

Abstract
Recent strategies for treating CML patients have focused on investigating new combinations of tyrosine kinase inhibitors (TKIs) as well as identifying novel translational research agents that can eradicate CML leukemia‐initiating cells (CML‐LICs). However, little is known about the therapeutic benefits such CML‐LIC targeting therapies might bring to CML patients. In this study, we investigated the therapeutic potential of EW‐7197, an orally bioavailable transforming growth factor‐β signaling inhibitor which has recently been approved as an Investigational New Drug (NIH, USA), to suppress CML‐LICs in vivo. Compared to TKI treatment alone, administration of TKI plus EW‐7197 to CML‐affected mice significantly delayed disease relapse and prolonged survival. Notably, combined treatment with EW‐7197 plus TKI was effective in eliminating CML‐LICs even if they expressed the TKI‐resistant T315I mutant BCR‐ABL1 oncogene. Collectively, these results indicate that EW‐7197 may be a promising candidate for a new therapeutic that can greatly benefit CML patients by working in combination with TKIs to eradicate CML‐LICs.
Keywords: ALK5 inhibitor, CML stem cells, relapse prevention, TGF‐β signaling, TKI resistance
The presence of the BCR‐ABL1 fusion oncogene in HSCs causes the stem cell disease CML.1, 2 Although the discovery of TKIs, such as the first‐generation TKI IM and the second‐generation TKIs dasatinib, nilotinib, and bosutinib, has dramatically improved the prognoses of CML patients, a cure remains elusive. For example, in the Stop IM trial, 61% of CML patients who achieved deep molecular response following 2 years of IM treatment suffered relapses of their disease after IM was stopped.3 Many other studies have shown that long‐term treatment with IM or second‐generation TKIs cannot completely cure CML patients.4, 5, 6, 7, 8, 9, 10
CML‐LICs are the cellular sources of the vast majority of differentiated CML cells and are reportedly responsible for the recurrence of CML disease following TKI therapy.2, 11, 12 Because TKIs target only actively dividing CML cells, quiescent CML‐LICs escape TKI‐mediated elimination. In addition, it seems that some of the remaining quiescent CML stem cells acquire TKI‐resistant mutations, such as the T315I mutation,13 in the BCR‐ABL1 and generate TKI‐resistant CML‐LICs. Although it has been expected that the third‐generation version of the pan‐Abl inhibitor AP24534 (ponatinib) would improve the survival of TKI‐resistant CML patients,14 it appears that CML‐LICs can acquire additional BCR‐ABL1 mutations that allow these cells to escape TKI‐mediated killing.13, 15 Thus, many oncologists believe that, to completely eradicate CML‐LICs and prevent the recurrence of CML disease, TKIs must be coupled with novel therapeutics targeting alternative molecular pathways.12
It has been reported that TGF‐β signaling plays both suppressive and supportive roles in normal hematopoiesis and leukemogenesis.16 In particular, Yamazaki et al. found that TGF‐β signaling is required for the maintenance of HSCs within their BM microenvironmental niche.17 In the same vein, we showed that intrinsic TGF‐β signaling maintains CML‐LICs in vivo.18 In contrast, Krause et al. showed that supraphysiological levels of TGF‐β1 secreted within the BM niche can decrease numbers of CML‐LICs.19 Thus, from a therapeutics point of view, both the promotion and suppression of TGF‐β signaling should be pursued for the development of new agents able to eliminate CML‐LICs.
EW‐7197 is a novel kinase inhibitor that targets TGF‐β type I receptor kinase ALK5 and can be given to animals by oral gavage.20, 21, 22 We hypothesized that EW‐7197 might be an attractive candidate for a new CML therapeutic by virtue of its ability to inhibit TGF‐β signaling in vivo and thereby potentially eliminate CML‐LICs. In this study, we take the first steps towards exploring whether EW‐7197 might be of therapeutic benefit to CML patients by evaluating the effects of combined treatment with EW‐7197 and TKIs on CML‐affected mice in vivo.
Materials and Methods
Transduction/transplantation‐based CML mouse models
Transduction/transplantation‐based CML mouse models used in this study were established by infection of HSCs isolated from C57BL/6 mice (Sankyo‐Lab Service, Tsukuba, Japan) with one of three types of retroviruses carrying human BCR‐ABL1 oncogenes: MSCV‐BCR‐ABL1‐ires‐GFP; MSCV‐BCR‐ABL1; and MSCV‐BCR‐ABL1‐T315I mutant‐ires‐GFP. In all three cases, retroviral packaging cells (Plat‐E) were transiently transfected with MSCV‐BCR‐ABL1 plasmids using FuGene6 (Roche, Mannheim, Germany).18 HSCs transduced with retroviruses were transplanted into irradiated (9 Gy) recipient C57BL/6 mice as described previously.18
Scl/Tal1‐tTA x TRE‐BCR‐ABL1 double tg‐CML mouse model
Scl/Tal1‐tTA (JAX strain 6209)23 and TRE‐BCR‐ABL1 (JAX strain 6202)24 tg‐mice (FVB/N background) were purchased from the Jackson Laboratory (Bay harbor, ME). These Scl/Tal1‐tTA and TRE‐BCR‐ABL1 animals were interbred to generate Scl/Tal1‐tTA × TRE‐BCR‐ABL1 double tg‐mice. These mutants were maintained in cages supplied with drinking water containing 20 μg/mL Dox (Sigma, St Louis, MO). At 5 weeks after birth, expression of the BCR‐ABL1 oncogene was induced by replacing the Dox‐containing drinking water with normal drinking water. Consistent with a previous report,25, 26 CML‐like disease developed in the double tg‐mutants approximately 2–5 weeks after Dox withdrawal. All animal care in our laboratory was in accordance with the guidelines for animal and recombinant DNA experiments of Kanazawa University (Kanazawa, Japan).
Cell subset terminology
We consider the most primitive murine “LT‐CML stem cells” to be CD150+CD135−CD48−cKit+Lin−Sca1+ cells in tg‐CML‐affected mice, and murine “CML‐MPPs” to be KLS cells in transduction/transplantation‐based CML‐affected mice and tg‐CML‐affected mice. We use the term “CML‐LICs” in the mouse context to refer collectively to both of these subpopulations. In the human context, we use “CML‐LICs” to refer to CD34+CD38−Lin− cells from CML patients.
Mouse survival
For “TKI‐insensitive” survival experiments using CML‐affected mice, IM (Novartis, Basel, Switzerland) was given to mice by oral gavage (200 mg/kg/day) on days 8–90 after transplantation of BCR‐ABL1 + CML‐MPPs.18 For “TKI‐resistant” recurrence experiments using T315I CML‐affected mice, ponatinib (AP24534; Selleck Chemicals, Houston, TX) was given to mice by oral gavage (15 mg/kg/day) on days 8–60 after transplantation of BCR‐ABL1‐T315I+ CML‐MPPs. In both cases, we accessed a stock solution of EW‐719720 that was prepared at 2 μg/mL in an artificial gastric fluid solution (900 mL ddH2O containing 2.0 g NaCl, 7 mL conc. HCl, and 3.2 g pepsin). Once every 3 days between days 15 to 90 post‐transplantation with BCR‐ABL1 + CML‐MPPs, or between days 15 to 60 post‐transplantation with BCR‐ABL1‐T315I + CML‐MPPs, a dilution of this mixture in artificial gastric fluid solution was given by oral gavage to CML‐affected mice to achieve a final concentration of 2.5 mg EW‐7197/kg body weight. Mouse survival was monitored for 125 days in “TKI‐insensitive” experiments and for 100 days in “TKI‐resistant” recurrence experiments.
Isolation of murine CML‐MPPs and LT‐CML stem cells
For experiments evaluating the effect of EW‐7197 on CML‐LICs cocultured on OP‐9 stromal cells in vitro, we isolated a CML‐MPP fraction containing retrovirus‐infected GFP/BCR‐ABL1‐positive and GFP/BCR‐ABL1‐T315I mutant‐positive KLS cells from CML‐affected mice, as previously described.18 For experiments evaluating the expression levels of TGF‐β target genes in CML‐LICs by quantitative real‐time RT‐PCR analysis, we isolated CML KLS cells from tetracycline‐inducible tg‐CML‐affected mice (Data S1).
For experiments evaluating the PD of EW‐7197 toward primitive LT‐CML stem cells in vivo, BM mononuclear cells were isolated from the two hind limbs of tetracycline‐inducible tg‐CML‐affected mice at 5 weeks after Dox withdrawal, as previously described.25 We stained these cells with anti‐Sca‐1(E13‐161.7)‐PE, anti‐CD4(L3T4)‐FITC, anti‐CD8(53‐6.7)‐FITC, anti‐B220(RA3‐6B2)‐FITC, anti‐TER119(Ly‐76)‐FITC, anti‐Gr‐1(RB6‐8C5)‐FITC, and anti‐Mac1(M1/70)‐FITC (all from BD Biosciences, Franklin Lakes, NJ), anti‐CD48(HM48‐1)‐APC‐Cy7 and anti‐CD150/SLAM(TC15‐12F12.2)‐Pacific blue (both from BioLegend, San Diego, CA), and anti‐cKit(ACK2)‐APC, and anti‐CD135/Flk2/Flt3(A2F10)‐biotin (both from eBiosciences, San Diego, CA) antibodies, followed by streptavidin‐PE‐Cy7 (BD Biosciences). We sorted these cells by flow cytometry using a FACS Aria III instrument (BD Biosciences) and isolated a cell fraction containing CD150/SLAM‐positive, CD135/Flk2/Flt3‐negative, CD48‐negative KLS (CD150+CD135−CD48−KLS) cells, which were deemed to be primitive LT‐CML stem cells.25
Colony‐forming capacity
To determine colony‐forming capacity after a combination treatment of EW‐7197 plus IM, or EW‐7197 plus ponatinib, we cocultured freshly isolated CML‐LICs on OP‐9 stromal cells in the presence of DMSO or EW‐7197 for 24 h.18 Cells were then treated with additional DMSO, 1 μM IM (LC Laboratories, Woburn, MA), or 1 μM ponatinib (AP24534; Selleck Chemicals) and cultured for another 2 days (total, 3 days). Colonies were counted 7 days later as previously described.18
Pharmacokinetics
To assess the PK of EW‐7197, tetracycline‐inducible tg‐CML‐affected mice were fasted overnight and given EW‐7197 (10 mg/kg) by oral gavage as described above. Blood samples were collected before treatment and at 30 min, 2 h, 4 h, and 8 h post‐treatment. Plasma concentrations of EW‐7197 were determined by liquid chromatography/tandem mass spectroscopy using an Agilent 1200 series HPLC and an Agilent 6410 triple quadrupole mass spectrometer equipped with an electrospray ionization source (Agilent Technologies Santa Clara, CA).
Pharmacodynamics using Duolink® in situ PLA technology
The PD of EW‐7197 was determined based on phosphorylation of Smad3. EW‐7197 (2.5 mg/kg) was given to tetracycline‐inducible tg‐CML‐affected mice by oral gavage. Then we isolated a cell fraction containing primitive LT‐CML stem cells (CD150+CD135−CD48−KLS cells) from tg‐CML‐affected mice by flow cytometry using a FACSAria III instrument (BD Biosciences).27 Phospho‐Smad3 was detected in cells using anti‐Smad3 (ab75512; Abcam, Cambridge, UK) and rabbit anti‐phospho‐Ser423/425 Smad3 (ab51451; Abcam) antibodies by the highly sensitive Duolink® in situ PLA technology approach.27
Determination of WBC numbers in PB
The CML‐affected mice received dasatinib (5 mg/kg/day) plus vehicle or EW‐7197 (2.5 mg/kg every third day) by oral gavage for 30 days. For blood cell counts, PB from the post‐orbital vein was collected in a heparinized microtube (Drummond Scientific, Broomall, PA) and analyzed on a CellTac (Nihonkoden, Tokyo, Japan).
Colony‐forming capacity of primary human CML‐LICs
Viable BM mononuclear cells from three human patients with CP‐CML were purchased from Allcells (#06‐255, #06‐620, and #147742, Alameda, CA). Cells were stained with anti‐CD34(8G12), anti‐CD38(HIT2), anti‐CD3(SK7), anti‐CD16(3G8), anti‐CD19(SJ25C1), anti‐CD20(L27), anti‐CD14(MϕP9), and anti‐CD56(NCAM16.2) antibodies (BD Biosciences). A mixture of mAbs recognizing CD3, CD16, CD19, CD20, CD14, and CD56 was used to identify Lin− cells, and CD34+CD38−Lin− cells were purified.18 To determine the effects of EW‐7197 alone or a combination of EW‐7197 plus dasatinib, CD34+CD38−Lin− cells were cultured on OP‐9 stromal cells under hypoxic conditions (3% O2). After harvesting and washing in PBS, the colony‐forming ability of primitive human CML‐LICs was evaluated by culture in semi‐solid medium (Methocult GF+H4435; Stem Cell Technologies, Vancouver, Canada).
Statistical analyses
Statistical differences were determined using unpaired Student's t‐test for P‐values, and the log–rank non‐parametric test for survival curves.
Results
Novel ALK5 kinase inhibitor EW‐7197 suppresses growth of CML‐LICs in vitro
We first examined whether the novel ALK5 inhibitor EW‐7197,20 which inhibits intracellular TGF‐β signaling,20, 21 could suppress the colony‐forming capacity of freshly isolated murine CML‐MPPs in vitro. Interestingly, EW‐7197 treatment dramatically reduced the colony‐forming capacity of CML‐MPPs in vitro in a dose‐dependent manner (Fig. 1a). When EW‐7197 treatment was combined with IM, the in vitro colony‐forming capacity of CML‐MPPs was much more efficiently suppressed than by treatment with IM alone (Fig. 1b, columns 4–6 vs. 3).
Figure 1.
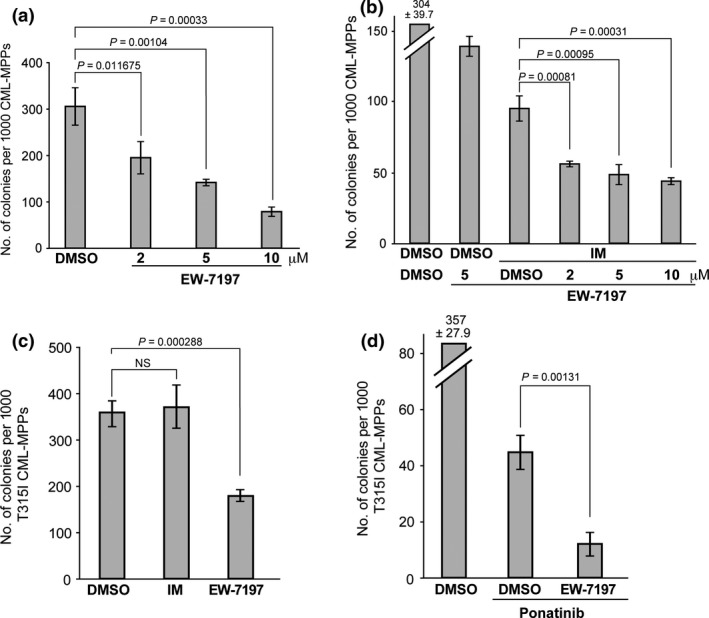
EW‐7197 inhibits the colony‐forming capacity of murine CML‐LICs in vitro. (a,b) Colony‐forming capacity of freshly isolated GFP/BCR‐ABL1+ cKit+Lineage−Sca1+ (KLS) CML multipotent progenitors (CML‐MPPs) that were cocultured on OP‐9 stromal cells under hypoxic conditions (3% O2) and treated for 3 days with either DMSO (control) or the indicated concentrations of EW‐7197 (a), or for 3 days with DMSO or EW‐7197, without or with 1 μM IM (b). Colony‐forming capacity was determined by culture in methylcellulose. Data shown are the mean colony number ± SD (n = 3). (c,d) Colony‐forming capacity of freshly isolated T315I‐BCR‐ABL1‐GFP + KLS CML‐MPPs that were treated for 3 days with either DMSO (vehicle control), 1 μM imatinib mesylate (IM), or 5 μM EW‐7197 (c), or for 3 days with DMSO or EW‐7197, with the addition (or not; DMSO) at 24 h post‐EW‐7197 treatment of 1 μM ponatinib for 48 h (d). Results were analyzed as for (a,b). NS, not significant.
To definitively show that EW‐7197 can suppress the colony‐forming capacity of TKI‐resistant CML‐MPPs in vitro, we further established a mouse model of TKI‐resistant CML by infecting HSCs with retrovirus expressing the TKI‐resistant T315I mutant BCR‐ABL1 oncogene.28 As expected, IM‐treated T315I+CML‐MPPs fully retained their colony‐forming capacity (Fig. 1c, column 2). However, treatment of these T315I+CML‐MPPs with EW‐7197 alone efficiently reduced their in vitro colony‐forming capacity despite their expression of the TKI‐resistant T315I mutant BCR‐ABL1 oncogene (Fig. 1c, column 3). Although ponatinib alone had an inhibitory effect on the in vitro colony‐forming capacity of T315I+CML‐MPPs, a substantial subpopulation of these cells clearly survived the treatment (Fig. 1d, column 2). Importantly, a combination of EW‐7197 plus ponatinib dramatically suppressed the in vitro colony‐forming capacity of residual T315I+CML‐MPPs (Fig. 1d, column 3). Moreover, treatment in vitro with EW‐7197 of CML‐MPPs isolated from tetracycline‐inducible CML‐affected mice suppressed the endogenous mRNA expression of TGF‐β target genes such as Smad7 and Cdkn1A (Fig. S1). These results supported our hypothesis that EW‐7197 might be a possible candidate for a novel CML therapeutic capable of killing CML‐LICs.
Pharmacokinetics and PD of EW‐7197 in tg‐CML‐affected mice
We next determined the PK of EW‐7197 in the bloodstream of tetracycline‐inducible tg‐CML‐affected mice by assessing its plasma concentration–time profile post‐treatment. We observed that EW‐7197 was rapidly absorbed into the bloodstream from the gastrointestinal tract of tg‐CML‐affected mice, and was then eliminated with a terminal phase half‐life (T1/2) of 3.26 ± 2.47 h (Fig. 2a). The maximum concentration (Cmax) of the drug in plasma was determined to be 625.0 ± 529.7 ng/mL (n = 7) at 30 min post‐administration.
Figure 2.
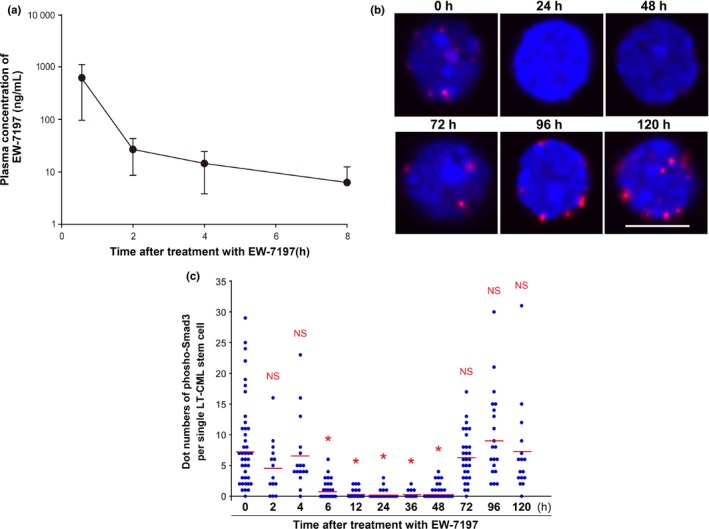
Pharmacokinetics and pharmacodynamics of EW‐7197 in tetracycline‐inducible transgenic (tg)‐CML‐affected mice. (a) Concentration–time profile of EW‐7197 in tetracycline‐inducible tg‐CML‐affected mice. EW‐7197 (10 mg/kg body weight) was given orally to Scl/Tal1‐tTA × TRE‐BCR‐ABL1 double tg mice at 5 weeks after doxycycline withdrawal. Blood samples were obtained from tg‐CML‐affected mice pre‐ and post‐administration of EW‐7197, and plasma concentrations of the drug were determined by liquid chromatography/tandem mass spectroscopy. Data are the mean concentration of EW‐7197 ± SD (n = 7). (b,c) Analyses of suppression of Smad3 phosphorylation by EW‐7197 in long‐term (LT)‐CML stem cells in vivo. (b) Duolink® in situ proximity ligation assay analysis. Tg‐CML‐affected mice received oral EW‐7197 (2.5 mg/kg). At the indicated time points after treatment, mice were killed and LT‐CML stem cells (CD150+ CD135− CD48− KLS) were isolated from hind limbs. Levels of phospho‐Smad3 (red) in LT‐CML stem cells were assessed by Duolink® in situ proximity ligation assay technology. Nuclei were visualized by DAPI (blue). Images shown are representative of three animals examined per time point. Scale bar = 10 μm. (c) Quantitation of the “dot number” of phospho‐Smad3 foci per single LT‐CML stem cell in the panels in (b) at the indicated times after treatment with EW‐7197. Values were determined using the Duolink® Image J software tool. Horizontal bars, cumulative mean values of three independent experiments. *P < 0.00005 versus 0 h. NS, not significant.
The PD of EW‐7197 were established by examining phosphorylation levels of Smad3, which is a direct target of TGF‐β type I receptor kinase ALK5 in the intracellular TGF‐β signaling pathway. To this end, we used the highly sensitive Duolink® in situ PLA technology approach using rabbit antibodies specific for phospho‐Ser423/425 Smad3 and mouse anti‐Smad3 antibody. This assay system is highly sensitive and detects miniscule amounts of a specific protein in fixed cells using an amplified fluorescent signal. As a positive control, we treated primitive LT‐CML stem cells in vitro with TGF‐β1 for 30 min and detected obvious fluorescent foci of phospho‐Smad3 using Duolink® in situ PLA (data not shown). To determine the effect of EW‐7197 on Smad3 phosphorylation, we gave 2.5 mg/kg EW‐7197 to tg‐CML‐affected mice, isolated LT‐CML stem cells from these animals at various time points up to 120 h after treatment, and immediately evaluated levels of Smad3‐Ser423/425 phosphorylation in these cells. We observed a decrease in numbers of fluorescent foci that began at 6 h after treatment and continued until 48 h after treatment with EW‐7197 (Fig. 2b,c), indicating that EW‐7197 effectively inhibits Smad3 phosphorylation in vivo. These data show that in vivo administration of EW‐7197 efficiently decreases ALK5 kinase activity in primitive LT‐CML stem cells of tg‐CML‐affected mice. Furthermore, this suppression is maintained for at least 48 h after treatment.
Prolonged survival of CML‐affected mice treated with a combination of TKI plus EW‐7197
Our observation that EW‐7197 decreased the colony‐forming capacity of CML‐MPPs in vitro prompted us to examine whether this orally bioavailable TGF‐β signaling inhibitor was of any therapeutic benefit to CML‐affected mice. Accordingly, we first evaluated the overall survival of CML‐affected mice that were treated with IM, or with EW‐7197 alone or in combination with IM. Although treatment with IM alone (200 mg/kg daily) delayed disease onset compared to the vehicle‐treated group, IM‐treated CML‐affected mice experienced recurrence of BCR‐ABL1 + disease well before the end of the 125‐day observation period (Fig. 3a, group 3 vs. 1). Treatment once every 3 days with EW‐7197 alone did not markedly extend the survival of CML‐affected mice beyond that of IM‐treated CML mice (Fig. 3a, group 2 vs. 1). However, the combined treatment of IM plus EW‐7197 significantly prolonged the survival of CML‐affected mice (Fig. 3a, group 4 vs. 3). Thus, these results indicated that EW‐7197 possesses therapeutic benefits in combination with a TKI in a CML‐affected mouse model.
Figure 3.

Combined treatment with tyrosine kinase inhibitor plus EW‐7197 prolongs the survival of CML‐affected mice. (a) Irradiated C57BL/6 recipient mice were transplanted with HSCs transduced with the BCR‐ABL1 oncogene. At 8 days post‐transplantation, recipient mice received either vehicle or imatinib mesylate (IM; 200 mg/kg/day). At 15 days post‐transplantation, these CML‐affected mice received additional vehicle alone (1), vehicle plus EW‐7197 (2.5 mg/kg every third day) (2), IM plus vehicle (3), or IM plus EW‐7197 (4). Mouse survival was monitored for up to 125 days. Results shown are cumulative data obtained from four independent experiments. (b,c) EW‐7197 (EW) treatment reduces the frequency of CML multipotent progenitors in vivo. (b) CML‐affected mice received dasatinib (5 mg/kg/day) plus vehicle or EW‐7197 (2.5 mg/kg every third day) by oral gavage for 30 days after bone marrow transplantation. Data shown are representative flow cytometric analyses highlighting GFP/BCR‐ABL1+ cKit+Lineage−Sca1+ (KLS) cells (red rectangles). Cells were gated on GFP +, Lineage−. (c) Mean frequency ± SD of GFP/BCR‐ABL1+ KLS cells among total GFP/BCR‐ABL1+ CML cells (n = 3) from the CML‐affected mice in (b).
Eradication of murine CML‐MPPs in vivo by a combination of TKI plus EW‐7197
To further evaluate the benefits and risks of EW‐7197 treatment, we examined the effects of this agent on fully differentiated and mature BCR‐ABL1‐GFP + CML cells in vivo. Surprisingly, treatment of CML‐affected mice with EW‐7197 alone appeared to increase WBC numbers in PB and to promote splenomegaly (Fig. S2, left). These data implied that EW‐7197 commits CML‐LICs to differentiate into mature CML cells and promotes their proliferation, and thus could pose a significant risk to patients if used as a single treatment. Strikingly, however, combined treatment of CML‐affected mice with the TKI dasatinib plus EW‐7197 completely blocked both the WBC elevation in PB and splenomegaly associated with EW‐7197 monotherapy (Fig. S2, right). Thus, in vivo, treatment with a TKI can overcome the unfavorable proliferation of differentiated CML cells induced by EW‐7197.
We next determined the effect of EW‐7197 on primitive CML‐KLS cells in CML‐affected mice in vivo. In contrast to its effects on the differentiation and proliferation of mature CML cells, the frequency of CML‐KLS cells among BCR‐ABL1‐GFP+CML cells isolated from BM of CML‐affected mice was dramatically decreased by EW‐7197 treatment (Fig. 3b,c). Significantly, although dasatinib alone also reduced the frequency of CML‐KLS cells, the combined treatment of dasatinib plus EW‐7197 had a much greater repressive effect on this population (Fig. 3b,c). These data show that EW‐7197 can inhibit the self‐renewal capacity of CML‐MPPs in CML‐affected mice. Collectively, our results suggest that combined treatment with EW‐7197 and TKI dasatinib may bring significant therapeutic benefits to CML patients by eradicating CML‐MPPs.
Suppression of disease recurrence in TKI‐resistant CML‐affected mice by treatment with EW‐7197 plus ponatinib
A serious problem for human CML patients is the generation during or post‐TKI therapy of CML‐LICs bearing a BCR‐ABL1 oncogene with a TKI‐resistant mutation (such as T315I).13 Given that EW‐7197 treatment suppressed the colony‐forming capacity of murine TKI‐resistant T315I+CML‐MPPs in vitro (as in Fig. 1c,d), we wondered whether this TGF‐β signaling inhibitor might block CML‐MPP function even after the acquisition of TKI resistance. To answer this question, we evaluated the overall survival of TKI‐resistant CML‐affected mice with BCR‐ABL1‐T315I+ disease that were treated with ponatinib alone (15 mg/kg daily), EW‐7197 alone (2.5 mg/kg every third day), or both in combination. Although treatment with ponatinib alone markedly delayed CML onset compared to vehicle‐treated controls, CML recurred in ponatinib‐treated CML‐affected mice after discontinuation of treatment (Fig. 4a, group 3 vs. 1). Treatment with EW‐7197 alone once every 3 days did not prolong the survival of T315I CML‐affected mice compared to the vehicle‐treated group (Fig. 4a, group 2 vs. 1). Neither was there an obvious decrease at 60 days post‐transplantation in the efficiency of induction of TKI‐resistant CML disease in mice that received BCR‐ABL1‐T315I +CML‐MPPs and were treated with ponatinib plus EW‐7197 compared to treatment with ponatinib alone. However, the rate of recurrence of TKI‐resistant CML in mice receiving ponatinib plus EW‐7197 was dramatically decreased after discontinuation of treatment compared to the rate in the group treated with ponatinib alone (Fig. 4a, group 4 vs. 3). These results indicate that oral co‐administration of EW‐7197 and ponatinib can reduce the disease‐relapsing capacity of T315I+CML‐MPPs in vivo.
Figure 4.
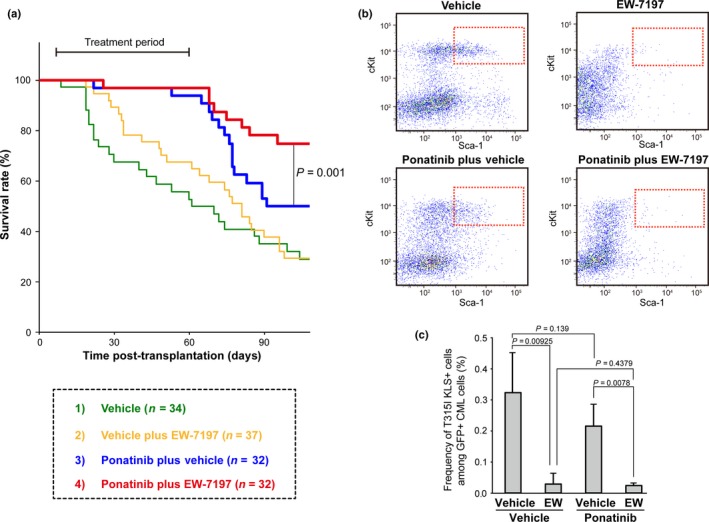
Combined treatment with ponatinib and EW‐7197 (EW) reduces disease relapse rate in mice with tyrosine kinase inhibitor (TKI)‐resistant BCR‐ABL1‐T315I+ CML. (a) Prolonged survival of TKI‐resistant T315I CML‐affected mice receiving ponatinib plus EW‐7197. Irradiated C57BL/6 recipient mice were transplanted with HSCs transduced with the BCR‐ABL1‐T315I mutant oncogene. At 8 days post‐transplantation, recipient mice received either vehicle or ponatinib (15 mg/kg/day). At 15 days post‐transplantation, these TKI‐resistant T315I CML‐affected mice received additional vehicle alone (1), vehicle plus EW‐7197 (2.5 mg/kg every third day) (2), vehicle plus ponatinib (15 mg/kg/day) (3), or ponatinib plus EW‐7197 (4) for 60 days post‐transplantation. Mouse survival and disease recurrence were monitored for up to 100 days. Results shown are cumulative data obtained from four independent experiments. (b) Reduced frequency of TKI‐resistant CML multipotent progenitors following treatment with ponatinib plus EW‐7197 in vivo. TKI‐resistant T315I CML‐affected mice received ponatinib (15 mg/kg/day) plus vehicle or EW‐7197 (2.5 mg/kg every third day) by oral gavage for 30 days. Data shown are representative flow cytometric analyses highlighting T315I BCR‐ABL1‐GFP + cKit+Lineage−Sca1+ (KLS) cells (red rectangles). Cells were gated on GFP +, Lineage−. (c) Mean frequency ± SD of BCR‐ABL1‐T315I‐GFP + KLS cells among total BCR‐ABL1‐T315I‐GFP + CML cells (n = 3) from the T315I CML‐affected mice in (b).
To evaluate the in vivo effects of EW‐7197 on TKI‐resistant CML‐MPPs in T315I CML‐affected mice, we focused on self‐renewal capacity. Consistent with our observations in Figure 3(b,c), EW‐7197 treatment effectively blocked the self‐renewal of T315I+CML‐KLS cells (Fig. 4b,c). Combined treatment with ponatinib and EW‐7197 led to even greater suppression of T315I CML‐KLS cells (Fig. 4b,c). Thus, EW‐7197 can efficiently block the self‐renewal of even TKI‐resistant CML‐MPPs in vivo.
EW‐7197 suppresses in vitro colony‐forming capacity of human CML‐LICs
To assess the relevance of our mouse results to the human situation, we examined whether EW‐7197 could suppress the in vitro colony‐forming capacity of primary human CML‐LICs obtained from CP‐CML patients. We used cell‐sorting to isolate the CD34+CD38−Lin− cell population, which includes CML‐LICs, from three CP‐CML patients and cocultured these cells on OP‐9 stromal cells under hypoxic (3% O2) conditions.18 Intriguingly, EW‐7197 significantly suppressed the in vitro colony‐forming capacity of all three samples of human CML‐LICs (Fig. 5). When EW‐7197 treatment was combined with dasatinib, the in vitro colony‐forming capacity of the human CML‐LICs was much more efficiently suppressed than by treatment with dasatinib alone (Fig. 5c). These data suggest that treatment with a combination of TKIs and EW‐7197 may be a promising strategy for eradicating primitive CML‐LICs in human CML patients.
Figure 5.
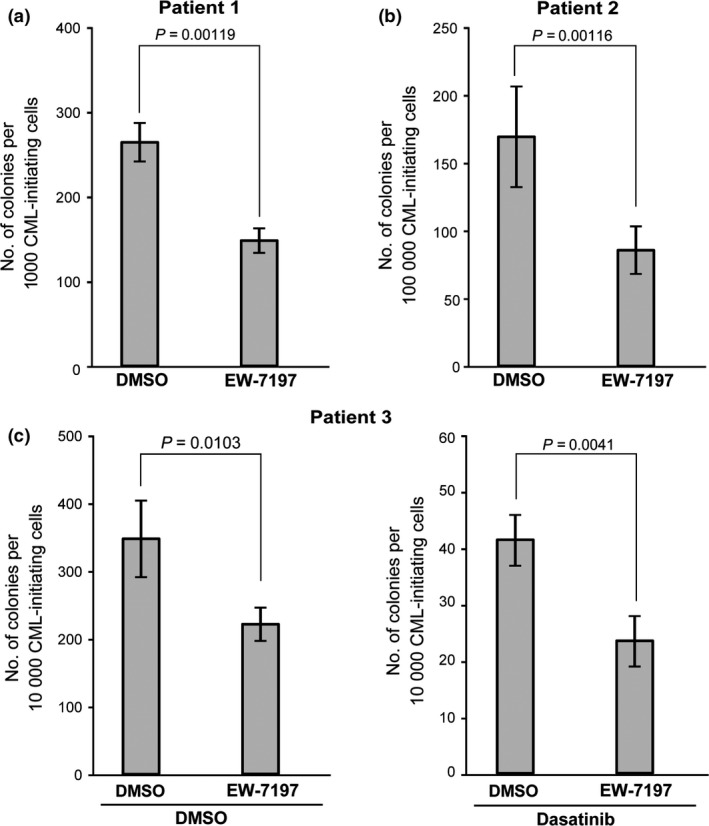
EW‐7197 inhibits the colony‐forming capacity of human CML leukemia‐initiating cells in vitro. Colony‐forming capacity of CD34+ CD38−Lin− cells isolated from bone marrow mononuclear cells obtained from three CP‐CML patients. CML leukemia‐initiating cells were cocultured on OP‐9 stromal cells under hypoxic conditions (3% O2) and treated for 3 days with either DMSO (vehicle control) or 5 μM EW‐7197 (a,b, and c, left panel), or for 3 days with DMSO or EW‐7197, with the addition (or not; DMSO) at 24 h post‐EW‐7197 treatment of 500 nM dasatinib for 48 h (c, right). Colony‐forming capacity was determined by culture in methylcellulose. Data shown are the mean colony number ± SD (n = 3).
Discussion
Because the intrinsic TGF‐β signaling pathway plays an essential role in the maintenance of immature CML‐LICs in vivo, 18 the inclusion of a TGF‐β inhibitor in the treatment of CML patients could be a promising therapeutic approach. In this study, we showed that the novel orally bioavailable TGF‐β inhibitor EW‐7197 can suppress murine CML‐LICs in vivo and human CML‐LICs in vitro. Notably, this strategy, which is based on inhibiting intrinsic TGF‐β signaling, also suppressed recurrence in mice of disease associated with the TKI‐resistant BCR‐ABL1 T315I mutation, the same alteration responsible for disease recurrence in CML patients. Our results therefore suggest that EW‐7197 may be an attractive candidate for a new therapy able to eradicate CML‐LICs in CP‐CML patients when used in combination with TKIs.
It has recently been reported that CML‐LICs cells are not oncogene‐addicted, and that a therapeutic strategy that targets only ABL1 kinase activity cannot completely eliminate CML‐LICs.29, 30, 31 Thus, it is important to develop novel therapeutics that do not depend on inhibiting BCR‐ABL1 kinase activity.12 In this study, EW‐7197 inhibited ALK5 and thus efficiently blocked intrinsic TGF‐β signal transduction in CML‐LICs in vitro and in vivo even after they had developed TKI resistance. These results suggest that TGF‐β signaling is an alternative molecular pathway that does not depend on BCR‐ABL1 activity to maintain CML‐LICs in vivo, and that combined treatment with EW‐7197 and TKIs may therefore be able to eradicate “TKI‐insensitive” and “TKI‐resistant” CML‐LICs in human CML patients.
Interestingly, Krause et al. have reported that CML‐affected mice transgenically overexpressing the receptor for parathyroid hormone on osteoblastic cells undergo increased bone remodeling associated with elevated TGF‐β1 expression and show prolonged overall survival.19 In contrast, the results of our study indicate that inhibition of intrinsic TGF‐β signaling may in fact “wake up” CML‐LICs so that they can become activated and thus vulnerable to killing by TKIs.18, 32 These opposing results are likely due to the well‐established differences in the effects of differing doses of TGF‐β1 on hematopoietic cells.16 Our work clearly revealed that inhibition of intrinsic TGF‐β signaling in CML‐LICs is a promising strategy for suppressing their function. We therefore believe that the intrinsic TGF‐β signaling pathway should be considered for the development of novel CML therapeutics.
In conclusion, we have shown that the novel orally bioavailable TGF‐β signaling inhibitor EW‐7197 can suppress the function of CML‐LICs in vitro and in vivo. EW‐7197 has already been approved by the FDA and certified as an Investigational New Drug. A phase I clinical trial for the use of EW‐7197 in the treatment of patients with advanced solid tumors has been initiated (NCT02160106, NIH). Our continuing investigation of EW‐7197 should contribute to the development of new therapeutics that can specifically suppress the effects of TGF‐β signaling on CML‐LICs, and so may provide concrete clinical benefits to CP‐CML patients.
Disclosure Statement
The authors have no conflict of interest.
Abbreviations
- BM
bone marrow
- CP
chronic phase
- Dox
doxycycline
- EW‐7197
[N‐((4‐([1,2,4]‐triazolo[1,5‐a]pyridin‐6‐yl)‐5‐(6‐methyl pyridin‐2‐yl)‐1H‐imidazol‐2‐yl) methyl)‐2‐fluoroaniline]
- HSC
hematopoietic stem cell
- IM
imatinib mesylate
- KLS
cKit+Lineage−Sca1+
- LIC
leukemia‐initiating cell
- Lin
Lineage
- LT
long‐term
- MPP
multipotent progenitors
- MSCV
murine stem cell virus
- PB
peripheral blood
- PD
pharmacodynamics
- PE
phycoerythrin
- PK
pharmacokinetics
- PLA
proximity ligation assay
- Tg
transgenic
- TGF‐β
transforming growth factor‐β
- TKI
tyrosine kinase inhibitor
- WBC
white blood cell
Supporting information
Fig. S1. Quantitative RT‐PCR of transforming growth factor‐β target genes.
Fig. S2. Effects on mature CML cells.
Data S1. Materials and methods.
Acknowledgments
We thank Drs. I.‐H. Ha, and K. Miyazono for their valuable input, D.G. Tenen for the Scl/Tal1‐tTA and TRE‐BCR‐ABL1 transgenic mouse strains, H. Honda for BCR‐ABL1 cDNA, T. Kitamura for Plat‐E retroviral packaging cells, and Ms. J.‐H. Park and I. Baek for administrative assistance. This work was supported by grants from the Yasuda Medical Foundation, the Mochida Memorial Foundation, the Naito Foundation, the Funding Program for Next Generation World‐Leading Researchers (LS050), Innovative Areas, Integrative Research on Cancer Microenvironment Network from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and a Grant‐in‐Aid for Scientific Research (B) from MEXT (26290038), Japan (K.N.). Support was also provided by the Bio‐Synergy Research Project (NRF‐2014M3A9C4048735) of Ministry of Science, ICT and Future Planning through the National Research Foundation, Korea (S.‐J.K.) and by a grant from the Ministry of Education, Science and Technology, Korea (20082005585) (D.‐K.K.).
Cancer Sci 107 (2016) 140–148
Funding Information
Yasuda Medical Foundation; Mochida Memorial Foundation; Naito Foundation; Ministry of Education, Culture, Sports, Science and Technology, Japan; Ministry of Science, ICT and Future Planning through the National Research Foundation, Korea; Ministry of Education, Science and Technology, Korea
References
- 1. Naka K, Hoshii T, Hirao A. Novel therapeutic approach to eradicate tyrosine kinase inhibitor resistant chronic myeloid leukemia stem cells. Cancer Sci 2010; 101: 1577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. O'Hare T, Zabriskie MS, Eiring AM, Deininger MW. Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat Rev Cancer 2012; 12: 513–26. [DOI] [PubMed] [Google Scholar]
- 3. Mahon FX, Rea D, Guilhot J et al Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: the prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol 2010; 11: 1029–35. [DOI] [PubMed] [Google Scholar]
- 4. Bhatia R, Holtz M, Niu N et al Persistence of malignant hematopoietic progenitors in chronic myelogenous leukemia patients in complete cytogenetic remission following imatinib mesylate treatment. Blood 2003; 101: 4701–7. [DOI] [PubMed] [Google Scholar]
- 5. Druker BJ, Guilhot F, O'Brien SG et al Five‐year follow‐up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med 2006; 355: 2408–17. [DOI] [PubMed] [Google Scholar]
- 6. Hochhaus A, O'Brien SG, Guilhot F et al Six‐year follow‐up of patients receiving imatinib for the first‐line treatment of chronic myeloid leukemia. Leukemia 2009; 23: 1054–61. [DOI] [PubMed] [Google Scholar]
- 7. Giles FJ, Rosti G, Beris P et al Nilotinib is superior to imatinib as first‐line therapy of chronic myeloid leukemia: the ENESTnd study. Expert Rev Hematol 2010; 3: 665–73. [DOI] [PubMed] [Google Scholar]
- 8. Kantarjian H, Shah NP, Hochhaus A et al Dasatinib versus imatinib in newly diagnosed chronic‐phase chronic myeloid leukemia. N Engl J Med 2010; 362: 2260–70. [DOI] [PubMed] [Google Scholar]
- 9. Ross DM, Branford S, Seymour JF et al Patients with chronic myeloid leukemia who maintain a complete molecular response after stopping imatinib treatment have evidence of persistent leukemia by DNA PCR. Leukemia 2010; 24: 1719–24. [DOI] [PubMed] [Google Scholar]
- 10. Ibrahim AR, Eliasson L, Apperley JF et al Poor adherence is the main reason for loss of CCyR and imatinib failure for chronic myeloid leukemia patients on long‐term therapy. Blood 2011; 117: 3733–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sinclair A, Latif AL, Holyoake TL. Targeting survival pathways in chronic myeloid leukaemia stem cells. Br J Pharmacol 2013; 169: 1693–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmed W, Van Etten RA. Alternative approaches to eradicating the malignant clone in chronic myeloid leukemia: tyrosine‐kinase inhibitor combinations and beyond. Hematology Am Soc Hematol Educ Program 2013; 2013: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gorre ME, Mohammed M, Ellwood K et al Clinical resistance to STI‐571 cancer therapy caused by BCR‐ABL gene mutation or amplification. Science 2001; 293: 876–80. [DOI] [PubMed] [Google Scholar]
- 14. O'Hare T, Shakespeare WC, Zhu X et al AP24534, a pan‐BCR‐ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation‐based resistance. Cancer Cell 2009; 16: 401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Khorashad JS, Kelley TW, Szankasi P et al BCR‐ABL1 compound mutations in tyrosine kinase inhibitor‐resistant CML: frequency and clonal relationships. Blood 2013; 121: 489–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blank U, Karlsson S. TGF‐β signaling in the control of hematopoietic stem cells. Blood 2015; 125: 3542–50. [DOI] [PubMed] [Google Scholar]
- 17. Yamazaki S, Ema H, Karlsson G et al Nonmyelinating Schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell 2011; 147: 1146–58. [DOI] [PubMed] [Google Scholar]
- 18. Naka K, Hoshii T, Muraguchi T et al TGF‐β‐FOXO signalling maintains leukaemia‐initiating cells in chronic myeloid leukaemia. Nature 2010; 463: 676–80. [DOI] [PubMed] [Google Scholar]
- 19. Krause DS, Fulzele K, Catic A et al Differential regulation of myeloid leukemias by the bone marrow microenvironment. Nat Med 2013; 19: 1513–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin CH, Krishnaiah M, Sreenu D et al Discovery of N‐((4‐([1,2,4]Triazolo[1,5‐α]pyridin‐6‐yl)‐5‐(6‐methylpyridin‐2‐yl)‐1H‐imidazol‐2‐yl)methyl)‐2‐ fluoroaniline (EW‐7197): a highly potent, selective, and orally bioavailable inhibitor of TGF‐β type 1 receptor kinase as cancer immunotherapeutic/antifibrotic agent. J Med Chem 2014; 57: 4213–38. [DOI] [PubMed] [Google Scholar]
- 21. Yoon JH, Jung SM, Park SH et al Activin receptor‐like kinase5 inhibition suppresses mouse melanoma by ubiquitin degradation of Smad4, thereby derepressing eomesodermin in cytotoxic T lymphocytes. EMBO Mol Med 2013; 5: 1720–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Son JY, Park S‐Y, Kim S‐J et al EW‐7197, a novel ALK‐5 kinase inhibitor, potently inhibits breast to lung metastasis. Mol Cancer Ther 2014; 13: 1704–16. [DOI] [PubMed] [Google Scholar]
- 23. Koschmieder S, Gottgens B, Zhang P et al Inducible chronic phase of myeloid leukemia with expansion of hematopoietic stem cells in a transgenic model of BCR‐ABL leukemogenesis. Blood 2005; 105: 324–34. [DOI] [PubMed] [Google Scholar]
- 24. Huettner CS, Zhang P, Van Etten RA, Tenen DG. Reversibility of acute B‐cell leukaemia induced by BCR‐ABL1. Nat Genet 2000; 24: 57–60. [DOI] [PubMed] [Google Scholar]
- 25. Reynaud D, Pietras E, Barry‐Holson K et al IL‐6 controls leukemic multipotent progenitor cell fate and contributes to chronic myelogenous leukemia development. Cancer Cell 2011; 20: 661–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pellicano F, Scott MT, Helgason GV et al The anti‐proliferative activity of kinase inhibitors in chronic myeloid leukaemia cells is mediated by FOXO transcription factors. Stem Cells 2014; 32: 2324–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Naka K, Jomen Y, Ishihara K et al Dipeptide species regulate p38MAPK‐Smad3 signalling to maintain chronic myelogenous leukaemia stem cells. Nat Commun 2015; 6: 8039. doi: 10.1038/ncomms9039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peng C, Brain J, Hu Y et al Inhibition of heat shock protein 90 prolongs survival of mice with BCR‐ABL‐T315I‐induced leukemia and suppresses leukemic stem cells. Blood 2007; 110: 678–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Corbin AS, Agarwal A, Loriaux M et al Human chronic myeloid leukemia stem cells are insensitive to imatinib despite inhibition of BCR‐ABL activity. J Clin Invest 2011; 121: 396–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamilton A, Helgason GV, Schemionek M et al Chronic myeloid leukemia stem cells are not dependent on Bcr‐Abl kinase activity for their survival. Blood 2012; 119: 1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ma L, Shan Y, Bai R et al A therapeutically targetable mechanism of BCR‐ABL‐independent imatinib resistance in chronic myeloid leukemia. Sci Transl Med 2014; 6: 252ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Møller GM, Frost V, Melo JV, Chantry A. Upregulation of the TGFβ signalling pathway by Bcr‐Abl: implications for haemopoietic cell growth and chronic myeloid leukaemia. FEBS Lett 2007; 581: 1329–34. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Quantitative RT‐PCR of transforming growth factor‐β target genes.
Fig. S2. Effects on mature CML cells.
Data S1. Materials and methods.