

Abstract
We previously identified receptor tyrosine kinase‐like orphan receptor 1 (ROR1) as a transcriptional target of the NKX2‐1/TTF‐1 lineage‐survival oncogene in lung adenocarcinoma. ROR1 consequently sustains a favorable balance between pro‐survival phosphatidylinositol 3‐kinase–protein kinase B and pro‐apoptotic apoptosis signal‐regulating kinase 1 (ASK1)‐p38MAPK signaling. In contrast to recent advances in understanding how ROR1 sustains pro‐survival signaling, the mechanism of ROR1 repression of pro‐apoptotic signaling remains rather elusive. In the present study, we investigated the underlying mechanism of ROR1‐mediated inhibition of the ASK1‐p38MAPK signaling pathway. Growth inhibition mediated by siROR1 was partially but significantly alleviated by ASK1 co‐knockdown in lung adenocarcinoma cell lines. Also, ASK1 phosphorylation at Thr845, which reflects its activated state, was clearly inhibited by ROR1 overexpression in both steady state and oxidative stress‐elicited conditions in MSTO‐211H cells. In addition, we found that ROR1 was physically associated with ASK1 at the C‐terminal serine threonine‐rich domain of ROR1. Furthermore, ROR1 kinase activity was shown to be required to repress the ASK1–p38 axis and oxidative stress‐induced cell death. The present findings thus support our notion that ROR1 sustains lung adenocarcinoma survival, at least in part, through direct physical interaction with ASK1 and consequential repression of the pro‐apoptotic ASK1–p38 axis in a ROR1 kinase activity‐dependent manner.
Keywords: Apoptosis, apoptosis signal‐regulating kinase 1, lung cancer, orphan receptor 1, thyroid transcription factor 1
Lung cancer has long been the leading cause of cancer death and lung adenocarcinoma is the most prevalent among the four major histologic types. We previously reported that NKX2‐1/TTF‐1, an indispensable transcription factor for peripheral lung development and physiological functions, plays a role as a lineage‐survival oncogene in lung adenocarcinoma.1 In addition, we have shown that neuroendocrine lung cancer cells, such as small‐cell lung cancer, have dependence on lineage‐survival signaling conferred by continued expression of ASH1, a transcription factor indispensable for pulmonary neuroendocrine cell development.2, 3, 4 Together, those findings suggested that a “lineage‐specific addiction” to signaling programmed for development of normal progenitor cells5 is involved in the molecular pathogenesis of lung cancer.6 The importance of NKX2‐1/TTF‐1 in lung adenocarcinoma development was further substantiated by subsequent identification of the NKX2‐1/TTF‐1 locus as a genomic region with frequent focal amplification.7, 8, 9 Notably, we identified the receptor tyrosine kinase‐like orphan receptor 1 (ROR1) as a direct transcriptional target of NKX2‐1/TTF‐1, which is crucial for maintaining a favorable balance between pro‐survival phosphatidylinositol 3‐kinase–protein kinase B (PI3K‐AKT) and pro‐apoptotic apoptosis signal‐regulating kinase 1 (ASK1)‐p38MAPK signaling for lung adenocarcinoma survival.10 Mechanistic insight into how ROR1 sustains epidermal growth factor receptor (EGFR)‐mediated PI3K‐AKT signaling in both kinase‐dependent and ‐independent manners has been well presented. In contrast, how ROR1 inhibits pro‐apoptotic signaling remains elusive.
A member of the MAPKKK family, ASK1 is activated by various stimuli such as oxidative and endoplasmic reticulum stress, as well as by tumor necrosis factor‐ and Fas‐elicited signals. In turn, ASK1 activates the MAPKK 4 (MKK4)‐JNK and MKK3/6‐p38 pathways, leading to induction of cellular apoptosis.11 ASK1 knockout mice were shown to be resistant to oxidative stress‐induced cell death,12 suggesting the functional importance of ASK1 for coping with cellular stress induced by reactive oxygen species (ROS). To date, some proteins have been identified as negatively regulating ASK1 activity through their interactions, including TRX,13 14‐3‐3,14 GSTM1‐1,15 Raf1,16 CIIA,17 and CIB1.18
In this study, we attempted to gain insight into the mechanism of ROR1 repression of pro‐apoptotic signaling to the ASK1–p38 axis. Our findings showed that ROR1 inhibits ASK1 activity in an ROR1‐kinase dependent manner through physical interaction at the C‐terminal serine threonine‐rich domain of ROR1.
Materials and Methods
Cell lines
NCI‐H1975 and NCI‐H441 cells were purchased from ATCC (Manassas, VA, USA), and PC‐9 cells from RIKEN Cell Bank (Tsukuba, Japan). The SK‐LC‐5 cell line was a generous gift from the late Lloyd J. Old (Memorial Sloan‐Kettering Cancer Center, New York, NY, USA). All cell lines were authenticated by short tandem repeat DNA profiling and confirmed to be free from mycoplasma contamination. A summary of NKX2‐1/TTF‐1 and ROR1 expression is provided in Table S1. Generations of wild‐type or kinase‐dead ROR1‐expressing MSTO‐211H cells were produced using a previously described technique.10 All cells were maintained in RPMI‐1640 with 10% FBS.
Antibodies
Recombinant MKK6 was obtained from Millipore (Watford, UK). Anti‐ROR1, anti‐ASK1, anti‐phospho‐ASK1 (T845), anti‐phospho‐ASK1 (S83), anti‐MKK3/6, anti‐phospho‐MKK3/6 (S189/S207), anti‐p38, anti‐phospho‐p38 (T180/Y182), anti‐mouse IgG, and anti‐rabbit IgG were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti‐c‐myc came from Santa Cruz Biotechnology (Santa Cruz, CA, USA), anti‐GST and anti‐maltose‐binding protein (MBP) from MBL (Nagoya, Japan), anti‐ROR1 and anti‐goat IgG from R&D Systems (Minneapolis, MN, USA), anti‐JNK and anti‐phospho‐JNK (T183/Y185) from BD Biosciences (Bedford, MA, USA), and anti‐α‐tubulin from Sigma (St. Louis, MO, USA).
RNA interference
The following RNA oligomers were obtained from Qiagen (Hilden, Germany) and Sigma‐Aldrich (Dorset, UK), and used for RNA interference: siROR1 #1, 5′‐CAGCAAUGGAUGGAAUUUCAA‐3′; siASK1 #1, 5′‐GGUAUACAUGAGUGGAAUU‐3′; and siASK1 #2, 5′‐CACUAUUGGAUGUUCUCUA‐3′.
AllStars Negative Control siRNA (siControl) was also obtained from Qiagen. Transfection of siRNAs (each at 20 nM) was carried out using Lipofectamine RNAiMAX (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions. Cells were harvested at 72 h after transfection and subjected to Western blotting (WB). Cell proliferation was determined 5 days after transfection by colorimetric assays with a Cell‐Counting kit‐8 (Dojindo Laboratories, Kumamoto, Japan).
Constructs
Construction of expression vectors of full‐length human ROR1 cDNA (pCMVpuro‐ROR1) as well as its various derivatives was carried out using a previously described method.10 Full‐length human ASK1 cDNA was purchased from RIKEN and inserted into a pCMV‐puro expression vector. The entire ASK1 ORF of the resultant construct (pCMVpuro‐ASK1) was thoroughly sequenced. In addition, a myc‐tagged derivative of pCMVpuro‐ASK1 (pCMVpuro‐ASK1‐myc) was also constructed by in vitro mutagenesis using KOD‐plus‐DNA polymerase (Toyobo, Osaka, Japan) and the following oligonucleotides: ASK1‐myc‐F, 5′‐TCCGAAGAGGACCTGTGACTGTTGCTCAATCTAATCTTC‐3′; and ASK1‐myc‐R, 5′‐GATCAGCTTCTGCTCAGTCTGTTTGTTTCGAAAGTCAAT‐3′.
Preparation of recombinant proteins
The GST‐tagged ROR1 (intracellular domain) was expressed in Sf9 insect cells using a Gateway system (Invitrogen) and purified by glutathione‐affinity chromatography, as previously described.10 An Escherichia coli‐expressing protein of MBP‐tagged kinase‐dead MKK6 was purchased from Millipore.
Western blotting and immunoprecipitation–WB analyses
Western blotting and immunoprecipitation (IP)‐WB analyses were carried out according to standard procedures using Immobilon‐P filters (Millipore) and an Amersham ECL Western Blotting Detection Reagent (GE Healthcare, Amersham, UK). For analysis of physical interactions between ROR1 and ASK1, pCMVpuro‐ASK1 was transfected with various ROR1 expression constructs, including wild‐type, ∆tyrosine kinase domain 1 (∆TK1), ∆TK2, ∆TK3, ∆ST1, ∆P, and ∆ST2. Cells were harvested 24 h after transfection with NP‐40 lysis buffer containing 20 mM Tris–HCl (pH 8.0), 137 mM NaCl, 2 mM EDTA, 1% NP‐40, 10% glycerol, and 1 mM NA3VO4.
Analysis of responses to ROS‐inducing conditions
Wild‐type and kinase‐dead ROR1‐expressing MSTO‐211H cells were plated into 6‐well plates. After 24 h, the cells were harvested for WB analysis after treatment with or without 10 mM hydrogen peroxide (H2O2) for 15 min. Colorimetric assays were also carried out to determine the influence on cell proliferation using a Cell‐Counting kit‐8 (Dojindo), following prolonged incubation in the presence of 100 μM H2O2 for 24 h. Oxidative stress‐generating ROS was also produced by treating cells with 100 μM tert‐butyl hydroperoxide (TBHP; Molecular Probes, Paisley, UK) for 120 min, after which the cells were harvested for WB analysis.
In vitro kinase assays
The 293T cells were transiently transfected with pCMVpuro‐ASK1‐myc, then myc‐tagged ASK1 was purified using a c‐Myc tagged Protein Mild Purification Kit (version 2; MBL). ASK1‐myc and MBP‐MKK6 cells were co‐incubated at 37°C for 1 h with GST or GST‐ROR1‐WT in phosphorylation buffer (25 mM Tris–HCl [pH 7.5], 5 mM MgCl2, 0.1 mM ATP), and subjected to SDS‐PAGE, followed by WB analysis with a specific antibody.
Flow cytometric analysis
Cell apoptosis was measured by flow cytometry after propidium iodide staining. Briefly, cells were treated with 100 μM H2O2 for 24 h, then harvested by trypsinization. All cells were collected and fixed with 70% ethanol and 30% PBS at −20°C, followed by treatment with RNase and propidium iodide (Sigma‐Aldrich), essentially as previously described.4 Using a FACSCalibur system, 1 × 104 cells were analyzed and sub‐G1 cells were quantitated with CellQuest Pro software (Becton, Dickinson and Company, Franklin Lakes, NJ, USA).
Results
Knockdown of ASK1 alleviates siROR1‐elicited growth inhibition
We examined the effects of co‐treatment with siASK1 and siROR1 in the lung adenocarcinoma cell lines PC‐9 and NCI‐H1975, which readily express both ROR1 and ASK1. Co‐knockdown of ASK1 significantly repressed siROR1‐induced phosphorylation of both MKK3/6 and p38 but not JNK in PC‐9 and NCI‐H1975 cells (Fig. 1). Co‐knockdown of ASK1 also significantly alleviated siROR1‐induced growth inhibition in PC‐9 cells (Fig. 1a), while similar but more modest effects were also observed in NCI‐H1975 cells (Fig. 1b, as well as Fig. S1a,b with siASK1 #2). These findings suggested that ASK1‐mediated signaling to the MKK3/6–p38 axis is involved, at least in part, in siROR1‐elicited growth inhibition in lung adenocarcinoma cells.
Figure 1.
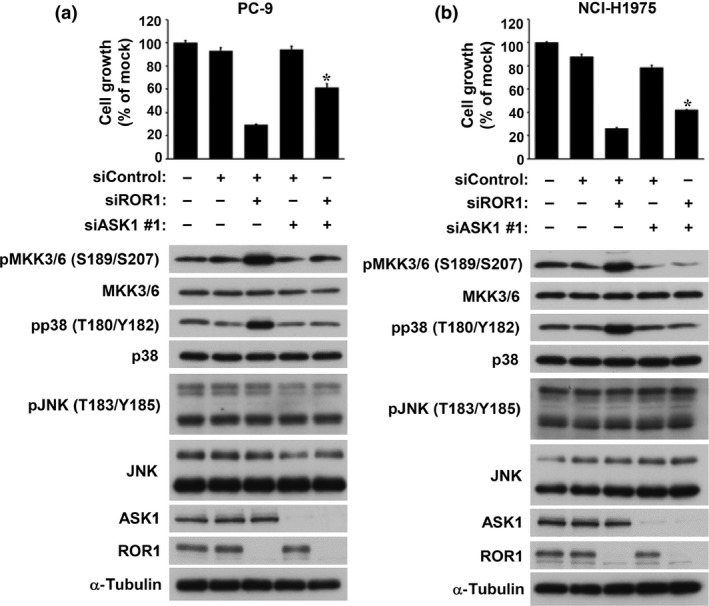
Apoptosis signal‐regulating kinase 1 (ASK1)‐mediated signaling is involved, at least in part, in receptor tyrosine kinase‐like orphan receptor 1 (ROR1) siRNA‐induced growth inhibition. Effects of co‐treatment with siASK1 and siROR1 in (a) PC‐9 and (b) NCI‐H1975 cells. Colorimetric (top panel) and Western blot (bottom panel) analyses were carried out at 5 and 3 days, respectively, after co‐transfection. Data are shown as the mean ± SEM (n = 3). *P < 0.05.
Repression of ROS‐induced ASK1 phosphorylation requires ROR1
In order to investigate whether ROR1 is involved in regulation of the ASK1‐p38 MAPK signaling pathway, we first compared steady state levels of phosphorylation of ASK1 and p38 in MSTO‐211H cells, which had been stably transfected with either a wt‐ROR1 expression construct or an empty vector. Significant decreases in autophosphorylation of ASK1 at Thr845 and p38 phosphorylation at T180 and Y182 were observed in ROR1‐transfected MSTO‐211H cells, suggesting ROR1‐mediated negative regulation of the ASK1–p38 axis (Fig. 2a). Phosphorylation of ASK1 and p38 was also analyzed by WB analysis in the presence or absence of H2O2. Consequently, we observed marked repression of oxidative stress‐induced, activation‐reflecting phosphorylation of ASK1 (T845) and p38 (F180/Y182) in MSTO‐211H cells overexpressing ROR1 (Fig. 2b). It was also noted that ROR1 overexpression significantly maintained ASK1 phosphorylation at serine 83, which is known to be phosphorylated by AKT and inhibits ASK1 function,19 even in the presence of hydrogen peroxide. Similar results were obtained with another oxidative stress inducer, TBHP (Fig. 2c). An in vitro kinase assay was also carried out using cellular ASK1 prepared from ASK1‐transfected 293T cells as well as recombinant MBP‐tagged kinase‐deficient MKK6. While readily detectable autophosphorylation of ASK1 was found along with phosphorylation of MKK6, which is a substrate of ASK1 and directly upstream of p38, co‐incubation of ASK1 with recombinant GST‐tagged ROR1 clearly diminished ASK1 autophosphorylation and MKK6 phosphorylation (Fig. 3). Taken together, these findings indicated that ROR1 negatively regulates pro‐apoptotic signaling of the ASK1–MKK6–p38 axis.
Figure 2.
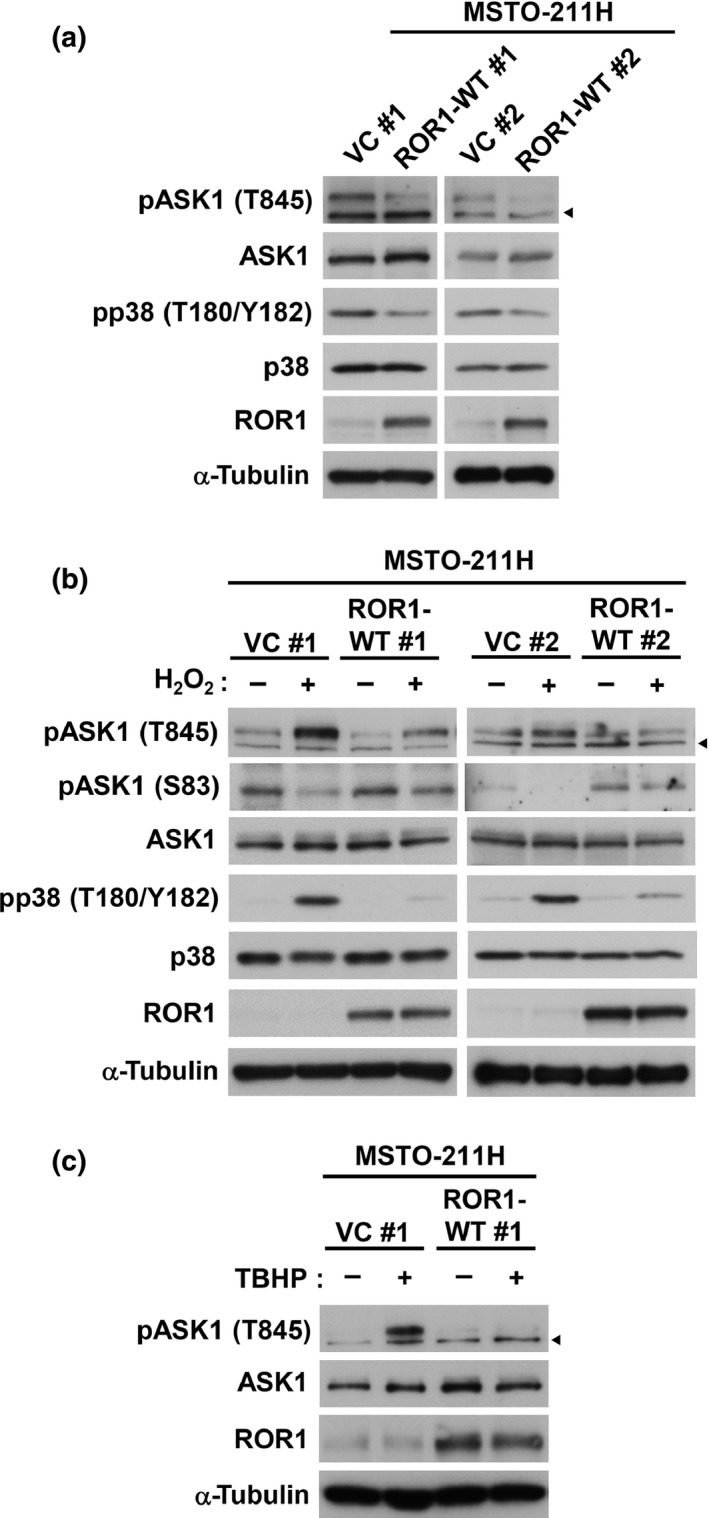
Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) suppresses reactive oxygen species‐induced apoptosis signal‐regulating kinase 1 (ASK1) and p38 phosphorylation. (a) Western blot (WB) analysis of ASK1 and p38 phosphorylation in MSTO‐211H cells stably transfected with an empty vector (VC #1 and VC #2), or an expression construct of wild‐type ROR1 (ROR1‐WT #1 and ROR1‐WT #2). (b) WB analysis of ASK1 and p38 phosphorylation in the presence or absence of H2O2 in MSTO‐211H cells stably transfected with an empty or ROR1 expression vectors. (c) WB analysis of tert‐butyl hydroperoxide (TBHP)‐induced ASK1 and p38 phosphorylation in vector control or ROR1‐introduced MSTO‐211H cells. Arrowheads indicate non‐specific bands.
Figure 3.
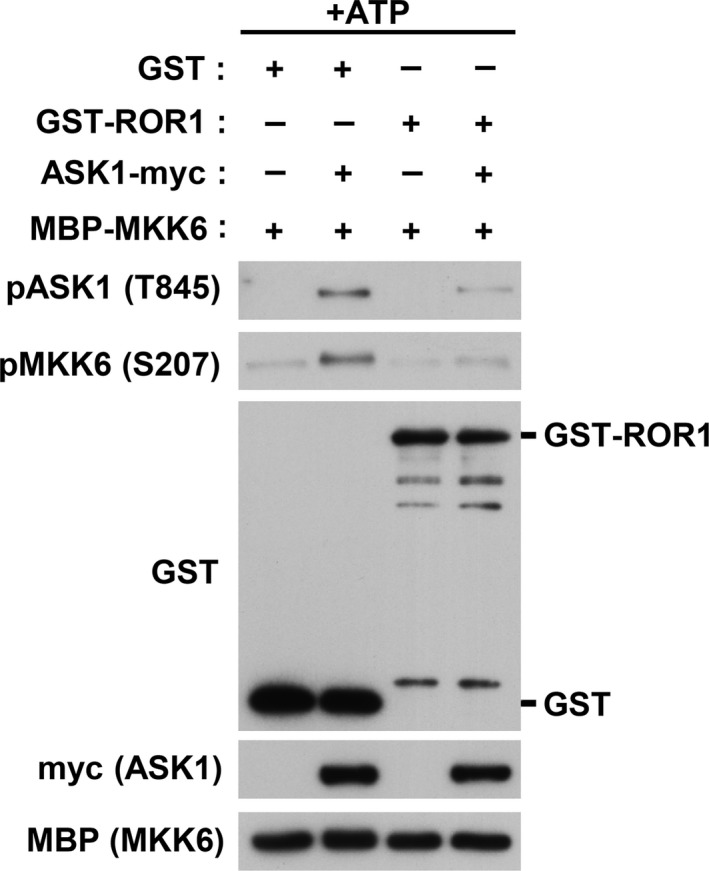
Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) inhibits apoptosis signal‐regulating kinase 1 (ASK1) kinase activity. Results of in vitro ASK1 kinase assays with use of myc‐tagged ASK1 immunoprecipitated from ASK1‐transfected 293T cells, as well as recombinant GST‐ROR1 and maltose‐binding protein–MAPKK 6 (MBP‐MKK6) proteins purified from Escherichia coli.
Physical interaction between ROR1 and ASK1
We next investigated whether ROR1 physically interacts with ASK1. 293T cells were co‐transfected with ROR1 and ASK1, then subjected to IP‐WB analysis. ASK1 was co‐immunoprecipitated with an anti‐ROR1 antibody, while an anti‐ASK1 antibody was conversely co‐immunoprecipitated with ROR1 (Fig. 4a). An association of transfected ROR1 and endogenous ASK1 was also observed in MSTO‐211H cells transfected with ROR1 (Fig. 4b). Interactions between endogenous ROR1 and ASK1 proteins were further analyzed by IP‐WB analysis using four different lung adenocarcinoma cell lines, which clearly demonstrated their physical associations (Fig. 4c for PC‐9 and NCI‐H1975; Fig. S2 for NCI‐H441 and SK‐LC‐5). We also found a consistent association between ASK1 and ROR1 regardless of the presence of H2O2 (Fig. 4d).
Figure 4.
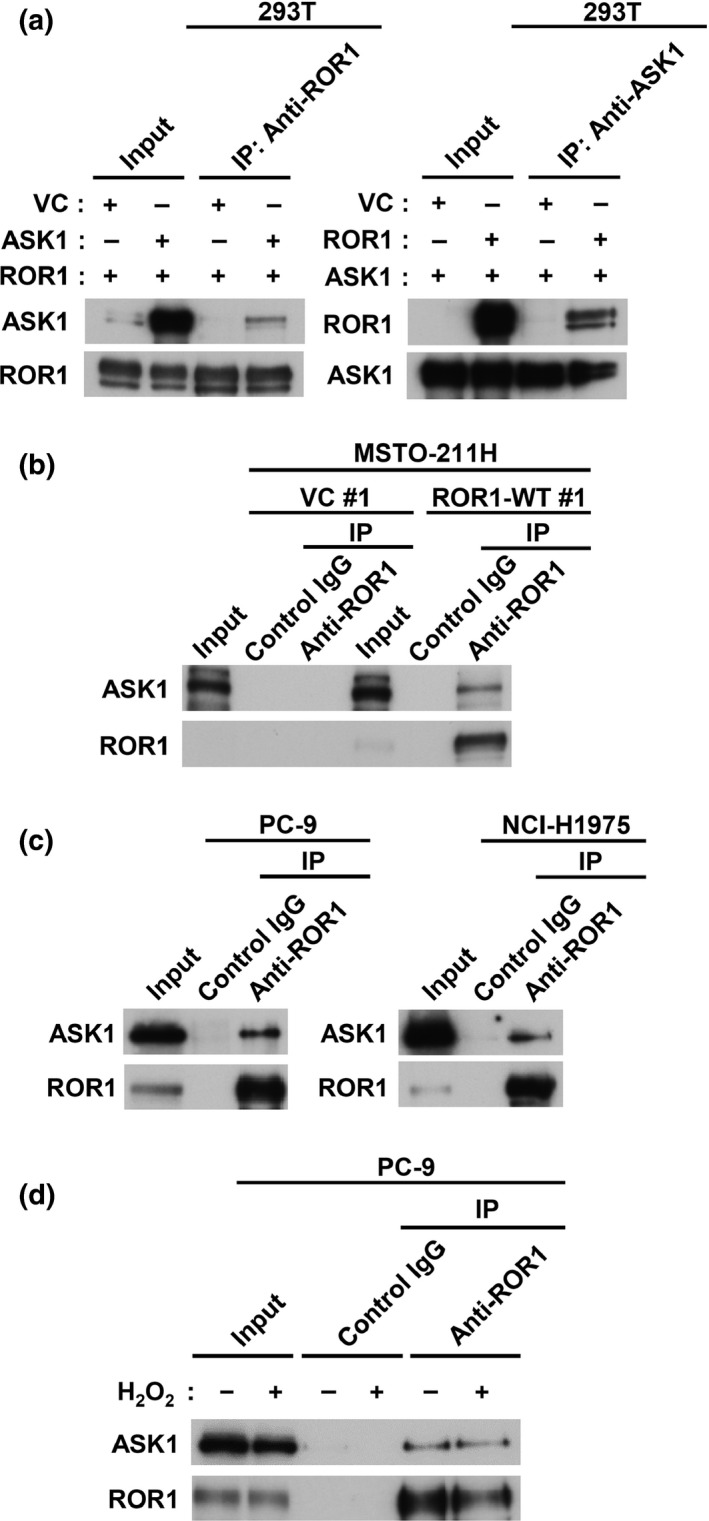
Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) physically interacts with apoptosis signal‐regulating kinase 1 (ASK1). (a) Immunoprecipitation–Western blot (IP‐WB) analysis of interaction between ROR1 and ASK1 co‐introduced into 293T cells. (b) IP‐WB analysis of interaction of endogenous ASK1 with ROR1 introduced into MSTO‐211H cells. (c) IP‐WB analysis of interaction between endogenous ROR1 and ASK1 using lysates of PC‐9 and NCI‐H1975 cells. (d) IP‐WB analysis of interaction of endogenous ROR1 and ASK1 in presence or absence of H2O2 in PC‐9 cells. VC, empty vector.
We then carried out detailed mapping of the ASK1 binding site of the ROR1 protein. Various deletion mutants of ROR1 were co‐transfected with ASK1 into COS‐7 cells (Fig. 5a), followed by IP‐WB analysis of their interactions. We found that the interaction between ROR1 and ASK1 required the presence of the C‐terminal serine/threonine‐rich domain of ROR1 (Fig. 5b). These results showed that ROR1 interacts with ASK1 at the C‐terminus in both steady state and ROS‐inducing conditions.
Figure 5.
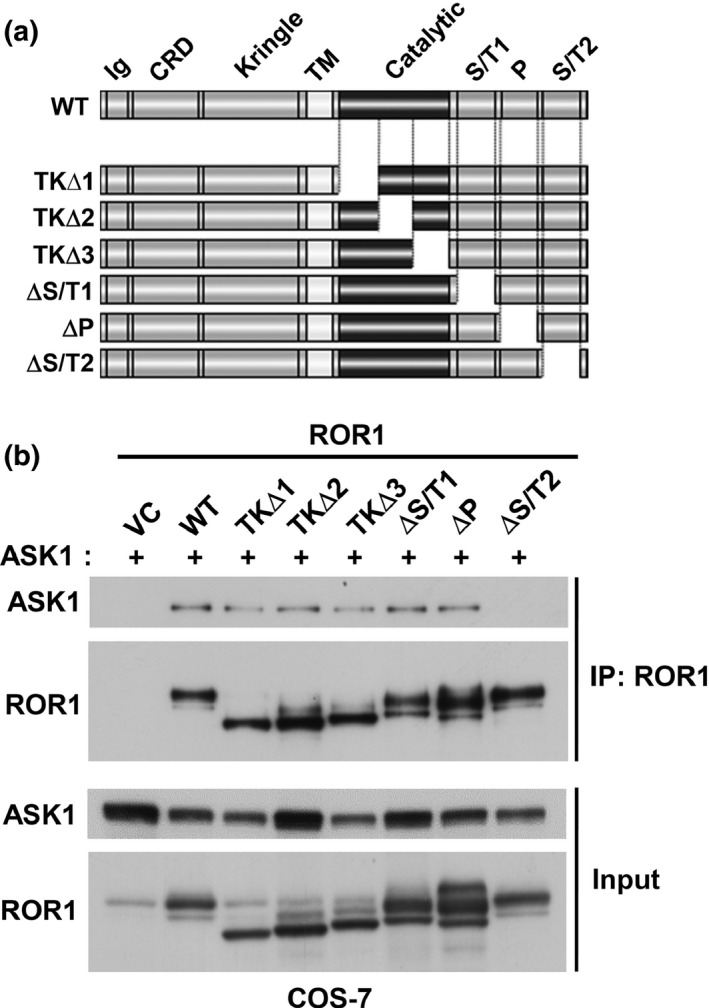
Fine mapping of apoptosis signal‐regulating kinase 1 (ASK1)‐interacting domain of receptor tyrosine kinase‐like orphan receptor 1 (ROR1). (a) Schematic diagram of deletion mutants of ROR1. Immunoprecipitation (IP)–Western blot analysis of COS‐7 cells co‐introduced with ASK1 and various forms of ROR1. ΔP, mutant lacking a proline‐rich region; ΔS/T1 and ΔS/T2, mutants lacking one of the two serine/threonine‐rich regions; TK∆1, TK∆2, and TK∆3, mutants lacking one‐third of the kinase domain; WT, wild‐type ROR1. (b) IP–Western blot analysis for fine mapping of ASK1‐interacting domain of ROR1.
Kinase activity of ROR1 is required for inhibition of ASK1 phosphorylation
Next, we examined whether the kinase activity of ROR1 is required for inhibition of ASK1 activity. MSTO‐211H cells, which had been stably transfected with either a wild‐type ROR1 (ROR1‐WT) or a kinase‐dead (ROR1‐KD) mutant, were treated with H2O2 and subjected to WB analysis. In contrast to ROR1‐WT, ROR1‐KD failed to significantly repress H2O2‐induced ASK1 phosphorylation as well as consequential p38 phosphorylation (Fig. 6a). In addition, we noted that H2O2‐induced cell growth inhibition was partly rescued by overexpression of ROR1‐WT but not by that of ROR1‐KD in MSTO‐211H cells (Fig. 6b). Consistent with these findings, flow cytometric analysis revealed a significant reduction in sub‐G1 cells among MSTO‐211H cells overexpressing ROR1‐WT but not ROR1‐KD (Fig. 6c). Together, the present findings showed that ROR1 physically interacts with ASK1 and that ROR1 kinase activity is required to reduce that activation of ASK1, leading to repression of pro‐apoptotic ASK1‐p38 signaling in lung adenocarcinoma cells.
Figure 6.
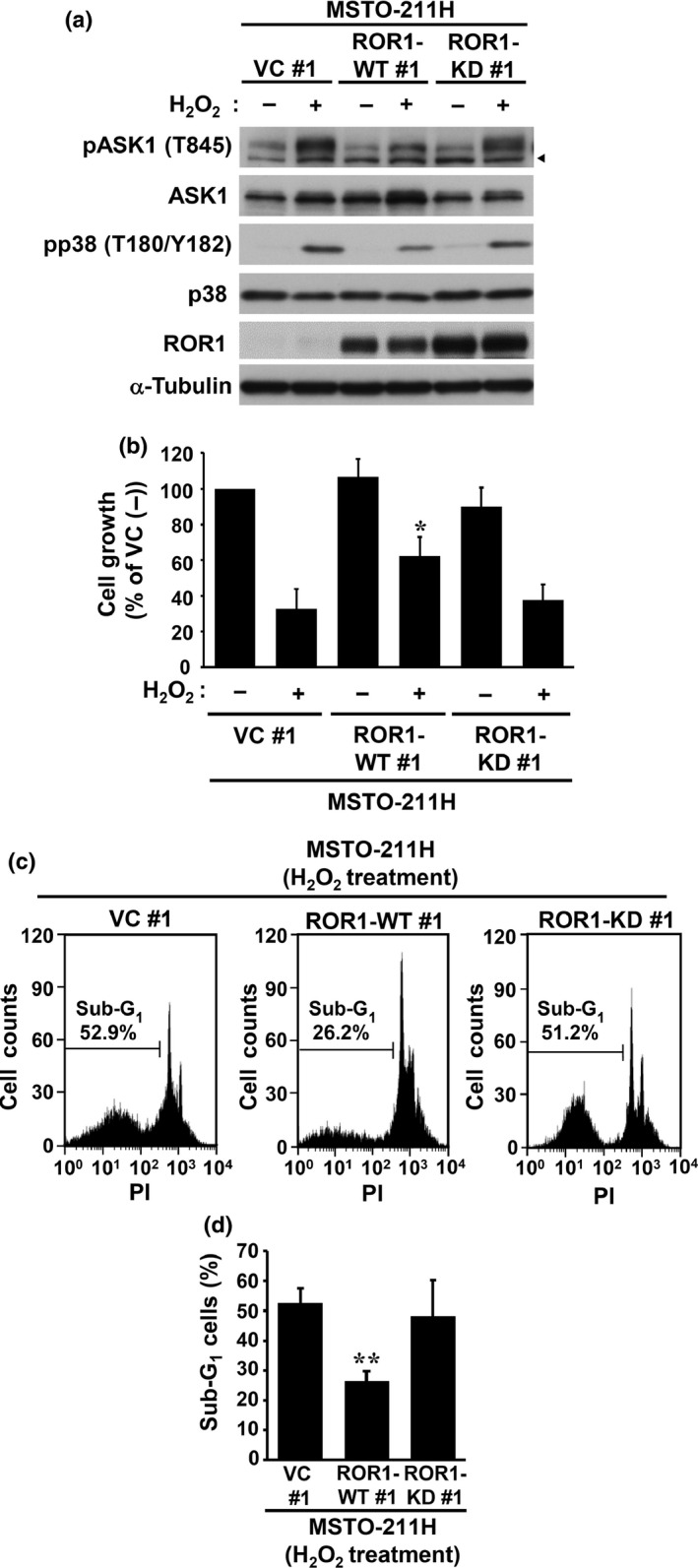
Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) kinase activity is required for inhibition of reactive oxygen species‐elicited phosphorylation of apoptosis signal‐regulating kinase 1 (ASK1) and p38. (a) Western blot analysis of ASK1 and p38 phosphorylation in the presence or absence of H2O2 in MSTO‐211H cells stably expressing wild‐type (WT) or kinase‐dead (KD) ROR1. Arrowheads indicate non‐specific bands. (b) Colorimetric assay of cell proliferation in MSTO‐211H transfectants in the presence or absence of H2O2. (c) Representative results of flow cytometric analysis of apoptosis induction in ROR1‐transfected MSTO‐211H cells treated with H2O2. (d) Flow cytometric analysis of sub‐G1 cells. Representative data from five independent experiments are shown as the mean ± SEM. *P < 0.05, **P < 0.01 vs empty vector (VC), as determined by Student's t‐test. PI, propidium iodide.
Discussion
We previously reported that ROR1 sustains a favorable balance between pro‐survival and pro‐apoptotic signaling, thus playing a role as a key downstream molecule of the NKX2‐1/TTF‐1 lineage‐survival oncogene.6 In contrast to significant advances in understanding of the molecular mechanisms related to ROR1 pro‐survival signaling, how ROR1 represses pro‐apoptotic signaling remains elusive. The present findings clearly show that ROR1 is involved in negative regulation of the pro‐apoptotic ASK1–p38 axis through its physical association with ASK1. Overexpression of ROR1 inhibited activation of ASK1 in response to oxidative stress in an ROR1 kinase‐dependent manner. Intriguingly, ASK1 phosphorylation was also repressed in a steady state conditions without exogenous ROS‐inducing stress, suggesting the existence of ROR1‐mediated repression of ASK1 activation in response to intrinsic cellular stresses. Thus, our results suggest that ROR1 may be part of an important cellular machinery for repression of irrelevant ASK1–p38 activation.
Activity of ASK1 is known to be tightly regulated by phosphorylation, as it has been observed at a representative Thr845 residue within the activation loop of the kinase domain,20 while there are additional residues phosphorylated by various kinases, including Ser83,21 Ser967,22 Ser1034,23 and Tyr718.24 Nevertheless, it remains to be elucidated which amino acid(s) of ASK1 among its more than 40 possible phosphorylation sites is affected either directly or indirectly by ROR1. As ASK1 is also known to be both positively25, 26, 27 and negatively13, 14, 15, 16, 17, 18 regulated through interaction with various other proteins, it is also possible that ASK1 activation may be inhibited by ROR1 by its effects on such ASK1‐interacting proteins, or a yet unidentified one.
The NKX2‐1/TTF‐1 oncogene is crucially involved in maintenance of physiological functions in normal lungs, thus it cannot be considered as a therapeutic target, even though its genetic aberrations are among the most frequent in lung adenocarcinoma.6 ROR1 is a crucial mediator of lineage‐survival signaling of NKX2‐1/TTF‐1 and ROR1 inhibition represses lung cancer cells, which even include EGFR tyrosine kinase inhibitor‐resistant ones due to the presence of T790M EGFR, MET amplification, and/or hepatocyte growth factor overexpression.10 Thus, ROR1 appears to be a promising molecular target for a future novel therapeutic method to treat this devastating cancer. As the present study indicates that ROR1 counteracts oxidative stress‐induced ASK1 activation and apoptosis, enhanced treatment efficacy of a ROS‐inducing chemotherapeutic agent when combined with ROR1 inhibition is anticipated. In addition to lung adenocarcinoma development, ROR1 has been suggested to be involved in the pathogenesis of various other human cancers, such as ovarian, colon, lung, skin, pancreatic, testicular, bladder, uterus, prostate, gastric cancers, melanoma, and lymphomas.28, 29, 30, 31, 32, 33, 34 In addition, onco‐fetal expression of ROR1 is another attractive feature.10, 28, 29
In conclusion, ROR1 directly interacts with ASK1, repressing ASK1 phosphorylation and resultant ASK1‐p38 pro‐apoptotic signaling. A future study is awaited for development of a therapeutic means to inhibit this attractive molecular target. It would also be interesting to investigate whether ROR1 has a role as a mediator of outside‐in signaling to the ASK1–p38 axis.
Disclosure Statement
The authors have no conflict of interest.
Supporting information
Fig. S1. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) interacts with apoptosis signal‐regulating kinase 1 (ASK1). Effects of co‐treatment with siASK1 and siROR1 in (a) PC‐9 and (b) NCI‐H1975 cells using siASK1 #2. Colorimetric analyses were carried out at 5 days after co‐transfection. Data are shown as the mean ± SEM (n = 3). *P < 0.05.
Fig. S2. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) interacts with apoptosis signal‐regulating kinase 1 (ASK1). Immunoprecipitation–Western blot analysis of endogenous ROR1 and ASK1 using lysates of NCI‐H441 and SK‐LC‐5 cells.
Table S1. Endogenous expression of NKX2‐1/TTF‐1 and receptor tyrosine kinase‐like orphan receptor 1 (ROR1) in cell lines used in this study.
Acknowledgments
This work was supported in part by a Grant‐in‐Aid for Scientific Research on Innovative Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan; Grants‐in‐Aid for Scientific Research (A) and (C) from the Japan Society for the Promotion of Science; the Program for Development of Innovative Research on Cancer Therapeutics (P‐Direct) from the Japan Agency for Medical Research and Development; and a grant from the Princess Takamatsu Cancer Research Fund.
Cancer Sci 107 (2016) 155–161
Funding Information
Ministry of Education, Culture, Sports, Science and Technology of Japan; Japan Society for the Promotion of Science; Japan Agency for Medical Research and Development; Princess Takamatsu Cancer Research Fund.
References
- 1. Tanaka H, Yanagisawa K, Shinjo K et al Lineage‐specific dependency of lung adenocarcinomas on the lung development regulator TTF‐1. Cancer Res 2007; 67: 6007–11. [DOI] [PubMed] [Google Scholar]
- 2. Osada H, Tatematsu Y, Yatabe Y, Horio Y, Takahashi T. ASH1 gene is a specific therapeutic target for lung cancers with neuroendocrine features. Cancer Res 2005; 65: 10680–5. [DOI] [PubMed] [Google Scholar]
- 3. Osada H, Tomida S, Yatabe Y et al Roles of achaete‐scute homologue 1 in DKK1 and E‐cadherin repression and neuroendocrine differentiation in lung cancer. Cancer Res 2008; 68: 1647–55. [DOI] [PubMed] [Google Scholar]
- 4. Nishikawa E, Osada H, Okazaki Y et al miR‐375 is activated by ASH1 and inhibits YAP1 in a lineage‐dependent manner in lung cancer. Cancer Res 2011; 71: 6165–73. [DOI] [PubMed] [Google Scholar]
- 5. Garraway LA, Sellers WR. From integrated genomics to tumor lineage dependency. Cancer Res 2006; 66: 2506–8. [DOI] [PubMed] [Google Scholar]
- 6. Yamaguchi T, Hosono Y, Yanagisawa K, Takahashi T. NKX2‐1/TTF‐1: an enigmatic oncogene that functions as a double‐edged sword for cancer cell survival and progression. Cancer Cell 2013; 23: 718–23. [DOI] [PubMed] [Google Scholar]
- 7. Kendall J, Liu Q, Bakleh A et al Oncogenic cooperation and coamplification of developmental transcription factor genes in lung cancer. Proc Natl Acad Sci USA 2007; 104: 16663–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Weir BA, Woo MS, Getz G et al Characterizing the cancer genome in lung adenocarcinoma. Nature 2007; 450: 893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kwei KA, Kim YH, Girard L et al Genomic profiling identifies TITF1 as a lineage‐specific oncogene amplified in lung cancer. Oncogene 2008; 27: 3635–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yamaguchi T, Yanagisawa K, Sugiyama R et al NKX2‐1/TITF1/TTF‐1‐Induced ROR1 is required to sustain EGFR survival signaling in lung adenocarcinoma. Cancer Cell 2012; 21: 348–61. [DOI] [PubMed] [Google Scholar]
- 11. Ichijo H, Nishida E, Irie K et al Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science 1997; 275: 90–4. [DOI] [PubMed] [Google Scholar]
- 12. Tobiume K, Matsuzawa A, Takahashi T et al ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep 2001; 2: 222–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Saitoh M, Nishitoh H, Fujii M et al Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 1998; 17: 2596–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang L, Chen J, Fu H. Suppression of apoptosis signal‐regulating kinase 1‐induced cell death by 14‐3‐3 proteins. Proc Natl Acad Sci USA 1999; 96: 8511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cho SG, Lee YH, Park HS et al Glutathione S‐transferase mu modulates the stress‐activated signals by suppressing apoptosis signal‐regulating kinase 1. J Biol Chem 2001; 276: 12749–55. [DOI] [PubMed] [Google Scholar]
- 16. Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf‐1 promotes cell survival by antagonizing apoptosis signal‐regulating kinase 1 through a MEK‐ERK independent mechanism. Proc Natl Acad Sci USA 2001; 98: 7783–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho SG, Kim JW, Lee YH et al Identification of a novel antiapoptotic protein that antagonizes ASK1 and CAD activities. J Cell Biol 2003; 163: 71–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yoon KW, Cho JH, Lee JK et al CIB1 functions as a Ca(2+)‐sensitive modulator of stress‐induced signaling by targeting ASK1. Proc Natl Acad Sci USA 2009; 106: 17389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell 2007; 129: 1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tobiume K, Saitoh M, Ichijo H. Activation of apoptosis signal‐regulating kinase 1 by the stress‐induced activating phosphorylation of pre‐formed oligomer. J Cell Physiol 2002; 191: 95–104. [DOI] [PubMed] [Google Scholar]
- 21. Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt phosphorylates and negatively regulates apoptosis signal‐regulating kinase 1. Mol Cell Biol 2001; 21: 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Q, Wilkins BJ, Lee YJ, Ichijo H, Molkentin JD. Direct interaction and reciprocal regulation between ASK1 and calcineurin‐NFAT control cardiomyocyte death and growth. Mol Cell Biol 2006; 26: 3785–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fujii K, Goldman EH, Park HR, Zhang L, Chen J, Fu H. Negative control of apoptosis signal‐regulating kinase 1 through phosphorylation of Ser‐1034. Oncogene 2004; 23: 5099–104. [DOI] [PubMed] [Google Scholar]
- 24. Yu L, Min W, He Y et al JAK2 and SHP2 reciprocally regulate tyrosine phosphorylation and stability of proapoptotic protein ASK1. J Biol Chem 2009; 284: 13481–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chang HY, Nishitoh H, Yang X, Ichijo H, Baltimore D. Activation of apoptosis signal‐regulating kinase 1 (ASK1) by the adapter protein Daxx. Science 1998; 281: 1860–3. [DOI] [PubMed] [Google Scholar]
- 26. Nishitoh H, Saitoh M, Mochida Y et al ASK1 is essential for JNK/SAPK activation by TRAF2. Mol Cell 1998; 2: 389–95. [DOI] [PubMed] [Google Scholar]
- 27. Noguchi T, Takeda K, Matsuzawa A et al Recruitment of tumor necrosis factor receptor‐associated factor family proteins to apoptosis signal‐regulating kinase 1 signalosome is essential for oxidative stress‐induced cell death. J Biol Chem 2005; 280: 37033–40. [DOI] [PubMed] [Google Scholar]
- 28. Fukuda T, Chen L, Endo T et al Antisera induced by infusions of autologous Ad‐CD154‐leukemia B cells identify ROR1 as an oncofetal antigen and receptor for Wnt5a. Proc Natl Acad Sci USA 2008; 105: 3047–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Baskar S, Kwong KY, Hofer T et al Unique cell surface expression of receptor tyrosine kinase ROR1 in human B‐cell chronic lymphocytic leukemia. Clin Cancer Res 2008; 14: 396–404. [DOI] [PubMed] [Google Scholar]
- 30. Daneshmanesh AH, Mikaelsson E, Jeddi‐Tehrani M et al Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int J Cancer 2008; 123: 1190–5. [DOI] [PubMed] [Google Scholar]
- 31. Zhang S, Chen L, Cui B et al ROR1 is expressed in human breast cancer and associated with enhanced tumor‐cell growth. PLoS ONE 2012; 7: e31127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhang S, Chen L, Wang‐Rodriguez J et al The onco‐embryonic antigen ROR1 is expressed by a variety of human cancers. Am J Pathol 2012; 181: 1903–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hojjat‐Farsangi M, Ghaemimanesh F, Daneshmanesh AH et al Inhibition of the receptor tyrosine kinase ROR1 by anti‐ROR1 monoclonal antibodies and siRNA induced apoptosis of melanoma cells. PLoS ONE 2013; 8: e61167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 34. Zhang S, Cui B, Lai H et al Ovarian cancer stem cells express ROR1, which can be targeted for anti‐cancer‐stem‐cell therapy. Proc Natl Acad Sci USA 2014; 111: 17266–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) interacts with apoptosis signal‐regulating kinase 1 (ASK1). Effects of co‐treatment with siASK1 and siROR1 in (a) PC‐9 and (b) NCI‐H1975 cells using siASK1 #2. Colorimetric analyses were carried out at 5 days after co‐transfection. Data are shown as the mean ± SEM (n = 3). *P < 0.05.
Fig. S2. Receptor tyrosine kinase‐like orphan receptor 1 (ROR1) interacts with apoptosis signal‐regulating kinase 1 (ASK1). Immunoprecipitation–Western blot analysis of endogenous ROR1 and ASK1 using lysates of NCI‐H441 and SK‐LC‐5 cells.
Table S1. Endogenous expression of NKX2‐1/TTF‐1 and receptor tyrosine kinase‐like orphan receptor 1 (ROR1) in cell lines used in this study.