

Abstract
Objective
To develop and implement a device and protocol for oocyte analysis at a single cell level. The device must be capable of high resolution imaging, temperature control, perfusion of media, drugs, sperm, and immunolabeling reagents all at defined flow-rates. Each oocyte and resultant embryo must remain spatially separated and defined.
Design
Experimental laboratory study
Setting
University and Academic Center for reproductive medicine.
Patients/Animals
Women with eggs retrieved for ICSI cycles, adult female FVBN and B6C3F1 mouse strains, sea stars.
Intervention
Real-time, longitudinal imaging of oocytes following fluorescent labeling, insemination, and viability tests.
Main outcome measure(s)
Cell and embryo viability, immunolabeling efficiency, live cell endocytosis quantitation, precise metrics of fertilization and embryonic development.
Results
Single oocytes were longitudinally imaged following significant changes in media, markers, endocytosis quantitation, and development, all with supreme control by microfluidics. Cells remained viable, enclosed, and separate for precision measurements, repeatability, and imaging.
Conclusions
We engineered a simple device to load, visualize, experiment, and effectively record individual oocytes and embryos, without loss of cells. Prolonged incubation capabilities provide longitudinal studies without need for transfer and potential loss of cells. This simple perfusion apparatus (SPA) provides for careful, precise, and flexible handling of precious samples facilitating clinical in vitro fertilization approaches.
Keywords: Oocyte, microfluidics, hydrodynamic trap, immunolabeling, cell viability assay, endocytosis activity
Introduction
In vitro fertilization (IVF) has emerged over the past 35 years as an important therapy for the treatment of human infertility. Clinical methods for IVF still rely heavily on manual manipulation of retrieved oocytes, which requires expert skills and the experience of trained embryologists. The use of microfluidic devices may one day effectively lower the extremely high cost of IVF, which has an average expense of $10,000 per cycle. A decrease in costs could lead to increased access worldwide to assisted reproductive technologies. This improvement is critically needed, as 7.4 million women (ages 15–44), or one in six couples, have utilized infertility services at some point in their lives (1). Currently, it is challenging for scientists and embryologists to handle individual oocytes, since they are typically few in number and a single lost cell is a significant percentage of available cells. Thus, lab-on-a-chip technologies might simplify laborious and delicate procedures with isolated human oocytes both in the clinic as well as in the research laboratory, and these technologies can additionally improve procedures common to many other cell types.
Several different approaches to micro-trap oocytes and embryos have appeared in the literature, as the interest in using microfluidics for reproductive science and the study of animal models have emerged (2, 3). Microfluidic technology has been utilized to facilitate gamete evaluation (4, 5) to remove cumulus cells and zona pellucida (6–8), for embryo culture (9), for transportation of embryos (10, 11), for monitoring of embryos (12, 13) and for studies on sperm selection and fertilization (14–16). Commonly used methods involve static culture methods (3), microwell designs in which the oocytes are confined (17, 18), and individual chamber based approaches (19). However, these types of devices are typically not compatible with high resolution imaging, such as confocal microscopy, they still require extensive manual handling, and they do not permit direct imaging without perturbation of the specimen.
Hydrodynamic trap methods for the manipulation of droplets have emerged as promising tools for conducting biochemical reactions using fluidic plugs and have launched this methodology as a fundamental lab-on-a-chip application (20, 21). However, the utility of hydrodynamic trapping is not limited to immiscible fluidic based reactions, and can be utilized for single-cell analysis (22, 23). This interest in cell trapping has expanded to include the culture of small organisms and hydrodynamic traps have been utilized for zebrafish and Drosophila embryos (24–26), but little work has been reported with mammalian gametes and embryos. Hydrodynamic trapping methods utilize a repeated sequence of looped channels, with a bypass channel and fluidic trap as upper and lower branches respectively. Our hydrodynamic trap array is modified from the work of Bithi et al. and utilizes a means of directly trapping oocytes for experimentation and real-time analysis (20, 21).
In addition to mammalian oocytes, researchers often work with diverse model organisms for the study of oogenesis and embryogenesis. Well studied models include zebrafish (Danio rerio), Caenorhabditis elegans, Drosophila melanogaster and Xenopus laevis, many of which have been utilized with microfluidic platforms for embryo evaluation (24, 25, 27–29). However, echinoderms are often a better choice, as they are closely related to chordates and comprise a major group of invertebrate deuterostomes. Each female accumulates millions of oocytes and eggs and in many species (such as Patiria miniata (P.miniata)), oocytes and eggs are optically clear, and easy to manipulate and image (30). We used sea star oocytes to develop iteratively advancements in a hydrodynamic trap – based microfluidic system. This Simple Perfusion Apparatus (SPA) permits easy experimentation and subsequent imaging using light or confocal microscopy of sea star oocytes and embryos, mouse and human oocytes. Our results show that this apparatus can be used for the analysis of single oocytes in different species for a wide variety of purposes, including longitudinal live cell development and high resolution imaging, as well as for drug screening assays.
Materials and Methods
Microfluidic chip design and fabrication
A hydrodynamic trapping array of eight traps was designed for oocytes in the size range of 60–200μm. The chips were cut to contain two separate trapping channels which are independent of one another to allow for simultaneous control or duplicate experiments when necessary. Channel dimensions can be found in Supplemental Table 1 and Supplementary Fig. 1. Photolithography masks were designed using AUTOCAD (San Rafael, CA) and purchased from CAD Art Services (Bandon, OR). Prototyping and fabrication was accomplished by making SU-8 silicon wafer molds using standard photolithography methods. SU-8 2100 photoresist (Microchem, Newton, MA) was used to create channels which were 200μm in depth. The SU-8 coated silicon wafer masters (Wafer World Inc, West Palm Beach, FL) were used for replica molding with polydimethylsiloxane (PDMS) as the fabrication material (Sylgard 184, Dow Corning Corp, Midland, MI). The PDMS was mixed at a 10:1 (w/w) ratio of elastomer to curing agent and degassed at 60 Torr to remove air bubbles. The PDMS was poured onto the SU-8 silicon wafer mold at approximately 2.5 mm thickness and cured at 70°C for at least one hour. The chips were cut and bonded to standard (24×60) microscope coverslips or slides (VWR, Radnor, PA) following plasma etching using a high frequency generator (Electrotechnic Products, Chicago, IL). Following fabrication, silicone tubing was attached to the inlet/outlet ports as reservoirs for adding solutions at the inlet, and for attachment to the syringe pump system at the outlet. Chips were primed and incubated overnight in appropriate buffer or media (artificial seawater at 4°C for sea stars, or PBS/0.05% Tween 20/1% BSA and at 37°C for mouse and human embryological media.
Loading of oocytes
Oocytes were loaded into the chip depending on size using two different methods. Large oocytes, like those from the sea star (180–200μm) were loaded into the inlet well with a standard pipette and were drawn through the chip into the traps by fluid flow. For smaller oocytes, like mouse and human (60–100μm, 100–150μm respectively) an additional loading well was designed and placed between the fluid inlet well and the hydrodynamic trap array. A 0.35 mm hole was punched in the PDMS prior to bonding above this well to allow for addition of oocytes using a manual oocyte micropipette (Drummond Scientific, Broomall, PA) with 175μm inner diameter stripper tips (ORIGIO Midatlantic Devices, Charlottesville, VA). After use of the loading port, it was sealed with a silicone aquarium sealant (All-Glass Aquarium Inc., Franklin, WI) to prevent leakage and evaporation from the device. This procedure was developed for human and mouse oocytes as they are commonly used experimentally but available in very low numbers and it is difficult to pipette with a standard pipette. This procedure also allows for precise addition of an exact number of oocytes to the device and minimizes any loss within the system.
Instrumentation and pump system
The fluids were driven and controlled using a PHD 2000 dual syringe pump (Harvard Apparatus, Holliston, MA), which supplies the buffers or media at an adjustable rate. Five-ml syringes (Becton Dickenson, Franklin Lakes, NJ) were utilized for each. In these experiments a fast flow rate of 10 μl/min was utilized for flushing the chip, and a slower flow rate of 50 μl/hr for development, live cell imaging and studies with fixed oocytes. The pump was run using the refill mode to pull fluid from the inlet well through the chip. The inlet reservoir is open and allows for easy addition of buffers, dyes, or media using a standard 200μl pipette. A one way check valve (Smart Products Inc., Morgan Hill, CA) was used at the outlet of the chip in order to minimize and eliminate any backflow through the device. For experiments requiring temperature control, a standard temperature control plate (Isotemp 3016S, Fisher Scientific, Pittsburgh, PA) for the microscope stage was utilized at 16°C for sea stars and 37°C for mammalian oocytes. Since the media was equilibrated at the same temperature on the stage, we noticed no bubble formation from temperature differentials and degassing was not required.
Multiphysics modeling
To calculate the fluid velocities within the hydrodynamic chip, the shear stress within the traps, and the dynamics of drug or dye transport within the device, two-dimensional models were created using COMSOL Multiphysics 4.3b (Stockholm, Sweden). The Navier-Stokes equations (Equation 1) for incompressible flow (Equation 2) and the convection-diffusion equations (Equation 3) were utilized and solved for using the microchip geometry and appropriate boundary conditions.
| (1) |
| (2) |
| (3) |
In Equations 1, 2, 3) the terms are defined as follows: u (velocity), t (time), p (pressure), C (concentration), ρ (density), μ (viscosity) and D (diffusion coefficient). The boundary condition utilized for the laminar flow module in COMSOL Multiphysics was laminar inflow, which assumes that the flow is fully developed at the inlet, and is appropriate for low Reynolds number systems. This was chosen based on calculating the Reynolds number for our system, from the standard relation, Re=ρud/μ, (where d is the depth of the channel), which was determined as Re = 0.35 for the initial condition flow rate (50 μl/hr) utilized during live-cell imaging and development. The outlet boundary condition was a pressure set to zero Pa and other surfaces of the chip were set to the no-slip condition. For the convection-diffusion equation (Equation 3), the boundary conditions were no flux at the walls, the velocity was solved from the Navier Stokes equations (Equations 1, 2) with an initial condition of C0=5.0 nM. The P_clet number Pe = ud/D, which is a dimensionless number that compares the advective transport rate to the diffusive transport rate was determined to be Pe~140, indicating that advective transport is dominant for this system.
Animals
Sea star oocytes
Patiria miniata were housed in aquaria with artificial seawater (ASW) at 16°C (Coral Life Scientific Grade Marine Salt, Carson, CA, USA). Gametes were acquired by gonad biopsy and full grown oocytes were collected in filtered seawater and sperm was collected dry. To obtain mature oocytes, the full grown, immature oocytes were incubated for two hours in filtered seawater containing 2 μM 1-methyladenine. After addition of sperm, fertilized eggs were cultured in filtered seawater at 16°C (30). Images were taken with a dissecting stereoscope (Olympus SZX16) connected to an Olympus X citep 120 lamp and to a digital camera (Nikon D90), managed by Pro 2 software.
Mouse oocytes
MII oocytes were obtained from superovulated ovaries, harvested from murine B6C3F1 oviducts with a tuberculin 25-gauge needle (Embryotech Haverhill, MA. This procurement protocol obviated IACUC.). A Drummond pipette was then used to denude oocytes, which were then separated and pooled for experimentation in M2 media.
Human oocytes
De-identified human oocytes deemed to be unsuitable for intracytoplasmic sperm injection because of GV or MI stage were collected from the IVF Laboratory at the Center for Reproduction and Infertility at Women & Infants Hospital of Rhode Island after obtaining Institutional Review Board approval for residual tissue studies. Oocytes were retained and washed in HTF (10%) buffered with Hepes and 10% LGPS supplement (Lifegroup).
In vitro endocytosis assay
Fertilized sea star eggs were loaded in the chip and were tested for individual rates of endocytosis in the chip. The fertilized eggs were incubated for 30 minutes in filtered sea water containing 15 μg/ml of FM1-43 (Molecular Probes, Grand Island, NY). The presence of endocytic vesicles was observed using a LSM 510 laser scanning confocal microscope (Carl Zeiss, Inc.; Thornwood, NY) while the cells remained within the SPA. The hydrodynamic trap provided excellent stability of the cells over time.
DNA labeling
To label DNA of individual oocytes and embryos, appropriately aged sea star embryos were fixed in 4% paraformaldehyde in ASW and washed with PBS/0.05% Tween 20. Embryos were loaded in the chip which was pre-incubated overnight in PBS/0.05% Tween 20/1% BSA at 4°C. The oocytes were washed for one hour at 20°C in blocking buffer: PBS/0.05% Tween 20/3% BSA (Sigma; St. Louis, MO). For DNA labeling, the cells were incubating for 4 hours in Hoechst (Life Technologies; Carlsbad, CA) at room temperature. Embryos were then washed with PBS/Tween 20/3% BSA for 3 hours. Images were captured in the chip using a LSM 510 laser scanning confocal microscope (Carl Zeiss, Inc.; Thornwood, NY).
Live/Dead assay
Live/Dead, Viability/Cytotoxicity Assay (Invitrogen) was used to test the viability of the mouse and human oocytes dynamically under SPA conditions. The oocytes were loaded in a chip and within 30 minutes were washed with PBS, with the temperature maintained at 37°C. The Live/Dead solution was added in the chip, and the fluorescence was monitored 20 minutes later. The cells were then washed for 30 minutes with PBS/PFA 4% (to induce death and fix the oocytes). After a PBS wash, a fresh Live/Dead solution was added in the SPA. The fluorescence was monitored 30 minutes later. Images were taken using a fluorescence dissecting stereoscope (Olympus SZX16) connected to an Olympus Xcite p120 lamp and to a camera (Nikon D90), managed by Pro 2 software. During the entire assay, the oocytes remained within the SPA and were monitored visually by microscopy, whereas all reagents were added sterilely to the entry port of the SPA and collected within a waste reservoir post-SPA.
Results
Hydrodynamic trapping chip design
Utilizing the principles described by Bithi et. al (20, 21), we designed a hydrodynamic trap array for trapping oocytes in the size range of 60–200 μm (Fig. 1A). Initial designs utilized two different ratios of the trap branch resistance (Rt) to the bypass branch resistance (Rb) of 1.56 and 0.8. These two ratios were chosen as they represent distinct trapping schemes, where a ratio of Rt/Rb >1 yields indirect trapping of cells and Rt/Rb <1 traps cells directly. Resistances were calculated using the following equation for Poiseuille flow in a rectangular channel,
| (4) |
where L, w, and d, are the length, width and depth of the channels respectively and μ is the fluid viscosity. The resistance for the trapping branch was calculated as a summation of parts, and the circular region was approximated as a square. The length of the bypass channel includes the perpendicular side channels. The hydrodynamic array design for the 1.56 ratio chip is displayed in Figure 1A, and a detailed view of one trap, highlighting the bypass and trapping arms is shown in Figure 1C. More information on the chip dimensions and calculations of the resistances is summarized in Supplementary Table 1. Oocytes load into the chip in the direction of flow from right to left, filling the traps one-by-one as they enter the chip (Fig. 1D). Double trapping can occur with oocytes of small size, as they do not increase the trap resistance enough to prevent another oocyte from entering, as seen in Fig. 1D in trap five. This does not impede imaging or analysis and can be prevented by size selecting oocytes prior to loading.
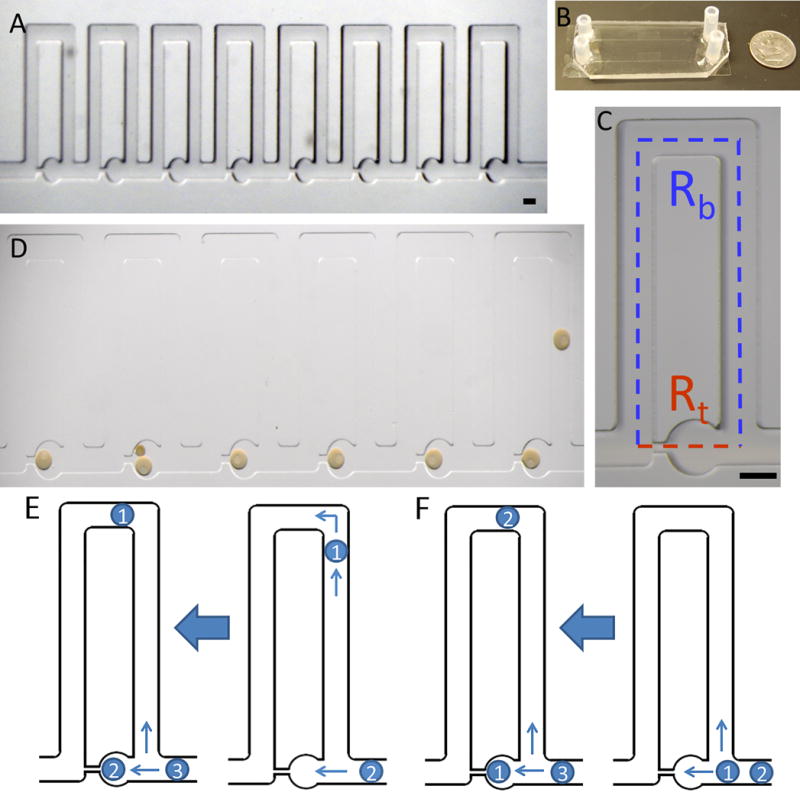
Figure 1. Hydrodynamic trap array images and trapping schemes.
(A) Images of the 8-trap hydrodynamic array: Oocytes flow in and load into the wells from right to left which is the direction of fluid flow. (B) Macroscale view of hydrodynamic trap arrays: Displays two parallel arrays bonded to a standard size microscope slide coverslip. (C) An individual trap: Consists of a trapping channel (red) and a bypass channel (blue). The resistances of each segment dictate the trapping behavior of the device. The trapping region consists of a circular trap (320 μm in diameter) and a high resistance channel (40 μm width) which connects to the ends of the bypass segment. (D) Immature sea star oocytes within the chip: Oocytes load into the chip from right to left for the six traps displayed. The nucleus and nucleolus are visible in each oocyte which is ~200 μm. Double trapping can occur with smaller oocytes, as seen in trap five. Scale bars are 200 μm. (E) Indirect trapping: Occurs when the trap resistance is greater than the bypass channel resistance (Rt/Rb=1.56) causing the first oocyte to be directed into the bypass channel. (F) Direct trapping: Occurs when the trap resistance is lower than the bypass resistance (Rt/Rb=0.8), directing the first oocyte into the trap. Subsequent oocytes can be directed into the trap causing double trapping, or directed into the bypass channel depending on the change in resistance due to the trapped oocytes for either regime.
The trapping schemes are represented in Figure 2. Indirect trapping occurs when the trap resistance is higher than the bypass channel (Fig. 2A), causing the first oocyte in the channel to move into the bypass channel. The higher resistance in the bypass channel from the presence of the oocyte then allows the next oocyte to move into the trap. It was found experimentally that this regime is less advantageous for trapping oocytes, as the spacing between subsequent cells needs to be small in order to trap the second cell. Thus, the chip with the ratio of Rt/Rb=1.56 was found to be less advantageous for minimizing loss of oocytes in the chip. The chip with the design Rt/Rb=0.8 was found to effectively trap the oocytes with a direct loading scheme (Fig. 2B). This occurs since the trap resistance is lower than the bypass and the first oocyte moves into the trap. The second will move into the bypass channel due to the increased resistance in the trapping channel from the first oocyte and load into the second trap. This regime allows for trapping of an exact number of oocytes with no loss of cells. Subsequent oocytes can be directed into the trap, causing double trapping, or directed into the bypass channel depending on the change in resistance due to the trapped oocytes for either regime. The high trapping yield of this regime is particularly important for trapping human oocytes where only a few are available at one time.
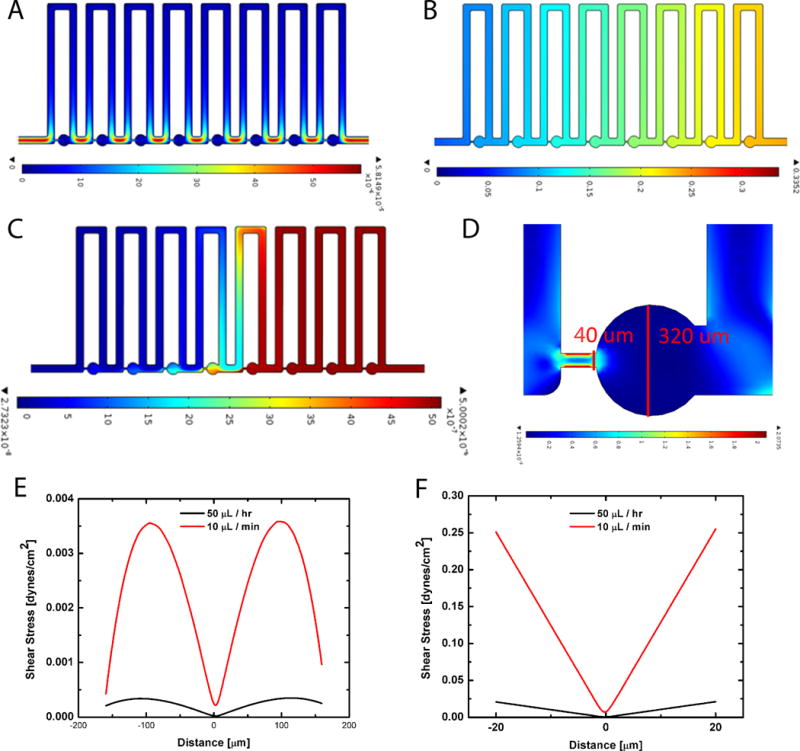
Figure 2. Numerical modeling of fluid velocity, pressure, concentration profiles and shear stresses.
(A) Velocity profile of fluidic environment: Shows that the fluid velocity is highest in the region bridging the traps and lowest within the trap regions for an inlet flow rate of 50 μl/hr; heat map is in m/s. (B) Pressure drop across the device: The pressure doesn’t exceed 0.332 Pa, with the first trap experiencing the highest pressure at the inlet; heat map is in Pascals (Pa). (C) Time dependent concentration profile: Results from the convection-diffusion study for a molecule of standard diffusivity after 600 seconds of perfusion, shows that nearly half the chip has reached the inlet concentration; heat map is concentration (mol/m3). (D) Strain distribution in a hydrodynamic trap: The red lines display the cut lines where the shear stress was calculated for two different flow rates; heat map is strain rate (1/s). (E) Shear stress across the diameter of the trap: shows results for a flow rate of 50 μl/hr and 10 μl/min. (F) Shear stress across the high resistance channel: shows that higher shear stresses occur at the boundary between the trap and the suction channel for both flow rates of 50 μl/hr and 10 μl/min, but especially for the faster flow rate, demonstrating maximum shear stresses of 0.25 dynes/cm2. These shear stresses are significantly below the threshold of 1.2 dynes/cm2.
Numerical modeling
Modeling of media exchange was accomplished using COMSOL Multiphysics, utilizing Navier Stokes equations for an incompressible Newtonian liquid for laminar flow simulations followed by convection diffusion analysis for transport of dilute species to understand the transport of small molecules within the chip for mixing time derivations. Modeling of the velocity profile through the hydrodynamic trap array for the selected design of Rt/Rb=0.8 was perfromed at two representative flow rates of 50 μl/hr and 10 μl/min. As the slower flow rate (50 μl/hr) was utilized for long term live-cell experiments and developmental studies, Figure 2 displays the velocity profile through the device for this condition. The fastest fluid velocity occurs within the high resistance channels and the lowest through the individual traps. From the fluid velocity model, we determined the pressure drop across the chip, (Fig. 2F) which doesn’t exceed 0.332 Pa, with the first trap experiencing the highest pressure at the inlet.
The faster flow rate of 10 μl/min was used to load dye and drugs to the system, and Figure 2E displays the transport profile through the chip. Numerical studies of the transport of drugs through the device indicate that mixing time for the faster flow rate of 10 μl/min is approximately 1400 seconds (23.3 minutes) for full perfusion of a molecule with a diffusivity of 5.0 × 10−10 m2/s which is typical for a drug or dye molecule. A snapshot of the perfusion animation is shown in Figure 2D after 600s of transport. The longer time scale for mixing underscores the trade-off between faster flow rates and higher shear stress on the oocytes and slower perfusion times. Additionally, the P_clet number for the slow flow rate Pe~140, while still >1 can be increased to Pe~1700, which for fast biochemical reactions results in fast transport and uptake (31). However, for slow biochemical reactions, like that of antibody-antigen binding, lower flow rates (and thus lower P_clet numbers) may prove more ideal but allowing for diffusive transport to the oocyte surface.
Overall, it is apparent that the device provides a wide range of inlet flow rate possibilities and corresponding transport schemes, which can be tailored to the specific needs of the user and application. We also analyzed the strain rate across the first trap (Fig. 2D). The strain rate was measured in two locations, across the diameter of the trapping region and at the entrance to the high resistance channel, as indicated by the red lines in Figure 2D. Due to the no-slip condition, the speed of the fluid at the boundary is zero, but the flow speed must equal that of the fluid at some height from the boundary. Thus, in a narrow channel, such as the 40 μm wide channel, the strain rate and shear stress will be the highest. We determined the shear stress, τ, for the two different flow rates, using the relation , where is the strain rate and μ is the viscosity of the fluid, which is 1.0 cP for water. The shear stress across the trap for each flow rate is shown in Figure 2E, and across the entrance to the high resistance channel in Figure 2F. We set a threshold of 1.2 dynes/cm2 as a limit to the amount of shear stress the fluid can impart, as adverse cell signaling events in blastomeres have been demonstrated above this limit (22, 32). It is evident from the data that for the slow flow rate of 50 μl/hr and the faster flow rate of 10 μl/min, the shear stress is between one and four orders of magnitude below the minimum value to cause adverse cellular events. We examined the upper limit of the flow rate that can be utilized and found that at a flow rate of 50 μl/min shear stresses still do not yet exceed 1.2 dynes/cm2. For this upper limit of the flow rate, the mixing time can be lowered to 400s (7.5 minutes) and still be utilized for live-cell studies. Thus, the working range of flow rates for live-cell studies and development is quite wide for a variety of applications and the speed at which certain assays are performed can be shortened.
Longitudinal culture for individual developmental analysis
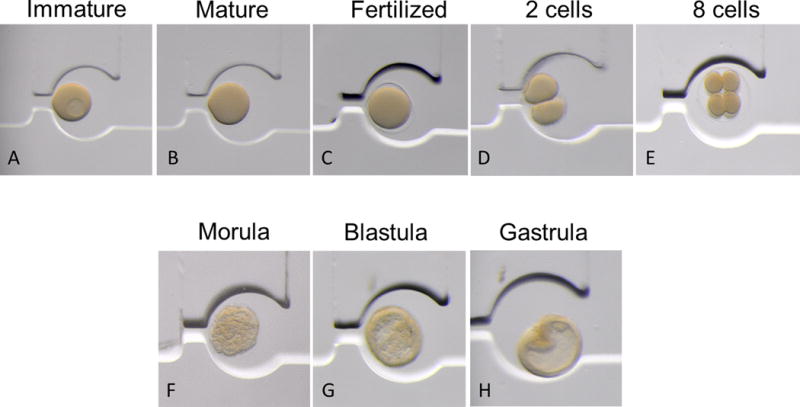
To determine the application of the chip for the dynamic recording of the development of oocytes to embryos, sea star oocytes were first added, maturation was stimulated, the oocytes were fertilized and development was monitored, all within the device. Dynamic recording of sea star development was followed for 30 hours within the microfluidic environment (Figure 3). In Figure 3, the detailed stages of development of the sea star oocytes and embryos were easily observed. Full-grown immature oocytes (that have an approximate diameter of 180 μm) are arrested in prophase of first meiotic division (Fig. 3A) and meiotic re-initiation was induced by the hormone 1-methyladenine (33) for maturation (Fig. 3B). Mature oocytes were then fertilized in the SPA with the addition of diluted sea star sperm to the entry port, and the resulting fertilization envelope was visible (Fig. 3C), indicating successful fertilization. After fertilization, embryos developed quickly through two-cell stage (Fig. 3D), eight-cell stage (Fig. 3E), and morula (Fig. 3F), reaching blastula (Fig. 3G), and they begin to gastrulate 24 hours after fertilization (Fig. 3H), all on schedule compared to their normally cultured siblings. Our results show that the sea star embryos developed normally in the chip, from an immature oocyte being stimulated to mature, through fertilization in the SPA, to gastrulation after 24 hours of development, all while being retained individually in each hydrodynamic trap, and while being monitored microscopically in the SPA.
Figure 3. In situ culture.
Sea star embryos develop normally inside the SPA. Full grown immature oocytes (A) were loaded in the chip, and perfused with 1-methyladenine to stimulate maturation and the media was then replaced by perfusion (B). Sperm were then perfused throughout the chip and the matured eggs each fertilized (C). Embryos developed synchronously and normally, and were readily followed with optical clarity within the SPA from two-cell (D), to eight-cell (E), to morula (F), blastula (G) and through gastrulation (H) within 24 hours. The temperature was maintained at 16°C.
Optical imaging of individual oocyte activities
FM1-43 is a lipophilic, membrane impermeant fluorescent probe that is non-toxic, water-soluble, and virtually non-fluorescent in aqueous medium. It becomes fluorescent after integrating into the outer leaflet of the plasma membrane and its subsequent membrane fates. It has been used in several species to look at the general endocytic activity, especially in sea urchin eggs (34), sea urchin oocytes (35), and sea star oocytes (36). To evaluate the dynamic and longitudinal imaging capability of the chip, the dye FM1-43 was utilized to visualize endocytic uptake and fluorescent labeling of the plasma membrane of live fertilized sea star embryos. Figure S2 displays the confocal microscope images of a fertilized sea star embryo incubated for 30 minutes in the dye diluted in filtered sea water. Bright green fluorescence is observed at the surface as a ring, indicative of endocytic uptake in the embryo (Supplemental Fig. 2A). Corresponding control experiments (media only) display no auto-fluorescence of the embryos or the chip (Supplemental Fig. 2C). The bright field images display healthy sea star embryos with a visible fertilization envelope contained within the traps (Supplemental Fig. 2B and 2D). The high resolution imaging capability of the chip, as well as the application of dynamic recording of oocytes-to-embryos and further development underscores the versatility of the device as a tool for multiple biological applications.
DNA labeling of fixed samples
In order to test a broader range of applications, we also took advantage of the sea star embryos to label the cellular DNA following fixation (Supplemental Fig. 3). Fixed morulas were loaded in the chip for DNA staining. Hoechst 33342 is a popular cell-permeant dye that emits blue fluorescence when bound to double-stranded DNA. Embryos washed in Hoechst diluted with PBST-BSA, showed a very strong staining of DNA as displayed in Supplemental Fig. 3A (20× magnification) and Supplemental Fig. 3C (40× magnification). The images taken using the objectives 20× and 40× displayed the remarkable resolution of the SPA for confocal imaging. The corresponding bright-field images of the fixed morula are shown in Supplemental Fig. 3B (20×) and Supplemental Fig. 3D (40×) and shows the cell placement within the trap. Altogether, the data in Supplemental Fig. 2 and 3 showed that the simple perfusion apparatus enabled high resolution imaging of live or fixed oocytes and embryos using different labeling methods. Further studies can explore more complex assays for fixed oocytes and embryos like FISH and immunofluorescence.
Versatility in application
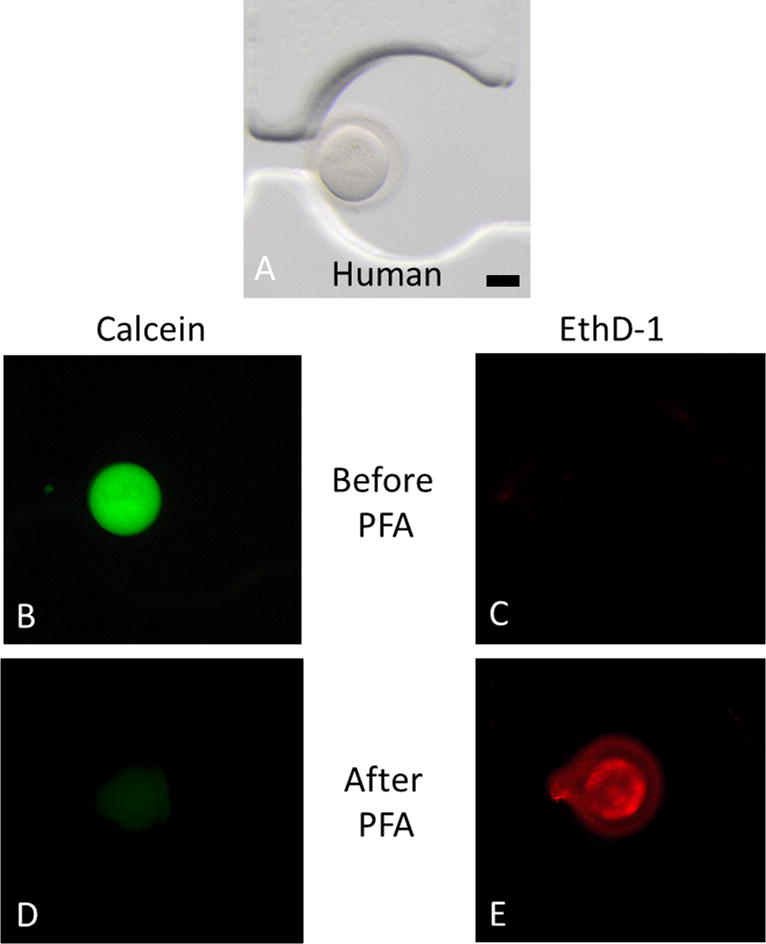
In order to evaluate the ability of the device for use with live mammalian cells, a simple Live/Dead assay with a select number of mouse and human oocytes was performed. Human oocytes (approximate diameter of 130 μm), at the maturation stage of MI, were loaded in the chip for the viability assay as shown in the bright-field image of Figure 4A. A control was performed by washing the living oocytes with the Live/Dead solution and imaging after 30 minutes of exposure. As expected (37), the oocytes displayed bright green fluorescence (calcein AM), and no red fluorescence (ethidium homodimer-1), which in conjunction is indicative of living cells (Figure 4B and 4C). As a control, a fixative (PFA) was utilized to induce cell death, the oocytes were washed and fresh dye was added. Following this procedure, the oocytes displayed almost no green fluorescence and instead fluoresce bright red, indicative of cell death (Fig. 4D and 4E). The strong signal from the ethidium homodimer-1 stain following fixation confirms the expected result of inducing death of the oocyte. Any residual green fluorescence from calcein AM was thus considered to be carry-over from the earlier positive control, and in contrast to the red fluorescence was not indicative of a living cell. Similar results were obtained using mouse oocytes that are approximately 60 μm at the stage MII (Supplementary Figure S4). This assay result verified the utility of the Live/Dead stain within the SPA as a positive and negative control, and created a benchmark for future experiments with chemotherapeutic drugs. In contrast to sea stars that provide several thousand oocytes at once, only few oocytes can be obtained from mouse and human ovaries for experimental purposes. Thus, we demonstrate that independent of the number or the size of the oocytes available for experimentation, the simple perfusion apparatus enables easy manipulation and imaging of individual oocytes without loss.
Figure 4. Live cell imaging of human oocytes.
Live/Dead assay on human oocytes in the chip. The Live/Dead assay was performed by simultaneously monitoring the fluorescence after addition of calcein AM and ethidium homodimer-1. Calcein AM (green) is retained in live cells, whereas ethidium homodimer-1 (red) enters dead cells. The bright field represents a human oocyte in the chip (A). Before addition of PFA, the oocyte is alive ((B) and (C)). After addition of PFA, the oocytes demonstrate cell death ((D) and (E)). Scale bar is 40 μm.
Discussion
We described and applied a hydrodynamic trap design that provides a perfusion platform to easily perform individual oocyte and embryo experimentation while retaining high resolution imaging. This device can be used to load, visualize, experiment, and effectively record individual oocytes and embryos, without loss of cells. The device uniquely provides prolonged incubation capabilities for longitudinal studies without need for transfer (with potential loss) of cells. Through the use of sea star oocytes and embryos, we have established the utility of the platform for single cell analysis with a wide array of biological applications. This simple perfusion apparatus (SPA) is optimized to conduct lengthy time course experiments (Figure 5) enabling live oocyte and even embryogenesis over extended times. The oocytes and embryos can be observed one-by-one as separable entities, allowing for cell-by-cell analysis. Utilizing an environmentally controlled temperature stage makes the device applicable for a variety of oocytes and embryos and multiple SPAs can be employed, side-by-side, for testing variables under exacting conditions.
SPA applications for low cost IVF and fertility preservation
Human oocytes and embryos are some of the rarest cells in the human female. This simple perfusion apparatus (SPA) uniquely provides for careful, precise, and flexible handling of precious cellular samples. Due to the precious nature of mammalian oocytes, experimentation and research with these cells is often problematic and better methods of handling, isolation and manipulation would facilitate improved biological and clinical discovery. This SPA has the potential to fill such needs.
The possible applications for the SPA include improved capability to analyze the effect of drugs, toxins and other factors on these precious cells on a cell-by-cell basis. We anticipate its application may also change clinical practice by allowing gamete manipulation without the personnel, overhead, cost, and space necessary for a standard IVF laboratory. Reiterations of the SPA could be designed also to mimic exact temperature and O2/CO2 conditions in incubators. In addition, a follow up study using the SPA for efficient and simpler oocyte and embryo vitrification could make gamete and embryo cryopreservation a more widely available opportunity for oncology patients facing gonadotoxic therapies. This SPA could also permit better understanding of the pathophysiology of gamete toxicity induced by chemotherapeutic drugs (37).
Supplementary Material
Acknowledgments
Thanks to Siva Vanapalli for discussions regarding the hydrodynamic trapping array. Thanks to Richard Hackett, Linda Sousa, Benning Cao, Sue Casey, Julie Labreche, Deborah Pierce, and Monique O′Brien of the Women & Infants Hospital of Rhode Island for the human oocytes and laboratory supplies.
References
- 1.CDC. CDC/National Center for Health Statistics. 2006–2010 [Google Scholar]
- 2.Krisher RL, Wheeler MB. Reproduction, Fertility and Development. 2010;22:32–39. doi: 10.1071/RD09219. [DOI] [PubMed] [Google Scholar]
- 3.Smith GD, Takayama S, Swain JE. Biology of reproduction. 2012;86:62. doi: 10.1095/biolreprod.111.095778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kricka LJ, Faro I, Heyner S, Garside WT, Fitzpatrick G, McKinnon G, Ho J, Wilding P. Journal of Pharmaceutical and Biomedical Analysis. 1997;15:1443–1447. doi: 10.1016/s0731-7085(96)02046-8. [DOI] [PubMed] [Google Scholar]
- 5.Kricka L, Nozaki O, Heyner S, Garside W, Wilding P. Clinical chemistry. 1993;39:1944–1947. [PubMed] [Google Scholar]
- 6.Zeringue H, Beebe D. In: Germ Cell Protocols SE - 22. Schatten H, editor. Vol. 254. Humana Press; 2004. pp. 365–373. [Google Scholar]
- 7.Zeringue HC, Rutledge JJ, Beebe DJ. Lab on a Chip. 2005;5:86–90. doi: 10.1039/b316494m. [DOI] [PubMed] [Google Scholar]
- 8.Zeringue HC, Wheeler MB, Beebe DJ. Lab on a Chip. 2005;5:108–110. doi: 10.1039/b406703g. [DOI] [PubMed] [Google Scholar]
- 9.Raty S, Walters EM, Davis J, Zeringue H, Beebe DJ, Rodriguez-Zas SL, Wheeler MB. Lab on a Chip. 2004;4:186–190. doi: 10.1039/b316437c. [DOI] [PubMed] [Google Scholar]
- 10.Glasgow IK, Zeringue HC, Beebe DJ, Choi SJ, Lyman JT, Chan NG, Wheeler MB. IEEE Transactions on Biomedical Engineering. 2001;48:570–578. doi: 10.1109/10.918596. [DOI] [PubMed] [Google Scholar]
- 11.Sadani Z, Wacogne B, Pieralli C, Roux C, Gharbi T. Sensors and Actuators A: Physical. 2005;121:364–372. [Google Scholar]
- 12.Date Y, Takano S, Shiku H, Ino K, Ito-Sasaki T, Yokoo M, Abe H, Matsue T. Biosensors & bioelectronics. 2011;30:100–6. doi: 10.1016/j.bios.2011.08.037. [DOI] [PubMed] [Google Scholar]
- 13.Dahan E, Bize V, Lehnert T, Horisberger J-D, Gijs MaM. Lab on a chip. 2008;8:1809–18. doi: 10.1039/b806404k. [DOI] [PubMed] [Google Scholar]
- 14.Clark SG, Haubert K, Beebe DJ, Ferguson CE, Wheeler MB. Lab on a Chip. 2005;5:1229–1232. doi: 10.1039/b504397m. [DOI] [PubMed] [Google Scholar]
- 15.Suh RS, Zhu X, Phadke N, Ohl DA, Takayama S, Smith GD. Human Reproduction. 2006;21:477–483. doi: 10.1093/humrep/dei323. [DOI] [PubMed] [Google Scholar]
- 16.Cho BS, Schuster TG, Zhu X, Chang D, Smith GD, Takayama S. Analytical chemistry. 2003;75:1671–5. doi: 10.1021/ac020579e. [DOI] [PubMed] [Google Scholar]
- 17.Ma R, Xie L, Han C, Su K, Qiu T, Wang L, Huang G, Xing W, Qiao J, Wang J, Cheng J. Analytical chemistry. 2011;83:2964–70. doi: 10.1021/ac103063g. [DOI] [PubMed] [Google Scholar]
- 18.Han C, Zhang Q, Ma R, Xie L, Qiu T, Wang L, Mitchelson K, Wang J, Huang G, Qiao J, Cheng J. Lab on a chip. 2010;10:2848–54. doi: 10.1039/c005296e. [DOI] [PubMed] [Google Scholar]
- 19.Esteves TC, van Rossem F, Nordhoff V, Schlatt S, Boiani M, Le Gac S. RSC Advances. 2013;3:26451. [Google Scholar]
- 20.Bithi SS, Vanapalli Sa. Biomicrofluidics. 2010;4:44110. doi: 10.1063/1.3523053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun M, Bithi SS, Vanapalli Sa. Lab on a chip. 2011;11:3949–52. doi: 10.1039/c1lc20709a. [DOI] [PubMed] [Google Scholar]
- 22.Wlodkowic D, Faley S, Zagnoni M, Wikswo JP, Cooper JM. Analytical chemistry. 2009;81:5517–23. doi: 10.1021/ac9008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlo DD, Wu LY, Lee LP. Lab on a Chip. 2006;6:1445–1449. doi: 10.1039/b605937f. [DOI] [PubMed] [Google Scholar]
- 24.Akagi J, Khoshmanesh K, Evans B, Hall CJ, Crosier KE, Cooper JM, Crosier PS, Wlodkowic D. PloS one. 2012;7:e36630. doi: 10.1371/journal.pone.0036630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levario TJ, Zhan M, Lim B, Shvartsman SY, Lu H. Nature protocols. 2013;8:721–36. doi: 10.1038/nprot.2013.034. [DOI] [PubMed] [Google Scholar]
- 26.Chung K, Kim Y, Kanodia JS, Gong E, Shvartsman SY, Lu H. Nature methods. 2011;8:171–6. doi: 10.1038/nmeth.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choudhury D, van Noort D, Iliescu C, Zheng B, Poon KL, Korzh S, Korzh V, Yu H. Lab on a chip. 2012;12:892–900. doi: 10.1039/c1lc20351g. [DOI] [PubMed] [Google Scholar]
- 28.Schaffhauser DF, Andrini O, Ghezzi C, Forster IC, Franco-Obreg_n A, Egli M, Dittrich PS. Lab on a chip. 2011;11:3471–8. doi: 10.1039/c0lc00729c. [DOI] [PubMed] [Google Scholar]
- 29.Wielhouwer EM, Ali S, Al-Afandi A, Blom MT, Riekerink MBO, Poelma C, Westerweel J, Oonk J, Vrouwe EX, Buesink W, VanMil HGJ, Chicken J, van’t Oever R, Richardson MK. Lab on a chip. 2011;11:1815–24. doi: 10.1039/c0lc00443j. [DOI] [PubMed] [Google Scholar]
- 30.Wessel GM, Reich AM, Klatsky PC. Syst Biol Reprod Med. 2010;56:236–245. doi: 10.3109/19396361003674879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Squires TM, Messinger RJ, Manalis SR. Nat Biotech. 2008;26:417–426. doi: 10.1038/nbt1388. [DOI] [PubMed] [Google Scholar]
- 32.Yin H, Zhang X, Pattrick N, Klauke N, Cordingley HC, Haswell SJ, Cooper JM. Analytical chemistry. 2007;79:7139–44. doi: 10.1021/ac071146k. [DOI] [PubMed] [Google Scholar]
- 33.Kanatani H, Shirai H, Nakanishi K, Kurokawa T. Nature. 1969;221:273–274. doi: 10.1038/221273a0. [DOI] [PubMed] [Google Scholar]
- 34.Whalley T, Terasaki M, Cho MS, Vogel SS. J Cell Biol. 1995;131:1183–1192. doi: 10.1083/jcb.131.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brooks JM, Wessel GM. Dev Biol. 2003;261:353–370. doi: 10.1016/s0012-1606(03)00301-4. [DOI] [PubMed] [Google Scholar]
- 36.Oulhen N, Onorato TM, Ramos I, Wessel GM. Dev Biol. 2014;388:94–102. doi: 10.1016/j.ydbio.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brayboy LM, Oulhen N, Witmyer J, Robins J, Carson S, Wessel GM. Fertil Steril. 2013;100:1428–1435. doi: 10.1016/j.fertnstert.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.