 |
| C-reactive protein | 1.414 | 1.166-1.714 | < 0.001 | 1.546 | 1.254-1.906 | < 0.001 |
Factors included in the HSI or NAFLD-LFS formula, such as sex, body mass index, diabetes, insulin, or metabolic syndrome components, were excluded from the analysis.
HSI = 8 × ALT/AST + body mass index (if diabetes mellitus, +2; if female, +2); 2NAFLD-LFS = -2.89 + 1.18 × metabolic syndrome (yes = 1, no = 0) + 0.45 × type 2 diabetes (yes = 2, no = 0) + 0.15 × insulin (mU/L) + 0.04 × AST (U/L) - 0.94 × AST/ALT. NAFLD: Nonalcoholic fatty liver disease; HSI: Hepatic steatosis index; NAFLD-LFS: Nonalcoholic fatty liver disease liver fat score.
DISCUSSION
In our study, current H. pylori infection was not a risk factor for NAFLD. Risk factors for NAFLD included male gender; higher BMI; the presence of diabetes, hypertension, or the metabolic syndrome; smoking; insulin resistance; and higher concentrations of total cholesterol, triglyceride, LDL-C, and CRP. HDL-C concentration was a preventive factor, whereas age had opposite effects in the 2 scoring systems. Our results showing a lack of association between H. pylori and NAFLD are in contrast with those of recent reports, which have linked H. pylori infection with NAFLD, insulin resistance, and the metabolic syndrome[16,27,28].
NAFLD is the most common liver disease worldwide and is especially prevalent in obese or diabetic people[2]. People with NAFLD have been reported to have higher overall mortality and increased risk of liver disease, cardiovascular disease, and malignancy[2,4]. Originally, the pathogenesis of NAFLD was thought to be a 2-hit process[29]. Recently, a multi-hit theory, which may involve the gut microbiota, has been suggested to explain the pathogenesis of NAFLD[30,31].
The gut microbiota is thought to contribute to the pathogenesis of NAFLD through various mechanisms. Small intestinal overgrowth resulting in increased gut permeability and involving interaction with Toll-like receptors induces inflammatory and fibrogenic responses in the liver[7]. Changes in the innate immune system induced through Toll-like receptors and increased energy absorption through production of short-chain fatty acids have also been implicated in the pathogenesis of NAFLD[8,30]. Other possible mechanisms include decreased glucagon-like-peptide-1 expression, modification of conjugated bile acid patterns, and production of endogenous ethanol[10-12]. Although these proposed mechanisms offer an attractive target for treating NAFLD, the current treatment is limited because the target bacterial strains and therapeutic modalities remain under investigation[8].
The link between H. pylori infection and NAFLD was first suggested when H. pylori 16S rDNA was discovered in a liver biopsy from an NAFLD patient[32]. Various clinical studies have reported further evidence about H. pylori infection and NAFLD[20,22,33,34]. The potential mechanisms include an association between insulin resistance and H. pylori infection, inflammation and production of proinflammatory cytokines, changes in lipid metabolism, and increased intestinal permeability[16,27,35]. However, evidence for an association between H. pylori infection and NAFLD remains limited and studies have produced contradictory results[20-22]. This may be explained by the limited numbers of subjects[19,20,22,36], use of serum immunoglobulin G as the H. pylori detection method, or publication bias (Table 4)[21,22].
Table 4.
Characteristics of Studies Investigating Helicobacter pylori and nonalcoholic fatty liver disease
| Ref. | Type of study | Country | No. of subjects (male) | H. pylori detection | H. pylori % (female/male) | NAFLD diagnosis | NAFLD % (female/male) | Association with H. pylori |
| Jamali et al[19], 2013 | RCT1 | Iran | 100 (49) | 13C UBT | 100% (N/A) | Ultrasonography and elevated liver enzyme levels | 100% | No association with H. pylori eradication |
| Polyzos et al[20], 2013 | Cross-sectional | Greece | 533 | Serum IgG, history of H. pylori eradication | 75.5% (N/A) | Liver biopsy | 52.8% (39.6/13.2) | Higher IgG seropositivity in NAFLD group |
| Okushin et al[21], 2015 | Cross-sectional | Japan | 5289 (1816) | Serum IgG | 27.4% (27.5/27.1) | Ultrasonography | 34.1% (25.4/50.7) | None |
| Sumida et al[22], 2015 | Cross-sectional | Japan | 130 (65) | Serum IgG | 40% (44.6/35.4) | Liver biopsy | 100% | Associated with hepatocyte ballooning |
| Polyzos et al[36], 2014 | Prospective2 | Greece | 12 (3) | 13C UBT | 50% (N/A) | NAFLD-LFS HSENSI MRI-HFF | 100% | Significant in HSENSI only |
Data compared between H. pylori eradication and lifestyle modification group vs lifestyle modification only group;
Data collected at baseline and 12 months after H. pylori eradication;
28 (7 males) NAFLD cases vs 25 (5 males) controls. NAFLD: Nonalcoholic fatty liver disease; RCT: Randomized controlled trial; UBT: Urea breath test; N/A: Not available; IgG: Immunoglobulin G; NAFLD-FS: Nonalcoholic fatty liver disease liver fat score; HSENSI: Homocysteine, serum glutamic oxaloacetic transaminase, erythrocyte sedimentation rate, nonalcoholic steatohepatitis index; MRI-HFF: Magnetic resonance imaging hepatic fat fraction.
Our results suggest that, in contrast to previous reports[20,22], H. pylori infection is not a risk factor for NAFLD. Our results are similar to those of a recent large-scale Japanese cross-sectional study that reported no significant relationship between NAFLD and H. pylori seropositivity[21]. However, the H. pylori-positive group in our study had a significantly higher percentage of hypertensive subjects and higher BMI, total cholesterol, triglyceride, and LDL-C levels, and lower HDL-C levels. These findings support the concept that H. pylori may affect components of the metabolic syndrome, as previously reported[28].
The reasons behind the discrepancy between metabolic syndrome components, NAFLD scores, and H. pylori infection are unclear. One possible mitigating mechanism may be the role of the commensal gut microbiota. A recent animal study reported that the commensal microbiota can attenuate the metabolic effects induced by H. pylori infection[37]. Another factor may relate to the stage of fatty liver disease. A recent Japanese study reported that people with nonalcoholic steatohepatitis who are positive for H. pylori serology are more likely to exhibit hepatocyte ballooning[22]. This suggests that H. pylori by itself may not be associated with NAFLD but may contribute to the progression to nonalcoholic steatohepatitis. The NAFLD formulas used in our study were designed to identify individuals with NAFLD rather than nonalcoholic steatohepatitis, which may help explain the lack of an association between H. pylori and NAFLD in our study.
This study has clinical significance because we included > 3600 people with 13C urea breath test data. To our knowledge, this is the largest study to investigate the association between H. pylori infection and NAFLD that did not rely on H. pylori serology. Serology testing for H. pylori has limited effectiveness because the diagnostic accuracy may be low, and testing cannot distinguish between current and past infections[38].
Our study has some limitations. First, this was a cross-sectional study with inherent limitations that allow us to draw conclusions only about the association between H. pylori infection and NAFLD. Second, we used 2 scoring formulas to determine NAFLD status instead of a liver biopsy or ultrasonography because of ethical and cost considerations. Although these 2 formulas showed statistically significant agreement, the lack of biopsy or ultrasonographic confirmation may have limited the accuracy of the NAFLD diagnosis. However, though these formula may be limited in differentiating the degree of NAFLD[39,40], studies have reported that they are fairly robust in discriminating NAFLD with an AUROC of 0.80[24,40,41]. Third, we did not account for secondary causes of steatosis such as some hepatic viral infections, autoimmune hepatitis, Wilson’s disease, α-1-antitrypsin deficiency, cystic fibrosis, hemochromatosis, and celiac disease. However, as these are either very rare in the Korean population[42-47], we believe that the inadvertent inclusion of these diseases would not have affected our study results. Fourth, though virulence factors of H. pylori such as cagA and VacA genes may have affected the degree of NAFLD, we could not investigate virulence factors as this was a retrospective study involving individuals who underwent routine health check-up.
Our large-scale, cross-sectional study showed that H. pylori infection was not a significant risk factor for NAFLD as defined by the HSI and NAFLD-LFS. Further prospective, large-scale studies involving liver biopsies and ultrasonography should be performed to determine more accurately whether there is a relationship between H. pylori infection and NAFLD.
ACKNOWLEDGMENTS
We would like to thank Kyung-Do Han from the Catholic University of Korea College of Medicine, Department of Biostatistics, for providing statistical support.
COMMENTS
Background
Nonalcoholic fatty liver disease (NAFLD) is a complex disorder that is influenced by diverse mechanisms, including genetic, environmental, and metabolic factors. Recent studies have focused on the microbiota of the gastrointestinal tract as a cause of NAFLD. Helicobacter pylori (H. pylori) is thought to contribute to the pathogenesis of NAFLD by increasing insulin resistance, stimulating the release of proinflammatory cytokines, and increasing intestinal permeability. However, clinical data linking H. pylori with NAFLD are limited.
Research frontiers
Though H. pylori has been implicated in the pathogenesis of metabolic syndrome, the relationship between H. pylori and NAFLD are limited. The authors have investigated this by analyzing the association between H. pylori and two noninvasive NAFLD formula.
Innovations and breakthroughs
In contrast to other reports which have linked H. pylori infection with insulin resistance and metabolic syndrome, the authors have found that H. pylori infection was not associated with NAFLD. This paper has merit in that H. pylori infection was tested through 13C urea breath testing which is more accurate that previously used H. pylori serum immunoglobulin.
Applications
Patients with H. pylori infection are not at higher risk of NAFLD. This serves to further fuel the controversy surrounding H. pylori infection and insulin resistance.
Peer-review
This is well presented report, providing the evidence for the lack of relationship between H. pylori infection and NAFLD. Considering that H. pylori has been blamed for most of maladies affecting the human race, the data provided are clearly going against the grain.
Footnotes
Institutional review board statement: This study was reviewed and approved by Seoul St. Mary’s Hospital Institutional Review Board, No. KC12RISI0317.
Informed consent statement: No consent statements were needed in carrying out this manuscript as this was a retrospective study using blinded records. The Institutional Review Board of The Catholic Medical Center approved the waiving of informed consent forms.
Conflict-of-interest statement: No conflicts of interest were reported for all authors.
Data sharing statement: The original, blinded data are available from the corresponding author at yoonsk@catholic.ac.kr.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: October 5, 2015
First decision: November 5, 2015
Article in press: December 14, 2015
P- Reviewer: Eshraghian A, Pacifico L, Papamichael KX, Slomiany BL S- Editor: Ma YJ L- Editor: A E- Editor: Zhang DN
References
- 1.Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- 2.Rinella ME. Nonalcoholic fatty liver disease: a systematic review. JAMA. 2015;313:2263–2273. doi: 10.1001/jama.2015.5370. [DOI] [PubMed] [Google Scholar]
- 3.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 4.Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10:330–344. doi: 10.1038/nrgastro.2013.41. [DOI] [PubMed] [Google Scholar]
- 5.Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature. 2012;482:179–185. doi: 10.1038/nature10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, Martin P, Philippe C, Walker F, Bado A, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut. 2013;62:1787–1794. doi: 10.1136/gutjnl-2012-303816. [DOI] [PubMed] [Google Scholar]
- 7.Miele L, Valenza V, La Torre G, Montalto M, Cammarota G, Ricci R, Mascianà R, Forgione A, Gabrieli ML, Perotti G, et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology. 2009;49:1877–1887. doi: 10.1002/hep.22848. [DOI] [PubMed] [Google Scholar]
- 8.Nieuwdorp M, Gilijamse PW, Pai N, Kaplan LM. Role of the microbiome in energy regulation and metabolism. Gastroenterology. 2014;146:1525–1533. doi: 10.1053/j.gastro.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 9.Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NM, Magness S, Jobin C, Lund PK. High-fat diet: bacteria interactions promote intestinal inflammation which precedes and correlates with obesity and insulin resistance in mouse. PLoS One. 2010;5:e12191. doi: 10.1371/journal.pone.0012191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cani PD, Possemiers S, Van de Wiele T, Guiot Y, Everard A, Rottier O, Geurts L, Naslain D, Neyrinck A, Lambert DM, et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut. 2009;58:1091–1103. doi: 10.1136/gut.2008.165886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claus SP, Tsang TM, Wang Y, Cloarec O, Skordi E, Martin FP, Rezzi S, Ross A, Kochhar S, Holmes E, et al. Systemic multicompartmental effects of the gut microbiome on mouse metabolic phenotypes. Mol Syst Biol. 2008;4:219. doi: 10.1038/msb.2008.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, Gill SR. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology. 2013;57:601–609. doi: 10.1002/hep.26093. [DOI] [PubMed] [Google Scholar]
- 13.Malfertheiner P, Venerito M, Selgrad M. Helicobacter pylori infection: selected aspects in clinical management. Curr Opin Gastroenterol. 2013;29:669–675. doi: 10.1097/MOG.0b013e328365d443. [DOI] [PubMed] [Google Scholar]
- 14.Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–490. doi: 10.1128/CMR.00054-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buzás GM. Metabolic consequences of Helicobacter pylori infection and eradication. World J Gastroenterol. 2014;20:5226–5234. doi: 10.3748/wjg.v20.i18.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Shen Z, Li YM. Potential role of Helicobacter pylori infection in nonalcoholic fatty liver disease. World J Gastroenterol. 2013;19:7024–7031. doi: 10.3748/wjg.v19.i41.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eshraghian A, Hashemi SA, Hamidian Jahromi A, Eshraghian H, Masoompour SM, Davarpanah MA, Eshraghian K, Taghavi SA. Helicobacter pylori infection as a risk factor for insulin resistance. Dig Dis Sci. 2009;54:1966–1970. doi: 10.1007/s10620-008-0557-7. [DOI] [PubMed] [Google Scholar]
- 18.Eshraghian A, Eshraghian H, Ranjbar Omrani G. Insulin resistance and metabolic syndrome: is Helicobacter pylori criminal? Minerva Gastroenterol Dietol. 2011;57:379–385. [PubMed] [Google Scholar]
- 19.Jamali R, Mofid A, Vahedi H, Farzaneh R, Dowlatshahi S. The effect of helicobacter pylori eradication on liver fat content in subjects with non-alcoholic Fatty liver disease: a randomized open-label clinical trial. Hepat Mon. 2013;13:e14679. doi: 10.5812/hepatmon.14679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Polyzos SA, Kountouras J, Papatheodorou A, Patsiaoura K, Katsiki E, Zafeiriadou E, Zavos C, Anastasiadou K, Terpos E. Helicobacter pylori infection in patients with nonalcoholic fatty liver disease. Metabolism. 2013;62:121–126. doi: 10.1016/j.metabol.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 21.Okushin K, Takahashi Y, Yamamichi N, Shimamoto T, Enooku K, Fujinaga H, Tsutsumi T, Shintani Y, Sakaguchi Y, Ono S, et al. Helicobacter pylori infection is not associated with fatty liver disease including non-alcoholic fatty liver disease: a large-scale cross-sectional study in Japan. BMC Gastroenterol. 2015;15:25. doi: 10.1186/s12876-015-0247-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sumida Y, Kanemasa K, Imai S, Mori K, Tanaka S, Shimokobe H, Kitamura Y, Fukumoto K, Kakutani A, Ohno T, et al. Helicobacter pylori infection might have a potential role in hepatocyte ballooning in nonalcoholic fatty liver disease. J Gastroenterol. 2015;50:996–1004. doi: 10.1007/s00535-015-1039-2. [DOI] [PubMed] [Google Scholar]
- 23.Lee JH, Kim D, Kim HJ, Lee CH, Yang JI, Kim W, Kim YJ, Yoon JH, Cho SH, Sung MW, et al. Hepatic steatosis index: a simple screening tool reflecting nonalcoholic fatty liver disease. Dig Liver Dis. 2010;42:503–508. doi: 10.1016/j.dld.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Kotronen A, Peltonen M, Hakkarainen A, Sevastianova K, Bergholm R, Johansson LM, Lundbom N, Rissanen A, Ridderstråle M, Groop L, et al. Prediction of non-alcoholic fatty liver disease and liver fat using metabolic and genetic factors. Gastroenterology. 2009;137:865–872. doi: 10.1053/j.gastro.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 25.Oh SW. Obesity and metabolic syndrome in Korea. Diabetes Metab J. 2011;35:561–566. doi: 10.4093/dmj.2011.35.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alberti KG, Eckel RH, Grundy SM, Zimmet PZ, Cleeman JI, Donato KA, Fruchart JC, James WP, Loria CM, Smith SC. Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation. 2009;120:1640–1645. doi: 10.1161/CIRCULATIONAHA.109.192644. [DOI] [PubMed] [Google Scholar]
- 27.Polyzos SA, Kountouras J, Zavos C, Deretzi G. The association between Helicobacter pylori infection and insulin resistance: a systematic review. Helicobacter. 2011;16:79–88. doi: 10.1111/j.1523-5378.2011.00822.x. [DOI] [PubMed] [Google Scholar]
- 28.Gunji T, Matsuhashi N, Sato H, Fujibayashi K, Okumura M, Sasabe N, Urabe A. Helicobacter pylori infection is significantly associated with metabolic syndrome in the Japanese population. Am J Gastroenterol. 2008;103:3005–3010. doi: 10.1111/j.1572-0241.2008.02151.x. [DOI] [PubMed] [Google Scholar]
- 29.Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114:842–845. doi: 10.1016/s0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- 30.Duseja A, Chawla YK. Obesity and NAFLD: the role of bacteria and microbiota. Clin Liver Dis. 2014;18:59–71. doi: 10.1016/j.cld.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 31.Dowman JK, Tomlinson JW, Newsome PN. Pathogenesis of non-alcoholic fatty liver disease. QJM. 2010;103:71–83. doi: 10.1093/qjmed/hcp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cindoruk M, Cirak MY, Unal S, Karakan T, Erkan G, Engin D, Dumlu S, Turet S. Identification of Helicobacter species by 16S rDNA PCR and sequence analysis in human liver samples from patients with various etiologies of benign liver diseases. Eur J Gastroenterol Hepatol. 2008;20:33–36. doi: 10.1097/MEG.0b013e3282efa4f2. [DOI] [PubMed] [Google Scholar]
- 33.Doğan Z, Filik L, Ergül B, Sarikaya M, Akbal E. Association between Helicobacter pylori and liver-to-spleen ratio: a randomized-controlled single-blind study. Eur J Gastroenterol Hepatol. 2013;25:107–110. doi: 10.1097/MEG.0b013e3283590c10. [DOI] [PubMed] [Google Scholar]
- 34.Takuma Y. [Helicobacter pylori infection and liver diseases] Gan To Kagaku Ryoho. 2011;38:362–364. [PubMed] [Google Scholar]
- 35.Gen R, Demir M, Ataseven H. Effect of Helicobacter pylori eradication on insulin resistance, serum lipids and low-grade inflammation. South Med J. 2010;103:190–196. doi: 10.1097/SMJ.0b013e3181cf373f. [DOI] [PubMed] [Google Scholar]
- 36.Polyzos SA, Nikolopoulos P, Stogianni A, Romiopoulos I, Katsinelos P, Kountouras J. Effect of Helicobacter pylori eradication on hepatic steatosis, NAFLD fibrosis score and HSENSI in patients with nonalcoholic steatohepatitis: a MR imaging-based pilot open-label study. Arq Gastroenterol. 2014;51:261–268. doi: 10.1590/s0004-28032014000300017. [DOI] [PubMed] [Google Scholar]
- 37.Khosravi Y, Seow SW, Amoyo AA, Chiow KH, Tan TL, Wong WY, Poh QH, Sentosa IM, Bunte RM, Pettersson S, et al. Helicobacter pylori infection can affect energy modulating hormones and body weight in germ free mice. Sci Rep. 2015;5:8731. doi: 10.1038/srep08731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burucoa C, Delchier JC, Courillon-Mallet A, de Korwin JD, Mégraud F, Zerbib F, Raymond J, Fauchère JL. Comparative evaluation of 29 commercial Helicobacter pylori serological kits. Helicobacter. 2013;18:169–179. doi: 10.1111/hel.12030. [DOI] [PubMed] [Google Scholar]
- 39.Meffert PJ, Baumeister SE, Lerch MM, Mayerle J, Kratzer W, Völzke H. Development, external validation, and comparative assessment of a new diagnostic score for hepatic steatosis. Am J Gastroenterol. 2014;109:1404–1414. doi: 10.1038/ajg.2014.155. [DOI] [PubMed] [Google Scholar]
- 40.Fedchuk L, Nascimbeni F, Pais R, Charlotte F, Housset C, Ratziu V. Performance and limitations of steatosis biomarkers in patients with nonalcoholic fatty liver disease. Aliment Pharmacol Ther. 2014;40:1209–1222. doi: 10.1111/apt.12963. [DOI] [PubMed] [Google Scholar]
- 41.Kahl S, Straßburger K, Nowotny B, Livingstone R, Klüppelholz B, Keßel K, Hwang JH, Giani G, Hoffmann B, Pacini G, et al. Comparison of liver fat indices for the diagnosis of hepatic steatosis and insulin resistance. PLoS One. 2014;9:e94059. doi: 10.1371/journal.pone.0094059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim BH, Kim YJ, Jeong SH, Tak WY, Ahn SH, Lee YJ, Jung EU, Lee JI, Yeon JE, Hwang JS, et al. Clinical features of autoimmune hepatitis and comparison of two diagnostic criteria in Korea: a nationwide, multicenter study. J Gastroenterol Hepatol. 2013;28:128–134. doi: 10.1111/j.1440-1746.2012.07292.x. [DOI] [PubMed] [Google Scholar]
- 43.Kim GH, Yang JY, Park JY, Lee JJ, Kim JH, Yoo HW. Estimation of Wilson’s disease incidence and carrier frequency in the Korean population by screening ATP7B major mutations in newborn filter papers using the SYBR green intercalator method based on the amplification refractory mutation system. Genet Test. 2008;12:395–399. doi: 10.1089/gte.2008.0016. [DOI] [PubMed] [Google Scholar]
- 44.Ko DH, Chang HE, Song SH, Yoon H, Park KU, Song J. Identification of compound heterozygous mutation in a Korean patient with alpha 1-antitrypsin deficiency. Korean J Lab Med. 2011;31:294–297. doi: 10.3343/kjlm.2011.31.4.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jung H, Ki CS, Koh WJ, Ahn KM, Lee SI, Kim JH, Ko JS, Seo JK, Cha SI, Lee ES, et al. Heterogeneous spectrum of CFTR gene mutations in Korean patients with cystic fibrosis. Korean J Lab Med. 2011;31:219–224. doi: 10.3343/kjlm.2011.31.3.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ. Global prevalence of putative haemochromatosis mutations. J Med Genet. 1997;34:275–278. doi: 10.1136/jmg.34.4.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gweon TG, Lim CH, Byeon SW, Baeg MK, Lee JY, Moon SJ, Kim JS, Choi MG. [A case of celiac disease] Korean J Gastroenterol. 2013;61:338–342. doi: 10.4166/kjg.2013.61.6.338. [DOI] [PubMed] [Google Scholar]