

Supplemental Digital Content is available in the text.
Keywords: endothelium, kidney, nitric oxide, pharmacology, prostaglandins
Abstract
Background—
Cardiovascular side effects associated with cyclooxygenase-2 inhibitor drugs dominate clinical concern. Cyclooxygenase-2 is expressed in the renal medulla where inhibition causes fluid retention and increased blood pressure. However, the mechanisms linking cyclooxygenase-2 inhibition and cardiovascular events are unknown and no biomarkers have been identified.
Methods and Results—
Transcriptome analysis of wild-type and cyclooxygenase-2−/− mouse tissues revealed 1 gene altered in the heart and aorta, but >1000 genes altered in the renal medulla, including those regulating the endogenous nitric oxide synthase inhibitors asymmetrical dimethylarginine (ADMA) and monomethyl-l-arginine. Cyclo-oxygenase-2−/− mice had increased plasma levels of ADMA and monomethyl-l-arginine and reduced endothelial nitric oxide responses. These genes and methylarginines were not similarly altered in mice lacking prostacyclin receptors. Wild-type mice or human volunteers taking cyclooxygenase-2 inhibitors also showed increased plasma ADMA. Endothelial nitric oxide is cardio-protective, reducing thrombosis and atherosclerosis. Consequently, increased ADMA is associated with cardiovascular disease. Thus, our study identifies ADMA as a biomarker and mechanistic bridge between renal cyclooxygenase-2 inhibition and systemic vascular dysfunction with nonsteroidal anti-inflammatory drug usage.
Conclusions—
We identify the endogenous endothelial nitric oxide synthase inhibitor ADMA as a biomarker and mechanistic bridge between renal cyclooxygenase-2 inhibition and systemic vascular dysfunction.
Cyclooxygenase (COX) catalyzes the conversion of arachidonic acid to prostaglandin H2, which is then further metabolized by downstream synthase enzymes to a range of eicosanoids. COX is expressed in 2 isoforms, constitutive COX-1 and inducible COX-2. Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used to treat inflammation and pain and show potential to prevent cancer. COX-2 is the therapeutic target of NSAIDs, although most members of this class of drugs inhibit both isoforms of COX1 and, as a consequence, are associated with gastrointestinal side effects.2 NSAID-induced gastrointestinal side effects are relatively common and can be serious, limiting the use of these drugs in some patients and proving fatal in others.3 COX-2 selective inhibitors such as Vioxx (rofecoxib) and Celebrex (celecoxib) were originally developed to target COX-2 at the site of inflammation while sparing COX-1 in the gut and so reducing the gastrointestinal side effects typical of NSAIDs.4 However, now, cardiovascular side effects associated with COX-2 inhibition dominate concern over NSAID usage5–7 and have stopped the development of new drugs in this class. The cardiovascular side effects caused by the inhibition of COX-2 are mainly myocardial infarctions,8,9 which implies increased thrombosis and atherosclerosis and have been considered to reflect local loss of the cardioprotective hormone prostacyclin by blood vessels. However, COX-2 is only sparsely detected in blood vessels, where, by contrast, COX-1 is readily detected. COX-2 is upregulated at sites of vascular inflammation, including atherosclerotic plaques, although, even at these areas, COX-1 drives prostacyclin release.10 Although COX-2 is largely absent from blood vessels,11–13 there are key areas in the body, including the kidney,14–17 where it is expressed constitutively. COX-2 in the kidney regulates natriuresis, diuresis, and vasodilation.16–18 NSAIDs, including COX-2 selective drugs, therefore have well-documented effects on the kidney and increase blood pressure by ≈1 to 2 mm Hg in normotensive individuals and by up to 14 mm Hg in patients with hypertension.19 In addition, they are known to dysregulate glomerular filtration and salt/water homeostasis.20–23 Given the well-established link between impaired renal function and cardiovascular events, including myocardial infarctions, it is logical to expect that these changes in renal function may contribute to the cardiovascular side effects associated with NSAID usage.24 However, the precise mechanism by which COX-2 regulates renal function and the relevance of COX-2 in the kidney in comparison with other cardiovascular tissues are not known, and the debate continues regarding the relative contribution of vascular prostacyclin versus renal effects to cardiovascular side effects of NSAIDs.25 Consequently, there are no biomarkers of NSAID-induced cardiovascular risk and no avenues to protect those that might be more susceptible than the general population.
Clinical Perspective on p 642
To address this issue, we have performed a transcriptomic analysis of the 4 key cardiovascular tissues, kidney, heart, aorta, and blood, from wild-type and COX-2–deficient mice. Pathway analysis revealed alterations in the kidney of genes associated with nitric oxide (NO) biology, particularly those regulating the synthesis and metabolism of the endogenous NO synthase (NOS) inhibitors asymmetrical dimethylarginine (ADMA) and monomethyl-l-arginine (l-NMMA). We went on to validate increased ADMA and l-NMMA levels in plasma of mice lacking COX-2 and in wild-type mice or healthy human volunteers taking COX-2 selective inhibitors. Our work fits well with and helps to explain recent observations showing that endothelial NO synthase (eNOS) responses are reduced in vessels from COX-2–deficient mice.26 Because NO release from endothelial cells performs the same protective role that prostacyclin performs on platelets and blood vessels, and because it synergizes powerfully with prostacyclin in platelets,27 our observations are the first to provide a rational explanation for how the inhibition of COX-2 in the kidney can remotely regulate vascular side effects throughout the vasculature.
Methods
Experimental Animals
Male or female COX-1−/− and COX-2−/− mice,11 back-crossed for >7 generations onto a C57Bl/6J background (Harlan, UK) were used at 10 to 12 weeks of age. For transcriptomic studies, littermate COX-2+/+ mice from COX-2−/− colonies were used. For other studies, wild-type mice maintained separately were used as controls. Animals were genotyped before use.11 Mice lacking prostacyclin receptors (IP−/−), supplied by Dr Rolf Nüsing, were on a C57BL/6 background as described.28 Animal studies were conducted in accordance with Animals (Scientific Procedures) Act 1986, which is a UK act after local ethical review.
Pharmacological COX-2 Inhibition and Blood Pressure Measurement
Parecoxib (Pfizer, USA; 100 mg·kg–1·d–1) in drinking water was administered for 4 days to wild-type mice. Mean carotid arterial blood pressure was measured by using radiotelemetry. In brief, a 1.4F Millar MikroTip pressure catheter was inserted in the right common carotid artery of spontaneously breathing mice anesthetized with inhaled isoflurane (Abbott, USA). Animals were allowed to recover from the anesthetic, and measurements were made in conscious, unrestrained animals 21 days later. Blood pressure traces were recorded continuously with the use of the PowerLab and Chart 5 software (AD Instruments Ltd, UK). Blood was obtained by tail nick in conscious animals into 3.2% citrate and plasma separated by centrifugation.
Mouse Tissue Collection and Processing
Mice were euthanized by CO2 narcosis. Blood collected from the inferior vena cava into heparin (10 U/mL final; Leo Laboratories, UK) or clotting-tubes (Sarstdet, UK) was centrifuged for serum/plasma. Urine was collected by amniocentesis. Serum urea was measured by a commercial veterinary diagnostics service (IDEXX Laboratories, UK).
Microarray Analysis
Tissues were snap frozen and homogenized, and total RNA was extracted by using a silica column–based kit (Nucleospin RNA II; Machery-Nagal, UK). Samples were converted to cDNA, fragmented, labeled, and hybridized to MouseRef-8v3 BeadChip arrays (Illumina, UK). Data were quantile normalized and analyzed by using the linear models for microarray data modified t test method and Benjamini-Hochberg false discovery rate correction by using GeneSpring GX 12.1 software (Agilent, USA). Differential expressed genes with a corrected P value of q<0.05 were considered statistically significant. Focused pathway analysis was performed by examination of genes altered >1.3-fold (q<0.05) featuring in the following human Gene Ontology (http://www.geneontology.org) pathways: (1) regulation of systemic arterial blood pressure (GO:0003073); (2) regulation of blood vessel size (GO:0050880); (3) NO biosynthetic process (GO:0006809); (4) prostaglandin biosynthetic process (GO:0001516); (5) regulation of angiotensin levels in blood (GO:0002002); (6) endothelin maturation (GO:0034959); (7) epinephrine biosynthetic process (GO:0042418); and (8) protein-arginine N-methyltransferase activity (GO:0090627).
Real-Time Quantitaive Polymerase Chain Reaction
RNA was converted to cDNA using the iScript cDNA synthesis kit (BioRad, CA, USA). Quantitative polymerase chain reaction was performed using a 7500 Fast Real-time polymerase chain reaction system (Applied Biosystem). Ddah1 (5′-CACAGAAGGCCCTCAAGATCA-3′, 5′-TCTCATAGACCTTTGCGCTTTC-3′), Ddah2 (5′-CCTGGTGCCACACCTTTCC-3′, 5′-AGGGTGACATCAGAGAGCTTCTG-3′) and Agxt2 (5′-GGCTTCCCCATGGCTGCAGTT-3′, 5′-CAATCACCTCAAGCACAGCAGATCC-3′). mRNA levels were determined busing the iTAQ fast SybrGreen supermix with ROX (BioRad, CA, USA) expression assays and normalized to levels of Actb (5′-CCAGGGTGTGATGGTGGGAATG-3′, 5′-CGCACGATTTCCCTCTCAGCTG-3′). For Ptgs2 (probe ID: Mm00478374_m1), Prmt1 (probe ID: Mm00480133_m1), Arg1 (probe ID: Mm00475988_m1), Arg2 (probe ID: Mm00477592_m1), 18S rRNA (probe ID: Mm03928990_g1), and Gapdh (probe ID: Mm99999915_g1) gene expression levels were determined using TaqMan expression assays. Genes were quantified relative to housekeeping genes (Actb or Gapdh/18S) by comparative Ct methods.
Liquid-Chromatography Tandem Mass-Spectrometry Measurements
Samples were analyzed as described previously29 with a Agilent 6400 series triple quadruple liquid-chromatography tandem mass-spectrometry using mobile phase (0.1% formic acid, 1% acetonitrile). The mass spectrum parameters for detection were as follows: ADMA, mass-to-charge ratio (m/z): 203.3 to 46.0, collision energy: 12; symmetrical dimethylarginine (SDMA), m/z: 203.3 to 70.2, collision energy: 24; l-NMMA, m/z: 189.3 to 70.2, collision energy: 24; d7-ADMA, m/z: 210.0 to 46.0, collision energy: 24. Concentrations of individual amino acids, l-NMMA, ADMA, SDMA, or creatinine were calculated from standard curves generated by using authentic standards.
Vasomotor Responses
Mice were euthanized by CO2 narcosis, and the thoracic aorta was cut into 2-mm aortic rings, mounted in organ baths of a Mulvany-Halpern myograph at a resting tension of 13 kPa in Krebs with or without l-arginine (100 μmol/L; Sigma, UK) and contracted with of endothelin (ET)-1 (30 pmol/L to 30 nmol/L; Sigma, UK) or the thromboxane mimetic U46619 (1 nmol/L to 1 μmol/L). After washing, tissues were contracted with an EC80 concentration of U46619 (30–100 nmol/L) followed by acetylcholine (1 nmol/L to 10 μmol/L) or sodium nitroprusside (SNP; 1 pmol/L to 10 μmol/L). For ET-1 studies, vessels were preincubated with the NOS inhibitor NG-nitro-l-arginine methyl ester (100 μmol/L; Sigma, UK) to exclude any effect of changes in the NO/ADMA pathway. Force was recorded via a PowerLab/800 (AD Instruments Ltd., UK) and analyzed by using Chart 6.0 acquisition system (AD Instruments Ltd, UK).
Clinical Study
Sixteen healthy young male volunteers aged 20 to 35 who had abstained from NSAID use for 14 days previously were recruited. Volunteers received standard doses of the COX-2 selective inhibitor Celebrex (celecoxib; 200 mg twice a day; Pfizer USA), or the nonselective COX-1/COX-2 inhibitor Naprosyn (naproxen; 500 mg twice a day; Roche Switzerland) for 7 days. After overnight fasting, on day 0, before the first dose, and day 7, 2 hours after the final dose, blood was collected into citrate Vacutainers (BD Diagnostics, UK) and separated by centrifugation. This study was approved by the St Thomas’s Hospital Research Ethics Committee (Ref. 07/Q0702/24) and conducted according to the Declaration of Helsinki. All volunteers gave written informed consent before entering the study.
Statistical Methods
Data are mean±standard error of the mean. Statistical significance (taken as P<0.05) was determined by using the following tests: Mann–Whitney (1 variable, 2 groups), Wilcoxon signed-rank (1 variable, 2 paired groups), Kruskal–Wallis with the Dunn post hoc test (1 variable, 3 groups), or Quades test (2 variables). All tests were performed using GraphPad Prism 6 software, with the exception of the Quades test, which was performed using R. Significance was assumed where P<0.05. No further correction has been made for multiple testing (typically <5 end points per animal). The exception is analysis of transcriptomic data where false discovery rate correction has been applied as described above.
Results
Expression of COX-2 in the Kidney
As others have shown,16 constitutive expression of Ptgs2 gene, which encodes the COX-2 protein, was seen in the medulla region of the kidney, with lower levels in the cortex (Figure 1A), which others have shown is relatively sparse unless animals are salt depleted.16 As seen by others, the loss of COX-2, but not of COX-1, resulted in renal compromise associated with increased serum urea and creatinine30 (Figure 1B and 1C).
Figure 1.
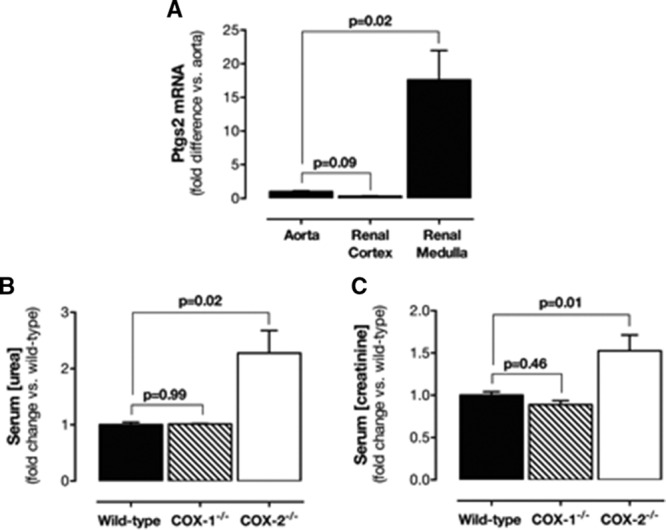
Renal COX-2 expression and effects on renal function. A, Expression of COX-2 in the aorta, renal cortex, and renal medulla of wild-type mice. Changes in levels of serum urea (B) and serum creatinine (C) in wild-type, COX-1−/−, and COX-2−/− mice. Data are mean±SEM for n=8 to 14 mice in each group. P values by Kruskal–Wallis with the Dunn post hoc test. COX indicates cyclooxygenase; and SEM, standard error of the mean.
Effect of COX-2 on the Transcriptome in Cardiovascular Tissues
In the blood, heart, and aorta, COX-1 activity drives prostanoid production with little or no contribution from COX-2.11,14 Consistent with this, no genes were altered by COX-2 deletion in the blood of COX-2−/− mice, and, in the heart and aorta, only 1 gene was altered/decreased, Rgl1, in both tissues (Table I in the online-only Data Supplement). By contrast, in the renal medulla, deletion of COX-2 altered the expression of 1018 genes by >1.5-fold (Figure 2A; Table I in the online-only Data Supplement). To explore the consequences of this finding, we applied focused pathway analysis to specifically examine changes in (1) blood pressure control, (2) vascular tone, and (3) vascular hormones and identified alterations in the expression of a number of genes involved in angiotensin, ET, and NO (Figure 2B). Changes in angiotensin genes (Anpep, Agt, Mme) were predictive of reduced activity, so they were unlikely to be implicated in the deleterious effects of COX-2 inhibitors. ET-1 and ET receptor gene expression (Edn1, Endra, Ednrb) was increased (Figure 2B). However, levels of ET-1 in the plasma of COX-2−/− mice were not altered (Figure IA in the online-only Data Supplement). Moreover, contractile responses in the aorta to ET-1 were not significantly altered in COX-2−/− mice (Figure IB in the online-only Data Supplement). We therefore focused on a cluster of genes (Agxt2, Ddah2, Prmt1) related to the turnover of methylarginines such as ADMA and l-NMMA, which are inhibitors of NOS enzymes. Using quantitative polymerase chain reaction, we confirmed that in COX-2−/− renal medulla expression of Prmt1 (Figure 2C), which drives methylarginine synthesis,29 was increased; Ddah2, which breaks down methylarginine, was nonsignificantly increased (Figure II in the online-only Data Supplement), whereas Agxt2, which degrades methylarginines,29 tended to be reduced (Figure 2D). Using quantitative polymerase chain reaction, we also observed reduced expression of Ddah1, which, like Agxt2, is responsible for methylarginine breakdown29 (Figure 2E).
Figure 2.
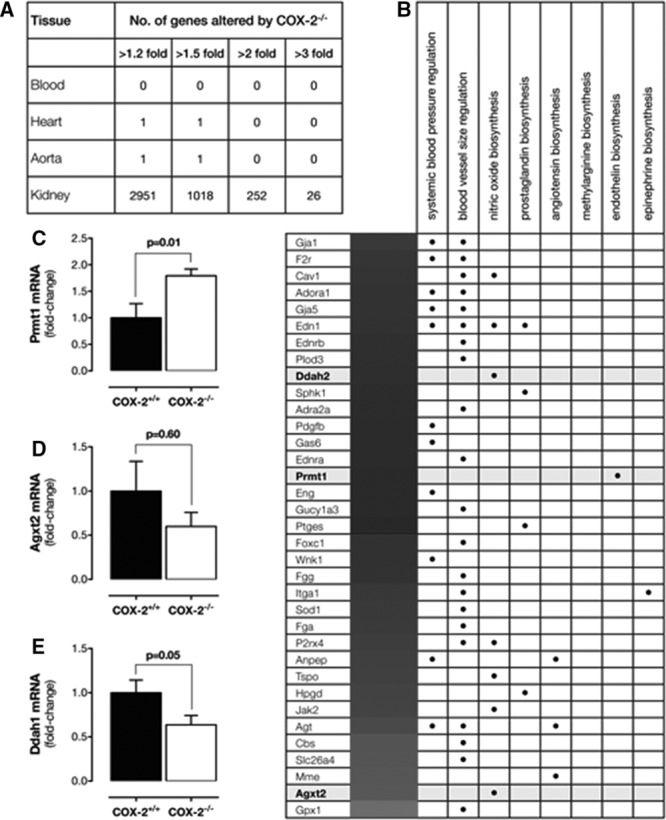
Transcriptome profiling in cardiovascular tissues from COX-2−/− mice. A, Number of genes altered in the transcriptome of blood, heart, aorta, and kidney (renal medulla) by >1.2- to >3-fold. B, Focused pathway analysis of genes altered >1.3-fold (q<0.05). qPCR validation of changes in expression of Prmt1 (C), Agxt2 (D), and Ddah1 (E) in the renal medulla of COX-2−/− mice. Data are from n=7 to 8 mice. Data in C through E are mean±SEM. P values by Mann–Whitney U test. COX indicates cyclooxygenase; qPCR, quantitative polymerase chain reaction; and SEM, standard error of the mean.
Effect of COX-2 on Plasma Methylarginines and eNOS Responses in Mouse Aorta
Plasma levels of ADMA and l-NMMA were increased in COX-2−/−, but not COX-1−/− mice (Figure 3A and 3B). Plasma SDMA levels were not changed in COX-2−/− mice (Table II in the online-only Data Supplement). Arginases, like NOS, use l-arginine as a substrate and can be altered in inflammatory conditions. However, in the renal medulla, Arg1 was not expressed and Arg2 was not altered in COX-2−/− mice (Figure IIIA in the online-only Data Supplement). In addition, in the aorta, Arg2 was not expressed in and Arg1 was not altered in COX-2−/− mice (Figure IIIB in the online-only Data Supplement). In line with this finding, l-arginine was not significantly altered in COX-2−/− mice (Figure IIIC in the online-only Data Supplement), resulting in ADMA:l-arginine (Figure IIID in the online-only Data Supplement) and l-NMMA:l-arginine (Figure IIIE in the online-only Data Supplement) ratios being increased. In agreement with these data, the pharmacological inhibition of COX-2 in wild-type mice with parecoxib (100 mg/kg for 4 days) blocked diurnal blood pressure decline (Figure 4A) and plasma ADMA (Figure 4B) and l-NMMA levels (Figure 4C), as well. l-Arginine levels (Figure IVA in the online-only Data Supplement) along with ADMA:l-arginine (Figure IVB in the online-only Data Supplement) and l-NMMA:l-arginine (Figure IVC in the online-only Data Supplement) ratios were increased by parecoxib.
Figure 3.
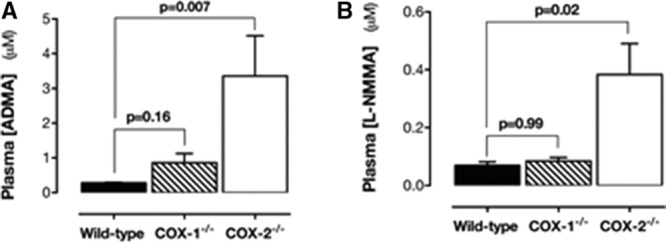
Effect of COX gene deletion ADMA and l-NMMA in plasma. Levels of ADMA (A) and l-NMMA (B) were increased in the plasma of COX-2−/− mice, but not in COX-1−/− mice. Data are from n=4 and presented as mean±SEM. P values by Kruskal–Wallis with the Dunn post hoc test. ADMA indicates asymmetrical dimethylarginine; COX, cyclooxygenase; l-NMMA, monomethyl-l-arginine; and SEM, standard error of the mean.
Figure 4.
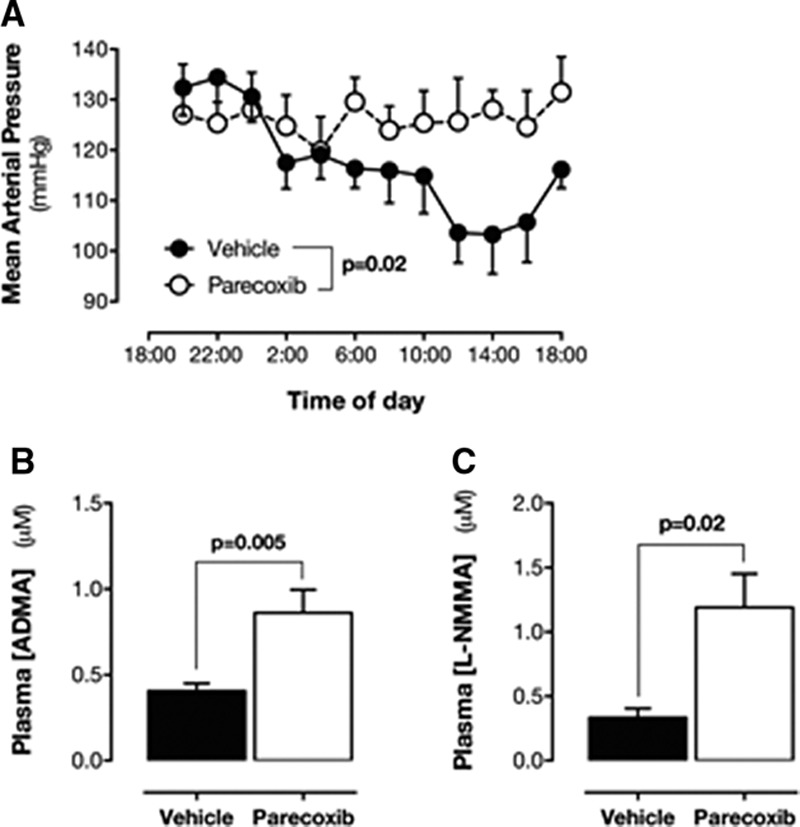
Effect of parecoxib on blood pressure and methylarginines. Effect of parecoxib on mean arterial blood pressure (A) and plasma ADMA (B) and l-NMMA (C) levels in mice. Data are mean±SEM for n=3 to 7. Data in A were analyzed by Quades 2-way ANOVA. Data in B and C were analyzed by the Mann–Whitney U test. ADMA indicates asymmetrical dimethylarginine; ANOVA, analysis of variance; l-NMMA, monomethyl-l-arginine; and SEM, standard error of the mean.
Increased plasma levels of methylarginines are associated in other models with reduced eNOS activity in vessels ex vivo31 and hypertension in vivo. Aorta from COX-2−/− mice had a reduced eNOS response to acetylcholine (Figure 5A). However, their response to the NO-donor SNP that relaxes vessels via NO independently of the endothelium was not altered (Figure 5B), nor was their contractile response to U46619 (Figure V in the online-only Data Supplement). The reduced eNOS responses in the aorta of COX-2−/− mice were reversed in the presence of l-arginine (100 μmol/L; Figure 5C). No effect of l-arginine (Figure 5D) was seen on response to SNP. These effects were not due to local changes in aortic Nos3, Ddah1, Ddah2, or Agxt2 gene expression (Figure VI in the online-only Data Supplement).
Figure 5.
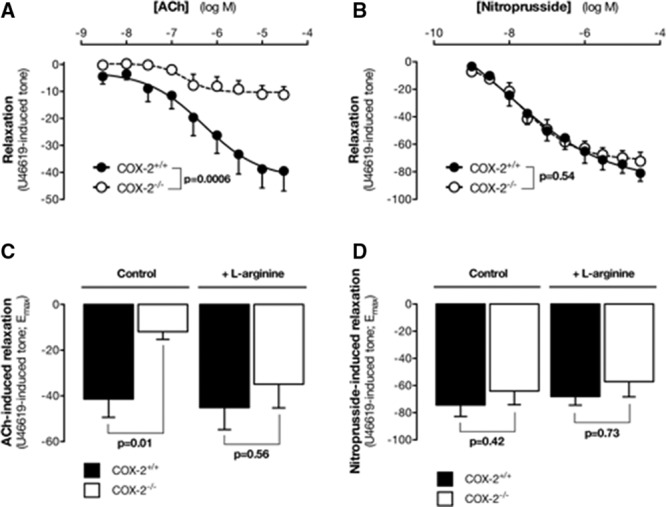
Effect of COX-2 gene deletion of eNOS responses in aorta. Acetylcholine (ACh; A) or sodium nitroprusside (SNP; B) induced the relaxation of aorta from wild-type and COX-2−/− mice. Maximum relaxation responses of the aorta from wild-type and COX-2−/− mice induced by Ach (C) or SNP (D) in the absence or presence of l-arginine (100 μmol/L). Data are the mean±SEM for n=6 to 10. P values for A and C were by Quades 2-way ANOVA and for B and D were by Mann–Whitney U test. ANOVA indicates analysis of variance; COX, cyclooxygenase; and eNOS, endothelial nitric oxide synthase.
Effect of Loss of the Prostacyclin Receptor (IP) on Renal Gene Expression and Plasma ADMA Levels
Prostacyclin, which acts via cell surface IP receptors, is an important COX-derived mediator in the cardiovascular system. To establish the relative role of prostacyclin, in comparison with the myriad of other COX products, in our study, we performed experiments using IP−/− mice. None of our genes of interest (Prmt1, Agxt2, Ddah1, Ddah2, or Arg2) were altered in the renal medulla from IP−/− mice (Figure VII in the online-only Data Supplement). In line, the increase seen in ADMA levels associated with the loss of IP gene was, although statistically significant (Figure VIIIA in the online-only Data Supplement), minor in magnitude in comparison with what we report in COX-2−/− mice (Figure VIIIB in the online-only Data Supplement). l-Arginine (Figure VIIIC in the online-only Data Supplement) and l-NMMA (Figure VIIID in the online-only Data Supplement) levels were decreased, whereas those of SDMA (Table II in the online-only Data Supplement) were not affected by the loss of IP receptors. These changes may reflect altered renal function in IP−/− mice, because creatinine levels were elevated in plasma (Figure VIIIE in the online-only Data Supplement).
Effect of Inhibition of COX-2 in Healthy Human Volunteers
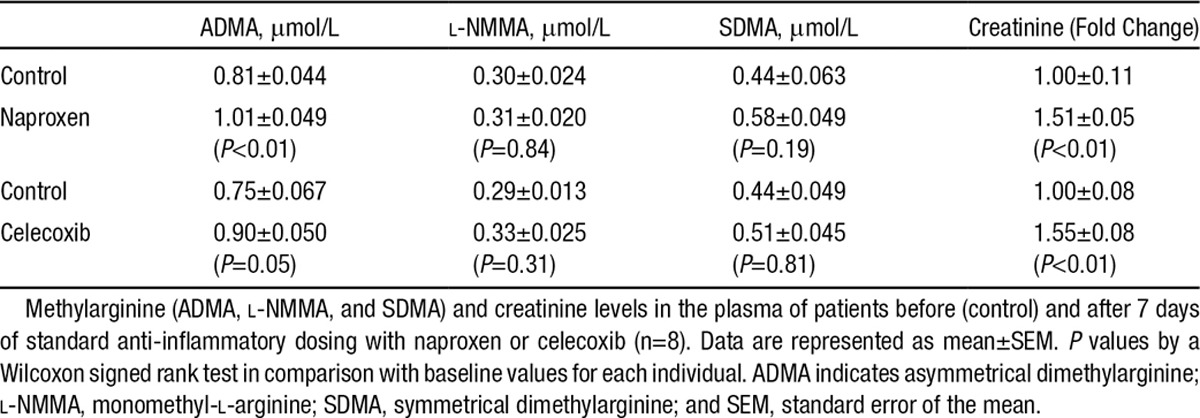
In direct corroboration of our findings in mice, the inhibition of COX-2 with standard anti-inflammatory doses of celecoxib (200 mg twice a day) for 7 days (n=8) increased ADMA levels in healthy human volunteers (Table). Naproxen (500 mg twice a day), which inhibits COX-2, but also inhibits COX-1, similarly increased ADMA levels in volunteers taking the drug for 7 days (n=8; Table).
Table.
Plasma Methylarginines In Healthy Volunteers Taking Naproxen or Celecoxib
Discussion
The notion that COX-2 is constitutively expressed in the kidney is not new and is, in fact, based on observations made as early as 1994,32 well before selective COX-2 inhibitors were introduced to the clinic. Similarly, the idea that the inhibition of COX-2 in the kidney could explain the cardiovascular side effects of NSAIDs has been suggested.18–20 However, this view has been somewhat sidelined by the idea that vascular COX-2 drives prostacyclin throughout the systemic circulation,7 and, until our recent studies,14,33 the relative importance of the kidney in comparison with large vessels had not been specifically addressed. Using a COX-2 luciferase knock-in mouse, we performed an unbiased systems-based analysis of COX-2 expression in an extensive panel of organs and tissues in the mouse and found that renal COX-2 was localized to an intense hot spot in the center of the kidney, aligned to the inner region of the renal medulla. Here, we validated our earlier report and used polymerase chain reaction to show that the COX-2 (Ptgs2) gene is highly enriched in the medulla in comparison with the renal cortex. This is in line with what is known about COX-2 in the kidney, which under normal or high-salt conditions is predominantly expressed in the medulla region, particularly in the interstitial fibroblasts.14,16
Although COX-2 expression and activity are low relative to those of COX-1 in other cardiovascular structures, such as the heart and major blood vessels, conditional knockout mice where either endothelial26 or cardiomyocyte34 COX-2 is deleted have been used to implicate a role of COX-2 in cardiovascular function in these structures; although, in the heart, the findings with conditional knockout mice were questioned and suggested to be confounded by the nonspecific effects of tamoxifen/cre toxicity.35 Nevertheless, to identify the gross effect of COX-2 at these sites in comparison with the renal medulla and to identify the novel mechanisms of NSAIDs, we investigated the effects of COX-2 gene deletion on the transcriptome. In line with constitutive COX-2 gene activity that we recently reported,14 we found that only 1 gene is altered in the heart and aorta of COX-2−/− mice, whereas >600 genes were changed by >1.5-fold in the renal medulla by COX-2 deletion. Clearly, these findings implicate a dominant role for constitutive COX-2 in the kidney in comparison with the heart or aorta. This may suggest that endothelial cells outside large vessels, potentially in the renal circulation, mediate the effects previously reported.26 Because of the large number of genes altered in the renal medulla by COX-2 deletion, we chose to apply a focused gene pathway analysis of transcripts relating to control of blood pressure, vascular tone, and vascular hormones to extract leads for understanding how renal COX-2 can regulate systemic cardiovascular homeostasis. Using this approach, we identified a number of groups of genes, for example, related to ET and angiotensin signaling. Angiotensin genes were altered in a way that would not predict cardiovascular side effects and so were not pursued in this study. Changes in ET-1 genes may well be important in the effects of NSAIDs in some settings, but, in our model using healthy mice, the changes we found in the renal medulla did not ultimately affect plasma ET-1 levels or vascular responses to ET-1 in the aorta ex vivo. We therefore focused our study around genes altered that regulate the synthesis and metabolism of the endogenous eNOS inhibitors ADMA and l-NMMA. The kidney is a major site of methylarginine synthesis and clearance from the circulation and changes to these pathways have the potential to systemically regulate vascular health. Specifically, Prmt1, first identified because of its association with the TIS21 gene,36 methylates arginine residues in proteins leading to the release of mature ADMA and l-NMMA after proteolysis, was increased by COX-2 deletion, whereas both Agxt237 and Ddah131, enzymes that metabolize methylarginines, were decreased in COX-2−/− renal medulla.
To define the interaction between COX-2 and the methylarginine pathways and to validate biologically our gene expression data, we measured the levels of ADMA, SDMA, and l-NMMA. ADMA and l-NMMA are both competitive inhibitors of eNOS, whereas SDMA is biologically inactive and not cleared by DDAH1. COX-1−/− mice exhibited normal renal, plasma, and urinary levels of each methylarginine. In contrast, the plasma levels of ADMA and l-NMMA were increased, whereas l-arginine was unchanged, in samples from COX-2−/− mice. These findings are in line with others showing increased methylarginines in models where the activity of PRMT1 is elevated and AGXT2 is reduced.29 We found that SDMA is not significantly increased in COX-2−/− mouse plasma, implicating an additional functional role for reduced DDAH1 in this model. It should be noted that nonrenal factors may act together with events in the kidney to regulate plasma ADMA in COX-2−/− mice.
After demonstrating that COX-2 deletion increases ADMA and l-NMMA, we next sought to establish if this could be recapitulated with a therapeutically relevant COX-2 selective drug in wild-type mice. We chose parecoxib, which is the prodrug of valdecoxib and a licensed therapy for acute pain. It is highly COX-2 selective,1 and, because it is water soluble, it can be easily administered to mice in drinking water. Similar to our findings in COX-2−/− mice, pharmacological COX-2 inhibition by parecoxib produced an increase in circulating levels of ADMA and l-NMMA. This further supports our findings, confirming that increased levels of endogenous NOS inhibitors is not a developmental consequence of COX-2 deletion; rather, it is an acute biochemical consequence of the loss of COX-2 enzyme activity.
We have shown previously that the global heterozygous deletion of the Ddah1 gene results in increased levels of plasma ADMA and l-NMMA and increased blood pressure31 similar in magnitude to that seen in the current study. Further, we have demonstrated that plasma increases of ADMA and l-NMMA in Ddah1-deficient mice, in the concentration range shown here in COX-2−/− mice, were associated with vascular dysfunction manifesting as reduced acetylcholine-induced vasodilator responses in endothelium intact segments of aorta.31 In the aorta, acetylcholine-induced vasodilation is mediated predominately by NO derived from eNOS, with no role for local prostacyclin release. Thus, in this work, the loss of acetylcholine-induced vasodilation was attributed to the inhibition of eNOS as a direct consequence of increased ADMA and l-NMMA.31 In the current study, we have taken a similar approach and found that aorta from COX-2−/− mice had a reduced eNOS-dependent response to acetylcholine, but a normal response to the endothelial-independent NO donor SNP. This observation is consistent with recent work from others showing reduced eNOS activity in the aorta of COX-2−/− mice,26 although, in that study, a role for natural eNOS inhibitors was not addressed. ADMA and l-NMMA are competitive inhibitors of eNOS, and, as such, their effects are reversed when the natural substrate, l-arginine, is increased in the system.38 To establish the specific role of ADMA and l-NMMA in the loss of eNOS function in the aorta, in our study, we assessed the ability of l-arginine to rescue the vascular dysfunction. In wild-type mouse aorta, l-arginine did not amplify acetylcholine-induced vasodilation, consistent with l-arginine availability not being rate limiting in healthy blood vessels.39 In contrast, l-arginine reversed the impairment in endothelial function present in aorta from COX-2−/− mice; returning acetylcholine-dependent vasodilation to wild-type levels, without affecting smooth muscle sensing of NO. It must be remembered that this effect cannot be explained by any loss of COX-2 activity locally within the vessel, because, in this tissue, COX-1, and not COX-2, is expressed.11,14 Rather, this is explained by changes in kidney function, leading to increased circulating ADMA and l-NMMA, which then act in a endocrine manner on the endothelium in the systemic circulation. Our findings that the reduced eNOS activity seen in vessels from COX-2−/− mice could be prevented by exogenous l-arginine clearly implicate a role for ADMA and l-NMMA and are in line with others showing that ADMA and l-NMMA specifically and actively accumulate in endothelial cells and that this results in decreased eNOS activity ex vivo, which is reversible by l-arginine.40
Finally, to establish if the phenomenon demonstrated in mice, ie, that loss/inhibition of COX-2 results in increased circulating levels of ADMA also occurs in humans, we performed a clinical study in which healthy male volunteers were treated with standard doses of the selective COX-2 inhibitor celecoxib or naproxen, a dual COX-1/COX-2 inhibitor.1 At therapeutic doses, both of these drugs inhibit COX-2 by ≈ 80%.1 Creatinine levels were increased to a similar level by celecoxib and naproxen treatment highlighting the effect of COX-2 inhibition on renal function. In direct corroboration of our findings in mice, both celecoxib and naproxen increased ADMA levels. These observations suggest that, in humans, after even short-term inhibition of COX-2, renal dysfunction, including the reduction in filtration drive, increases the levels of ADMA. It is important to note, however, that ADMA, l-NMMA, and SDMA are similarly cleared by the kidney. Thus, the finding that ADMA is increased without a concomitant increase in other methylarginines suggests that some additional specific pathways may be altered by COX-2 inhibition. We recognize that the relative change in ADMA levels in our human study was much lower than seen in either COX-2−/− mice or in wild-type mice treated with a COX-2 inhibitor. We do not suggest that, in this small sample size with healthy volunteers, the increase would be sufficient to adversely affect cardiovascular function, simply that, it provides a proof-of-concept study and that, in a much larger cohort, such as the Framingham study41 and in patients with inflammation or cardiovascular disease, this small but significant increase could account for the equally small but significant increase in cardiovascular events seen in patients taking anti-inflammatory drugs, including COX-2 inhibitors. Indeed, as reported in the Framingham study41 in large populations, even relatively small changes in ADMA levels are associated with significant increases in the risk of cardiovascular events, and the plasma levels of ADMA and l-NMMA, which others have shown specifically can accumulate in endothelial cells,40 are highly correlated with cardiovascular disease in a range of human clinical studies.42–46 This is relevant because COX-2 selective drugs, including celecoxib and rofecoxib, increase the risk of cardiovascular events even in healthy individuals – that is to say, in those people without overt evidence of cardiovascular disease, requiring medication, or recent previous events, because these individuals were excluded from trials. Furthermore, although the efficacy trials for COX-2 inhibitors were performed in patients with arthritis,47 who are commonly in a category of increased risk of cardiovascular disease owing to general inflammation and age, the Adenomatous Polyp Prevention on Vioxx (APPROVE)48 (rofecoxib) and Adenoma Prevention With Celecoxib (APC)49 (celecoxib) trials were conducted on volunteers without arthritis. In both the APPROVE and APC trials, increased cardiovascular events were noted, which in the case of rofecoxib lead to the withdrawal of the drug.
The differences seen in ADMA in COX-2−/− mice and human volunteers taking NSAIDs for 1 week are probably explained by the duration and extent of loss of COX-2 activity. The relatively small increase in ADMA seen in humans versus mice administered inhibitors is less obviously explained, but it could be due to species differences and the fact that humans are outbred, and so responses are likely to be more variable. In addition, mice treated with parecoxib were chronically instrumented, which may also influence responses.
Methylarginines inhibit eNOS, reducing NO released across the entire vascular tree. NO performs functions similar to prostacyclin in vessels and platelets. Moreover, NO and prostacyclin synergize strongly to inhibit platelet activation.27 Our data provide the first explanation for how the inhibition of COX-2 in the kidney can lead to reduced vascular function and firmly establish a COX-2/eNOS axis involving ADMA. Our results also show that ADMA and other methylarginines are potential biomarkers for the cardiovascular risk in patients taking traditional NSAIDs, and COX-2 selective inhibitors, as well. In addition, if endogenous inhibitors of NOS are mechanistically involved in the cardiovascular toxicity associated with COX-2 inhibitor use in humans, then there is the very clear potential for l-arginine to be used as a preventative treatment in at-risk patients.50 Moreover, the potential for genetic mutations in genes such as PRMT1, DDAH1, and AGXT2 to be associated with increased risk of cardiovascular side effects in patients taking NSAIDs should be considered.
Acknowledgments
We thank Hime Gashaw, Ivana Vojnovic, Mathew Delahaye, Olga Boruc, and Maxim Freydin for technical assistance.
Sources of Funding
This work was supported by the Wellcome Trust (0852551Z108/Z), the BHF (FS/12/53/29643), and MRC Intramural funding.
Disclosures
Dr Mitchell is a member of the scientific advisory board for Antibe Therapeutics, and Drs Mitchell and Warner have been expert witnesses for cases associated with Pfizer products. The other authors report no conflicts.
Supplementary Material
Footnotes
Drs Ahmetaj-Shala and Kirkby contributed equally.
Drs Leiper and Mitchell contributed equally.
The online-only Data Supplement is available with this article at http://circ.ahajournals.org/lookup/suppl/doi:10.1161/CIRCULATIONAHA.114.011591/-/DC1.
CLINICAL PERSPECTIVE
Nonsteroidal anti-inflammatory drugs are the most common over-the-counter medications worldwide and the mainstay therapy for those with chronic inflammatory conditions such as arthritis. Cyclooxygenase (COX)-2 is the therapeutic target of nonsteroidal anti-inflammatory drugs, but these drugs also inhibit the constitutive form COX-1, potentially causing serious gastrointestinal side effects. COX-2 selective drugs, such as celecoxib and rofecoxib, were introduced to reduce gastrointestinal side effects. However, it is now clear that COX-2 selective anti-inflammatory drugs and older-style nonsteroidal anti-inflammatory drugs like ibuprofen and diclofenac increase the risk of cardiovascular events. These side effects are relatively rare, but concern over them has meant that there has been a return to prescribing older style drugs, which generally carry a similar risk of heart attacks and strokes but with the added disadvantage of increased gastrointestinal side effects. Currently there is no accepted unifying mechanism to explain how COX-2 inhibitors cause cardiovascular side effects, and, consequently, there are no biomarkers or rescue strategies to identify or treat those few individuals who might be at risk. Our study shows methylarginines, which block cardioprotective endothelial nitric oxide synthase in vessels, are elevated when COX-2 is inhibited. Methylarginines are elevated in a range of cardiovascular diseases and as such are considered risk factors. Our work suggests methylarginines may also be biomarkers of the risk associated with COX-2 inhibition. If these findings are validated in a larger patient group, a simple test for methylarginines could be used to identify those individuals most at risk for cardiovascular events while taking COX-2 inhibitors.
References
- 1.Warner TD, Giuliano F, Vojnovic I, Bukasa A, Mitchell JA, Vane JR. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc Natl Acad Sci U S A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wallace JL, McKnight W, Reuter BK, Vergnolle N. NSAID-induced gastric damage in rats: requirement for inhibition of both cyclooxygenase 1 and 2. Gastroenterology. 2000;119:706–714. doi: 10.1053/gast.2000.16510. [DOI] [PubMed] [Google Scholar]
- 3.Wallace JL. Prostaglandins, NSAIDs, and gastric mucosal protection: why doesn’t the stomach digest itself? Physiol Rev. 2008;88:1547–1565. doi: 10.1152/physrev.00004.2008. doi: 10.1152/physrev.00004.2008. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell JA, Warner TD. Cyclo-oxygenase-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy. Br J Pharmacol. 1999;128:1121–1132. doi: 10.1038/sj.bjp.0702897. doi: 10.1038/sj.bjp.0702897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Antman EM, Bennett JS, Daugherty A, Furberg C, Roberts H, Taubert KA American Heart Association. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–1642. doi: 10.1161/CIRCULATIONAHA.106.181424. doi: 10.1161/CIRCULATIONAHA.106.181424. [DOI] [PubMed] [Google Scholar]
- 6.Warner TD, Mitchell JA. COX-2 selectivity alone does not define the cardiovascular risks associated with non-steroidal anti-inflammatory drugs. Lancet. 2008;371:270–273. doi: 10.1016/S0140-6736(08)60137-3. doi: 10.1016/S0140-6736(08)60137-3. [DOI] [PubMed] [Google Scholar]
- 7.Funk CD, FitzGerald GA. COX-2 inhibitors and cardiovascular risk. J Cardiovasc Pharmacol. 2007;50:470–479. doi: 10.1097/FJC.0b013e318157f72d. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 8.McGettigan P, Henry D. Cardiovascular risk with non-steroidal anti-inflammatory drugs: systematic review of population-based controlled observational studies. PLoS Med. 2011;8:e1001098. doi: 10.1371/journal.pmed.1001098. doi: 10.1371/journal.pmed.1001098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kohli P, Steg PG, Cannon CP, Smith SC, Jr, Eagle KA, Ohman EM, Alberts MJ, Hoffman E, Guo J, Simon T, Sorbets E, Goto S, Bhatt DL REACH Registry Investigators. NSAID use and association with cardiovascular outcomes in outpatients with stable atherothrombotic disease. Am J Med. 2014;127:53–60.e1. doi: 10.1016/j.amjmed.2013.08.017. doi: 10.1016/j.amjmed.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Wong E, Huang JQ, Tagari P, Riendeau D. Effects of COX-2 inhibitors on aortic prostacyclin production in cholesterol-fed rabbits. Atherosclerosis. 2001;157:393–402. doi: 10.1016/s0021-9150(00)00756-5. [DOI] [PubMed] [Google Scholar]
- 11.Kirkby NS, Lundberg MH, Harrington LS, Leadbeater PD, Milne GL, Potter CM, Al-Yamani M, Adeyemi O, Warner TD, Mitchell JA. Cyclooxygenase-1, not cyclooxygenase-2, is responsible for physiological production of prostacyclin in the cardiovascular system. Proc Natl Acad Sci U S A. 2012;109:17597–17602. doi: 10.1073/pnas.1209192109. doi: 10.1073/pnas.1209192109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu B, Luo W, Zhang Y, Li H, Zhu N, Huang D, Zhou Y. Involvement of cyclo-oxygenase-1-mediated prostacyclin synthesis in the vasoconstrictor activity evoked by ACh in mouse arteries. Exp Physiol. 2012;97:277–289. doi: 10.1113/expphysiol.2011.062034. doi: 10.1113/expphysiol.2011.062034. [DOI] [PubMed] [Google Scholar]
- 13.Zidar N, Odar K, Glavac D, Jerse M, Zupanc T, Stajer D. Cyclooxygenase in normal human tissues–is COX-1 really a constitutive isoform, and COX-2 an inducible isoform? J Cell Mol Med. 2009;13(9B):3753–3763. doi: 10.1111/j.1582-4934.2008.00430.x. doi: 10.1111/j.1582-4934.2008.00430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirkby NS, Zaiss AK, Urquhart P, Jiao J, Austin PJ, Al-Yamani M, Lundberg MH, MacKenzie LS, Warner TD, Nicolaou A, Herschman HR, Mitchell JA. LC-MS/MS confirms that COX-1 drives vascular prostacyclin whilst gene expression pattern reveals non-vascular sites of COX-2 expression. PLoS One. 2013;8:e69524. doi: 10.1371/journal.pone.0069524. doi: 10.1371/journal.pone.0069524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harris RC., Jr Cyclooxygenase-2 inhibition and renal physiology. Am J Cardiol. 2002;89(6A):10D–17D. doi: 10.1016/s0002-9149(02)02232-4. [DOI] [PubMed] [Google Scholar]
- 16.Qi Z, Hao CM, Langenbach RI, Breyer RM, Redha R, Morrow JD, Breyer MD. Opposite effects of cyclooxygenase-1 and -2 activity on the pressor response to angiotensin II. J Clin Invest. 2002;110:61–69. doi: 10.1172/JCI14752. doi: 10.1172/JCI14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan KN, Paulson SK, Verburg KM, Lefkowith JB, Maziasz TJ. Pharmacology of cyclooxygenase-2 inhibition in the kidney. Kidney Int. 2002;61:1210–1219. doi: 10.1046/j.1523-1755.2002.00263.x. doi: 10.1046/j.1523-1755.2002.00263.x. [DOI] [PubMed] [Google Scholar]
- 18.Harris RC. Physiologic and pathophysiologic roles of cyclooxygenase-2 in the kidney. Trans Am Clin Climatol Assoc. 2013;124:139–151. [PMC free article] [PubMed] [Google Scholar]
- 19.Snowden S, Nelson R. The effects of nonsteroidal anti-inflammatory drugs on blood pressure in hypertensive patients. Cardiol Rev. 2011;19:184–191. doi: 10.1097/CRD.0b013e31821ddcf4. doi: 10.1097/CRD.0b013e31821ddcf4. [DOI] [PubMed] [Google Scholar]
- 20.Brater DC. Renal effects of cyclooxygyenase-2-selective inhibitors. J Pain Symptom Manage. 2002;23(4 suppl):S15–S20; discussion S21. doi: 10.1016/s0885-3924(02)00370-6. [DOI] [PubMed] [Google Scholar]
- 21.Carmichael J, Shankel SW. Effects of nonsteroidal anti-inflammatory drugs on prostaglandins and renal function. Am J Med. 1985;78(6 pt 1):992–1000. doi: 10.1016/0002-9343(85)90223-2. [DOI] [PubMed] [Google Scholar]
- 22.Kömhoff M, Grone HJ, Klein T, Seyberth HW, Nüsing RM. Localization of cyclooxygenase-1 and -2 in adult and fetal human kidney: implication for renal function. Am J Physiol. 1997;272(4 pt 2):F460–F468. doi: 10.1152/ajprenal.1997.272.4.F460. [DOI] [PubMed] [Google Scholar]
- 23.Catella-Lawson F, McAdam B, Morrison BW, Kapoor S, Kujubu D, Antes L, Lasseter KC, Quan H, Gertz BJ, FitzGerald GA. Effects of specific inhibition of cyclooxygenase-2 on sodium balance, hemodynamics, and vasoactive eicosanoids. J Pharmacol Exp Ther. 1999;289:735–741. [PubMed] [Google Scholar]
- 24.Singh G, Miller JD, Huse DM, Pettitt D, D’Agostino RB, Russell MW. Consequences of increased systolic blood pressure in patients with osteoarthritis and rheumatoid arthritis. J Rheumatol. 2003;30:714–719. [PubMed] [Google Scholar]
- 25.Salvo F, Antoniazzi S, Duong M, Molimard M, Bazin F, Fourrier-Réglat A, Pariente A, Moore N. Cardiovascular events associated with the long-term use of NSAIDs: a review of randomized controlled trials and observational studies. Expert Opin Drug Saf. 2014;13:573–585. doi: 10.1517/14740338.2014.907792. doi: 10.1517/14740338.2014.907792. [DOI] [PubMed] [Google Scholar]
- 26.Yu Y, Ricciotti E, Scalia R, Tang SY, Grant G, Yu Z, Landesberg G, Crichton I, Wu W, Puré E, Funk CD, FitzGerald GA. Vascular COX-2 modulates blood pressure and thrombosis in mice. Sci Transl Med. 2012;4:132ra54. doi: 10.1126/scitranslmed.3003787. doi: 10.1126/scitranslmed.3003787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kirkby NS, Lundberg MH, Chan MV, Vojnovic I, Solomon AB, Emerson M, Mitchell JA, Warner TD. Blockade of the purinergic P2Y12 receptor greatly increases the platelet inhibitory actions of nitric oxide. Proc Natl Acad Sci U S A. 2013;110:15782–15787. doi: 10.1073/pnas.1218880110. doi: 10.1073/pnas.1218880110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schuh CD, Pierre S, Weigert A, Weichand B, Altenrath K, Schreiber Y, Ferreiros N, Zhang DD, Suo J, Treutlein EM, Henke M, Kunkel H, Grez M, Nusing R, Brune B, Geisslinger G, Scholich K. Prostacyclin mediates neuropathic pain through interleukin 1beta-expressing resident macrophages. Pain. 2014;155:545–555. doi: 10.1016/j.pain.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Caplin B, Leiper J. Endogenous nitric oxide synthase inhibitors in the biology of disease: markers, mediators, and regulators? Arterioscler Thromb Vasc Biol. 2012;32:1343–1353. doi: 10.1161/ATVBAHA.112.247726. doi: 10.1161/ATVBAHA.112.247726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dinchuk JE, Car BD, Focht RJ, Johnston JJ, Jaffee BD, Covington MB, Contel NR, Eng VM, Collins RJ, Czerniak PM. Renal abnormalities and an altered inflammatory response in mice lacking cyclooxygenase II. Nature. 1995;378:406–409. doi: 10.1038/378406a0. doi: 10.1038/378406a0. [DOI] [PubMed] [Google Scholar]
- 31.Leiper J, Nandi M, Torondel B, Murray-Rust J, Malaki M, O’Hara B, Rossiter S, Anthony S, Madhani M, Selwood D, Smith C, Wojciak-Stothard B, Rudiger A, Stidwill R, McDonald NQ, Vallance P. Disruption of methylarginine metabolism impairs vascular homeostasis. Nat Med. 2007;13:198–203. doi: 10.1038/nm1543. doi: 10.1038/nm1543. [DOI] [PubMed] [Google Scholar]
- 32.Harris RC, McKanna JA, Akai Y, Jacobson HR, Dubois RN, Breyer MD. Cyclooxygenase-2 is associated with the macula densa of rat kidney and increases with salt restriction. J Clin Invest. 1994;94:2504–2510. doi: 10.1172/JCI117620. doi: 10.1172/JCI117620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stichtenoth DO, Frölich JC. COX-2 and the kidneys. Curr Pharm Des. 2000;6:1737–1753. doi: 10.2174/1381612003398717. [DOI] [PubMed] [Google Scholar]
- 34.Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J, Zhang H, Hui Y, Cheng Y, Petrenko N, Yu Y, FitzGerald GA. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009;106:7548–7552. doi: 10.1073/pnas.0805806106. doi: 10.1073/pnas.0805806106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papanicolaou KN, Streicher JM, Ishikawa TO, Herschman H, Wang Y, Walsh K. Preserved heart function and maintained response to cardiac stresses in a genetic model of cardiomyocyte-targeted deficiency of cyclooxygenase-2. J Mol Cell Cardiol. 2010;49:196–209. doi: 10.1016/j.yjmcc.2010.04.002. doi: 10.1016/j.yjmcc.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lin WJ, Gary JD, Yang MC, Clarke S, Herschman HR. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem. 1996;271:15034–15044. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 37.Rodionov RN, Murry DJ, Vaulman SF, Stevens JW, Lentz SR. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J Biol Chem. 2010;285:5385–5391. doi: 10.1074/jbc.M109.091280. doi: 10.1074/jbc.M109.091280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc Natl Acad Sci U S A. 1989;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swierkosz TA, Mitchell JA, Sessa WC, Hecker M, Vane JR. L-glutamine inhibits the release of endothelium-derived relaxing factor from the rabbit aorta. Biochem Biophys Res Commun. 1990;172:143–148. doi: 10.1016/s0006-291x(05)80184-6. [DOI] [PubMed] [Google Scholar]
- 40.Cardounel AJ, Cui H, Samouilov A, Johnson W, Kearns P, Tsai AL, Berka V, Zweier JL. Evidence for the pathophysiological role of endogenous methylarginines in regulation of endothelial NO production and vascular function. J Biol Chem. 2007;282:879–887. doi: 10.1074/jbc.M603606200. doi: 10.1074/jbc.M603606200. [DOI] [PubMed] [Google Scholar]
- 41.Böger RH, Sullivan LM, Schwedhelm E, Wang TJ, Maas R, Benjamin EJ, Schulze F, Xanthakis V, Benndorf RA, Vasan RS. Plasma asymmetric dimethylarginine and incidence of cardiovascular disease and death in the community. Circulation. 2009;119:1592–1600. doi: 10.1161/CIRCULATIONAHA.108.838268. doi: 10.1161/CIRCULATIONAHA.108.838268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mügge A, Hanefeld C, Böger RH CARDIAC study investigators. Plasma concentration of asymmetric dimethylarginine and the risk of coronary heart disease: rationale and design of the multicenter CARDIAC study. Atheroscler Suppl. 2003;4:29–32. doi: 10.1016/s1567-5688(03)00031-x. [DOI] [PubMed] [Google Scholar]
- 43.Surdacki A, Nowicki M, Sandmann J, Tsikas D, Boeger RH, Bode-Boeger SM, Kruszelnicka-Kwiatkowska O, Kokot F, Dubiel JS, Froelich JC. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetric dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol. 1999;33:652–658. doi: 10.1097/00005344-199904000-00020. [DOI] [PubMed] [Google Scholar]
- 44.Lundman P, Eriksson MJ, Stühlinger M, Cooke JP, Hamsten A, Tornvall P. Mild-to-moderate hypertriglyceridemia in young men is associated with endothelial dysfunction and increased plasma concentrations of asymmetric dimethylarginine. J Am Coll Cardiol. 2001;38:111–116. doi: 10.1016/s0735-1097(01)01318-3. [DOI] [PubMed] [Google Scholar]
- 45.Gorenflo M, Zheng C, Werle E, Fiehn W, Ulmer HE. Plasma levels of asymmetrical dimethyl-L-arginine in patients with congenital heart disease and pulmonary hypertension. J Cardiovasc Pharmacol. 2001;37:489–492. doi: 10.1097/00005344-200104000-00016. [DOI] [PubMed] [Google Scholar]
- 46.Miyazaki H, Matsuoka H, Cooke JP, Usui M, Ueda S, Okuda S, Imaizumi T. Endogenous nitric oxide synthase inhibitor: a novel marker of atherosclerosis. Circulation. 1999;99:1141–1146. doi: 10.1161/01.cir.99.9.1141. [DOI] [PubMed] [Google Scholar]
- 47.Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, Day R, Ferraz MB, Hawkey CJ, Hochberg MC, Kvien TK, Schnitzer TJ VIGOR Study Group. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. 2 p following 1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- 48.Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A, Konstam MA, Baron JA Adenomatous Polyp Prevention on Vioxx (APPROVe) Trial Investigators. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. doi: 10.1056/NEJMoa050493. doi: 10.1056/NEJMoa050493. [DOI] [PubMed] [Google Scholar]
- 49.Bertagnolli MM, Eagle CJ, Zauber AG, Redston M, Solomon SD, Kim K, Tang J, Rosenstein RB, Wittes J, Corle D, Hess TM, Woloj GM, Boisserie F, Anderson WF, Viner JL, Bagheri D, Burn J, Chung DC, Dewar T, Foley TR, Hoffman N, Macrae F, Pruitt RE, Saltzman JR, Salzberg B, Sylwestrowicz T, Gordon GB, Hawk ET APC Study Investigators. Celecoxib for the prevention of sporadic colorectal adenomas. N Engl J Med. 2006;355:873–884. doi: 10.1056/NEJMoa061355. doi: 10.1056/NEJMoa061355. [DOI] [PubMed] [Google Scholar]
- 50.Lerman A, Burnett JC, Jr, Higano ST, McKinley LJ, Holmes DR., Jr Long-term L-arginine supplementation improves small-vessel coronary endothelial function in humans. Circulation. 1998;97:2123–2128. doi: 10.1161/01.cir.97.21.2123. [DOI] [PubMed] [Google Scholar]