

Abstract
Immune-based therapies have been in use for decades but recent work with immune checkpoint inhibitors has now changed the landscape of cancer treatment as a whole. While these advances are encouraging, clinicians still do not have a consistent biomarker they can rely on that can accurately select patients or monitor response. Molecular imaging technology provides a noninvasive mechanism to evaluate tumors and may be an ideal candidate for these purposes. This review provides an overview of the mechanism of action of varied immunotherapies and the current strategies for monitoring patients with imaging. We then describe some of the key researches in the preclinical and clinical literature on the current uses of molecular imaging of the immune system and cancer.
Keywords: immunotherapy, molecular imaging, biomarker
Overview
Significant strides have been made in the treatment of cancer over the past several decades. Refinements have been made in surgical techniques allowing for more complex tumors to be resected with reduced morbidity and mortality. New radiation therapy technologies and strategies have been developed, which have led to more precise delivery of treatment, sparing normal tissues, and delivering much more potent cytotoxic effects to cancer. Systemic therapy has also overcome huge hurdles and made remarkable advances during this time. The tolerability of standard cytotoxic chemotherapies has improved with more modern agents and improved supportive measures. The era of molecular medicine is also upon us with options for a more precise treatment directed at molecular aberrations pathologically identified in tumors on an individual patient level.1–7 More recently, a new type of systemic therapy, immunotherapy, has made its mark on the management of multiple cancers.8–15 Attempts have been made at harnessing the immune system as cancer therapy for decades, but immune-based therapies had a noteworthy impact only in the past few years.16–21
One of the challenges of immunotherapy is that an accurate and reproducible biomarker that would allow treating physicians to select the patients most likely (or least likely) to respond has yet to be identified.22 In addition, standard anatomic imaging may not provide the concrete representation of initial response or progression to which the medical community has become accustomed when working with cytotoxic chemotherapy.23,24 The goal of this article is to review the advances of immunotherapy in cancer as well as to find how anatomic imaging has been applied for use in this field. The potential of molecular imaging as a biomarker for use in identifying patients and interpreting their response to this novel treatment option will also be highlighted.
Immune System in Cancer
For a cancer to develop, tumor cells have to succeed in evading the immune system.25,26 One of the tasks of the immune system is to rid the body of threats both “foreign”, such as bacterial and viral pathogens and “domestic”, such as cancer cells. Multiple mechanisms have been identified by which cancer cells cloak themselves from the immune system. Cancer cells, by downregulation of the major histocompatibility complex (MHC) I, present antigens on their surface less effectively making detection by immune cells less likely.26 Cancer cells secrete immunosuppressive factors such as transforming growth factor-beta (TGF-β) and interleukin (IL)-10, which downregulate cytotoxic immune cells.26 One of the additional benefits of secreting immunosuppressive cytokines is the recruitment of immunosuppressive cells such as regulatory T-cells into the tumor microenvironment.26 Regulatory T-cells are key in the process of moderating the immune system and tolerizing self-antigens. Lastly, the tumor cells upregulate surface proteins such as programmed cell death-ligand 1 (PD-L1), which is a key component of normal cells preventing autoimmune phenomenon.26 When PD-L1 interacts with programmed cell death (PD-1) receptor-1 on cytotoxic T-cells, the T-cells define these cells as a part of normal self, and despite previous identification of a foreign surface antigen, the T-cells do not proceed to eradicate the cell.26 Each of these unique mechanisms is part of the complex strategy cancer employs to grow unchecked by the immune system’s defenses.
Targeting the Immune System for Cancer Therapy
For decades, scientists have strived to enhance the body’s immune attack against cancer.27 Various techniques have been attempted. One early type of immunotherapy is the administration of immunostimulatory cytokines.27 Interferon and IL-2 have both been used as treatment strategies in various cancers.19,20 To date, the main success has been in cancers that have historically been defined as sensitive to immune attack, such as renal cell carcinoma and melanoma.19,20 Broad application has not been successful. Another strategy has been tumor vaccines. Tumor vaccines allow for administration of a large quantity of tumor antigen into the system with the hope of focusing the patient’s immune system against that specific target. Currently, the only commercially available tumor vaccine is sipuleucel-T, a vaccine for castrate-resistant prostate cancer, but uptake has not been widespread.18 Technologies are also actively being developed to increase the activity and the number of immune cells in a patient. Genetically modified lymphocytes, such as chimeric antigen receptor (CAR) T-cells, are one of the examples of this strategy, but this type of treatment is not available clinically to date.28
The most recent strategy to amplify the potency of the immune system is immune checkpoint inhibitors.8–10,12–15 Checkpoints regulate the immune attack and are found at multiple phases in the development of the immune response.29,30 One checkpoint is found during the priming phase of the immune system – the time when naïve immune cells are being shown a target by antigen-presenting cells.30 When an antigen is presented to a naïve T-cell, if the costimulatory interaction between B7 and CD28 occurs, that T-cell becomes activated and amplifies.30 In contrast, if B7 interacts with cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), the T-cell activation is not completed and that immune attack is stifled.30 Monoclonal antibodies to CTLA-4 have been developed, which prevent downregulation of the immune system in the priming phase and allow a larger number of activated cytotoxic T-cells to be produced.31,32 This treatment strategy has proven successful in the treatment of melanoma and has changed the paradigm of management of this disease.10,14,15
Another checkpoint is found during the effector phase of the immune system.30 When an activated cytotoxic T-cell identifies the antigen it is seeking, secondary, either stimulatory or inhibitory, interactions between the two cells can occur.30 One of the key inhibitory interactions is between PD-1 and PD-L1.30 As discussed earlier, one of the mechanisms that tumor cells use to evade the immune system is to increase the amount of PD-L1 expressed on their surface. If PD-L1 on the tumor cells interacts with PD-1 on the T-cell, despite antigen recognition, the killer effector function of the T-cell will not be triggered.30 Several monoclonal antibodies have been developed, which interrupt the interaction between PD-1 and PD-L1, increasing the likelihood of the needed stimulatory interaction to promote T-cell killing.33 This class of monoclonal antibodies has also shown benefit in melanoma, a known immune sensitive cancer, and other cancers such as non-small cell lung cancer and bladder cancer, which are not classically viewed as responsive to immune attack.8,9,11,12,15,34
New therapies are always of interest in cancer care, but these immunotherapies have several unique characteristics. One is the tolerability of the therapy. The side effect profile of both CTLA-4 and PD-1/PD-L1 inhibitors is milder than that of many standard cytotoxic chemotherapies available.8,9 The side effect profile is also unique in comparison with chemotherapy.35 Inflammation and autoimmune phenomenon are the adverse events of clinical relevance.35 The most common adverse events are dermatologic and gastrointestinal (diarrhea) but only occur at a serious level in <25% of patients.35 The biggest draw to this type of treatment is the durability of the response. When the patients respond, the response tends to be long lasting. For example, single-agent CTLA-4 inhibition in melanoma haŝ20% 10-year survival in stage IV melanoma.14 PD-1 inhibitors have less mature survival data, but have shown impressive 1-year median survivals of ~70%.34 Stage IV non-small cell lung cancer also has a dismal prognosis but, despite this, >20% of patients who received a PD-1 inhibitor in randomized phase III trials have lived 2 years, despite progression after the best current treatment, a platinum doublet.8,9 We are now seeing our first 5-year survivors from the initial first in human phase I trials of PD-1 inhibitors in stage IV lung cancer as the data from these trials mature.
These drugs hold the promise of hope for patients. They also provide significant challenges. First, we do not have a robust biomarker to identify the patients who will most likely benefit from this class of treatments. Without a predictive biomarker, a considerable number of patients may receive treatment without benefit but yet at significant cost to the healthcare system. Second, monitoring the effects of this treatment is a challenge. When the immune system is stimulated, immune cells infiltrate the tumor that lead to the cytotoxic effects of the therapy. These immune infiltrates can make interpretation of imaging for response a challenge. The following sections outline the current ways in which imaging is used as a biomarker in oncology and how physicians have adapted their interpretation of imaging to respond to the unique challenges of immunotherapy.
Current Use of Anatomic Imaging in Immunotherapy
Although treatment effectiveness is often measured in terms of overall patient survival, this can be time-consuming and expensive. As such, there is significant interest in developing practical imaging markers for predicting long- and short-term therapy benefits. Various approaches have been considered, including anatomic and functional imaging. In recent years, the most common imaging response assessment in clinical oncology is based on anatomic change.
Response Evaluation Criteria in Solid Tumors
The first widely adopted anatomic imaging criteria for assessing tumor response was published in 1979 by the World Health Organization (WHO).36,37 Disease response was classified into four categories, namely, complete response (CR), partial response (PR), no response (NR), and progressive disease (PD). The classification was based on the product of perpendicular bidirectional tumor measurements summed over several tumor sites pre- and posttherapy using a four-week interval for follow-up. CR was defined by tumor disappearance, PR by a decrease of at least 50%, and NR by the spectrum of a decrease of <50% through an increase of <25%, while PD was defined by an increase of at least 25%. However, concerns with the WHO criteria were raised. For example, the number of disease sites to be measured was not defined.
In 2000, the response evaluation criteria in solid tumors (RECIST) was proposed.38 RECIST specified that the number of disease sites to be assessed should be up to 10, with up to 5 per organ. It also simplified implementation by suggesting that a sum of unidimensional measurements could be used as the imaging metric for determining response, rather than the product of perpendicular bidirectional tumor measurements. In this case, the unidimensional tumor measurement using the longest axis was suggested as the basis of disease measurement. CR was classified as the disappearance of the disease. PR was classified as a decrease of at least 30%. Stable disease (SD) was classified as a decrease of <30% through an increase of <20%, while PD was classified as an increase of at least 20%. PR and CR had to be confirmed by a second image performed at least four weeks after the first documentation of achieving a response. RECIST was criticized on several points. First, it was thought that the number of lesions measured had been determined arbitrarily. Also, using tumor’s long axis was considered to be impractical for cases of an irregular disease such as mesothelioma. Further, RECIST was limited in the assessment of pediatric malignancy and disease with central necrosis/cystic change.
In 2008, RECIST 1.1 (Table 1) was published in order to address issues raised with RECIST and simplify further implementation in clinical practice.39 First, the number of lesions to be measured was decreased to ≤5, with at most 2 per organ. Furthermore, it was specified that lymph nodes needed to be ≥1.5 cm in short axis measured, and nonlymph node lesions needed to be ≥1 cm in long axis measurable. Also, in addition to the increase of at least 20% required for PD, the requirement was added that there must be an increase of at least 5 mm in the sum of disease sites or the presence of new lesions. Unfortunately, clinical experience suggests that RECIST 1.1 may be insufficient to cover the spectrum of imaging response to immunotherapy. For example, although an increase in tumor size and/or the appearance of new lesions often suggests treatment failure, this might not always be the case. Indeed, immunotherapy often shows initial anatomic imaging changes that would be classified as PD using RECIST 1.1, followed by SD, PR, and/or CR, ultimately suggesting therapy effectiveness.
Table 1.
Summary of immune-related response criteria (irRC) guidelines compared with WHO handbook and response evaluation criteria in solid tumors (RECIST 1.1).24,37,39
| Complete response (CR) | |
| mWHO | Disappearance of all lesions in two consecutive observations ≥4 weeks apart. |
| RECIST 1.1 | Disappearance of all target lesions. Any pathological lymph nodes (whether target or non-target) must have reduction in short axis to <10 mm. Disappearance of all non-target lesions and normalization of tumor marker level. All lymph nodes must be non-pathological in size (<10 mm short axis). |
| irRC | Disappearance of all lesions in two consecutive observations ≥4 weeks apart. |
| Partial response (PR) | |
| mWHO | ≥50% decrease in the sum of the products of the two largest perpendicular diameters (SPD) of all index lesions vs. baseline in two observations at least 4 weeks apart, in absence of new lesions or unequivocal progression of non-index lesions. |
| RECIST 1.1 | At least a 30% decrease in the sum of diameters of target lesions, taking as reference the baseline sum diameters. |
| irRC | ≥50% decrease in tumor burden vs. baseline in two observations at least 4 weeks apart. |
| Stable disease (SD) | |
| mWHO | 50% decrease in SPD vs. baseline cannot be established nor 25% increase vs. nadir, in absence of new lesions or unequivocal progression of non-index lesions. |
| RECIST 1.1 | Neither sufficient shrinkage to qualify for PR nor sufficient increase to qualify for PD, taking as reference the smallest sum diameters while on study. Persistence of one or more non-target lesion(s) and/or maintenance of tumor marker level above the normal limits. |
| irRC | 50% decrease in tumor burden vs. baseline cannot be established nor 25% increase vs. nadir. |
| Progressive disease (PD) | |
| mWHO | At least 25% increase in SPD vs. nadir and/or unequivocal progression of non-index lesions and/or appearance of new lesions (at any single time point). |
| RECIST 1.1 | At least a 20% increase in the sum of diameters of target lesions, taking as reference the smallest sum on study (this includes the baseline sum if that is the smallest on study). In addition to the relative increase of 20%, the sum must also demonstrate an absolute increase of at least 5 mm. Unequivocal progression of existing non-target lesions. The appearance of one or more new lesions is also considered progression.* |
| irRC | At least 25% increase in tumor burden vs. nadir (at any single time point) in two consecutive observations at least 4 weeks apart. |
| Non-index lesions (non-measurable or over allowed number) | |
| mWHO | Changes contribute to defining best overall response of complete or partial response and stable or progressive disease. |
| RECIST 1.1 | Changes contribute to defining best overall response of complete or partial response and stable or progressive disease. |
| irRC | Contribute to defining immune-related complete response (complete disappearance required). |
| New measurable lesions (≥5 × 5 mm) | |
| mWHO | Always represent progressive disease. |
| RECIST 1.1 | Always represent progressive disease. |
| irRC | Incorporated in tumor burden. |
| New non-measurable lesions (≤5 × 5 mm, bone metastases, effusions) | |
| mWHO | Always represent progressive disease. |
| RECIST 1.1 | Always represent progressive disease. |
| irRC | Do not define progression (but preclude immune-related complete response). |
Note:
Non-CR/non-PD is preferred over SD when assessing nontarget lesion disease.
Abbreviations: irRC, immune-related response criteria; mWHO, modified World Health Organization; RECIST, response evaluation criteria in solid tumors; SPD, sum of the products of the two largest perpendicular diameters.
As the treatment of cancer evolves to include new agents, attention has been given to how a specific treatment might impact imaging findings and confound the results of anatomic imaging criteria such as RECIST. There is an increasing effort to personalize imaging response evaluation to therapy.40
Practical Considerations for Immunotherapy (Immune-related Response Criteria)
Initial discoveries that cancer provokes an immune response were followed by the knowledge that host reactions rarely impeded disease progression, possibly due to weak immunogenicity of nascent tumors and T-cell activation attenuation.41,42 Subsequent work on immunotherapy in melanoma suggested that the administration of a CTLA-4 blocking antibody such as ipilimumab could elicit significant antitumor effect; however, ipilimumab monotherapy was found to confound the results of classification with anatomic imaging criteria such as RECIST.10,43 In 2004 and 2005, a series of workshops were convened to explore the experience with immunotherapy in oncology.44 Four distinct response patterns on imaging were found, all of which were associated with favorable survival, as follows: (1) shrinkage of baseline lesions without new lesions; (2) durable SD (followed by a slow, steady decline in total tumor burden in some patients); (3) response after initial increase in total tumor burden; and (4) response in the presence of new lesions.24 The immune response criteria (Table 1) were defined as a way to incorporate imaging patterns observed with immunotherapy into the response assessment criteria and were evaluated in a series of patients with advanced melanoma who received ipilimumab.24 Further prospective evaluations of these criteria, particularly including their association with overall survival, are ongoing.
Anatomic or Metabolic Imaging for Response Assessment: Where are We?
Changes in tumor size on anatomic imaging may be related to overall survival; however, cytostatic treatment rather than cytocidal treatment can result in a good outcome without significant tumor shrinkage suggesting the need for both anatomic and metabolic response criteria. At the same time, as the anatomic criteria for disease response assessment to therapy were being developed, criteria using metabolic imaging were also being developed. In 1999, the European Organization for Research and Treatment of Cancer published recommendations on the measurement of clinical and subclinical tumor response using 18F-fluorodeoxyglucose (18F-FDG) and positron emission tomography (PET).45 Further suggestions were made by Hicks et al in 2001 and, subsequently, Juweid et al in 2005 for specific diseases such as lung cancer and lymphoma.46–48 In 2009, the PET response criteria in solid tumors were published.49 Although disease response was still classified as CR, PR, SD, and PD, a combination of metabolic and anatomic criteria were suggested to establish disease response. A maximum of 5 tumor sites, 2 sites per organ, with the highest FDG avidity were to be measured for comparison before and after therapy. It was recommended that patients should undergo PET at least 10 days after a chemotherapy cycle to maximize the prognostic value of the scan and minimize the effect of FDG-avid inflammation caused by chemotherapy and radiation. However, criticism was leveled at the known intrinsic variability in quantitative assessment between the studies performed at different centers.
We live in an era where there is ongoing discussion of the need for a personalized approach to medicine. Several imaging criteria have been proposed to evaluate disease burden at baseline and following therapy to determine treatment response. Recently, there has been a drive to incorporate both anatomic and metabolic information since disease response to therapy can produce both anatomic and metabolic changes and using imaging that takes this into account could provide the best method for assessing response. It has also been suggested that, perhaps, the imaging criteria applied should be modified based on specific disease histology and therapy. For example, the assessment of certain tumor types may benefit from the use of criteria that take into account the change in tumor density on computerized tomography (CT) scan (Choi criteria). Other disease types might benefit from the assessment of contrast enhancement patterns after use of vascular interventional therapies (European Association for the Study of the Liver criteria). All this progress notwithstanding the question remains: can a single practical system be developed for the imaging assessment of disease response or is there a need for a more personalized approach?
Overview of Molecular Imaging Approaches to Monitoring Therapy
The current state of knowledge of tumor response assessment recognizes that changes in tumor metabolic processes precede structural changes in anatomic imaging. There are a number of molecular imaging techniques available to monitor changes in tumor function reflective of therapeutic response, including immunotherapy. Fluorescence and bioluminescence are optical imaging techniques, which to date, have been used mainly in preclinical small animal models of human cancer lines due to the poor depth of tissue penetration. Many of these investigations are focused on tumor detection with an expanding number of studies in therapy assessment, particularly for drug development in animal models.50,51 An example is a study investigating cyclophosphamide treatment response in a firefly luciferase hepatic hepatocellular carcinoma tumor model in mice.52 A growing area of interest in fluorescence and bioluminescence imaging is focused on protease sensing probes and beacons to assess tumor response.53
In addition to providing high-contrast, high-resolution anatomic images, magnetic resonance imaging (MRI) can provide a range of functional measures. These include evaluation of tumor perfusion and cell membrane permeability through the use of dynamic contrast-enhanced MRI and assessment of parameters associated with the rate and distance of water molecule diffusion that may reflect drug access (diffusion-weighted MRI).54 More specific to evaluation of immunotherapy, MRI-based cell tracking, using cells labeled with either superparamagnetic iron oxide particles or perfluorocarbon nanoemulsions, has been used in a small number of pilot studies to assess the delivery of therapy.55
Single-photon emission computed tomography (SPECT) and PET are mature imaging technologies that have been used to detect in vivo differences in metabolic activity and differential expression of tumor-associated biochemical markers. Advantages of PET over SPECT are high-resolution images, the ability to quantify metabolic activity for accurate assessment of therapeutic effect. In the past decade, there has been an improved access to PET imaging in most jurisdictions with improving cost structure. Within the realm of molecular imaging techniques, 18F-FDG-PET imaging of cancer is the most studied and prevalent modality in oncologic nuclear imaging and an emerging molecular imaging standard for disease response assessment for both lymphoma and solid tumors.49,56,57 Current challenges with 18F-FDG-PET in tumor assessment include the differentiation between neoplasm and infectious or inflammatory processes. This proves particularly problematic when attempting to evaluate response to therapy in the midst of immune-related adverse events after treatment with immunotherapy agents.58,59
SPECT provides another avenue for the evaluation of tumor response, which, for some time, had been superseded by PET imaging. Recent innovations in SPECT imaging have increased the sensitivity of this modality, improved imaging resolution to similar levels to PET, and introduced the ability to accurately quantify disease activity as in PET.60–62 Advantages of SPECT imaging include the ubiquity of SPECT cameras, reduced cost structure compared with PET, improved logistics for imaging due to longer half-lives of the radionuclides, and a greater number of radionuclides available for labeling. The most frequently used SPECT agent today for treatment assessment in cancer is 99mTc-methyldiphosphonate (99mTc-MDP) for the imaging of osteoblastic bone metastases. However, bone scans do not detect actual metastases, but rather the reaction of the skeleton to metastases and the treatment of metastases. Therefore, the interpretation can be confounded, leading to an inaccurate assessment of the effects of therapy.63 Several other agents are in use according to specific tumor characteristics including, but not limited to, 123I-metaiodobenzylguanidine (123I-MIBG) for neuroblastoma, 111In-pentreotide for neuroendocrine tumors, and 123I-sodium iodide (NaI) for thyroid cancer.
Differentiation between expected postimmunotherapeutic effects and viable tumor require either radiopharmaceuticals targeted to a specific tumor type or novel agents that are increasingly specific for cancer cells. An example of a PET agent that has provided some advancement in this regard is 18F-fluorothymidine (18F-FLT), a marker of cell proliferation, which was developed to identify viable tumor while reducing the false positive rate related to infection or inflammation.64 Some limitations of this agent include lower signal to background ratio compared with 18F-FDG-PET, uptake in background structures including bone marrow, which can limit detection and quantification of tumor activity, and more recent evidence that 18F-FLT can accumulate at sites of infection and inflammation, although to a lesser degree than 18F-FDG.65
Survey of the Current Status of Imaging Biomarkers in Immunotherapy Trials
In an effort to establish how imaging biomarkers are being used currently in monitoring immunotherapy treatment, a search of the clinicaltrials.gov database was conducted from June to August 2015 searching for immunotherapy clinical trials meeting the following inclusion criteria:
any immunotherapy trials registered in 2010 or later, including those completed or in progress, and which included imaging outcome measures;
all phase I, II, III, and IV trials; and
all solid tumor and metastatic solid tumor clinical trials.
The following immunotherapy trials were excluded:
those that only concerned hematologic malignancies;
any trial that did not indicate imaging as an outcome measure; and
any terminated or withdrawn study, not related to adverse outcome.
A total of 484 clinical trials were identified and reviewed for cancer type, immunotherapy type, response criteria, and imaging methods. Melanoma, lung, prostate, and breast cancers were most frequently under study, although a substantial proportion of trials (10%) included multiple types of solid tumors (Fig. 1). The vast majority (455 of 484 trials, 94%) clearly stated outcomes dependent on CT, MRI, bone scan, or ultrasound measures of tumor mass or size. Approximately 60% of trials stated outcomes dependent on measureable disease, using the RECIST 1.1 criteria, and a further of trials 20% using the modified RECIST with immune-related response criteria (irRC). The remaining 20% of trials did not make reference to RECIST, but typically had measures of tumor mass. Immunotherapy trials for melanoma had the highest frequency (35%) of outcomes using the irRC and account for 50% of all trials that used irRC.
Figure 1.
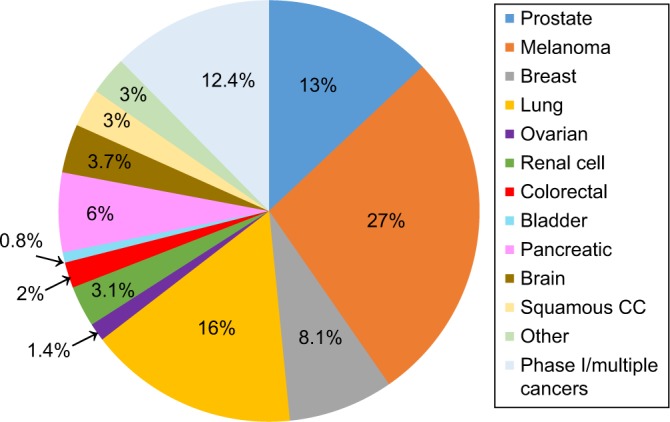
Distribution of cancer immunotherapy clinical trials by Cancer Site, total trials: 484.
Only 15 trials involving immunotherapy utilized some form of molecular imaging excluding bone scans (Table 2). Seven trials involve imaging agents that target metabolic or other markers on tumors, combined with PET/CT imaging. 18F-FDG was the dominant tracer among the trials using molecular imaging of tumor metabolism (six of seven trials). Of the remaining eight trials, five trials are testing markers of immune cells, such as T-cells or macrophages, including imaging of autologous lymphokine-activated killer cells, tumor-infiltrating lymphocytes (TILs), and CAR T-cells. This information suggests that molecular imaging has not been broadly incorporated into trials of immunotherapy representing an opportunity for future research.
Table 2.
Imaging agents in current immunotherapy trials.
| AGENT | CANCER | IMMUNOTHERAPY (IT) AND IMAGING TARGET | NATIONAL CLINICAL TRIALS (NCT) NUMBER | IMAGING TECHNOLOGY |
|---|---|---|---|---|
| 18F-FDG99,100 | Melanoma, renal cell, lung | IT: anti-CTLA-4, anti-PD-1 Target: tumor metabolism | NCT01666353 | PET/CT |
| 18F-FDG | Cervical, squamous cell | IT: anti-CTLA-4 Target: tumor metabolism | NCT01711515 | PET/CT |
| 18F-FDG | Multiple Ca | IT: CAR-T, anti-CTLA-4, IL-2 Target: tumor metabolism | NCT02070406 | PET |
| 18F-FDG | Renal cell | IT: IL-2 (plus chemo) Target: tumor metabolism | NCT01038778 | PET/CT |
| 18F-FDG | Multiple Ca | IT: CAR-T, IL-2, DC vaccine Target: tumor metabolism | NCT01697527 | PET |
| 18F-FDG or Na18F | Prostate | IT: DC vaccine with GM-CSF Target: tumor metabolism | NCT02042053 | PET/CT PET/MRI |
| 18F-FET | Brain melanoma metastases | IT: anti-PD-1, anti-CTLA-4 Target: tumor metabolism | NCT02374242 | PET/MRI |
| 11C-PBR28a | Brain | IT: various IT treatments, Target: tumor benzodiazepine receptor | NCT02431572 | PET |
| 18F-HBG71 | Glioma | IT: CAR-T, IL-2 Target: CAR-T cells | NCT01082926 | PET |
| 89Zr-MPDL3280A74 | Multiple cancers | IT: anti-PD-L1 Target: PD-L1 on tumor or other cells | NCT02453984 | PET |
| 99Tc-IL-268 | Melanoma | IT: anti-CTLA-4, anti-PD-1, IL-2 Target: TIL expressing IL-2 receptor | NCT01789827 | SPECT |
| 18F-L-FAC70 | Healthy volunteers and multiple cancers | IT: various immunotherapies Target: activated T-cells in tumor |
NCT01180868 NCT01180907 |
PET |
| 89Zr-GC100872 | Brain glioma | IT: anti-TGF-β Target: TGF-β | NCT01472731 | PET |
| Ferumoxytol73 (iron nanoparticles) | Brain | IT: various immunotherapies Target: macrophage in tumors | NCT02452216 | MRI |
| 18F F-AraG69 | Healthy subjects | IT: prior to various cancer IT trials Target: activated T-cells | NCT02323893 | PET |
Notes: All listed clinical trials involving molecular imaging are in phase I or II only. NCT numbers were obtained from www.clinicaltrials.gov as searched on September 30, 2015.
Abbreviations: 11C, carbon 11; CAR-T, chimeric antigen receptor T-cell; CT, computerized tomography; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; DC, dendritic cell; 18F, fluorine 18; FDG, fluorodeoxyglucose; F-AraG, fluoro-9-β-d-arabinofuranosylguanine; FET, fluoro-ethyl-tyrosine; GC1008, fresolimumab; GM-CSF, granulocyte-macrophage colony-stimulating factor; HBG, hydroxymethyl-butyl guanine; IL-2, interleukin-2; IT, immunotherapy; l-FAC, 1-l-(2 deoxy-2,-18 fluoroarabinofuranosyl) cytosine; MPDL3280A, atezolizumab; MRI, magnetic resonance imaging; Na18F, sodium fluoride; NCT, National Clinical Trials; PD-1, programmed cell death 1; PD-L1, programmed cell death-ligand 1; PET, positron emission tomography; PBR28, peripheral benzodiazepine receptor 28a; SPECT, single-photon emission computed tomography; 99Tc, technetium 99; TGF-β, transforming growth factor-beta; TIL, tumor-infiltrating lymphocyte; 89Zr, zirconium 89.
Probes to Image Immune Response within Tumors in Current Clinical Trials
There are relatively few current clinical trials involving immunotherapy that also include molecular imaging, but approximately half of these feature imaging of immune cells or immune functional markers within the tumors studied (Table 2). There could be substantial clinical benefit from the development of new molecular imaging probes that could either predict benefit from immunotherapy pretreatment or allow measurement of cell-mediated immune response on treatment. For instance, patients with cancers with high levels of TILs may be ideal candidates for PD-1 or PD-L1 inhibitors.66 Identifying TILs currently requires a biopsy, which is invasive and hampered by issues with sampling error. Obtaining biopsies and their pathologic results also institutes significant delays in instituting therapy. Molecular imaging could profile the whole primary tumor as well as metastases providing a more global view of the tumor and immune system interaction potentially aiding our ability to select patients in an expedited manner.67 Similar imaging technology could be used for patients on treatment with immunotherapy to monitor for increased activated T-lymphocyte or natural killer cell activity in tumors. Documentation of this early immune activity may precede documented anatomic imaging changes, allow for discrimination between true progression and pseudoprogression, and predict longer term benefits.
Among those probes already in clinical trial testing, nearly half (4/10) of them are designed to image cytotoxic lymphocytes (CTL) among the TILs.68–71 These include probes that identify activated T-cells or markers on activated T-cells, such as the IL-2 receptor. Another three probes target immunosuppressive factors (PD-L1 and TGF-β) or cells such as macrophages that can act as myeloid-derived suppressor cells.72–74 Several of the imaging studies in current trials are agents that are designed as direct labels of therapeutic, anti-tumor lymphocytes (CAR-T or lymphokine-activated killer) and that are given to patients as treatment.69–71 The localization of the labeled cells in the patient tumor can then be verified and monitored. Although these are important imaging methods to monitor specific cell-mediated immunotherapies, they have limited application.
Preclinical Studies of Molecular Imaging Probes to Visualize Immune Response within Tumors
The complex nature of the tumor microenvironment and the changing composition of immune cell infiltrates limit the effectiveness of metabolic markers (such as 18F-FDG) for imaging of immune cells within tumors.47 Immuno-PET, the use of antibodies or antibody fragments to target PET radionuclides, has the potential to detect T-cell subsets within tumors or lymphoid tissues and noninvasively monitor changes in TILs, which result from cancer immunotherapies.75 Immuno-PET could also be used to monitor immune activity within tumors. Two of the most obvious targets around which to develop molecular imaging probes to monitor immunotherapy of cancer are CD8, a phenotypic marker of cytotoxic T-lymphocytes, and CD4 expressed by T-helper cells that participate in the inflammation and cytotoxic attack within tumors. Other targets could include markers of dendritic cells and macrophages, which may alternatively enhance or regulate T-cell responses within tumors, and known immunosuppressive markers such as PD-L1 and CD47. Work is ongoing to develop novel molecular imaging probes for a range of different immunotherapy-relevant biomarkers. Table 3 lists a selection of recent reports that focus on targeted agents for PET, SPECT, or ultrasound use that are in preclinical development. This selection is far from comprehensive as publications on new probes are appearing with increasing frequency. We focus on representative examples notably those with potential for wide application to imaging immune cells in various solid tumors, rather than those with labeled single-cell tracking or reporter gene approaches that are also in development. When evaluating a new probe, key features that lead to successful clinical translation include high target affinity as demonstrated by high tumor to off target ratios, low off target binding (such as spleen and liver), and early clearance from the blood pool.
Table 3.
Imaging agents for antitumor immune function in published preclinical studies.
| IMAGING AGENT | TARGETING CONCEPT | IMAGING TECHNOLOGY |
|---|---|---|
| 18F-/64Cu anti-CD11b or MHC-II81 | Labeled antibody fragments binding to CD11b or MHC II on tumor macrophage or myeloid cells | PET |
| 64Cu-anti-CD876 | Labeled antibody fragments binding to CD8 on tumor infiltrating cytotoxic T lymphocytes | PET |
| 89Zr-anti-CD878 | Labeled antibody fragments binding to CD8 on tumor infiltrating cytotoxic T lymphocytes | PET |
| 18F-FEAU101 | Labeled ligand identifies viral transgene in activated CAR-T that are present in tumor | PET |
| 111I-anti-PD-L184 | Labeled monoclonal antibody binds to PD-L1 expressed on macrophage and tumor cells | SPECT |
| 89Zr-anti-CD4783 | Labeled monoclonal antibody binds to CD47 expressed on cells within tumor | PET |
| 64Cu-Anti-CTLA-480 | Labeled monoclonal antibody binds to CTLA-4 expressed on cytotoxic T lymphocytes within tumor | PET |
| MB-anti-B7-H385 | Ultrasound microbubbles labeled with monoclonal antibody against B7-H3. Identifies cells expressing B7-H3 on macrophage and tumor cells | US |
| 64Cu-SPION102 | CAR-T cells loaded with 64Cu-SPION (iron nanoparticles). Image accumulation of therapeutic CAR-T | PET |
| DiR labeled T cells103 | DiR fluorophore, activated by near-Infrared light, is used to label T cells. T cells that located in tumor are imaged | Fluorescence imaging |
Abbreviations: CAR-T, chimeric antigen receptor T-cell; CD8, cluster of differentiation 8; CD11b, integrin alpha M; CD47, integrin-associated protein; CTLA-4, cytotoxic T-lymphocyte-associated protein 4; 64Cu, copper-64; DiR, 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindotricarbocyanine iodide; 18F, fluorine 18; FEAU, 1-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-ethyluridine; 111I, indium 111; MB, minibody; MHC II, major histocompatibility complex 2; PD-L1, programmed cell death-ligand 1; PET, positron emission tomography; SPECT, single-photon emission computed tomography; SPION, super paramagnetic iron oxide nanoparticles; US, ultrasound; 89Zr, zirconium 89.
One of the early approaches involved developing a CD8 probe to specifically target cytotoxic T-lymphocytes in mouse tumor models. Researchers utilized two rat anti-CD8 monoclonal antibodies that were engineered as mini-bodies, having truncated Fc regions.76 The minibodies were then derivatized with 64Cu using a 1,4,7-triazacyclononane-N,N′,N″-triacetic acid (NOTA) chelator. While intact antibodies have been used preciously to image T-cells in mice and human beings, the labeled minibody showed a high uptake in the spleen (75 ± 8.5%ID/g), lymph nodes (27 ± 7.9%ID/g), and liver (57 ± 11%ID/g) of antigen positive B/6 mice, with rapid clearance from the blood. Images could be acquired at early time points (four hours), because the agent cleared the blood rapidly.77 When target negative (CD8 allotype) strains of mice were studied, the liver uptake remained the same, but there was a significant decrease in the percent of injected dose per gram of tissue (%ID/g) in the spleen and lymph nodes, indicating target specificity. In addition, treatment of mice with excess unlabeled minibody or intact anti-CD8 antibody, prior to the 64Cu-NOTA-anti-CD8 minibody, resulted in low uptake by the spleen and lymph nodes, but no change in the liver. Analysis of target localization data indicated an estimate of 70,000–120,000 CD8+ cells, in a mouse lymph node. While this CD8 targeting agent showed excellent PET imaging and target specificity, and importantly did not deplete CD8 T-cells (due to the absence of Fc function), it did show high nonspecific liver uptake, possibly due to 64Cu transchelation, aggregation, or dimerization of the protein.
Wu et al have continued to develop probes for CD8, building on their minibody studies in mice. They recently developed two anti-CD4 and anti-CD8 cys-diabodies (cDb), labeled with 89Zr, and used PET to detect CD4+ and CD8+ T-cells in vivo.78 A maleimide derivative of deferoxamine (desferrioxamine B, DFO), a well-established chelate for 89Zr, was linked site-specifically to a cysteine thiol, following treatment of the cDb with a reducing agent, which did not alter the size-exclusion profile of the conjugated protein.79 89Zr labeling was achieved in high (>96%) yield, and the product was obtained in a specific activity range from 59 to 203 kBq/µg. Protein doses of the CD4 targeting construct of ~12 µg were administered intravenously to mice, and PET images were acquired at 4, 8, and 22 hours. The agent showed uptake in the kidney, lymph nodes, and spleen using PET, and this was verified by tissue biodistribution of 89Zr radioactivity, at 22 hours postinjection. Blocking with unlabeled, anti-CD4 cDb (3 mg/kg) or depletion of CD4 positive cells resulted in clearance from the blood at four hours and low uptake in lymph nodes and spleen. The 89Zr-labeled cDb that targeted CD8 showed high uptake in multiple lymph nodes (inguinal, axillary, mesenteric, and cervical) and spleen, and this was blocked by unlabeled anti-CD8-cDb and had very high lymph node-to-blood ratio of 78:1 and spleen-to-blood ratio of 67:1. The 89Zr-labeled cysdiabody agents showed higher levels in the kidney and lower levels in the liver compared with the previous minibody agents76 due to its smaller size. Importantly, both the CD8 and CD4 targeting agents were successfully used to monitor T-cell repopulation in lymph nodes and spleen, after hematopoietic stem cell transplantation, in lethally irradiated mice. The limitation noted was that the agents showed low uptake in the thymus.
Higashikawa et al reported the labeling of an anti-mouse CTLA-4 monoclonal antibody with 64Cu DOTA, which was subsequently evaluated in CT26 tumor-bearing BALB/c mice.80 CTLA-4 is expressed on the surface of two major subsets of CD4+ T-cells and on activated CD8-expressing CTL. CTLA-4 is a regulatory molecule, whose blockade by antibody (ipilimumab) provides a highly effective immunotherapy in various cancers.15 The derivatized antibody had an average of 4.2 chelators per antibody and was prepared using a DOTA-mono-N-hydroxysuccinimide (NHS) ester that enabled production of a product having a specific activity of 0.25 MBq/µg. The selected monoclonal antibody recognized the extracellular domain of mouse CTLA-4, similar to the epitope for human CTLA-4 recognized by ipilimumab. PET results indicated a clear expression of CTLA-4 within the tumor tissues, but no expression by the culture tumor cell line, when tested in vitro, indicating that the PET image was primarily from infiltrating CTLA-4 T-cells. The labeled anti-CTLA-4 antibody had slightly reduced binding to CTLA-4 in vitro, and the mean ratio of the maximum standardized uptake value (SUVmax) for PET images in the CT26 tumor model (compared with control immunoglobulin) was 1.29, similar to the ex vivo ratio. While the tumors were evident in the images presented, the biodistribution data indicated that %ID/g of tumor was modest, at ~8% (control IgG was 6%), and there was significant activity in the liver, kidneys, and blood at 48 hours. The tumor-to-blood ratio never exceeded 1, which could hinder further usage of this type of construct.
Recently, Rashidian et al developed murine-based antibody fragments to target cells expressing major histocompatibility complex (MHC) II and CD11b cells, in mice.81 The authors used single heavy-chain structure, variable regions (VHHs), which are much smaller than whole antibodies and smaller than a number of commonly used engineered immunoglobulin fragments. The specificities for MHC II and CD11b were chosen to noninvasively image macrophages, dendritic cells, and neutrophils in both tumor and inflammatory models. This approach employed a two-step radiolabeling process involving 18F and an inverse electron-demand Diels–Alder reaction. They were able to produce their product in 61% yield (decay corrected) using a two-step procedure, in quantities suitable for preclinical studies. Administration of 7.4 MBq (200 µCi, 10 µg) of the anti-MHC II radiolabeled antibody fragments to C57BL/6 wild-type mice resulted in visualization of lymph nodes, spleen, and thymus. Both the anti-MHC II and anti-CD11b agents were evaluated in xenograft tumor models. Detection of MHC II and CD11b positive expression in the margins of the xenograft tumors indicated likely infiltrate of macrophages or myeloid cells, in the early inflammatory response. The authors showed that the tracers could be used to detect tumor-associated inflammation, at early time points prior to the tumor palpation. Additional studies showed that the agents could be used to detect infiltrating inflammatory cells, in mice with a murine melanoma (B16) allografts, as well as accumulation of myeloid (CD11b positive) neutrophils, in tissue sites, after administration of Freund’s adjuvant. Work to humanize VHHs has been described and is a critical step in translating clinical studies.82 Another explored target was CD47, the integrin-associated protein that functions as an innate marker of “self” and inhibits phagocytosis of cells through an interaction with SIRPα expressed by phagocytes and particularly macrophages. CD47 is an interesting target for an immunotherapy molecular imaging probe because overexpression of CD47 on some types of tumor cells has been identified as a mechanism for evasion of immune surveillance. In one study, the authors radiolabeled (89Zr) two anti-CD47 antibodies: B6H12, which recognizes the extracellular domain of human CD47, and αM-CD47, which binds the extracellular portion of mouse CD47.83 They then evaluated the labeled probes in mouse xenografts of human ovarian carcinoma cells (OV10) and murine B16F10 melanoma allograft models. The chelate DFO was introduced using an isothiocyanate (NCS)-based modification of lysine residues, and the labeled products were produced with specific activities ranging from 0.9 to 1.6 µCi/µg. Approximately 50 µCi per animal was used for PET imaging studies. Tumor-associated image intensity of the labeled B6H12 antibody was significantly greater (2.8-fold) in the CD47+ tumor, compared with tumors that did not express CD47. Uptake of either probe was also evident in the liver, lungs, spleen, and kidney, with similar levels observed irrespective of the animal-tumor model. In the B16F10 allograft-bearing model, the labeled αM-CD47 probe showed high liver uptake, as much as 168 hours postadministration, and similarly in the spleen and bone. The tumor-to-blood ratio was ~3.0:168 hours, but the total uptake was modest. The use of both antibodies with allo- versus xenografts was important because the data showed that the distribution in the allograft model was different than that for the xenograft, indicating the potential impact of endogenous CD47 expression in the mouse models. Nevertheless, the study shows that targeting CD47 is feasible and further work is pending.
Heskamp et al radiolabeled an anti-PD-L1 antibody (PD-L1.3.1) with 111In as an imaging biomarker, in order to determine the level of PD-L1 expression in vivo.84 This SPECT agent was shown to have high affinity for PD-L1 (Kd = 1 nM), but tumor uptake in the PD-L1-positive, MD-MB-231 xenograft model was highly sensitive to the specific activity of the labeled agent. The optimal amount of protein was determined to be ≤1 µg for the 111In-diethylenetriaminepentaacetic acid-labeled construct, which was produced with specific activities up to 10 MBq/µg having an immunoreactive fraction of 82%. The labeled antibody was slowly internalized over 24 hours in MD-MB-231 cells in vitro, and tumor uptake in vivo reached an impressive 37%ID/g that was blocked using the excess of unlabeled antibody. Uptake in the tumors were observed as early as one day postinjection and increased to seven days. Tumor-to-blood ratios were very high (~4:1 at seven days), and tumor images had excellent contrast between tumor and other tissues. This agent has the potential for imaging human PD-L1 in various cancers, provided that a humanized form of the antibody can show similar results.
Ultrasound is being used increasingly as a molecular imaging technique as a result of the advent of targeted micro-bubbles (MB). Ultrasound with MB is an attractive approach because the technology is relatively inexpensive, is widely accessible, and does not require the use of ionizing radiation. Willmann et al reported a new MB construct that targets B7-H3 (CD276) using an anti-human B7-H3 antibody.85 The anti-B7-H3 antibodies are directly linked to the surface of the MB using a streptavidin–biotin linkage. B7-H3 is a member of the B7 family of ligands for T-cell coregulatory receptors and is implicated in the suppression of cell-mediated immunity. B7-H3 has been identified in human dendritic cells and some activated T-cells. A range of solid tumors has also been shown to express B7-H3.86–88 In a transgenic mouse model of human breast cancer, Willmann et al reported an ultrasound imaging signal from identified tumors, using the anti-B7-H3 MB, which was four times greater than the signal using the control MBs.85 The signal with the targeting MBs was reduced to background, when blocked by the administration of the unlabeled targeting antibody (anti-B7-H3). This MB targeting approach to imaging of B7-H3 provides an interesting tool for in vivo studies to image tumors, but its utility in human beings will require humanization of the MB constructs.
Imaging During Immunotherapy Using Tumor-specific T-cells
Although the majority of imaging studies in immunotherapy provide molecular probes to assess enhanced immune function or tumor regression, the specific cellular immunotherapies using chimeric antigen receptor bearing T-cells (CAR-T) allow for direct cell labeling to monitor their location during therapy. It is possible to assess CAR-T migration to lymph nodes or tumors by direct labeling of the cells89 using various imaging agents, including 111In labeling for SPECT imaging.90 In other preclinical work, electrotransfer of 64Cu-labeled gold nanoparticles allowed tracking of T-cells, which was correlated with bioluminescence imaging.91 More frequently, however, the ability to genetically modify T-cells has been used to express specific transgenes (typically viral) as unique molecular markers that allow PET and SPECT to assess T-cell trafficking and residency at tumor sites. This includes preclinical imaging of herpes simplex virus type 1 thymidine kinase (HSV1tk) via 18F-fluoro-29-deoxy-1-β-d-arabinofuranosyl-5-ethyluracil (18F-FEAU).92 Eissenberg et al recently described an early clinical work in which T-cells were modified with a suicide gene (CD34-HSV1tk) whose distribution could be tracked over time using 9-[4-(18 F)fluoro-3-hydroxymethyl-butyl] guanine ([18F]FHBG).93 A similar approach had been reported by Dubey et al using 9-[4-[18F]fluoro-3-(hydroxymethyl) butyl]guanine ([18F]FHBG) to image splenic T-cells bearing the HSV1-sr39tk PET reporter gene.94 Earlier precursor work in 2001 by Ponomarev et al used [124I]-(2′-deoxy-2′-fluoro-β-d-arabinofuranosyl)-5-iodouracil to detect T-cell activation in vivo using an HSV1tk/green fluorescent protein reporter system in rats.95 In an alternative approach, the human norepinephrine transporter was used as a transgenic molecular marker that was imaged with the approved SPECT tracer 123I-MIBG or the PET analog 124I-MIBG.96 This approach has several advantages, in that it uses an approved and accessible radio-tracer and avoids immunogenicity concerns because the cells do not express virus-derived reporter molecules. Clearly given the intensity of work on CAR-T-based treatments, there will be an expanding role for molecular imaging, both to provide spatial and temporal tracking of cells and assessment of treatment response to CAR-T treatment.
Regulatory Environment
Clinical trials with investigational agents, whether they are experimental drugs or imaging biomarkers, are subject to Part C, Division 5 of the Canadian Food and Drug Act and Regulations. These are designed to ensure that patients receiving an investigational drug are protected. Good clinical practices set out the requirements of clinical trial conduct, with a focus on the protection of the research subjects and ensuring data integrity. The manufacture of drug products must be in compliance with good manufacturing practices, which require the product to be manufactured according to the established conditions and fully tested against preapproved specifications.
Regulatory obligations include the submission of a clinical trial application for review by Health Canada. There must be sufficient evidence to support the safety and efficacy of the investigational agent, based on clinical and/or preclinical data. For radiopharmaceuticals, safety also encompasses an estimate of dosimetry, ie, the estimated radiation exposure to the subject. Initial data derived from preclinical biodistribution testing can be used to calculate dosimetry estimates for human beings, and in the early stages of clinical development, more appropriate estimates can be obtained from human biodistribution. Manufacturing details and data must be provided to demonstrate that the manufacturing process is adequately controlled to reliably produce a safe product meeting the predetermined quality attributes. The information provided is evaluated by the regulators in the context of the proposed clinical trial.
While there are no specific regulations pertaining to the development of biomarkers in Canada, in any submission sound scientific data should be presented to demonstrate that the use of the biomarker is qualified for the intended use, ie, “fit for purpose.” The mechanism of action of an in vivo bio-marker must be well understood, and while this can be established initially in a preclinical setting, adequate confirmation should be obtained clinically. For example, 18F-FLT is an investigational molecular imaging marker of cellular proliferation that has potential in diagnosis, prognosis, prediction of response, and monitoring of response to treatment in patients with cancer. Thus, detailed characterization of the mechanism of action, metabolism, and pharmacokinetics has been obtained from preclinical and clinical studies, and a validated kinetic model has been developed.97 Uptake of FLT in a number of tumor types has been found to correlate with an independent marker of proliferation (Ki-67).98 Reproducibility of images has also been demonstrated in non-small cell lung cancer, which has provided an estimate of the expected errors, thereby providing parameters for assessing treatment response. A recent review of FLT-PET proposes to a this biomarker could serve as an early negative predictor after the first cycle of treatment, thus providing an early go/no-go signal in drug development. However, further research is needed to better characterize and optimize imaging parameters to determine the full potential of FLT-PET in drug development.97 In addition, further well-controlled clinical studies are needed to validate the clinical utility of FLT-PET for the diagnosis and treatment management of various cancers. The development of FLT-PET serves as an example of the questions to address in order to fully qualify imaging biomarkers for these intended applications.
Health Canada recognizes that positron-emitting radiopharmaceuticals (PERs) are often used to investigate basic research questions under circumstances where there is minimal additional risk to research subjects. PERs that have an acceptable predetermined safety profile and an acceptable dosimetry and have no pharmacodynamic effect at the planned dose are considered to be eligible. Research subjects must be at least 18 years old, no more than 30 subjects can be enrolled, and approval from research ethics board must have been obtained prior to the application. Any concomitant drugs used in such a research must have market authorization. In order to reduce the regulatory burden in such cases, a simplified application process has been established, with a 15-day review period by Health Canada. This mechanism provides a foundation that facilitates an important research with PERs and is now being actively used by Canadian research groups. The regulatory aspects of developing and commercializing a radiopharmaceutical cannot be overlooked when considering this class of agents as a putative biomarker.
Conclusions
Immuno-oncology is an exciting field within cancer treatment with the potential to impact the management of numerous malignancies. The current system using anatomic imaging as the main imaging biomarker needs further improvement despite ongoing refinements. Molecular imaging technology is a powerful tool that may provide a means to not only predict the patients who are most likely to respond to immune-based treatments but also monitor their response. Molecular imaging biomarkers provide a more global look at the characteristics of tumors in patients and do not suffer from sampling error unlike biopsies. In addition, imaging biomarkers are routinely minimally invasive and can be interpreted quickly allowing more prompt impact on patient care. Even with the significant cost routinely associated with molecular imaging, the technique can become cost-effective if we could refine the patient population most likely to benefit or shorten the duration of therapy given the estimated cost of immuno-oncology treatment per year (~$100,000 USD).
Although molecular imaging is just starting to be used to monitor therapy in immuno-oncology, in preclinical models, as we have reviewed, the potential ability to monitor very specific cellular activity within tumors is evident. Our understanding of the interplay between cancer and the immune system continues to grow, and with this knowledge, more options will become available for relevant molecular imaging targets. The field of molecular imaging in immuno-oncology is a prime area for novel research with real opportunity for clinical translation and commercialization. In order to bring this type of technology to the clinic, a robust mechanism not only to evaluate but also to validate the effectiveness needs to be implemented. Effort needs to be directed at selecting the best probes for translation into the clinic so that the community can optimize the changes needed for meaningful clinical impact.
Footnotes
ACADEMIC EDITOR: Barbara Guinn, Editor in Chief
PEER REVIEW: Three peer reviewers contributed to the peer review report. Reviewers’ reports totaled 387 words, excluding any confidential comments to the academic editor.
FUNDING: This study was supported by Ontario Institute for Cancer Research and Boris Family Foundation. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE)
Author Contributions
Conceived and designed the manuscript: RAJ, JFV, and KYG. Analyzed the data: RAJ, DPS, JFV, and KYG. Wrote the first draft of the manuscript: RAJ, KAZ, AS, DPS, JFV, and KYG. Contributed to the writing of the manuscript: RAJ, KAZ, AS, DPS, JFV, and KYG. Agreed with manuscript results and conclusions: RAJ, KAZ, AS, DPS, JFV, and KYG. Jointly developed the structure and arguments for the paper: RAJ, KAZ, AS, DPS, JFV, and KYG. Made critical revisions and approved the final version: RAJ, KAZ, AS, DPS, JFV, and KYG. All the authors reviewed and approved the final manuscript.
REFERENCES
- 1.Baselga J, Cortes J, Kim SB, et al. CLEOPATRA Study Group Pertuzumab plus trastuzumab plus docetaxel for metastatic breast cancer. N Engl J Med. 2012;366(2):109–119. doi: 10.1056/NEJMoa1113216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Corcoran RB, Atreya CE, Falchook GS, et al. Combined BRAF and MEK inhibition with dabrafenib and trametinib in BRAF V600-mutant colorectal cancer. J Clin Oncol. 2015;33(34):4023–4031. doi: 10.1200/JCO.2015.63.2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Demetri GD, von Mehren M, Blanke CD, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347(7):472–480. doi: 10.1056/NEJMoa020461. [DOI] [PubMed] [Google Scholar]
- 4.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 5.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361(10):947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 6.O’Brien SG, Guilhot F, Larson RA, et al. IRIS Investigators Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348(11):994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 7.Shaw AT, Kim DW, Nakagawa K, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–2394. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 8.Borghaei H, Paz-Ares L, Horn L, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung cancer. N Engl J Med. 2015;373(17):1627–1639. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373(2):123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motzer RJ, Escudier B, McDermott DF, et al. CheckMate 025 Investigators Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373(19):1803–1813. doi: 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–562. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 13.Ribas A, Puzanov I, Dummer R, et al. Pembrolizumab versus investigator-choice chemotherapy for ipilimumab-refractory melanoma (KEYNOTE-002): a randomised, controlled, phase 2 trial. Lancet Oncol. 2015;16(8):908–918. doi: 10.1016/S1470-2045(15)00083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schadendorf D, Hodi FS, Robert C, et al. Pooled analysis of long-term survival data from phase ii and phase iii trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. 2015;33(17):1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coley WB. The treatment of inoperable sarcoma by bacterial toxins (the mixed toxins of the Streptococcus erysipelas and the Bacillus prodigiosus) Proc R Soc Med. 1910;3(Surg Sect):1–48. doi: 10.1177/003591571000301601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Coley WB. The treatment of malignant tumors by repeated inoculations of erysipelas. With a report of ten original cases. 1893. Clin Orthop Relat Res. 1991;(262):3–11. [PubMed] [Google Scholar]
- 18.Kantoff PW, Higano CS, Shore ND, et al. IMPACT Study Investigators Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 19.Mocellin S, Pasquali S, Rossi CR, Nitti D. Interferon alpha adjuvant therapy in patients with high-risk melanoma: a systematic review and meta-analysis. J Natl Cancer Inst. 2010;102(7):493–501. doi: 10.1093/jnci/djq009. [DOI] [PubMed] [Google Scholar]
- 20.Rosenberg SA, Yang JC, Topalian SL, et al. Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA. 1994;271(12):907–913. [PubMed] [Google Scholar]
- 21.Sylvester RJ, van der MA, Lamm DL. Intravesical Bacillus Calmette–Guerin reduces the risk of progression in patients with superficial bladder cancer: a meta-analysis of the published results of randomized clinical trials. J Urol. 2002;168(5):1964–1970. doi: 10.1016/S0022-5347(05)64273-5. [DOI] [PubMed] [Google Scholar]
- 22.Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol Cancer Ther. 2015;14(4):847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- 23.Pennock GK, Waterfield W, Wolchok JD. Patient responses to ipilimumab, a novel immunopotentiator for metastatic melanoma: how different are these from conventional treatment responses? Am J Clin Oncol. 2012;35(6):606–611. doi: 10.1097/COC.0b013e318209cda9. [DOI] [PubMed] [Google Scholar]
- 24.Wolchok JD, Hoos A, O’Day S, et al. Guidelines for the evaluation of immune therapy activity in solid tumors: immune-related response criteria. Clin Cancer Res. 2009;15(23):7412–7420. doi: 10.1158/1078-0432.CCR-09-1624. [DOI] [PubMed] [Google Scholar]
- 25.Jadus MR, Natividad J, Mai A, et al. Lung cancer: a classic example of tumor escape and progression while providing opportunities for immunological intervention. Clin Dev Immunol. 2012;2012:160724. doi: 10.1155/2012/160724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vinay DS, Ryan EP, Pawelec G, et al. Immune evasion in cancer: mechanistic basis and therapeutic strategies. Semin Cancer Biol. 2015;35(suppl):S185–S198. doi: 10.1016/j.semcancer.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 27.Shahani L, Singh S, Khardori NM. Immunotherapy in clinical medicine: historical perspective and current status. Med Clin North Am. 2012;96(3):421–431. ix. doi: 10.1016/j.mcna.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 28.Davila ML, Bouhassira DC, Park JH, et al. Chimeric antigen receptors for the adoptive T cell therapy of hematologic malignancies. Int J Hematol. 2014;99(4):361–371. doi: 10.1007/s12185-013-1479-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol. 2015;42(3):363–377. doi: 10.1053/j.seminoncol.2015.02.015. [DOI] [PubMed] [Google Scholar]
- 30.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipson EJ, Drake CG. Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res. 2011;17(22):6958–6962. doi: 10.1158/1078-0432.CCR-11-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tarhini AA. Tremelimumab: a review of development to date in solid tumors. Immunotherapy. 2013;5(3):215–229. doi: 10.2217/imt.13.9. [DOI] [PubMed] [Google Scholar]
- 33.Sunshine J, Taube JM. PD-1/PD-L1 inhibitors. Curr Opin Pharmacol. 2015;23:32–38. doi: 10.1016/j.coph.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Robert C, Schachter J, Long GV, et al. KEYNOTE-006 Investigators Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med. 2015;372(26):2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 35.Weber JS, Yang JC, Atkins MB, Disis ML. Toxicities of immunotherapy for the practitioner. J Clin Oncol. 2015;33(18):2092–2099. doi: 10.1200/JCO.2014.60.0379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47(1):207–214. doi: 10.1002/1097-0142(19810101)47:1<207::aid-cncr2820470134>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 37.World Health Organization . WHO Handbook for Reporting Results of Cancer Treatment. Geneva: World Health Organization; 1979. p. 45. [Google Scholar]
- 38.Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst. 2000;92(3):205–216. doi: 10.1093/jnci/92.3.205. [DOI] [PubMed] [Google Scholar]
- 39.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45(2):228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 40.Nishino M, Jagannathan JP, Krajewski KM, et al. Personalized tumor response assessment in the era of molecular medicine: cancer-specific and therapy-specific response criteria to complement pitfalls of RECIST. AJR Am J Roentgenol. 2012;198(4):737–745. doi: 10.2214/AJR.11.7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boon T, van der Bruggen P. Human tumor antigens recognized by T lymphocytes. J Exp Med. 1996;183(3):725–729. doi: 10.1084/jem.183.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sahin U, Tureci O, Schmitt H, et al. Human neoplasms elicit multiple specific immune responses in the autologous host. Proc Natl Acad Sci U S A. 1995;92(25):11810–11813. doi: 10.1073/pnas.92.25.11810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hodi FS, Mihm MC, Soiffer RJ, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U S A. 2003;100(8):4712–4717. doi: 10.1073/pnas.0830997100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoos A, Parmiani G, Hege K, et al. Cancer Vaccine Clinical Trial Working Group A clinical development paradigm for cancer vaccines and related biologics. J Immunother. 2007;30(1):1–15. doi: 10.1097/01.cji.0000211341.88835.ae. [DOI] [PubMed] [Google Scholar]
- 45.Young H, Baum R, Cremerius U, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer. 1999;35(13):1773–1782. doi: 10.1016/s0959-8049(99)00229-4. [DOI] [PubMed] [Google Scholar]
- 46.Hicks RJ, Kalff V, MacManus MP, et al. The utility of (18)F-FDG PET for suspected recurrent non-small cell lung cancer after potentially curative therapy: impact on management and prognostic stratification. J Nucl Med. 2001;42(11):1605–1613. [PubMed] [Google Scholar]
- 47.Juweid ME, Stroobants S, Hoekstra OS, et al. Imaging Subcommittee of International Harmonization Project in Lymphoma Use of positron emission tomography for response assessment of lymphoma: consensus of the imaging subcommittee of international harmonization project in lymphoma. J Clin Oncol. 2007;25(5):571–578. doi: 10.1200/JCO.2006.08.2305. [DOI] [PubMed] [Google Scholar]
- 48.Juweid ME, Wiseman GA, Vose JM, et al. Response assessment of aggressive non-Hodgkin’s lymphoma by integrated international workshop criteria and fluorine-18-fluorodeoxyglucose positron emission tomography. J Clin Oncol. 2005;23(21):4652–4661. doi: 10.1200/JCO.2005.01.891. [DOI] [PubMed] [Google Scholar]
- 49.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: evolving considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50(suppl 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Farrell AC, Shnyder SD, Marston G, Coletta PL, Gill JH. Non-invasive molecular imaging for preclinical cancer therapeutic development. Br J Pharmacol. 2013;169(4):719–735. doi: 10.1111/bph.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roda A, Guardigli M. Analytical chemiluminescence and bioluminescence: latest achievements and new horizons. Anal Bioanal Chem. 2012;402(1):69–76. doi: 10.1007/s00216-011-5455-8. [DOI] [PubMed] [Google Scholar]
- 52.Ma X, Liu Z, Yang X, et al. Dual-modality monitoring of tumor response to cyclophosphamide therapy in mice with bioluminescence imaging and small-animal positron emission tomography. Mol Imaging. 2011;10(4):278–283. doi: 10.2310/7290.2010.00041. [DOI] [PubMed] [Google Scholar]
- 53.Keyser KT, Strang CE, Namati E, et al. Optical Imaging of Cancer: Clinical Applications. New York, NY: Springer; 2009. [Google Scholar]
- 54.Workman P, Aboagye EO, Chung YL, et al. Cancer Research UK Pharmacodynamic/Pharmacokinetic Technologies Advisory Committee Minimally invasive pharmacokinetic and pharmacodynamic technologies in hypothesis-testing clinical trials of innovative therapies. J Natl Cancer Inst. 2006;98(9):580–598. doi: 10.1093/jnci/djj162. [DOI] [PubMed] [Google Scholar]
- 55.Ahrens ET, Bulte JW. Tracking immune cells in vivo using magnetic resonance imaging. Nat Rev Immunol. 2013;13(10):755–763. doi: 10.1038/nri3531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Basu S, Kumar R, Ranade R. Assessment of treatment response using PET. PET Clin. 2015;10(1):9–26. doi: 10.1016/j.cpet.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 57.Cheson BD, Fisher RI, Barrington SF, et al. Alliance, Australasian Leukaemia and Lymphoma Group. Eastern Cooperative Oncology Group. European Mantle Cell Lymphoma Consortium. Italian Lymphoma Foundation. European Organisation for Research. Treatment of Cancer/Dutch Hemato-Oncology Group. Grupo Español de Médula Ósea. German High-Grade Lymphoma Study Group. German Hodgkin’s Study Group. Japanese Lymphorra Study Group. Lymphoma Study Association. NCIC Clinical Trials Group. Nordic Lymphoma Study Group. Southwest Oncology Group. United Kingdom National Cancer Research Institute Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014;32(27):3059–3068. doi: 10.1200/JCO.2013.54.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilardi L, Colandrea M, Vassallo S, Travaini LL, Paganelli G. Ipilimumab-induced immunomediated adverse events: possible pitfalls in (18)F-FDG PET/CT interpretation. Clin Nucl Med. 2014;39(5):472–474. doi: 10.1097/RLU.0b013e31828da691. [DOI] [PubMed] [Google Scholar]
- 59.Sogge SM, Fotos JS, Tulchinsky M. Bacillus Calmette–Guerin injections for melanoma immunotherapy: potential for a false-positive PET/CT. Clin Nucl Med. 2015;40(4):368–369. doi: 10.1097/RLU.0000000000000718. [DOI] [PubMed] [Google Scholar]
- 60.Bailey DL, Willowson KP. An evidence-based review of quantitative SPECT imaging and potential clinical applications. J Nucl Med. 2013;54(1):83–89. doi: 10.2967/jnumed.112.111476. [DOI] [PubMed] [Google Scholar]
- 61.Seo Y, Mari C, Hasegawa BH. Technological development and advances in single-photon emission computed tomography/computed tomography. Semin Nucl Med. 2008;38(3):177–198. doi: 10.1053/j.semnuclmed.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slomka PJ, Pan T, Berman DS, Germano G. Advances in SPECT and PET hardware. Prog Cardiovasc Dis. 2015;57(6):566–578. doi: 10.1016/j.pcad.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 63.Sartor O, Eisenberger M, Kattan MW, Tombal B, Lecouvet F. Unmet needs in the prediction and detection of metastases in prostate cancer. Oncologist. 2013;18(5):549–557. doi: 10.1634/theoncologist.2013-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanghera B, Wong WL, Sonoda LI, et al. FLT PET-CT in evaluation of treatment response. Indian J Nucl Med. 2014;29(2):65–73. doi: 10.4103/0972-3919.130274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan Y, Liang J, Liu D, et al. F-FLT PET/CT imaging in a Wister rabbit inflammation model. Exp Ther Med. 2014;8(1):69–72. doi: 10.3892/etm.2014.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nameth DJ, Goldman JW, Wolf BR, et al. Evaluating causes of screen failure (SF) in non-small cell lung cancer (NSCLC) clinical trials requiring specific biomarker (BioM) results for enrollment. ASCO Meet Abstr. 2015;33(15_suppl):7517. [Google Scholar]
- 68.Glaudemans AW, Bonanno E, Galli F, et al. In vivo and in vitro evidence that (9)(9)mTc-HYNIC-interleukin-2 is able to detect T lymphocytes in vulnerable atherosclerotic plaques of the carotid artery. Eur J Nucl Med Mol Imaging. 2014;41(9):1710–1719. doi: 10.1007/s00259-014-2764-0. [DOI] [PubMed] [Google Scholar]
- 69.Namavari M, Chang YF, Kusler B, Yaghoubi S, Mitchell BS, Gambhir SS. Synthesis of 2′-deoxy-2′-[18F]fluoro-9-beta-D-arabinofuranosylguanine: a novel agent for imaging T-cell activation with PET. Mol Imaging Biol. 2011;13(5):812–818. doi: 10.1007/s11307-010-0414-x. [DOI] [PubMed] [Google Scholar]
- 70.Radu CG, Shu CJ, Nair-Gill E, et al. Molecular imaging of lymphoid organs and immune activation by positron emission tomography with a new [18F]-labeled 2′-deoxycytidine analog. Nat Med. 2008;14(7):783–788. doi: 10.1038/nm1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yaghoubi SS, Jensen MC, Satyamurthy N, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6(1):53–58. doi: 10.1038/ncponc1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.den Hollander MW, Bensch F, Glaudemans AW, et al. TGF-beta antibody uptake in recurrent high-grade glioma imaged with 89Zr-fresolimumab PET. J Nucl Med. 2015;56(9):1310–1314. doi: 10.2967/jnumed.115.154401. [DOI] [PubMed] [Google Scholar]
- 73.Iv M, Telischak N, Feng D, Holdsworth SJ, Yeom KW, Daldrup-Link HE. Clinical applications of iron oxide nanoparticles for magnetic resonance imaging of brain tumors. Nanomedicine. 2015;10(6):993–1018. doi: 10.2217/nnm.14.203. [DOI] [PubMed] [Google Scholar]
- 74.Lamberts LE, Williams SP, Terwisscha van Scheltinga AG, et al. Antibody positron emission tomography imaging in anticancer drug development. J Clin Oncol. 2015;33(13):1491–1504. doi: 10.1200/JCO.2014.57.8278. [DOI] [PubMed] [Google Scholar]
- 75.Knowles SM, Wu AM. Advances in immuno-positron emission tomography: antibodies for molecular imaging in oncology. J Clin Oncol. 2012;30(31):3884–3892. doi: 10.1200/JCO.2012.42.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tavare R, McCracken MN, Zettlitz KA, et al. Engineered antibody fragments for immuno-PET imaging of endogenous CD8+ T cells in vivo. Proc Natl Acad Sci U S A. 2014;111(3):1108–1113. doi: 10.1073/pnas.1316922111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Malviya G, Galli F, Sonni I, Pacilio M, Signore A. Targeting T and B lymphocytes with radiolabelled antibodies for diagnostic and therapeutic applications. Q J Nucl Med Mol Imaging. 2010;54(6):654–676. [PubMed] [Google Scholar]
- 78.Tavare R, McCracken MN, Zettlitz KA, et al. Immuno-PET of murine T cell reconstitution postadoptive stem cell transplantation using anti-CD4 and anti-CD8 Cys-diabodies. J Nucl Med. 2015;56(8):1258–1264. doi: 10.2967/jnumed.114.153338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Holland JP, Divilov V, Bander NH, Smith-Jones PM, Larson SM, Lewis JS. 89Zr-DFO-J591 for immunoPET of prostate-specific membrane antigen expression in vivo. J Nucl Med. 2010;51(8):1293–1300. doi: 10.2967/jnumed.110.076174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Higashikawa K, Yagi K, Watanabe K, et al. 64Cu-DOTA-anti-CTLA-4 mAb enabled PET visualization of CTLA-4 on the T-cell infiltrating tumor tissues. PLoS One. 2014;9(11):e109866. doi: 10.1371/journal.pone.0109866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rashidian M, Keliher EJ, Bilate AM, et al. Noninvasive imaging of immune responses. Proc Natl Acad Sci U S A. 2015;112(19):6146–6151. doi: 10.1073/pnas.1502609112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–3284. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 83.Zheleznyak A, Ikotun OF, Dimitry J, Frazier WA, Lapi SE. Imaging of CD47 expression in xenograft and allograft tumor models. Mol Imaging. 2013;12(8):1–10. doi: 10.2310/7290.2013.00069. [DOI] [PubMed] [Google Scholar]
- 84.Heskamp S, Hobo W, Molkenboer-Kuenen JD, et al. Noninvasive imaging of tumor PD-L1 expression using radiolabeled anti-PD-L1 antibodies. Cancer Res. 2015;75(14):2928–2936. doi: 10.1158/0008-5472.CAN-14-3477. [DOI] [PubMed] [Google Scholar]
- 85.Bachawal SV, Jensen KC, Wilson KE, Tian L, Lutz AM, Willmann JK. Breast cancer detection by B7-H3-targeted ultrasound molecular imaging. Cancer Res. 2015;75(12):2501–2509. doi: 10.1158/0008-5472.CAN-14-3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kang FB, Wang L, Li D, Zhang YG, Sun DX. Hepatocellular carcinomas promote tumor-associated macrophage M2-polarization via increased B7-H3 expression. Oncol Rep. 2015;33(1):274–282. doi: 10.3892/or.2014.3587. [DOI] [PubMed] [Google Scholar]
- 87.Wang L, Kang FB, Shan BE. B7-H3-mediated tumor immunology: friend or foe? Int J Cancer. 2014;134(12):2764–2771. doi: 10.1002/ijc.28474. [DOI] [PubMed] [Google Scholar]
- 88.Wang L, Zhang Q, Chen W, et al. B7-H3 is overexpressed in patients suffering osteosarcoma and associated with tumor aggressiveness and metastasis. PLoS One. 2013;8(8):e70689. doi: 10.1371/journal.pone.0070689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Leech JM, Sharif-Paghaleh E, Maher J, et al. Whole-body imaging of adoptively transferred T cells using magnetic resonance imaging, single photon emission computed tomography and positron emission tomography techniques, with a focus on regulatory T cells. Clin Exp Immunol. 2013;172(2):169–177. doi: 10.1111/cei.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parente-Pereira AC, Burnet J, Ellison D, et al. Trafficking of CAR-engineered human T cells following regional or systemic adoptive transfer in SCID beige mice. J Clin Immunol. 2011;31(4):710–718. doi: 10.1007/s10875-011-9532-8. [DOI] [PubMed] [Google Scholar]
- 91.Bhatnagar P, Li Z, Choi Y, et al. Imaging of genetically engineered T cells by PET using gold nanoparticles complexed to copper-64. Integr Biol (Camb) 2013;5(1):231–238. doi: 10.1039/c2ib20093g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dobrenkov K, Olszewska M, Likar Y, et al. Monitoring the efficacy of adoptively transferred prostate cancer-targeted human T lymphocytes with PET and bioluminescence imaging. J Nucl Med. 2008;49(7):1162–1170. doi: 10.2967/jnumed.107.047324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eissenberg LG, Rettig MP, Ritchey JK, et al. [(18)F]FHBG PET/CT imaging of CD34-TK75 transduced donor T cells in relapsed allogeneic stem cell transplant patients: safety and feasibility. Mol Ther. 2015;23(6):1110–1122. doi: 10.1038/mt.2015.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dubey P, Su H, Adonai N, et al. Quantitative imaging of the T cell antitumor response by positron-emission tomography. Proc Natl Acad Sci U S A. 2003;100(3):1232–1237. doi: 10.1073/pnas.0337418100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ponomarev V, Doubrovin M, Lyddane C, et al. Imaging TCR-dependent NFAT-mediated T-cell activation with positron emission tomography in vivo. Neoplasia. 2001;3(6):480–488. doi: 10.1038/sj.neo.7900204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Doubrovin MM, Doubrovina ES, Zanzonico P, Sadelain M, Larson SM, O’Reilly RJ. In vivo imaging and quantitation of adoptively transferred human antigen-specific T cells transduced to express a human norepinephrine transporter gene. Cancer Res. 2007;67(24):11959–11969. doi: 10.1158/0008-5472.CAN-07-1250. [DOI] [PubMed] [Google Scholar]
- 97.Soloviev D, Lewis D, Honess D, Aboagye E. [(18)F]FLT: an imaging biomarker of tumour proliferation for assessment of tumour response to treatment. Eur J Cancer. 2012;48(4):416–424. doi: 10.1016/j.ejca.2011.11.035. [DOI] [PubMed] [Google Scholar]
- 98.Chalkidou A, Landau DB, Odell EW, Cornelius VR, O’Doherty MJ, Marsden PK. Correlation between Ki-67 immunohistochemistry and 18F-fluorothymidine uptake in patients with cancer: a systematic review and meta-analysis. Eur J Cancer. 2012;48(18):3499–3513. doi: 10.1016/j.ejca.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 99.Schoder H, Larson SM. Positron emission tomography for prostate, bladder, and renal cancer. Semin Nucl Med. 2004;34(4):274–292. doi: 10.1053/j.semnuclmed.2004.06.004. [DOI] [PubMed] [Google Scholar]
- 100.Specht JM, Tam SL, Kurland BF, et al. Serial 2-[18F] fluoro-2-deoxy-D-glucose positron emission tomography (FDG-PET) to monitor treatment of bone-dominant metastatic breast cancer predicts time to progression (TTP) Breast Cancer Res Treat. 2007;105(1):87–94. doi: 10.1007/s10549-006-9435-1. [DOI] [PubMed] [Google Scholar]
- 101.Dotti G, Tian M, Savoldo B, et al. Repetitive noninvasive monitoring of HSV1-tk-expressing T cells intravenously infused into nonhuman primates using positron emission tomography and computed tomography with 18F-FEAU. Mol Imaging. 2009;8(4):230–237. [PMC free article] [PubMed] [Google Scholar]
- 102.Bhatnagar P, Alauddin M, Bankson JA, et al. Tumor lysing genetically engineered T cells loaded with multi-modal imaging agents. Sci Rep. 2014;4:4502. doi: 10.1038/srep04502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Youniss FM, Sundaresan G, Graham LJ, et al. Near-infrared imaging of adoptive immune cell therapy in breast cancer model using cell membrane labeling. PLoS One. 2014;9(10):e109162. doi: 10.1371/journal.pone.0109162. [DOI] [PMC free article] [PubMed] [Google Scholar]