

Abstract
MAP1LC3B, an ortholog of yeast Atg8 and a member of the family of proteins formerly also known as ATG8 in mammals (LC3B henceforth in the text), functions in autophagosome formation and autophagy substrate recruitment. LC3 exists in both a soluble (autophagosome-independent) form as well as a lipid modified form that becomes tightly incorporated into autophagosomal membranes. Although LC3 is known to associate with tens of proteins, relatively little is known about soluble LC3 aside from its interactions with the LC3 lipid conjugation machinery. In previous studies we found autophagosome-independent GFP-LC3B diffuses unusually slowly for a protein of its size, suggesting it may constitutively associate with a high molecular weight complex, form homo-oligomers or aggregates, or reversibly bind microtubules or membranes. To distinguish between these possibilities, we characterized the size, stoichiometry, and organization of autophagosome-independent LC3B in living cells and in cytoplasmic extracts using fluorescence recovery after photobleaching (FRAP) and fluorescence polarization fluctuation analysis (FPFA). We found that the diffusion of LC3B was unaffected by either mutational disruption of its lipid modification or microtubule depolymerization. Brightness and homo-FRET analysis indicate LC3B does not homo-oligomerize. However, mutation of specific residues on LC3B required for binding other proteins and mRNA altered the effective hydrodynamic radius of the protein as well as its stoichiometry. We conclude that when not bound to autophagosomes, LC3B associates with a multicomponent complex with an effective size of ~500 kDa in the cytoplasm. These findings provide new insights into the nature of soluble LC3B and illustrate the power of FRAP and FPFA to investigate the emergent properties of protein complexes in the autophagy pathway.
Keywords: MAP1LC3B, FRAP, FPFA, fluorescence, FRET, diffusion, autophagy, FCS, hydrodynamic radius, ATG8
Introduction
Autophagy is a major pathway for the capture and degradation of cytosolic materials via the lysosome. A distinctive feature of the pathway is the formation of a double-membrane-bound vesicle called the autophagosome.1 Autophagosome membrane expansion, fusion, and substrate selection are performed, in part, by a family of ATG8 proteins.2-10 Despite these proposed major roles for ATG8 proteins in autophagy, there are still many open questions in regard to how ATG8 proteins carry out these tasks.11
ATG8 proteins share high structural (but not sequence) homology with ubiquitin, and thus have been termed ubiquitin-like proteins.12 Interestingly, unlike ubiquitin, which is conjugated to other proteins, ATG8 proteins are conjugated to the head group of the lipid phosphatidylethanolamine.13 In mammals, the best-studied ATG8 family member, MAP1LC3B (LC3B), exists in both soluble (LC3-I), as well as lipid-modified form (LC3-II).14 Under conditions of stress or starvation, LC3 lipid modification becomes upregulated, in turn targeting the protein to phagophore membranes.15 LC3 is released from autophagosome membranes by the cysteine protease activity of ATG4B.16
LC3 and other members of the ATG8 protein family have numerous interacting proteins, suggestive of their possible participation in multiprotein complexes.17-25 In addition, many LC3-interacting proteins contain a consensus LC3-interacting motif (W/F/Y-X-X-L/I/V) for efficient binding to a hydrophobic surface on LC3 in the region of residues F52 and L53, as well as R70.6,10,26 Mutations to these key residues on LC3′s hydrophobic protein interacting surface disrupt binding to tens of proteins, highlighting the importance of this region for engaging other proteins in productive binding.6,17,18,25,27,28 Importantly, it is unknown if soluble LC3 interacts with its protein partners in a binary fashion or if instead it constitutively associates with multiprotein or other multicomponent complexes to accomplish its varied functions in autophagy.
Using confocal FRAP, our group recently showed that the soluble form of LC3B diffuses unusually slowly for a cytoplasmic protein of its size under basal conditions.29,30 Because the diffusion coefficient of a soluble, freely diffusing molecule is inversely related to its radius, this slower-than-expected-diffusion of LC3B could potentially reflect its association with macromolecular complexes comprised of LC3B and other components of the autophagy pathway. Alternatively, the slow diffusion of soluble LC3B could potentially arise from reversible binding to microtubules in cells31 or the incorporation of LC3B into homo-oligomers, aggregates, or possibly membranes.2,32,33
To address these possibilities, in the current study, we investigated the size, stoichiometry, and organization of LC3B-containing protein complexes using 2 complementary fluorescence-based approaches, confocal fluorescence recovery after photobleaching (FRAP) and fluorescence polarization fluctuation analysis (FPFA). The first of these methods, FRAP, is a powerful and versatile tool for measuring the ensemble dynamics of molecules. The instrumentation for FRAP is now widely accessible in many laboratories, and by analyzing FRAP data with recently described models it is now possible to accurately measure diffusion coefficients, as well as quantify reversible interactions.34-36 FRAP also has the unique ability to quantify the extent of irreversible interactions and very slow turnover on the timescale of the measurements as reflected in the mobile fraction. The second approach, FPFA, is a newly developed experimental method combining fluorescence correlation spectroscopy (FCS) and time-resolved fluorescence anisotropy (TRFA).37 Like FRAP, FCS reports on the diffusional mobility of molecules. However, FCS has exceptional sensitivity enabling measurements of the concentrations of fluorescent proteins in the pM to nM range, and is capable of resolving the diffusion coefficients of multiple diffusing species and their molecular brightness.38 By combining FCS and TRFA into a single measurement, FPFA enables the simultaneous measurement of translational and rotational mobility, concentration, brightness, and homo-FRET.37 Thus, FPFA is a tool that can be used to simultaneously investigate a protein’s mobility, stoichiometry, and organization.
Our FRAP and FPFA measurements reveal that under basal conditions, soluble Venus-LC3B diffuses as if it associates with a complex with an effective molecular weight of approximately 500 kDa in size, and each complex contains on average a single LC3B. Similar complexes were observed in living cells and in cytoplasmic extracts depleted of intact microtubules. In addition, we show that the size and stoichiometry of complexes containing LC3B are altered by mutational disruption of LC3B’s hydrophobic binding surface, but not mutational disruption of LC3B’s lipid modification. The effective size of the LC3B-associated complexes only modestly changes after perturbations to the autophagy pathway with rapamycin or chloroquine treatments. These findings provide new insights into the nature of soluble LC3B and illustrate the power of confocal FRAP and FPFA to provide novel insights into the emergent properties of protein complexes in the autophagy pathway.
Results
Effect of mutations of the hydrophobic-binding interface and lipid modification site on the subcellular distribution of LC3B
In previous studies, we found GFP-LC3B and Venus-LC3B diffuse more slowly in the cytoplasm of cells than expected for a monomer using confocal FRAP, suggesting LC3B may associate with a high molecular weight complex.29,30 Given that many LC3-interacting proteins interact with a hydrophobic interaction surface on LC3 using a W/F/Y-X-X-L/I/V motif,10 we asked whether disruption of specific residues in this region on LC3B will disrupt its association with the putative high molecular weight complexes. To address this question, we focused on 2 sets of mutations, R70A, as well as F52A and L53A. These mutations disrupt binding to at least 18 and 9 binding partners, respectively.25 Several of the disrupted binding partners are involved in activities ranging from LC3′s lipid modification to autophagy substrate specificity.17-19,25 For comparison, we also examined a G120A mutant of LC3B, which is unable to undergo lipid modification.33
We first examined the effects of the R70A, G120A, as well as the F52A and L53A mutations on the subcellular distribution of LC3B in HeLa cells. As previously described, Venus-LC3B is found in a diffuse cytoplasmic form as well as in discrete 0.5 to 2 µm puncta in the cytoplasm corresponding to LC3B labeled autophagosomes, autophagosome independent substrates, and aggregates.32 Some diffuse Venus-LC3B is also observed in the nucleus, consistent with our previous findings quantifying the amount of GFP tagged LC3B in the nucleus (Fig. 1A).29

Figure 1. Venus-LC3BF52A L53A and Venus-LC3BR70A show a reduced association with cytoplasmic puncta compared with wild-type Venus-LC3B. (A) Venus-LC3B, (B) Venus-LC3BF52A L53A, (C) Venus-LC3BR70A, and (D) Venus-LC3BG120A were expressed individually in HeLa cells and imaged live under basal conditions (complete media, 37 °C) using confocal microscopy. Scale bar: 10 µm. (E) The numbers of ~0.5 to 2 µm punctate spots per cell cytoplasm were counted for each of the indicated constructs. Bars represent the median. One-way ANOVA test P < 1 × 10−4. Pairwise comparisons between all of the constructs were made using Bonferonni-corrected t tests as described in the text.
Like wild-type Venus-LC3B, a substantial fraction of diffuse Venus-LC3BF52A L53A, Venus-LC3BR70A, and Venus-LC3BG120A was present in both the cytoplasm and nucleoplasm of live HeLa cells (Fig. 1B–D). However, on average about 2-fold fewer puncta were present in cells expressing Venus-LC3BR70A than in cells expressing Venus-LC3B (P ≤ 0.008; Bonferonni-corrected t test), and even fewer puncta were positive for Venus-LC3BF52A L53A or Venus-LC3BG120A (P ≤ 0.008; t tests) (Fig. 1E). These findings are consistent with the idea that LC3B's hydrophobic binding surface regulates protein-protein interactions, including SQSTM1/p62 (sequestosome 1) binding, which facilitate targeting LC3B to autophagy-independent puncta.27 In addition, they suggest that the F52A and L53A, as well as the G120A mutants are more strongly defective in their ability to bind to cytoplasmic puncta than is the R70A mutant of LC3B.
Since the Venus-LC3BF52A L53A, Venus-LC3BR70A, and Venus-LC3BG120A mutants have defects in the numbers puncta present under steady-state conditions, we wanted to determine if these mutants retain their ability to associate with autophagosomal membranes. To test this, we monitored their accumulation on autophagosomal membranes after inhibition of lysosomal acidification with chloroquine. After treatment with chloroquine, both Venus-LC3BF52A L53A and Venus-LC3BR70A accumulated on autophagosomal membranes, similar to wild-type Venus-LC3B (Fig. 2A–F). This result is consistent with what was previously reported for a GFP-LC3F52A mutant after treatment with both starvation and bafilomycin A1.28 In contrast, no changes to the localization of Venus-LC3BG120A were observed after treatment with chloroquine (Fig. 2G and H). This is consistent with the inability of the G120A mutant to be targeted to autophagosomes by lipid modification of this residue.33

Figure 2. Venus-LC3BF52A L53A and Venus-LC3BR70A, like wild-type Venus-LC3B, accumulate on lysosomal membranes after treatment with chloroquine. Cells expressing (A and B) Venus-LC3B, (C and D) Venus-LC3BR70A, (E and F) Venus-LC3BF52A L53A, or (G and H) Venus-LC3BG120A were imaged under basal conditions (A,C,E, and G) or after a 2 h incubation with 100 μM chloroquine (B,D,F, and H). Scale bar: 10 µm. The steady-state localization of LC3B, R70A, as well as F52A and L53A on autophagosomal membranes is upregulated after blocking lysosome function with chloroquine. In contrast, G120A was not targeted to autophagosomal membranes under these conditions. (I) Individual spots as seen in (B,D,F and H) were selectively photobleached using a 1 μm radius Bleach ROI revealing a large immobile fraction for membrane-associated LC3B, R70A, as well as F52A and L53A puncta in contrast with the smaller immobile fraction of substrate-associated G120A puncta. The differences in the recovery of membrane-associated LC3 vs. substrate-associated LC3 are reflected in the mobile fractions quantified in (J). Symbols for the R70A recovery are underneath the symbols for the F52A and L53A recovery. (K and L) SDS-PAGE and western blot for LC3-I vs. LC3-II under basal conditions and after chloroquine treatment. (K) Antibodies against LC3 show there are no major defects in the formation of GFP-LC3-II for LC3B, R70A, or F52A and L53A, in contrast with the negative control G120A. (L) Similar to (K), except the electrophoresis was performed for a longer period of time in order to obtain better resolution of the higher molecular weight GFP-LC3-I vs. GFP-LC3-II bands. Blotting was performed using an anti-GFP antibody. Antibodies against tubulin were used as loading controls.
It was recently shown, using selective photobleaching of GFP-LC3-associated puncta, that LC3 incorporated into autophagosomal membranes turns over much more slowly than GFP-LC3 associated with autophagy substrates.39 We used this method to further characterize the nature of the LC3B-positive puncta present after incubation with chloroquine (Fig. 2I). The FRAP analysis revealed that while Venus-LC3B, Venus-LC3BR70A and Venus-LC3BF52A L53A puncta had very large immobile fractions, Venus-LC3BG120A had a relatively high mobile fraction (Fig. 2J). This is consistent with the notion that the R70A, as well as the F52A and L53A mutants of LC3B retain their ability to become lipid modified and subsequently incorporated into the membranes of autophagosomes, whereas the LC3BG120A mutant does not. In order to further verify that there are no defects in the LC3-I to LC3-II processing of the R70A, as well as the F52A and L53A mutants, we performed SDS-PAGE and western blotted for LC3 (Fig. 2K) and GFP (Fig. 2L) both under basal conditions and after treatment with chloroquine. This experiment showed that there are no defects in the LC3-I to LC3-II processing of Venus-LC3B, Venus-LC3BF52A L53A, or Venus-LC3BR70A in stark contrast with the GFP-LC3BG120A negative control.33
Disruption of LC3B’s hydrophobic-binding interface changes the effective diffusion of LC3B-associated complexes in living cells
Next, we tested the effect of the R70A, as well as the F52A and F53A mutations, on the diffusional mobility of soluble cytoplasmic LC3B in living cells using a quantitative confocal FRAP assay.34,35 As shown in Figure 3A, this experiment was aimed toward quantifying the diffusion of the diffuse pool of LC3B, and excludes any LC3B associated with puncta. As an internal control, we performed FRAP experiments of Venus, a soluble, freely diffusing protein. The Venus FRAP curve was well fit by a single-component model for Brownian motion with a diffusion coefficient of 41 ± 4 µm2/s and a mobile fraction of 100% in HEK 293 cells (Fig. 3B and C; Table 1). Next, we performed FRAP experiments on Venus-LC3B in the cytoplasm. Consistent with our previous findings,29,30 we found Venus-LC3B had an approximately 2.5-fold slower D compared with that of Venus, much slower than the predicted diffusion coefficient for monomeric Venus-LC3B (P ≤ 0.008; t test) (dashed line in Fig. 3C). This corresponds to an effective molecular weight of ~500 kDa (Table 1). We found similar results in HeLa cells (P ≤ 0.008; t test) (Fig. 3D; Table 2).

Figure 3. Mutations to LC3B’s hydrophobic protein interaction-surface at residues R70 or F52 and L53 change the diffusional mobility of Venus-LC3B in the cytoplasm of living cells. (A) Cells expressing Venus, Venus-LC3B, Venus-LC3BR70A, Venus-LC3BF52A L53A, or Venus-LC3BG120A were photobleached using a 1 μm radius Bleach ROI placed in the cytoplasm such that puncta were avoided. Dashed lines designate the typical Imaging ROI and Bleach ROI used in these experiments. (B) Comparison of the mean FRAP data from approximately 30 HeLa cells expressing either Venus or Venus-LC3B (symbols) were well fit by a Brownian diffusion model (solid lines). The data are normalized between 0 and 1 using (F(t) − F(0)) / (F(∞) − F(0)) to more clearly demonstrate the differences in their recovery rates. (C–F) Box plots of the diffusion coefficients obtained from FRAP experiments on either HEK 293 cells or HeLa cells. While experiments summarized in (C and D) were performed under basal conditions those in (E) were obtained from HeLa cells pretreated for 2 h with 100 μM chloroquine, and those in (F) were obtained from HeLa cells pretreated for 2 h with 200 nM rapamycin. The ratio of D for the monomeric Venus control to LC3B, F52A and L53A, R70A, or G120A is a quantitative indicator of their effective sizes. Whiskers represent the minimum and maximum. N can be found in Table 1. The dashed horizontal lines in (C–E) are the predicted diffusion coefficients for a 45 kDa Venus-LC3B monomer assuming both Venus and Venus-LC3B are spherical. For (C–E) One-way ANOVA test P < 1 × 10−4. Pairwise comparisons between all of the constructs were made using Bonferonni-corrected t tests as described in the text.
Table 1. Predicted molecular weights and mobile fractions for Venus, Venus-LC3B, and Venus-LC3B mutants based on the FRAP diffusion measurements in live HEK 293 cells under basal conditions.
| Construct | Monomer MW (kDa) | FRAP predicted MW (kDa)* | FRAP mobile fraction (%) |
|---|---|---|---|
| Venus | 27 | N/A | 100 ± 1 (34) |
| Venus-LC3B | 45 | 500 ± 200 (30) | 99 ± 1 (30) |
| Venus-LC3B (F52A/L53A) | 45 | 1200 ± 400 (30) | 100 ± 1 (30) |
| Venus-LC3B (R70A) | 45 | 130 ± 50 (30) | 102 ± 2 (30) |
*Calculated assuming diffusing species has a spherical shape; mean ± 95% CI (N, # cells).
Table 2. Predicted molecular weights and mobile fractions for Venus, Venus-LC3B, and Venus-LC3B mutants based on the FRAP diffusion measurements in live HeLa cells under basal conditions, after incubation with 100 μM chloroquine (CQ) for 2 h, and after incubation with 200 nM rapamycin (Rp) for 2 h.
| Construct | Monomer MW (kDa) | Basal predicted MW (kDa)* | CQ predicted MW (kDa)* | Rap predicted MW (kDa)* | NZ predicted MW (kDa)* | Basal mobile fraction (%) | CQ mobile fraction (%) | Rap mobile fraction (%) |
|---|---|---|---|---|---|---|---|---|
| Venus | 27 | N/A | N/A | N/A | N/A | 99.5 ± 0.7 (30) | 99.2 ± 0.7 (80) | 99.5 ± 0.8 (30) |
| Venus-LC3B | 45 | 500 ± 100 (30) | 900 ± 200 (60) | 800 ± 200 (30) | 500 ± 100 (20) | 100.5 ± 0.8 (30) | 99 ± 1 (60) | 99 ± 1 (30) |
| Venus-LC3B (F52A/L53A) | 45 | 1200 ± 300 (35) | 1400 ± 300 (60) | 1600 ± 500 (30) | 900 ± 300 (20) | 98 ± 1 (35) | 99 ± 1 (60) | 100 ± 1 (30) |
| Venus-LC3B (R70A) | 45 | 110 ± 40 (30) | 190 ± 40 (60) | 160 ± 40 (30) | 80 ± 30 (20) | 99.2 ± 0.8 (30) | 99 ± 1 (60) | 100 ± 1 (30) |
| Venus-LC3B (G120A) | 45 | 400 ± 100 (30) | 1000 ± 200 (40) | 900 ± 200 (30) | 700 ± 200 (20) | 99 ± 1 (30) | 99 ± 1 (40) | 100.4 ± 0.7 (30) |
*Calculated assuming diffusing species has a spherical shape; mean ± 95% CI (N, # cells); CQ, chloroquine; Rap, rapamycin; NZ, nocodazole.
After establishing baselines for diffusion comparisons, next, we measured the diffusion of soluble Venus-LC3BF52A L53A and Venus-LC3BR70A by confocal FRAP. We found that their mobile fractions were also both approximately 100% (Tables 1 and 2), similar to that of soluble Venus-LC3B itself (ns; P > 0.008; t tests). However, their diffusion coefficients differed from that of one another as well as from that of Venus-LC3B. Interestingly, in HEK 293 cells, D for Venus-LC3BF52A L53A (11.6 ± 0.7 µm2/s) was slower than that of wild-type Venus-LC3B (15 ± 2 µm2/s) (P ≤ 0.008; t test), while D for Venus-LC3BR70A (24 ± 2 µm2/s) was faster than Venus-LC3B, but still not to the level expected for a Venus-LC3B monomer (P ≤ 0.008; t test) (Fig. 3). Similar results were obtained in HeLa cells (Fig. 3D). Assuming the slow diffusion of the proteins reflects their association with spherical complexes, their predicted molecular weights would be ~1.2 MDa for Venus-LC3BF52A L53A and ~130 kDa for Venus-LC3BR70A (Tables 1 and 2).
The slow diffusion of LC3B is not due to its lipid modification or association with autophagosome membranes
Although our FRAP experiments were directed toward the soluble pool of LC3B, it is formally possible that LC3B’s slow diffusion is a result of a small fraction of lipid modified LC3B incorporated into sub-diffraction vesicles or autophagosomes. This seemed unlikely, because, theoretically, ~250 nm radius vesicles would be expected to diffuse ~100 fold slower than Venus (D1/D2 = 2 nm/250 nm), whereas, experimentally, Venus-LC3B diffused only ~2.5-fold slower. However, our FRAP measurements do not have the resolution to adequately test this possibility without additional experimental information.40 Therefore, to address this question we performed FRAP measurements on Venus-LC3BG120A, since this mutation of LC3 prevents its lipid modification and subsequent incorporation into autophagosomal membranes.41 As shown in Figure 3D, Venus-LC3BG120A diffuses similarly to wild-type Venus-LC3B (ns; P > 0.005; t test). These data support the notion that the slow diffusion of LC3B is not the result of its association with autophagosomes or interactions with other molecules that depend on lipid modification of the protein. Rather, the slow diffusion could reflect the association of LC3B with other molecules.
The effective size of putative LC3B-associated complexes is modestly affected by autophagy modulators
In order to gain further insights into how these complexes are modulated by perturbations to the autophagy pathway, we extended our analysis of the diffusion of LC3B under basal conditions to 2 additional conditions known to modulate the autophagy pathway. The first condition was to inhibit autophagy by incubating the cells with 100 μM chloroquine for 2 h, and the second condition was to stimulate autophagy by incubating the cells with 200 nM rapamycin for 2 h before performing FRAP measurements. We found that rapamycin and chloroquine treatments produced similar changes in the diffusion of the constructs, as shown in Figure 3. At first glance, the trends between the constructs appear to be maintained. However, after normalization to the diffusion of Venus, using the Stokes Einstein relationship as before, we found that the effective molecular weights for the LC3B-associated complexes were modestly upregulated approximately 2-fold compared with basal conditions (P ≤ 0.005; t tests; see Table 2 for a summary). These data suggest that the effective sizes of the soluble LC3B-associated complexes are only modestly affected by perturbations to the autophagy pathway with rapamycin and chloroquine in vivo.
Disruption of LC3B’s hydrophobic-binding interface but not of its lipidation site changes the effective diffusion of LC3B-associated complexes in cytoplasmic extracts
In order to test if the complexes observed in cells are intrinsically stable, or if they require cellular components, such as microtubules, we next examined the properties of the LC3B constructs in cytoplasmic extracts. In principle, it is possible to measure the diffusion of LC3 in cytoplasmic extracts using confocal FRAP; however, we wished to obtain additional information about the putative complexes, such as whether multiple types of complexes were present and how many LC3B molecules were present in each complex. Therefore, we turned to another technique that is sensitive to protein mobility and stoichiometry, FPFA,37 to analyze the LC3B-associated complexes in cytoplasmic extracts. To carry out FPFA measurements on the fluorescently labeled proteins in our cytoplasmic extracts, we utilized a recently described custom-built FPFA instrument with 2-photon excitation to define the focal volume.37 HEK cells expressing Venus or Venus-LC3B were extracted using passive lysis buffer and clarified by a brief high speed spin before the FPFA measurements were performed.
In order to determine the diffusivity and molecular brightness of the species, FPFA macro-time measurements of orthogonally polarized fluorescence emission were cross-correlated. The cross-correlation curves for both Venus and Venus-LC3B were well fit by a single-component model for Brownian motion suggesting the absence of multiple diffusing species with large differences in their sizes (see Fig. 4A for an example of 1- vs. 2-component models). The diffusion coefficients from the fits to Venus and Venus-LC3B were 67 ± 2 and 25 ± 2 µm2/s, respectively (Fig. 4B). Thus, the diffusion coefficient for Venus-LC3B in cell extracts was much slower than Venus alone (approximately 2.5-fold) as seen previously using FRAP in live cells, and much slower than predicted for a freely diffusing Venus-LC3B monomer (P ≤ 0.003; t tests). Making the simplifying assumption that the complexes containing Venus-LC3B are spherical, we calculated their predicted size is about 500 kDa (Table 3).
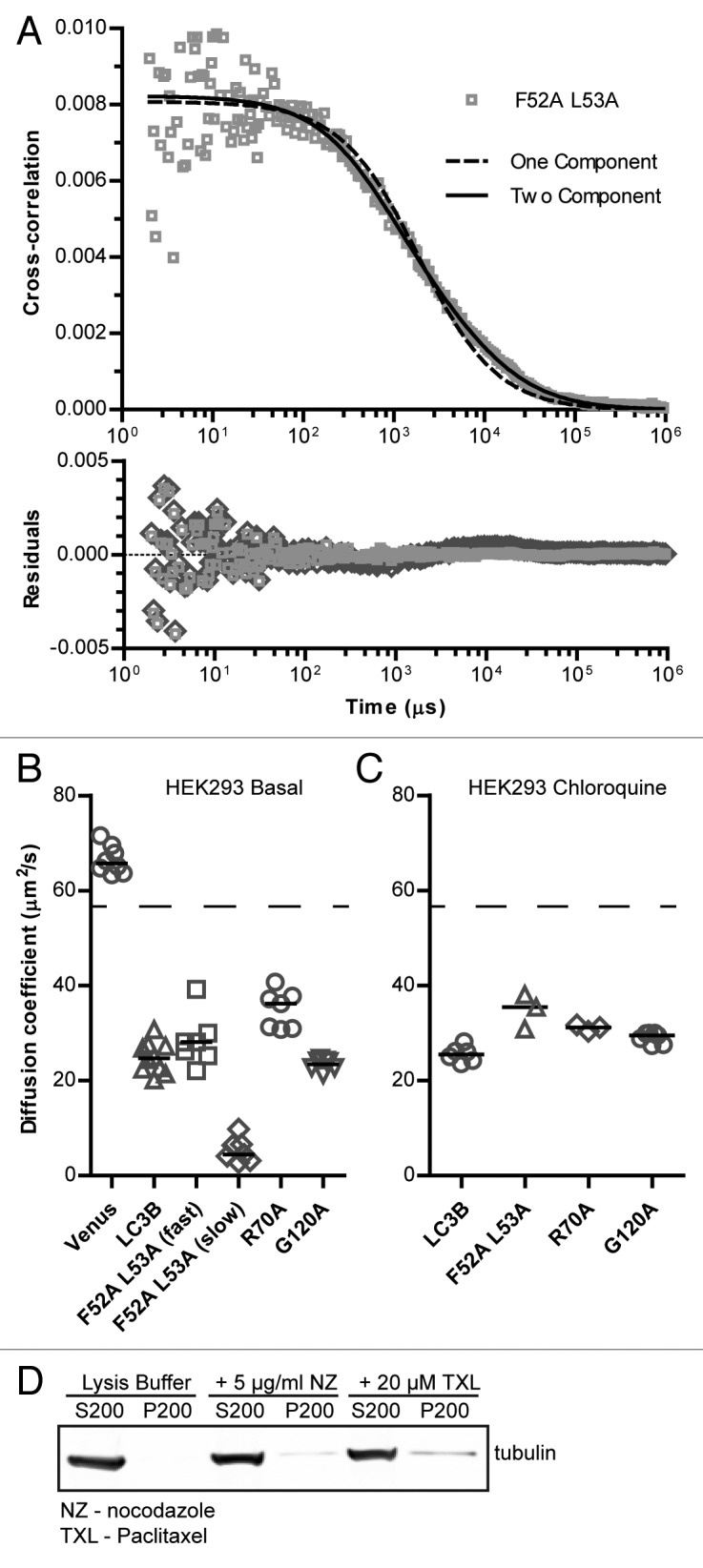
Figure 4. Mutations to LC3B’s hydrophobic protein interaction-surface at residues R70 or F52 and L53 change the diffusional mobility of Venus-LC3B in cytoplasmic extracts. FPFA measurements were collected from cytoplasmic extracts of HEK 293 cells individually expressing Venus, Venus-LC3B, Venus-LC3BF52A L53A, Venus-LC3BR70A, or Venus-LC3BG120A; the macro time fluorescence fluctuations were cross-correlated and fit with diffusion models. See (A) for an example of one and 2-component fits to the Venus-LC3BF52A L53A data. In this case, the 2-component model was a better fit than the 1-component model. (B and C) Diffusion coefficients obtained from the fits to the data collected under basal conditions or after the cells were incubated for 2 h in 100 μM chloroquine respectively. Compare the ratio of the Venus control to each of the constructs as an indicator of their effective sizes. The dashed lines are the predicted diffusion coefficients for a 45 kDa Venus-LC3B monomer assuming both Venus and Venus-LC3B are spherical. Bars represent the median. Samples are replicate measurements from 4 to 5 extract preparations. One-way ANOVA test P < 1 × 10−4. Pairwise comparisons between all of the constructs were made using Bonferonni-corrected t tests as described in the text. (D) Assay to quantify the amount of polymerized microtubules in the extract preparations. Unpolymerized tubulin remains in the supernatant (S200) after centrifugation at 200,000 x RCF over a sucrose cushion, while polymerized microtubules pellet (P200). As controls, extracts were treated with nocodazole (NZ) to depolymerize microtubules or taxol (TXL) to stabilize microtubules. Note the buffer that was used for the extract preparations is not suitable for stabilizing polymerized microtubules.
Table 3. Predicted molecular weights and rotational correlation times for Venus, Venus-LC3B, and Venus-LC3B mutants based on the FPFA diffusion measurements in HEK 293 cell extracts under basal conditions, and after incubation with 100 μM chloroquine (CQ) for 2 h.
| Construct | Monomer MW (kDa) | Basal predicted MW (kDa)* | CQ predicted MW (kDa)* | Basal percent species (%) | Basal rotation correlation time (ns) | CQ rotation correlation time (ns) |
|---|---|---|---|---|---|---|
| Venus | 27 | N/A | N/A | N/A | 12.3 ± 0.2 (4) | N/A |
| Venus-LC3B | 45 | 500 ± 100 (10) | 480 ± 90 (6) | N/A | 24.9 ± 0.6 (10) | 25.0 ± 0.3 (6) |
|
Fast Venus-LC3B (F52A/L53A) |
45 | 300 ± 100 (7) | 190 ± 70 (3) | 70 ± 4 (4) | 25.6 ± 0.3 (7) | 25.5 ± 0.1 (4) |
|
Slow Venus-LC3B (F52A/L53A) |
45 | 5 × 104 ± 5 × 104 (7) | N/A | 30 ± 4 (4) | N/A | N/A |
| Venus-LC3B (R70A) | 45 | 190 ± 50 (7) | 260 ± 30 (3) | N/A | 24.3 ± 0.6 (7) | 23.9 ± 0.1 (3) |
| Venus-LC3B (G120A) | 45 | 600 ± 90 (7) | 330 ± 40 (7) | N/A | 24.8 ± 0.3 (7) | 25.3 ± 0.5 (7) |
*Calculated assuming diffusing species has a spherical shape; mean ± 95% CI (N, # extracts); CQ, chloroquine.
After establishing baselines for diffusion comparisons, next, we performed FPFA measurements of the Venus-LC3BR70A and Venus-LC3BF52A L53A mutants in cytoplasmic extracts. Interestingly, the cross-correlation curve for Venus-LC3BF52A L53A was not well described by a single-component model (Fig. 4A). It was, however, fit well by a 2-component diffusion model where approximately 3/4 of the molecules have a diffusion coefficient of 28 ± 4 µm2/s while the remainder have a diffusion coefficient of 5 ± 2 µm2/s (Fig. 4A and B). The diffusion coefficient for the faster diffusing Venus-LC3BF52A L53A species was similar to that of Venus-LC3B (P > 0.003; t test), while the diffusion coefficient for the slower diffusing species was much slower than Venus-LC3B (P ≤ 0.003; t test). The cross-correlation curve for Venus-LC3BR70A on the other hand was well fit by a single-component diffusion model with a diffusion coefficient of 35 ± 3 µm2/s, which was statistically faster than Venus-LC3B (P ≤ 0.003; t test) (Fig. 4B). Assuming the complexes have a spherical geometry, the predicted sizes of the Venus-LC3BF52A L53A-associated complexes are about 300 kDa and 50 MDa (note, however, that we observed relatively high variability in the measurements of the F52A and L53A construct), and the predicted size of Venus-LC3BR70A-associated complexes is about 190 kDa (Table 3). These values are in agreement with those measured by confocal FRAP in live cells, assuming the D measured for Venus-LC3BF52A L53A by FRAP reflects the contributions of 2 diffusing species detected by FPFA.
In order to directly test if LC3B’s incorporation into protein complexes is regulated by its lipid modification we performed FPFA measurements on Venus-LC3BG120A. The diffusion coefficient for Venus-LC3BG120A, 23.4 ± 0.9 µm2/s, was identical to wild-type Venus-LC3B (ns; P > 0.003; t test), further supporting our conclusion based on the FRAP results that the slow diffusion of Venus-LC3B is not due to its lipid modification (Fig. 4B). In addition, these data further suggest that LC3B’s lipid modification is not a major factor in regulating the effective size of the LC3B-associated complexes.
Our in vivo results revealed the effective sizes of complexes are modestly upregulated by perturbations to the autophagy pathway. We next explored the possibility that the effective sizes of these soluble LC3B-associated complexes under chloroquine conditions are maintained in vitro. We prepared cytoplasmic extracts from cells incubated in 100 μM chloroquine for 2 h before cell lysis. We found that in cells treated with chloroquine, there was little effect on the sizes of the wild-type Venus-LC3B, Venus-LC3BR70A, and Venus-LC3BG120A-associated complexes compared with basal conditions (ns; P > 0.003; t test) (Fig. 4C). The diffusion of the Venus-LC3BF52A L53A complex under chloroquine conditions was primarily fit with a single-component model. These results differed slightly from what we observed in living cells, and suggest that some of the components of the soluble LC3B-associated complexes present after treatment with chloroquine were not maintained in vitro. These results further support the conclusion that the complexes we are detecting are independent of lipid modified LC3B incorporated in autophagosomal membranes, as this would have shown up as a component with a diffusion coefficient approximately 2 orders of magnitude slower than Venus.
Interactions of LC3B with polymerized microtubules are not responsible for the slow diffusion of soluble LC3B
It is formally possible that LC3B’s slow diffusion could be due to interactions with microtubules given that LC3B was originally identified as a microtubule-associated protein.31 To address this possibility we specifically tested for the presence of polymerized microtubules in the cytoplasmic extracts by sedimentation at a relatively high centrifugal force.42 We were unable to detect significant levels of polymerized microtubules under the conditions of our experiments (Fig. 4D). This suggests that the slow diffusion of Venus-LC3B detected by FPFA is not the result of reversible binding to polymerized microtubules. We further tested this possibility in vivo, by performing FRAP experiments on cells depleted of polymerized microtubules by treatment with nocodazole for 15 min on ice followed by 1 h at 37 °C. We observed that the effective sizes of the complexes were maintained in live cells treated with nocodazole (Table 2), further supporting the notion that reversible interaction with polymerized microtubules is not responsible for LC3B's slow diffusion.
There is no evidence of homo-FRET between Venus-LC3B, Venus-LC3BF52A L53A, Venus-LC3BR70A, or Venus-LC3BG120A
Our diffusion measurements indicate soluble LC3B is constitutively associated with a high molecular weight complex; however, it is possible the complexes we detect by diffusion may correspond to small aggregates of LC3B. We previously tested for oligomerization of puncta-independent LC3B in vivo using Cerulean-LC3B and Venus-LC3B as FRET donors and acceptors. FRET microscopy failed to detect significant levels of energy transfer, suggesting soluble LC3B is unlikely to homo-oligomerize or aggregate.30 However, unfavorable dipole-dipole orientation may have prevented energy transfer, even between fluorophores in close proximity.43,44 Thus, LC3B’s oligomerization state remains unclear.
In order to test if there were multiple LC3B molecules within FRET proximity in our cytoplasmic extracts, next we examined our FPFA measurements for evidence of homo-FRET. Homo-FRET analysis provides information about the proximity of Venus fluorophores, and thus may be able to detect homo-oligomerization of Venus-LC3B or the Venus-LC3B mutants. For the purpose of performing an analysis of homo-FRET, our FPFA micro-time measurements of orthogonally polarized fluorescence emission were used to calculate time-resolved fluorescence anisotropy decay curves. The decay of fluorescence anisotropy can be due to several factors including rotational diffusion and the presence or absence of FRET.43 In the absence of FRET, we expect the fluorescence anisotropy curve will exhibit a single exponential decay as was previously measured for Venus, and in the presence of FRET we expect the fluorescence anisotropy curves will exhibit a multicomponent exponential decay as has been previously measured for a series of positive controls consisting of Venus molecules separated by short linkers.37
We found the fluorescence anisotropy decay curves for both Venus and Venus-LC3B were well fit using a single exponential decay model, indicating little, if any, homo-FRET was occurring in these samples (Fig. 5A). This result is consistent with a previous study in which we have found that no significant FRET occurs between soluble Cerulean-LC3B and Venus-LC3B in the cytoplasm of living cells.30 The single exponential decay rates of the fluorescence anisotropies for Venus and Venus-LC3B are related to the rotational diffusion of the Venus fluorophore attached to LC3B by a flexible linker (Table 3).45 However, because of the flexible nature of the linker it is not possible to use these values to calculate the size of the Venus-LC3B-associated complexes based on their rotational correlation times alone.

Figure 5. The majority of complexes contain one soluble Venus-LC3B, and mutations to LC3B’s hydrophobic surface at residues F52 and L53 but not R70 alter the stoichiometry of LC3B in complexes. TRFA analysis was performed using the microtime FPFA measurements collected from cytoplasmic extracts of HEK 293 cells individually expressing Venus, Venus-LC3B, Venus-LC3BF52A/L53A, Venus-LC3BR70A, or Venus-LC3BG120A under (A) basal conditions, or (C) after incubation in 100 μM chloroquine for 2 h. The decays are single exponential (linear on a semilog plot) indicating no detectable homo-FRET in all cases. Brightness analysis was performed using the macrotime from the FPFA measurements under (B) basal conditions or (D) after incubation in 100 μM chloroquine for 2 h. Anisotropy correlation times from replicate measurements can be found in Table 3. Brightness data are normalized to the average brightness of Venus. Bars represent the median. One-way ANOVA test P < 1 × 10−4 for the brightness measurements from either condition. Pairwise comparisons between all of the constructs under either condition were made using Bonferonni-corrected t tests as described in the text.
Next, we examined our FPFA measurements of Venus-LC3BF52A L53A, Venus-LC3BR70A, and Venus-LC3BG120A for evidence of homo-FRET in order to determine if these mutations resulted in a change in the proximity of multiple LC3B molecules. In both cases, the anisotropy decay curves were fit by a single exponential, with similar rotational correlation times to that of wild-type Venus-LC3B (Fig. 5A; Table 3). This suggests the Venus proteins attached to the LC3B mutants are also, to some extent, free to rotate independently of LC3B, and that no detectable homo-FRET occurs in either case.
There is on average only one soluble Venus-tagged LC3B protein per complex, and disruption of LC3B’s hydrophobic binding interface changes the stoichiometry of LC3B-associated complexes
A negative FRET result cannot conclusively rule out the presence of multiple Venus-LC3B’s per complex, as the Venus fluorophores may be positioned such that their proximity or their average relative dipole orientations does not satisfy the FRET requirements.43 Fortunately, FPFA measurements provide an additional opportunity to gain insights into the stoichiometry of a protein by quantifying its molecular brightness. Fluorescence brightness is extracted from the statistics of fluorescence intensity fluctuations using a method that has been termed brightness analysis.38 The principle behind the interpretation of our data lies in the idea that a Venus labeled molecule with a stoichiometry of 2 will have twice the brightness compared with Venus alone, and it follows that higher order oligomerization states will have quantized brightness values of Venus as was previously measured using a series of positive controls.37
After establishing the Venus baseline for brightness comparisons, next, we investigated the brightness of Venus-LC3B using our FPFA measurements. We found Venus-LC3B has a normalized brightness of 1.16 ± 0.09, which is approximately 16% brighter than Venus (P ≤ 0.008; t test) (Fig. 5B). These data strongly suggest the majority of Venus-LC3B does not extensively oligomerize or aggregate in solution. Instead, they suggest Venus-LC3B primarily has a stoichiometry of close to one in cytoplasmic complexes.
Finally, in order to gain additional information about the stoichiometry of the mutant LC3B proteins, we performed brightness analysis using our FPFA measurements of Venus-LC3BF52A L53A, Venus-LC3BR70A, and Venus-LC3BG120A. Assuming the brightness values of the 2 Venus-LC3BF52A L53A species with distinct diffusion times are the same, the normalized brightness of the Venus-LC3BF52A L53A complexes is about 1.8 ± 0.1. This brightness value was approximately twice that of Venus, suggesting either one or both of the Venus-LC3BF52A L53A complexes has a stoichiometry of 2 (P ≤ 0.008; t test) (Fig. 5B). On the other hand, the brightness values obtained from the Venus-LC3BR70A, and the Venus-LC3BG120A FPFA data, were both similar to Venus-LC3B. Venus-LC3BR70A had a normalized brightness of 1.1 ± 0.1, or about 10% brighter than Venus (P > 0.008; t test), and Venus-LC3BG120A had a normalized brightness of 1.0 ± 0.1 which was indistinguishable from that of Venus (ns; P > 0.008; t test) (Fig. 5B). Thus, like wild-type Venus-LC3B, these data suggest only one Venus-LC3BR70A and only one Venus-LC3BG120A molecule is associated with a given complex.
Given the effective sizes of complexes appear to change upon treatment with chloroquine, we reasoned there may also be changes in LC3B's stoichiometry or organization under these conditions. We tested for these changes by examining the time-resolved anisotropy and the molecular brightness from our FPFA measurements of the LC3B constructs, after treatment with chloroquine, in cytoplasmic extracts. However, we found the anisotropy decays for the constructs were similar to basal conditions (Fig. 5C), and the molecular brightness values were also similar to basal conditions (Fig. 5D). On the other hand, the Venus-LC3BF52A L53A construct’s brightness became more similar to Venus at 1.0 ± 0.2. These data suggest soluble LC3B does not have a propensity to aggregate or homo-oligomerize either under basal conditions or after treatment with chloroquine.
Discussion
In this study, we analyzed the size and stoichiometry of puncta-independent LC3B in the cytoplasm of living cells and in cytoplasmic extracts and examined how these properties are regulated by specific residues on LC3B's hydrophobic protein binding surface as well its G120 residue required for lipid modification. On the basis of our findings, we suggest the following working model for the basal organization of LC3B (Fig. 6). A single soluble Venus-LC3B protein associates with a cytoplasmic complex with a molecular weight about an order of magnitude greater than expected for a spherical monomer. Similarly slow diffusion for puncta-independent LC3B was detected in HeLa, COS7, and HEK 293 cells (29,30 and current study), suggesting this is a general feature of LC3B in the cytoplasm.

Figure 6. Working model of the size, stoichiometry, and organization of cytoplasmic LC3B-associated complexes. On average one soluble Venus-LC3B is constitutively bound to an approximately 500 kDa complex of unknown composition. Mutating LC3B residue R70 to an alanine disrupts LC3B’s association with the large complex. Mutating LC3B residues F52 and L53 to alanines results in altered stoichiometry and binding to a second, ~50 MDa, complex. Mutating LC3B residue G120 to an alanine does not change its effective size or stoichiometry.
We also find that disruption of specific residues on LC3B's hydrophobic protein binding surface alters both the hydrodynamic radius and stoichiometry of LC3B-associated complexes (Fig. 6). We studied 2 mutants known to disrupt the interaction of multiple proteins with LC3. The first is an F52A and L53A double mutation to LC3B that has been reported to disrupt the interaction of LC3B with several important protein binding partners including SQSTM1 and NBR1 (neighbor of BRCA1 gene 1), cargo adaptor proteins involved in targeting substrates for autophagic degradation.17,18,25,27,28 The second, an R70A mutation to LC3B, is also known to disrupt interactions with important binding partners such as SQSTM1 and NBR1, but also additional proteins including FYCO1, a RAB7 effector mediating microtubule plus-end directed vesicular transport.17-19,25
Both Venus-LC3BF52A L53A and Venus-LC3BR70A were poorly targeted to puncta in the cytoplasm under steady-state conditions, but showed no dramatic defects in their accumulation on autophagosomal membranes, consistent with the idea that these residues are important for the interactions of LC3 with cargo adaptors, but not autophagosomal membranes.17,18,28 Remarkably, however, the R70A, as well as the F52A and L53A mutations had dramatically different effects on LC3B’s rate of diffusion. Venus-LC3BF52A L53A diffused more slowly than Venus-LC3B, indicating the complexes it associates with become larger, rather than smaller. The predicted size of the larger Venus-LC3BF52A L53A -associated complex is exceptionally large, with a predicted molecular weight on the order of 50 MDa. This corresponds to an effective Stoke's radius of ~25 nm, although we note there is large variability in this estimate. For comparison, the measured hydrodynamic radius of the 70S ribosome is between ~12 to 15 nm.46,47 Furthermore, the Venus-LC3BF52A L53A mutant appears to associate with at least 2 discrete complexes, in which one or both contain multiple copies of LC3BF52A L53A. The Venus-LC3BF52A L53A data provide an excellent example of how FPFA can overcome the limitations of FRET by simultaneously providing information about molecular brightness. In particular, we found no evidence of homo-FRET, but the brightness indicated a stoichiometry of approximately 2 suggesting the 2 Venus-LC3BF52A L53A proteins are part of the same complex, but further than 10 nm apart. Although it is appealing to imagine the larger Venus-LC3BF52A L53A complex revealed by FPFA might serve as a sink for many LC3B proteins, this is not supported by the molecular brightness data.
In contrast to the F52A and L53A double mutation, the R70A mutant largely disrupted LC3B's slow diffusion. In addition, the measured stoichiometry of LC3B in complexes containing the R70A mutant is one, similar to Venus and wild-type Venus-LC3B. Thus, the R70A mutation decreases the size of LC3B-associated complexes, while the F52A and L53A mutations increase the size of LC3B-associated complexes as well as the number of LC3B molecules in the complexes (Fig. 6). This was unexpected, since both mutants are less efficient at binding to numerous important LC3 interacting proteins compared with wild type.25 If the residues on LC3B’s hydrophobic protein interaction surface were all required for its association with a high molecular weight complex, we would have predicted that both mutants would have smaller hydrodynamic radii compared with wild-type LC3B. The data taken together raise the possibility that the F52A and L53A mutations to LC3B may change the properties of the protein such that it is sequestered in an entirely new complex, whereas the R70A mutation to LC3B nearly completely disrupts its ability to associate with the complex altogether. Furthermore, the complexes appeared to change upon treatment of cells with rapamycin or chloroquine, suggesting they could be modulated by perturbations to the autophagy pathway.
The identity of the components of these soluble LC3B-associated cytoplasmic complexes and their function(s) in the regulation of autophagy or autophagy-independent processes remain to be determined. Given that the F52A and L53A, as well as the R70A mutations interfere with the binding of LC3 to SQSTM1 and NBR1, it seems unlikely that binding to cytoplasmic cargo adaptors, and thus sub-diffraction aggregates could account for the composition of the complexes. Instead, we speculate that these complexes may consist of other LC3B interacting proteins or LC3B interacting protein complexes that may be disrupted due to the R70A mutation. At first glance, the LC3 lipid conjugation machinery is an obvious candidate given that ATG7 is known to homo-oligomerize and form a complex with ATG3 to carry out E1- and E2-like LC3 processing reactions.48 The ATG12, ATG5, and ATG16L1 proteins form complex as large as ~800 kDa,49 which is thought to function as an E3-like ubiquitin ligase for LC3 lipid modification50 and direct LC3 to the site of autophagosome formation.51 However, our observation that the diffusion of the LC3BG120A mutant is similar to the wild-type protein, despite the fact that it fails to interact with the LC3 ubiquitin-like conjugation machinery,33 suggests the possibility that puncta-independent LC3B constitutively associates with the ATG12–ATG5-ATG16L1 complex is unlikely. There are, however, several other proteins whose binding to LC3B is completely disrupted by mutation of LC3B’s residue R70 that would make for prime candidates.25 Future work will be required to determine whether the ~500 kDa LC3B-associated complexes consist of these other known autophagy-related proteins, or perhaps include others of the recently identified LC3 interacting proteins.25 One approach that can be used in the future to test for the presence of specific multiprotein complexes in cells is through the combined use of FRET and FRAP, as we have illustrated previously for the case of complexes containing LC3B and ATG4BC74A.30 The study of the interaction of LC3B and ATG4BC74A validated these approaches as a useful means for characterizing the properties of protein complexes under physiological conditions. Intriguingly, LC3 has also been reported to associate with the 3′ UTR of FN1 (fibronectin) mRNA,52 and this interaction depends on a triple arginine motif (residues 68 to 70) which overlaps with the R70A mutant analyzed in our current study. Thus, LC3B-associated complexes detected in our experiments could also potentially represent mRNA-containing complexes.
Our findings also allow us to rule out several alternative models that could account for the slow diffusion of Venus-LC3B. First, we found that the measured diffusion of Venus-LC3BG120A was indistinguishable from wild-type LC3B. This implies that neither lipidation of LC3B nor its ability to bind to autophagosomal membranes contribute to its slow diffusion. Second, we compared the diffusion of Venus-LC3B in live cells under control conditions and following microtubule disruption, as well in cytoplasmic extracts under conditions that are unfavorable for the maintenance of polymerized microtubules in order to determine if its diffusion is slowed as the result of reversible binding to microtubules. We observed no difference in LC3B’s diffusion in cells following microtubule disruption, and very close agreement between our in vivo measurements and our in vitro measurements for the diffusion of LC3B. This suggests that microtubule binding is not responsible for the slow diffusion of LC3B, at least under basal conditions. Finally, our homo-FRET and brightness results strongly suggest that the slowly diffusing complexes of LC3B do not represent either homo-oligomers2 or aggregates32 of LC3B. These findings imply that if LC3B homo-oligomerization is involved in contributing energy to membrane fusion events in autophagy,2 this event must be limited to autophagosomal membranes.
In this study we utilized 2 complementary fluorescence-based approaches, FRAP and FPFA, to characterize the properties of soluble LC3B-associated complexes. The FPFA and FRAP results were in excellent agreement, lending confidence in the conclusions of our studies. However, like any method, each approach has some limitations. First, both FRAP and FPFA rely on measurements of exogenous, fluorescently tagged versions of the protein of interest. This could have several possible consequences on the interpretation of our findings. Our experiments were performed under conditions where Venus-LC3B is overexpressed, and thus other LC3B-binding partners could potentially become limiting for complex formation. Under these conditions one might expect to find excess monomeric Venus-LC3B, but our data suggest this is unlikely given the combination of our FRAP and FCS measurements failed to show any evidence for such a scenario. On the other hand, one advantage of overexpressing Venus-LC3B is that it is in excess over the endogenous protein, and thus it is likely that the complexes we detect contain primarily the tagged form of the protein. This increases the likelihood that our measurements accurately report the effective size, stoichiometry, and organization of LC3B-associated complexes.
Second, both FRAP and FPFA interpretation requires the fitting of models which carry with them certain a priori assumptions. Often, several competing models can fit a particular data set equally well, e.g., a small percentage of LC3B associating with membranes or a fraction of LC3B reversibly binding to a microtubule network. Therefore, additional experiments are required to distinguish between competing models, such as the use of mutants, drug treatments, or comparisons of in vivo vs. in vitro results as we have done here. Even with such information in hand, there are still circumstances where different models cannot be resolved by an analysis of diffusion alone. For example, although our FRAP and FPFA data were well fit by single-component diffusion models, we cannot conclusively rule out the possibility that there are multiple distinct complexes of similar size present. Similar assumptions are inherent in our modeling of the FPFA brightness measurements; in the case of a mixture of complexes, as we detected for F52A and L53A, we cannot rule out the possibility that the 2-fold increased brightness may have originated from one of the complexes in particular and not both.
Third, although FPFA can potentially provide information about rotational diffusion, and thus yield insights into the size and shape of the diffusing species, our current data cannot be quantitatively analyzed in this way. This is because in our experiments, the Venus label is not rigidly attached to LC3B allowing it to undergo rotational motion independently of LC3B or the LC3B-associated complexes. In addition, the lifetime of the Venus fluorophore (on the order of a few nanoseconds) is not long enough to allow accurate quantitation of rotational diffusion for slowly rotating complexes.45 Thus, our rotational diffusion measurements should not be expected to match our translational diffusion measurements. A rigidly attached fluorophore with a much longer lifetime would be a better choice for measuring the rotational mobility of large complexes.
Lastly, the details of our experimental design should be considered carefully as they also constrain our conclusions in several ways. Our FRAP measurements were specifically designed to avoid LC3B in puncta, and instead focus on the diffuse pool of LC3B independent of bright puncta. Under certain conditions, upregulation of the autophagy pathway for example, the cytoplasm may become full with large numbers of puncta making it difficult or impossible to avoid puncta with a 1 μm radius bleach region. Although this was not a problem under the conditions examined in our study, in the future, methods of analysis may need to be devised in order to extend the range of conditions which can be analyzed by FRAP, for example, the application of reaction and diffusion models36 to accurately quantify autophagy protein turnover rates on puncta. Similarly, our method of cellular extract preparation should be taken into consideration when interpreting our FPFA findings; we prepared them in such a way that LC3B-II incorporated into autophagosomal membranes was either solubilized by detergent and/or excluded from the extract by centrifugation. In the future, FPFA measurements could be performed on LC3-II in autophagosomes using an alternative extraction procedure or using an in vivo configuration in order to test if lipid modified LC3 homo-oligomerizes on autophagosomal membranes and to follow up on previous stoichiometry determinations for ATG proteins on autophagosomes.53
In summary, our data suggest that in the cytoplasm, individual soluble LC3B molecules associate with a ~500-kDa complex and that residues on LC3B’s hydrophobic protein interaction surface are important for regulating its association with these complexes. In the future, we anticipate FRAP and FPFA will become valuable methods for uncovering the emergent properties of protein complexes in the autophagy pathway.
Materials and Methods
cDNA constructs
GFP-LC3B was the kind gift of T Yoshimori.8 Cerulean and Venus tagged versions of LC3B were as previously described.30 Venus-LC3BR70A, Venus-LC3BF52A L53A, and Venus-LC3BG120A mutations were constructed using Stratagene’s QuikChange site-directed mutagenesis kit (Agilent Technologies, Inc., 200523). For LC3BR70A the forward and reverse primers were 5′-CATCAAGATA ATTAGAAGGGC CCTGCAGCTCA ATGCTAAC-3′ and 5′-GTTAGCATTG AGCTGCAGGG CCCTTCTAAT TATCTTGATG-3′ respectively. For LC3BF52A L53A the forward and reverse primers were 5′-GTCCTGGACA AGACCAAGGC CCTTGTACCT GATCACGT-3′ and 5′-ACGTGATCAG GTACAAGGGC CTTGGTCTTG TCCAGGAC-3′ respectively. For LC3BG120A the forward and reverse primers were 5′-CAGGAGACGT TCGCGACAGC ACTGGCT-3′ and 5′-AGCCAGTGCT GTCGCGAACGT CTCCTG-3′ respectively.
Cell culture and transfections
HeLa cells and HEK 293T cells (American Type Culture Collection, CCL-2 and CRL-1573) were maintained in Dulbecco Modified Eagle’s Medium (DMEM) containing 10% fetal bovine serum (Life Technologies, 10437028), 1% PenStrep, and phenol red or high glucose DMEM containing L-glutamine, sodium pyruvate, 10% fetal bovine serum, 1% PenStrep, and phenol red respectively at 37 °C, 5% CO2.
For our live-cell imaging experiments, on the day prior to transfection, HeLa cells were plated in MatTek 35 mm No. 1.5 glass bottom culture dishes (Ashland, P35G-1.5-10-C). On the following day the cells (50 to 80% confluent monolayer) were transfected with described mammalian expression constructs using FuGENE 6 transfection reagent according to the manufacturer’s recommended protocol (Promega Corp., E2691). HEK 293T cells were plated and transfected similarly, except the MatTek chambers were first coated with poly-d-lysine (Sigma-Aldrich, P7886). On the day of the experiment (24 h after transfection) cell culture medium was rinsed and replaced with phenol red-free DMEM supplemented with 10% fetal calf serum, 1% PenStrep, and 25 mM HEPES. The cells were allowed to come to equilibrium at 37 °C for ~5 min before transferring to the temperature-controlled microscope stage and objective set to 37 °C.
For our FPFA measurements in cytoplasmic extracts, on the day prior to measurements, plasmid DNA (typically 1 µg/250,000 cells) were transfected using electroporation (Digital Bio/BTX MicroPorator). On the following day, the cells were harvested and lysed using passive lysis buffer (Promega, E1941) containing 1% Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific Inc., 78440). The homogenates were centrifuged at 20,000 RCF for 15 min, and the supernatants were diluted ~20-fold with purified water for FPFA to yield a photon count rate between ~25 kcps and 100 kcps (> 30× the dark count rate) to avoid TCSPC pile-up artifacts.54 The clarified homogenates were then loaded into 35 mm glass bottom dishes for measurements at 25 °C.
Analysis of LC3-I to LC3-II processing
The LC3-I to LC3-II processing assay was performed by lysing cells plated in 6-well culture dishes using CellLytic M reagent (Sigma-Aldrich Co. LLC, C2978) as per the manufacturer’s instructions, spinning down at 13,000 × RCF for 20 min, followed by SDS-PAGE of the supernatant fraction. We blotted using antibodies against TUBB (β-tubulin) (Developmental Studies Hybridoma Bank, E7), GFP (JL-8) (Clontech, 632380), and LC3 (Novus Biologicals, NB100-2220).
Drug treatments
In order to block lysosomal degradation of LC3 or upregulate the autophagy pathway, we incubated the cells with either 100 μM chloroquine (Sigma-Aldrich Co. LLC, C6628) or 200 nM rapamycin (Sigma-Aldrich, R8781) for 2 h at 37 °C before performing live-cell imaging or preparing cell extracts. In order to disrupt microtubules in live cells, we incubated the cells on ice with 5 μg/ml nocodazole (Sigma-Aldrich Co. LLC, M1404) for 15 min before transferring to 37 °C for an additional 1 h. The cells were imaged in the continued presence of nocodazole.
Microtubule pelleting assay
We examined the extent of polymerized microtubules in our cytoplasmic extracts using a microtubule pelleting assay.42 The cytoplasmic extracts were loaded onto a 40% sucrose cushion and spun at 200,000 × RCF for 20 min at 25 °C. Fractions from the pellet and supernatant were analyzed by SDS-PAGE. Although the conditions of the passive lysis buffer are not suitable for maintenance of polymerized microtubules in vitro, we attempted to further depolymerize microtubules or repolymerize tubulin by incubating our extracts with nocodazole (5 μg/ml) for 15 min on ice or with paclitaxel (20 μM) at 37 °C for 15 min before the pelleting assay.
Laser-scanning confocal microscopy and quantification of LC3-associated puncta
Cells were imaged live using a Zeiss LSM 510 confocal microscope (Carl Zeiss Microscopy LLC) equipped with an Argon/2 30 mW laser (458, 488, 514 nm) using an oil immersion 40 × 1.3 N.A. Zeiss Plan-Neofluar objective. The total numbers of bright, Venus-LC3B labeled spots (~0.5–2 µm) obtained in single confocal sections (1 Airy unit) were manually counted in the cytoplasm of each cell and reported as the mean number of cytoplasmic spots per cell.
FRAP methods
Confocal FRAP measurements were performed using a Zeiss LSM 510 confocal microscope (Carl Zeiss MicroImaging Inc.) equipped with an Argon/2 30 mW laser (458, 488, 514 nm) using an oil immersion 40 × 1.3 N.A. Zeiss Plan-Neofluar objective. Confocal FRAP data were obtained using 512 × 45 pixel images at 4× digital zoom using a 1 Airy unit pinhole at a rate of 42 frames per second. Venus was irreversibly photobleached by iteratively scanning a 514 nm laser (30 mW nominal power) 10 times in a circular ROI with a nominal radius rn of 0.99 µm, centered in the imaging window. Imaging of the prebleach steady-state fluorescence and the postbleach recovery was performed using a much lower nominal laser power of 0.06 mW. In order to separate excitation and emission light sources we employed several standard filter sets for the Zeiss LSM 510 (HFT 458/514 and LP 530). A total of 600 images were acquired for each recovery (~15 s), and the bleaching event required 129 ms before acquisition of the postbleach image under our conditions.
To analyze the confocal FRAP data we utilized a recently developed analytical equation for simple Brownian motion to extract an instrument-independent diffusion coefficient.35 The first step in the approach is to plot the mean fluorescence intensity within the bleach region as a function of time to yield a FRAP curve, I(t). We next normalize I(t) by dividing by the prebleach steady-state fluorescence and correct for any unintentional photobleaching encountered during normal imaging. To do this we fit the last 50 data points, at a time sufficiently long enough after the bleach to reach diffusional steady-state, of I(t) to a first order decay process,
I(t) = exp(−kdecayt)
(1)
where t is time, and kdecay is the unintentional photobleaching rate constant. I(t) is corrected by dividing by the exponential decay.55 Next, the diffusion coefficient D and mobile fraction Mf were found by fitting the data to a 1-component FRAP model for Brownian motion with infinite boundary conditions,
| (2) |
where I0 is 1 for a normalized FRAP curve, and rn is the nominal radius of the bleaching ROI.35 This is a modified form of the Axelrod equation56 where the laser is assumed to be a Gaussian, and the parameters re and K take into account the initial conditions for the solution of the diffusion equation. The FRAP model assumes a homogenous distribution of molecules were bleached along the Z-axis resulting in diffusional exchange along the X and Y axes. We determined re and K by fitting the normalized radial post-bleach profile, I(x;t = 0), to an analytical approximation,
| (3) |
where I0 is 1 for a normalized post-bleach profile, and x is the radial distance from the center of the bleaching ROI.57
For comparison, we also analyzed a subset of the data using a newly described simplified FRAP equation for Brownian motion.34 In this approach, I(x;t = 0) was further approximated as,
| (4) |
resulting in the simplified FRAP model,
| (5) |
A comparative analysis of these 2 analytical FRAP models (Eqns. 2 and 5) revealed no significant differences in D or Mf with the level of noise present in our data. This means using Equation 5 is the preferred method of extracting diffusion coefficients and mobile fractions, as it greatly reduces the complexity in programming a nonlinear fitting routine. Furthermore, if there is a priori knowledge that the fluorescence recovery is dominated by Brownian motion rather than reaction kinetics, and the system was allowed to relax back to a steady-state one could use linear interpolation to find the half time of the recovery (τ1/2) and resultant D without any fitting required using
| (6) |
Where indicated, D values were used to calculate the Stokes radius using the Stokes-Einstein equation:
| (7) |
where kB is Boltzmann constant, T is temperature, r is the Stokes radius, and η is viscosity. The apparent molecular weight MW was then calculated as follows (assuming a spherical geometry):
| (8) |
where MWGFP is the molecular weight of GFP (27 kDa) and DGFP is the measured diffusion coefficient of the GFP control.
In principle, a freely diffusing control such as Venus should be 100% mobile on the time scale of our experiments as indicated by Mf. However, under the conditions of our experiments a significant fraction of molecules in the cytoplasm were irreversibly destroyed during the experiment resulting in Mf values less than 1. In order to correct for the significant loss of fluorescence in the compartment due to the bleaching event, we calculated the true mobile fraction (Mfcorrect) by,
Mfcorrect = 1−(Iadjacent(t)−I(t))
(9)
where Iadjacent(t) is the intensity inside an ROI placed adjacent to the bleaching ROI.
FPFA methods
The experimental setup and validation are described elsewhere.37 Briefly, an 80-MHz, 200-fs mode locked Ti:sapphire laser (Coherent Chameleon Ultra-2, Santa Clara, CA) operated at 950 nm provided pulsed 2-photon excitation. The excitation beam, passed through a power attenuator, a spatial filter system (Thorlabs, KT310), a near-IR linear polarizer (100,000:1 extinction ratio, Thorlabs), and finally a multiphoton short-pass dichroic beamsplitter (Semrock, FF670-SDi01-25x36) was used to reflect the excitation beam to a Zeiss 63X-1.2NA water objective (back aperture slightly overfilled). This setup focused the beam to a diffraction-limited spot (~0.4 μm in diameter). The excitation power was kept low (typically ~10.2 mW at the focal point) to avoid bleaching during acquisition (150 to 200 s).
Fluorescence collected from the observation volume in the sample was guided through a BG39 filter (to block residual near-IR photons), a high throughput band-pass filter (Semrock, FF01-540/50-25), and finally a polarizing beam splitter (Thorlabs) augmented with 2 orthogonally oriented linear polarizers (Thorlabs) to increase the extinction ratio. At the beam splitter, parallel and perpendicular emitted photons were separated and focused onto 2 HPM-100-40 hybrid detectors (Becker and Hickle GmbH, Berlin, Germany). The dark count rate for these detectors was typically 200 to 750 cps at 25 °C. Photons detected by each detector were processed by a SPC-132 TCSPC card (Becker and Hickle). For synchronization between excitation pulses and detected photons, a small fraction of the excitation beam was extracted and focused onto a high-speed photodetector (DET10A, Thorlabs) powered by a battery to avoid crosstalk. Note that all optics used in the excitation pathway were selected to minimize group delay dispersion.
For each homogenate, 3 to 5 replicate measurements were performed and these were averaged for each point. All measurements were performed at room temperature.
SPCM software (Becker and Hickle) running in FIFO mode was used to calculate time-resolved fluorescence and cross-correlation functions. Time-resolved anisotropy was calculated based on fluorescence decay of parallel and perpendicular channels using the following equation:45,58
| (10) |
where I//(t) and I⊥(t) are the fluorescence intensity of parallel and perpendicular channels (dark noise subtracted) respectively, and g is an instrument correction factor which for our microscope had a value of 0.96 as determined by tail fitting calibration measurements of fluorescein.
For FPFA, independently measured emission events detected in the parallel and perpendicular light paths were cross-correlated, G(τ). The time dependence of these curves were initially fit using a single-component 3-dimensional (3D) diffusion equation38 to estimate the value of <N> , the average number of fluorescent molecules in the excitation volume, and τD, the correlation time (the average time that a molecule spends in the detection volume) for each sample:
| (11) |
where ω and z, are the radial and axial beam waists respectively calculated from calibration experiments, and the constant γ has a value of 0.35 for a 3-dimensional Gaussian PSF.38
The molecular brightness b is the average number of photon emitted per second per molecule (cpsm)
| (12) |
where <k> is the average photon count rate recorded during data acquisition.
To model a mixture of 2 species having the same brightness but independent diffusion times:
| (13) |
In this case,
| (14) |
With 2-photon excitation the relationship between correlation time τD and D is given by:59
| (15) |
FPFA calibrations
The instrument correction factor g for calculating time-resolved anisotropy (Eqn. 10) was measured using tail-fitting45 of fluorescein samples and found to be 0.96. The value of ω/z for our microscope (used in Eqns. 11 and 13) was 0.1 at an excitation power of 10.2 mW (at 950 nm) using calibration standards of fluorescein in water at pH 9.5.37 Assuming fluorescein has a diffusion coefficient of 300 μm2/s, using the measured correlation time of 75 ± 9 μs, the value of ω was 424 ± 30 nm. The validity of this calibration procedure was confirmed by measuring the diffusion coefficient of Venus monomers under identical conditions. Using ω/z = 0.1, the measured correlation time for Venus monomers with 10.2 mW excitation power was 343 ± 17 µs (n = 3). This corresponds to a diffusion coefficient for Venus monomers in solution of 65.5 ± 8.6 µm2/s (n = 3), in good agreement with the diffusion coefficient measured for GFP.60
Curve fitting and statistics
IGOR Pro software (Version 6.22) was used for fitting of time-resolved anisotropy, cross-correlation curves, and for global fitting of calibration standards. FRAP data was fit using custom routines written in MATLAB based on the trust-region-reflective algorithm (The MathWorks; R2010B). The errors reported throughout the text are 95% confidence intervals unless otherwise stated. To test the hypothesis that 2 group means are not different, under circumstances where there were multiple groups being compared, we used the Bonferonni method to control the overall type-I error rate such that for all pairs of comparisons the familywise significance level was 0.05. For example, if 4 groups are being compared the total number of comparisons is 6, thus the P value cutoff is 0.05/6 = 0.008.
Glossary
Abbreviations:
- ATG8
autophagy-related 8
- ATG4B
autophagy-related 4B
- CI
95% confidence interval
- D
diffusion coefficient
- F52A L53A
MAP1LC3B with mutations F52A and L53A
- FCS
fluorescence correlation spectroscopy
- FPFA
fluorescence polarization fluctuation analysis
- FRAP
fluorescence recovery after photobleaching
- FRET
Förster resonance energy transfer
- G120A
MAP1LC3B with mutation G120A
- GFP
green fluorescent protein
- HEK
human embryonic kidney
- LC3
microtubule-associated protein 1 light chain 3
- LC3-I
soluble microtubule-associated protein 1 light chain 3
- LC3-II
lipid modified microtubule-associated protein 1 light chain 3
- LC3B
microtubule-associated protein 1 light chain 3 beta
- MAP1LC3B
microtubule-associated protein 1 light chain 3 beta
- Mf
mobile fraction
- NBR1
neighbor of BRCA1 gene 1
- R70A
MAP1LC3B with mutation R70A
- RCF
relative centrifugal force
- ROI
region of interest
- SQSTM1
sequestosome 1
- TRFA
time-resolved fluorescence anisotropy
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful for the constructive criticism and advice from the other members of the Kenworthy laboratory and to Dr Christian Behrends for helpful discussions. This work was funded by grant NSF/DMS 0970008 from the National Science Foundation, by the intramural program of the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD, and utilized the core(s) of the Vanderbilt Diabetes Research and Training Center funded by grant DK020593 from the National Institute of Diabetes and Digestive and Kidney Disease. The funding sources had no role in the study design, collection, analysis or interpretation of data, writing the report, or the decision to submit the paper for publication.
References
- 1.Chen Y, Klionsky DJ. . The regulation of autophagy - unanswered questions. J Cell Sci 2011; 124:161 - 70; http://dx.doi.org/ 10.1242/jcs.064576; PMID: 21187343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakatogawa H, Ichimura Y, Ohsumi Y. . Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130:165 - 78; http://dx.doi.org/ 10.1016/j.cell.2007.05.021; PMID: 17632063 [DOI] [PubMed] [Google Scholar]
- 3.Xie Z, Nair U, Klionsky DJ. . Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell 2008; 19:3290 - 8; http://dx.doi.org/ 10.1091/mbc.E07-12-1292; PMID: 18508918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shintani T, Huang W-P, Stromhaug PE, Klionsky DJ. . Mechanism of cargo selection in the cytoplasm to vacuole targeting pathway. Dev Cell 2002; 3:825 - 37; http://dx.doi.org/ 10.1016/S1534-5807(02)00373-8; PMID: 12479808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. . p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603 - 14; http://dx.doi.org/ 10.1083/jcb.200507002; PMID: 16286508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. . Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008; 13:1211 - 8; http://dx.doi.org/ 10.1111/j.1365-2443.2008.01238.x; PMID: 19021777 [DOI] [PubMed] [Google Scholar]
- 7.Weidberg H, Shpilka T, Shvets E, Abada A, Shimron F, Elazar Z. . LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev Cell 2011; 20:444 - 54; http://dx.doi.org/ 10.1016/j.devcel.2011.02.006; PMID: 21497758 [DOI] [PubMed] [Google Scholar]
- 8.Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. . An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651 - 9; http://dx.doi.org/ 10.1091/mbc.E08-03-0312; PMID: 18768752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. . LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 2010; 29:1792 - 802; http://dx.doi.org/ 10.1038/emboj.2010.74; PMID: 20418806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansen T, Lamark T. . Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279 - 96; http://dx.doi.org/ 10.4161/auto.7.3.14487; PMID: 21189453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shpilka T, Mizushima N, Elazar Z. . Ubiquitin-like proteins and autophagy at a glance. J Cell Sci 2012; 125:2343 - 8; http://dx.doi.org/ 10.1242/jcs.093757; PMID: 22736434 [DOI] [PubMed] [Google Scholar]
- 12.Shpilka T, Weidberg H, Pietrokovski S, Elazar Z. . Atg8: an autophagy-related ubiquitin-like protein family. Genome Biol 2011; 12:226; http://dx.doi.org/ 10.1186/gb-2011-12-7-226; PMID: 21867568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichimura Y, Kirisako T, Takao T, Satomi Y, Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi M, et al. . A ubiquitin-like system mediates protein lipidation. Nature 2000; 408:488 - 92; http://dx.doi.org/ 10.1038/35044114; PMID: 11100732 [DOI] [PubMed] [Google Scholar]
- 14.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. . LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720 - 8; http://dx.doi.org/ 10.1093/emboj/19.21.5720; PMID: 11060023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. . LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805 - 12; http://dx.doi.org/ 10.1242/jcs.01131; PMID: 15169837 [DOI] [PubMed] [Google Scholar]
- 16.Tanida I, Sou YS, Ezaki J, Minematsu-Ikeguchi N, Ueno T, Kominami E. . HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem 2004; 279:36268 - 76; http://dx.doi.org/ 10.1074/jbc.M401461200; PMID: 15187094 [DOI] [PubMed] [Google Scholar]
- 17.Kirkin V, Lamark T, Sou Y-S, Bjørkøy G, Nunn JL, Bruun J-A, Shvets E, McEwan DG, Clausen TH, Wild P, et al. . A role for NBR1 in autophagosomal degradation of ubiquitinated substrates. Mol Cell 2009; 33:505 - 16; http://dx.doi.org/ 10.1016/j.molcel.2009.01.020; PMID: 19250911 [DOI] [PubMed] [Google Scholar]
- 18.Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. . Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem 2008; 283:22847 - 57; http://dx.doi.org/ 10.1074/jbc.M802182200; PMID: 18524774 [DOI] [PubMed] [Google Scholar]
- 19.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjørkøy G, Johansen T. . FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 2010; 188:253 - 69; http://dx.doi.org/ 10.1083/jcb.200907015; PMID: 20100911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, et al. . Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J 2012; 31:3691 - 703; http://dx.doi.org/ 10.1038/emboj.2012.225; PMID: 22885598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I. . Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 2012; 32:1733 - 44; http://dx.doi.org/ 10.1128/MCB.06717-11; PMID: 22354992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, Carrier A, Iovanna JL, Dusetti NJ. . TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ 2012; 19:1525 - 35; http://dx.doi.org/ 10.1038/cdd.2012.30; PMID: 22421968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sancho A, Duran J, García-España A, Mauvezin C, Alemu EA, Lamark T, Macias MJ, DeSalle R, Royo M, Sala D, et al. . DOR/Tp53inp2 and Tp53inp1 constitute a metazoan gene family encoding dual regulators of autophagy and transcription. PLoS One 2012; 7:e34034; http://dx.doi.org/ 10.1371/journal.pone.0034034; PMID: 22470510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nowak J, Archange C, Tardivel-Lacombe J, Pontarotti P, Pébusque M-J, Vaccaro MI, Velasco G, Dagorn JC, Iovanna JL. . The TP53INP2 protein is required for autophagy in mammalian cells. Mol Biol Cell 2009; 20:870 - 81; http://dx.doi.org/ 10.1091/mbc.E08-07-0671; PMID: 19056683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Behrends C, Sowa ME, Gygi SP, Harper JW. . Network organization of the human autophagy system. Nature 2010; 466:68 - 76; http://dx.doi.org/ 10.1038/nature09204; PMID: 20562859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. . ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275 - 90; http://dx.doi.org/ 10.1074/jbc.M112.378109; PMID: 23043107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shvets E, Elazar Z. . Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy 2008; 4:1054 - 6; PMID: 18776740 [DOI] [PubMed] [Google Scholar]
- 28.Shvets E, Fass E, Scherz-Shouval R, Elazar Z. . The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci 2008; 121:2685 - 95; http://dx.doi.org/ 10.1242/jcs.026005; PMID: 18653543 [DOI] [PubMed] [Google Scholar]
- 29.Drake KR, Kang M, Kenworthy AK. . Nucleocytoplasmic distribution and dynamics of the autophagosome marker EGFP-LC3. PLoS One 2010; 5:e9806; http://dx.doi.org/ 10.1371/journal.pone.0009806; PMID: 20352102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kraft LJ, Kenworthy AK. . Imaging protein complex formation in the autophagy pathway: analysis of the interaction of LC3 and Atg4B(C74A) in live cells using Förster resonance energy transfer and fluorescence recovery after photobleaching. J Biomed Opt 2012; 17:011008; http://dx.doi.org/ 10.1117/1.JBO.17.1.011008; PMID: 22352642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mann SS, Hammarback JA. . Molecular characterization of light chain 3. A microtubule binding subunit of MAP1A and MAP1B. J Biol Chem 1994; 269:11492 - 7; PMID: 7908909 [PubMed] [Google Scholar]
- 32.Kuma A, Matsui M, Mizushima N. . LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 2007; 3:323 - 8; PMID: 17387262 [DOI] [PubMed] [Google Scholar]
- 33.Tanida I, Yamaji T, Ueno T, Ishiura S, Kominami E, Hanada K. . Consideration about negative controls for LC3 and expression vectors for four colored fluorescent protein-LC3 negative controls. Autophagy 2008; 4:131 - 4; PMID: 18000393 [DOI] [PubMed] [Google Scholar]
- 34.Kang M, Day CA, Kenworthy AK, DiBenedetto E. . Simplified equation to extract diffusion coefficients from confocal FRAP data. Traffic 2012; 13:1589 - 600; http://dx.doi.org/ 10.1111/tra.12008; PMID: 22984916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kang M, Day CA, Drake K, Kenworthy AK, DiBenedetto E. . A generalization of theory for two-dimensional fluorescence recovery after photobleaching applicable to confocal laser scanning microscopes. Biophys J 2009; 97:1501 - 11; http://dx.doi.org/ 10.1016/j.bpj.2009.06.017; PMID: 19720039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang M, Day CA, DiBenedetto E, Kenworthy AK. . A quantitative approach to analyze binding diffusion kinetics by confocal FRAP. Biophys J 2010; 99:2737 - 47; http://dx.doi.org/ 10.1016/j.bpj.2010.09.013; PMID: 21044570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen TA, Sarkar P, Veetil JV, Koushik SV, Vogel SS. . Fluorescence polarization and fluctuation analysis monitors subunit proximity, stoichiometry, and protein complex hydrodynamics. PLoS One 2012; 7:e38209; http://dx.doi.org/ 10.1371/journal.pone.0038209; PMID: 22666486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Müller JD, Chen Y, Gratton E. . Fluorescence correlation spectroscopy. Methods Enzymol 2003; 361:69 - 92; http://dx.doi.org/ 10.1016/S0076-6879(03)61006-2; PMID: 12624907 [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Chen M, Yang J, Zhang Z. . LC3 fluorescent puncta in autophagosomes or in protein aggregates can be distinguished by FRAP analysis in living cells. Autophagy 2013; 9:756 - 69; http://dx.doi.org/ 10.4161/auto.23814; PMID: 23482084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mai J, Trump S, Ali R, Schiltz RL, Hager G, Hanke T, Lehmann I, Attinger S. . Are assumptions about the model type necessary in reaction-diffusion modeling? A FRAP application. Biophys J 2011; 100:1178 - 88; http://dx.doi.org/ 10.1016/j.bpj.2011.01.041; PMID: 21354390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanida I, Ueno T, Kominami E. . Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem 2004; 279:47704 - 10; http://dx.doi.org/ 10.1074/jbc.M407016200; PMID: 15355958 [DOI] [PubMed] [Google Scholar]
- 42.Archuleta TL, Du Y, English CA, Lory S, Lesser C, Ohi MD, Ohi R, Spiller BW. . The Chlamydia effector chlamydial outer protein N (CopN) sequesters tubulin and prevents microtubule assembly. J Biol Chem 2011; 286:33992 - 8; http://dx.doi.org/ 10.1074/jbc.M111.258426; PMID: 21841198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogel SS, Thaler C, Koushik SV. . Fanciful FRET. Sci STKE 2006; 2006:re2; PMID: 16622184 [DOI] [PubMed] [Google Scholar]
- 44.Piston DW, Kremers GJ. . Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci 2007; 32:407 - 14; http://dx.doi.org/ 10.1016/j.tibs.2007.08.003; PMID: 17764955 [DOI] [PubMed] [Google Scholar]
- 45.Vogel SS, Thaler C, Blank PS, Koushik SV. Time Resolved Fluorescence Anisotropy. In: Periasamy A, Clegg RM, eds. FLIM Microscopy in Biology and Medicine. Boca Raton: Taylor & Francis, 2010:245-90 [Google Scholar]
- 46.Lavalette D, Amand B, Pochon F. . Rotational relaxation of 70S ribosomes by a depolarization method using triplet probes. Proc Natl Acad Sci U S A 1977; 74:1407 - 11; http://dx.doi.org/ 10.1073/pnas.74.4.1407; PMID: 323851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koppel DE. . Study of Escherichia coli ribosomes by intensity fluctuation spectroscopy of scattered laser light. Biochemistry 1974; 13:2712 - 9; http://dx.doi.org/ 10.1021/bi00710a009; PMID: 4603215 [DOI] [PubMed] [Google Scholar]
- 48.Taherbhoy AM, Tait SW, Kaiser SE, Williams AH, Deng A, Nourse A, Hammel M, Kurinov I, Rock CO, Green DR, et al. . Atg8 transfer from Atg7 to Atg3: a distinctive E1-E2 architecture and mechanism in the autophagy pathway. Mol Cell 2011; 44:451 - 61; http://dx.doi.org/ 10.1016/j.molcel.2011.08.034; PMID: 22055190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mizushima N, Kuma A, Kobayashi Y, Yamamoto A, Matsubae M, Takao T, Natsume T, Ohsumi Y, Yoshimori T. . Mouse Apg16L, a novel WD-repeat protein, targets to the autophagic isolation membrane with the Apg12-Apg5 conjugate. J Cell Sci 2003; 116:1679 - 88; http://dx.doi.org/ 10.1242/jcs.00381; PMID: 12665549 [DOI] [PubMed] [Google Scholar]
- 50.Hanada T, Noda NN, Satomi Y, Ichimura Y, Fujioka Y, Takao T, Inagaki F, Ohsumi Y. . The Atg12-Atg5 conjugate has a novel E3-like activity for protein lipidation in autophagy. J Biol Chem 2007; 282:37298 - 302; http://dx.doi.org/ 10.1074/jbc.C700195200; PMID: 17986448 [DOI] [PubMed] [Google Scholar]
- 51.Fujita N, Itoh T, Omori H, Fukuda M, Noda T, Yoshimori T. . The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell 2008; 19:2092 - 100; http://dx.doi.org/ 10.1091/mbc.E07-12-1257; PMID: 18321988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou B, Rabinovitch M. . Microtubule involvement in translational regulation of fibronectin expression by light chain 3 of microtubule-associated protein 1 in vascular smooth muscle cells. Circ Res 1998; 83:481 - 9; http://dx.doi.org/ 10.1161/01.RES.83.5.481; PMID: 9734470 [DOI] [PubMed] [Google Scholar]
- 53.Geng J, Klionsky DJ. . Determining Atg protein stoichiometry at the phagophore assembly site by fluorescence microscopy. Autophagy 2010; 6:144 - 7; http://dx.doi.org/ 10.4161/auto.6.1.10249; PMID: 20131413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Becker W. Advanced Time-Correlated Single Photon Counting Techniques. Berlin, Heidelberg, New York: Springer, 2005 [Google Scholar]
- 55.Mueller F, Wach P, McNally JG. . Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys J 2008; 94:3323 - 39; http://dx.doi.org/ 10.1529/biophysj.107.123182; PMID: 18199661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Axelrod D, Koppel DE, Schlessinger J, Elson E, Webb WW. . Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys J 1976; 16:1055 - 69; http://dx.doi.org/ 10.1016/S0006-3495(76)85755-4; PMID: 786399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braga J, Desterro JM, Carmo-Fonseca M. . Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Mol Biol Cell 2004; 15:4749 - 60; http://dx.doi.org/ 10.1091/mbc.E04-06-0496; PMID: 15292455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thaler C, Koushik SV, Puhl HL 3rd, Blank PS, Vogel SS. . Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proc Natl Acad Sci U S A 2009; 106:6369 - 74; http://dx.doi.org/ 10.1073/pnas.0901913106; PMID: 19339497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berland KM, So PT, Gratton E. . Two-photon fluorescence correlation spectroscopy: method and application to the intracellular environment. Biophys J 1995; 68:694 - 701; http://dx.doi.org/ 10.1016/S0006-3495(95)80230-4; PMID: 7696520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen Y, Müller JD, Ruan Q, Gratton E. . Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys J 2002; 82:133 - 44; http://dx.doi.org/ 10.1016/S0006-3495(02)75380-0; PMID: 11751302 [DOI] [PMC free article] [PubMed] [Google Scholar]