

Abstract
The presence of apoptotic cells and loss of extracellular matrix (ECM) are common characteristics of degenerated cartilage endplates (CEPs). In addition, therapeutic efficacy is hampered by an incomplete understanding regarding the mechanisms underlying CEP homeostasis and degeneration. The CCN proteins have recently emerged as important regulators of cell-ECM interactions, and have been identified as key mediators of nucleus pulposus ECM composition and tissue homeostasis. However, whether CCN3 is associated with CEP homeostasis has yet to be elucidated. The present study aimed to investigate the effects of CCN3 on the apoptosis and ECM synthesis of CEP cells cultured under serum deprivation. Rat CEP cells were confirmed to be of the chondrocytic phenotype by toluidine blue staining. The mRNA expression levels of CCN3 were markedly increased, and a dose-dependent increase of apoptotic rate was detected under serum deprivation conditions following treatment with recombinant CCN3, whereas CCN3 did not exert a proapoptotic effect on cells cultured under normal conditions. Furthermore, CCN3-treated cells exhibited a decrease in the expression levels of aggrecan and collagen II in both groups. These results suggested that CCN3 may act as a regulator, rather than an initiator, of serum deprivation-induced cellular apoptosis, and that CCN3 has a catabolic effect on the mediation of ECM synthesis under both normal and serum deprivation conditions. Therefore, CCN3 may represent a novel therapeutic target for the prevention of CEP degeneration.
Keywords: intervertebral disc degeneration, CCN3, apoptosis, serum deprivation, cartilage endplate, extracellular matrix
Introduction
Intervertebral disc (IVD) degeneration has a critical role in the pathogenesis of spinal disorders. In addition, IVD degeneration is the main cause of lower back pain, and presents a serious socioeconomic burden (1,2). The treatment and prevention of degenerative disc disease is hampered by a limited understanding regarding the mechanisms that regulate IVD development, maintenance and degeneration. The cartilage endplate (CEP) is the predominant source of nutrients for the IVD (3,4). CEP degeneration markedly decreases the biomechanical integrity and nutrition of IVDs, resulting in a breakdown of the metabolic equilibrium of the extracellular matrix (ECM) and subsequent acceleration of disc degeneration (5–8). The prevention of CEP degeneration is a potential therapeutic strategy for maintaining IVD health and preventing spondylopathy. It is well known that the presence of apoptotic cells and loss of ECM are characteristics of degenerated CEP; however, much remains to be elucidated regarding the regulation of matrix protein biosynthesis in cells of both the normal and degenerated disc.
The CCN family proteins, also known as extracellular proteins, are dynamically expressed and have critical roles in cardiovascular and skeletal development, injury repair, fibrotic disease and cancer (9,10). The acronym CCN is derived from the first three members of the family described, namely cysteine-rich 61/CCN1, connective tissue growth factor/CCN2, and nephroblastoma overexpressed/CCN3. CCN proteins interact with various types of molecules, including growth factors, ECM components and cell-surface receptors, in order to regulate cell adhesion, migration, proliferation, gene expression, differentiation, apoptosis and survival (11). The expression of CCN3 has previously been assessed in notochord neonatal and mature rat IVDs, the results of which indicated that CCN3 is involved in IVD development (12). In addition, recombinant CCN3 (rCCN3) has been demonstrated to act as a negative regulator; when nucleus pulposus (NP) cells were treated with exogenous CCN3 in vitro, cell proliferation was reduced, and the expression levels of ECM components were decreased (12). A previous study reported that CCN3 expression was markedly upregulated in CCN2 deletion mice, which was associated with impaired development of IVDs and markedly accelerated age-associated IVD degeneration (13). The antiproliferative activities of CCN3 have also been documented in embryonic fibroblasts, chondrocytes, vascular smooth muscle cells, osteogenic mesenchymal cells and NP cells (12,14,15). Furthermore, CCN proteins are able to interact with tumor necrosis factor (TNF) family cytokines to promote cell apoptosis (16–18). CCN3 can unmask the cytotoxic potential of TNFα and lymphotoxin-α (LTα), and promote the apoptotic activity of Fas ligand (FasL) and TNF-related apoptosis-inducing ligand (TRAIL) in normal fibroblasts (18). In addition, CCN2 can increase the expression of ECM proteins (12), whereas CCN3 appears to be expressed reciprocally and to act antagonistically to CCN2 (13,14,19). The most prominent phenotype of CCN3 knockout mice involves enhanced chondrogenesis and osteogenesis (20), whereas CCN2 mutant mice exhibit severe chondrodysplasia (21). However, whether CCN3 is associated with CEP homeostasis has yet to be elucidated.
In the present study, a serum deprivation-induced experimental model of apoptosis was used to explore the biological effects of CCN3 on CEP cells (22), which mimicked the low nutrient conditions in IVDs.
Materials and methods
Cell isolation and culture
All animal experiments were approved by the Ethics Committee on Animal Experiments of Fudan University (Shanghai, China). Two male Sprague-Dawley rats (age, ~12 weeks; weight, 400 g) were used in the present study and were supplied by the Chinese Academy of Sciences (Shanghai, China). Animals were housed with free access to food and water, under a 12-h light/dark cycle with a constant temperature (20–23°C) and humidity (55±5%). The rats were euthanized by cervical dislocation following anesthesia with pelltobarbitalum natricum (50 mg/kg; Shanghai New Asia Pharmaceutical Co., Ltd., Shanghai, China). All lumbar spines were obtained within 1 h of sacrifice, and the discs were carefully dissected under a microscope, in order to obtain only the CEP, which was minced into small pieces (<0.3 mm3) under aseptic conditions. To isolate the chondrocytes, the tissues were sequentially treated with 0.25% trypsin (Sigma-Aldrich, St. Louis, MO, USA) at 37°C for 120 min followed by 0.02% collagenase (Sigma-Aldrich) at 37°C for 24 h. Following enzymatic digestion, the tissues were filtered through a 70-µm mesh cell strainer (Beyotime Institute of Biotechnology, Nantong, China) and were washed with phosphate-buffered saline (PBS). Subsequently, the cells were released from the matrix by centrifugation at 200 × g for 5 min at room temperature, seeded into six-well plates at 2×104 cells/well, and maintained in Dulbecco's modified Eagle medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin (Beyotime Institute of Biotechnology) at 37°C in a humidified incubator containing 5% CO2. Primary chondrocytes were maintained in a high-density monolayer culture for 1 week, and toluidine blue staining (Shanghai Haoran Biotechnology Co., Ltd., Shanghai, China) was used to confirm the chondrocytic phenotype of the cells. The cells were then trypsinized and subcultured into six-well plates, which were used in the subsequent experiments as secondary cells.
Detection of apoptosis
After being cultured in either 1 or 10% FBS for 24 h, the cells were treated with a concentration gradient of recombinant CCN3 (rCCN3; Sino Biological Inc., Beijing, China; 0, 0.5, 1 and 2 µg/ml) for 24 h. Subsequently, apoptosis was determined by staining with annexin V fluorescein isothiocyanate (FITC) and propidium iodide (PI) (BD Pharmingen, San Diego, CA, USA), according to the manufacturer's protocol. To quantify apoptosis, the cells were washed with cold PBS and suspended in binding buffer. The cells were then stained with 5 µl annexin V-FITC and 5 µl PI at 4°C for 15 min, and were analyzed using FACScan flow cytometry (BD Biosciences, San Jose, CA, USA) (22). The cells were quantified as follows: i) Annexin V negative/PI negative (viable cells); ii) annexin V positive/PI negative (cells in the initial stages of apoptosis); iii) annexin V positive/PI positive (cells in the advanced stages of apoptosis); and iv) annexin V negative/PI positive (necrotic cells).
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to detect the mRNA expression levels of CCN3 in cells cultured with 1 or 10% FBS at 24, 36 and 48 h. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal standard control. In addition, after treating cells with 1 µg/ml rCCN3, the mRNA expression levels of aggrecan, collagen II and CCN2 were assessed by RT-PCR at 8 and 24 h. Total RNA was extracted from the cells using TRIzol® reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. Single-strand cDNA templates were prepared from 1 µg total RNA using the RT-for-PCR kit (Invitrogen; Thermo Fisher Scientific, Inc.), according to the manufacturer's instructions. Specific cDNAs were subsequently amplified by PCR using the following primers (Shanghai R&S Biotechnology Co., Ltd., Shanghai, China): Aggrecan, forward CAGAAAAACAGACTGAATGGGA, reverse GCAAGTGAAGGTGTGTATGGC; collagen II, forward GCAAGAATCCCGCTCGCA, reverse TGGGTTGGGGTAGACGCA; CCN3, forward GCCCTTCCAGCCTACAGACC, reverse GGTGGATGGATTTCAGGGACT; CCN2, forward GCTAATGGTGGACCGCAA, reverse CCAAGGTAACGCCAGGAAT; and GAPDH, CCCCAATGTATCCGTTGTG, and reverse CTCAGTGTAGCCCAGGATGC. PCR amplification from cDNA was performed using the Takara TP800 Thermal Cycler Dice (Takara Bio Inc., Shiga, Japan) in a final volume of 20 µl [2X SYBR Green mix (10 µl; Toyobo Co., Ltd., Tokyo, Japan) 1 µl primer mix, 1 µl template DNA and 8 µl diethypyrocarbonate water] under the following cycling conditions: Initial denaturation at 95°C for 2 min, denaturation at 95°C for 15 sec, annealing at 59°C for 20 sec, elongation at 72°C for 20 sec and final extension at 72°C for 10 min; the optimal cycle number was 40 cycles. PCR products were subjected to amplification curve analysis and were quantified using SYBR Green (Invitrogen; Thermo Fisher Scientific, Inc.). The data were normalized to GAPDH and were presented as ΔΔCt (22).
Western blot analysis
Cells cultured in 1% FBS were treated with 0 or 2 µg/ml rCCN3 for 24 h, and control group cells were cultured in 10% FBS only. The protein expression levels of Fas, B-cell lymphoma 2 (Bcl-2) and Bcl-2-associated X protein (Bax) were detected in the three groups by western blot analysis, according to the manufacturer's protocol. Total protein was extracted from the cells using protein loading buffer, and the total protein concentration was determined by bicinchoninic acid assay (Sigma-Aldrich). Protein extracts were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). The membranes were then blocked with 5% non-fat dry milk in Tris-buffered saline with 0.1% Tween (TBST) for 1 h at 37°C, and incubated overnight at 4°C in TBST with polyclonal goat anti-Fas (dilution 1:200; Sigma-Aldrich; cat. no. SAB2501317), rabbit anti-Bcl-2 (dilution 1:500; cat. no. AB112; Beyotime Institute of Biotechnology), mouse anti-Bax (dilution 1:500; cat. no. AB026; Beyotime Institute of Biotechnology) and mouse monoclonal anti-β-actin (dilution 1:2,000; cat. no. AF0003; Beyotime Institute of Biotechnology) antibodies. Following further incubation with a horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (dilution 1:5,000; cat. no. A0208; Beyotime Institute of Biotechnology) for 1 h at room temperature, the membranes were treated with Electrochemiluminescence Plus (Tanon Science & Technology Co., Ltd., Shanghai, China), according to the manufacturer's protocol. β-actin was used as a control to verify equal protein loading.
Statistical analysis
All measurements were carried out using the same instrument under the same experimental conditions, and were independently performed at least three times to ensure consistency. Statistical analyses were performed using SPSS 16.0 software (SPSS, Inc., Chicago, IL, USA). Data are expressed as the mean ± standard deviation. Significant differences were analyzed by one-way analysis of variance among the groups and Student's t-test. P<0.05 was considered to indicate a statistically significant difference.
Results
Cells cultured from the CEP maintain a chondrocytic phenotype
Toluidine blue staining was used to confirm the phenotype of cells cultured from the CEP. The cells were observed to be viable and possessed characteristic features of chondrocytes, as evidenced by typical cell morphology and ECM staining (Fig. 1A).
Figure 1.
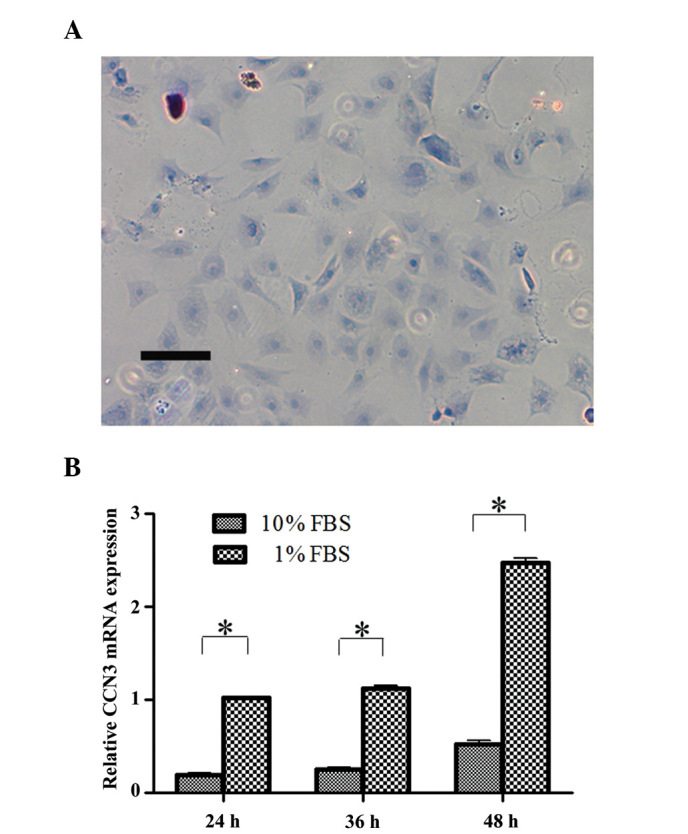
Cell identification and CCN3 expression in cells cultured under serum deprivation. (A) Typical chondrocytic phenotype was confirmed by toluidine blue staining (magnification, ×100). (B) Quantitative analysis of CCN3 mRNA expression, as determined by reverse transcription-quantitative polymerase chain reaction. CCN3 was markedly increased in cells under serum deprivation conditions at 24, 36 and 48 h. Data are presented as the mean ± standard deviation. *P<0.05. Scale bars represent 25 mm. FBS, fetal bovine serum.
Serum deprivation increases CCN3 expression
To investigate whether serum deprivation regulates CCN3 expression, RT-qPCR was used to quantify the mRNA expression levels of CCN3 in cells cultured in 1 or 10% FBS. As shown in Fig. 1B, the mRNA expression levels of CCN3 in the serum deprivation group were markedly increased, as compared with in the control group at 24, 36 and 48 h. A marked increase was detected at 48 h in the serum deprivation group; however, no obvious difference in the expression of CCN3 was detected in the serum-deprived cells between 24 and 36 h.
CCN3 is involved in serum deprivation-induced cellular apoptosis
To determine whether exogenous rCCN3 had an effect on the apoptosis of CEP cells, the apoptotic rate of cells stained with annexin V-FITC and PI were quantitatively assessed following treatment with a concentration gradient of rCCN3 for 24 h. As shown in Fig. 2, a dose-dependent increase of apoptosis was detected in the 1% FBS groups (13.28±0.52, 15.86±0.58, 18.34±0.63 and 23.78±0.64%, respectively), whereas rCCN3 did not exert a proapoptotic effect on cells cultured in 10% FBS (4.22±0.24, 4.35±0.34, 4.26±0.30 and 4.75±0.42%, respectively).
Figure 2.

Evaluation of the apoptosis of cartilage endplate cells following treatment with various doses of recombinant (r)CCN3 for 24 h. (A) Representative graphs of flow cytometric analysis. (B) Dose-dependent increase in apoptosis was detected in the 1% fetal bovine serum (FBS) groups, whereas rCCN3 had no proapoptotic effect on cells cultured in 10% FBS. The results are presented as the percentage of cell apoptosis. Data are presented as the mean ± standard devaition. *P<0.05. FITC, fluorescein isothiocyanate.
To explore the mechanism underlying the proapoptotic effects of rCCN3, Fas, Bax and Bcl-2 expression levels were detected by western blot analysis in cells cultured under 1 or 10% FBS with or without rCCN3 (Fig. 3). The results demonstrated that Fas expression was markedly upregulated following serum deprivation with or without rCCN3 at 24 h, and no obvious variation was detected following treatment with 2 µg/ml rCCN3. Furthermore, Bax expression was also markedly increased, whereas Bcl-2 expression levels were decreased in the cells under serum deprivation conditions, as compared with the cells in the 10% FBS group. Notably, the results also demonstrated that Bax and Bcl-2 were affected by exogenous rCCN3 treatment. The differences in Bax and Bcl-2 were investigated and found to be consistent with the effects on apoptosis following treatment with rCCN3. These results suggest that rCCN3 may regulate cellular apoptosis under serum deprivation conditions, and the mitochondrial apoptotic pathway may be responsible for increased cellular apoptosis under serum deprivation conditions.
Figure 3.
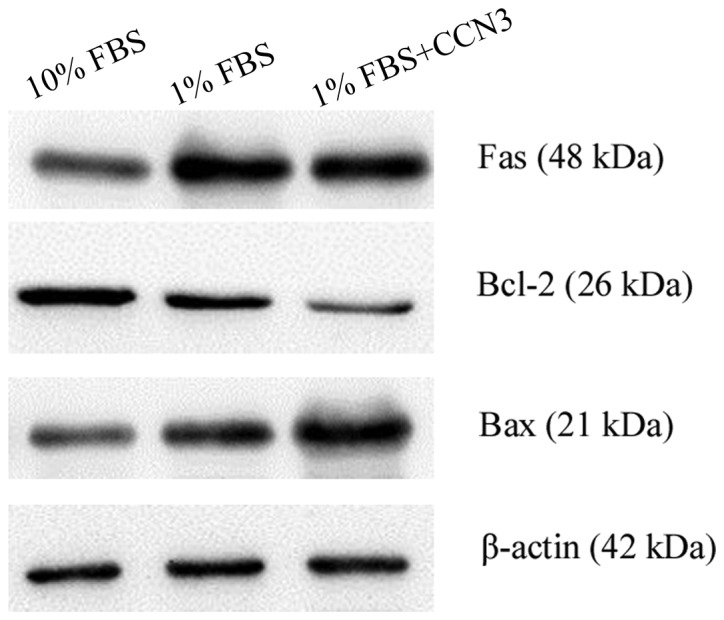
Expression of apoptosis-associated proteins, as determined by western blotting. Fas and B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax) expression levels were markedly increased under serum deprivation conditions with or without recombinant (r)CCN3 treatment, whereas Bcl-2 expression was decreased. A markedly different trend in Bax and Bcl-2 expression was detected following treatment with rCCN3. FBS, fetal bovine serum.
CCN3 modulates the expression of critical ECM genes
Finally, the effects of rCCN3 treatment on the expression levels of critical ECM genes were determined by RT-qPCR in CEP cells cultured under normal or serum deprivation conditions. Treatment with rCCN3 decreased the expression levels of aggrecan and collagen II under both normal and serum deprivation conditions at 8 and 24 h, and a more obvious difference was detected under serum deprivation conditions (Fig. 4A and B). Furthermore, the mRNA expression levels of CCN2 were decreased following treatment with rCCN3, whereas a decrease in CCN2 expression was not as apparent in the 1% FBS group (Fig. 4C). These findings indicate that CCN2 expression may also be influenced by serum deprivation.
Figure 4.
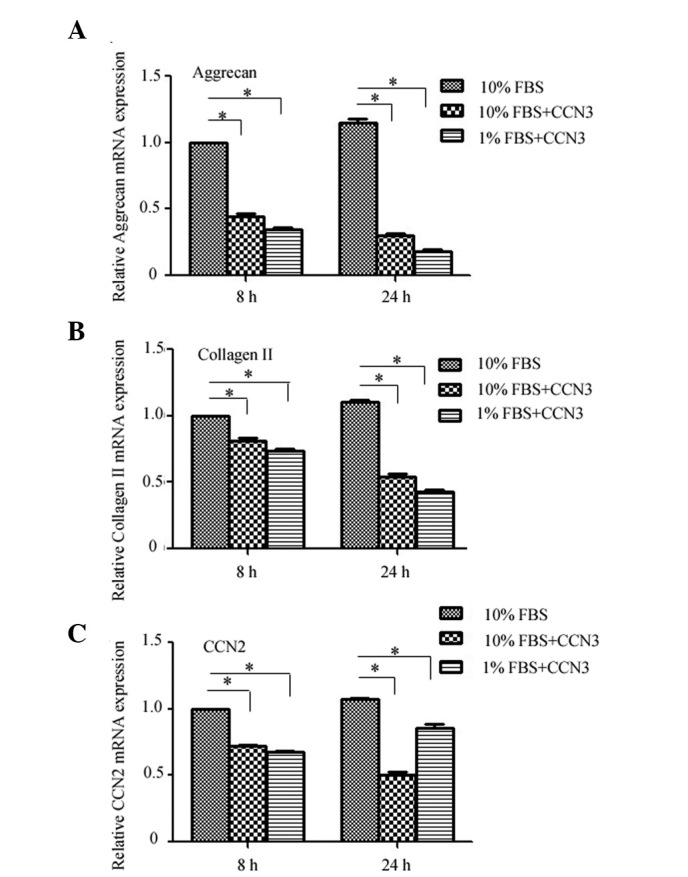
Expression levels of critical extracellular matrix genes. (A) Aggrecan, (B) collagen II and (C) CCN2 mRNA expression levels were markedly suppressed following treatment with recombinant CCN3 under both normal and serum deprivation conditions, as determined by reverse transcription-quantitative polymerase chain reaction. Data are presented as the mean ± standard deviation. *P<0.05. FBS, fetal bovine serum.
Discussion
The CCN family of proteins has emerged as dynamically expressed, multifunctional regulators of cellular behavior, which maintain tissue homeostasis (11). Previous studies have demonstrated that the CCN proteins are key mediators in the regulation of IVD cell behavior and disc tissue homeostasis (13,23–25). Bedore et al (13) revealed that deletion of CCN2, accompanied by upregulated CCN3 expression, induced a decrease in aggrecan and type II collagen expression in NP cells, resulting in accelerated IVD degeneration. Transforming growth factor-β may also modulate CCN expression in NP cells, thus inhibiting CCN3 expression and increasing CCN2 expression, in order to promote ECM deposition (12). Of the CCN family, CCN2 appears to possess anabolic expression patterns and activities, whereas CCN3 appears to be expressed reciprocally and to act antagonistically to CCN2 (12). However, whether CCN3 may regulate CEP cell behavior has yet to be explored.
The expression of CCN genes is particularly sensitive to environmental perturbations, including the availability of growth factors, hormones, cytokines, and exposure to oxygen deprivation, UV and mechanical forces (26,27). However, whether serum deprivation may regulate CCN3 expression has yet to be reported. In the present study, rat CEP cells were successfully isolated, cultured and confirmed to be of the chondrocytic phenotype by toluidine blue staining. The mRNA expression levels of CCN3 were markedly upregulated in response to serum deprivation, particularly at 48 h. These results indicated that CCN3 expression is sensitive to serum deprivation and that the low nutrition condition of IVDs may regulate CCN3 expression, in order to participate in the process of degeneration.
Western blot analysis demonstrated that high expression levels of Fas could be detected under serum deprivation conditions, with or without CCN3 treatment. A previous study (16) reported that CCN proteins are able to synergize with TNF family cytokines, including TNFα, LTα, FasL and TRAIL, and enhance their cytotoxicity by binding their receptors to promote the accumulation of reactive oxygen species (ROS). In the present study, CCN3 and Fas expression were simultaneously induced by serum deprivation, allowing them the opportunity to interact with each other. As a result, it may be hypothesized that CCN3 acts synergistically with Fas/FasL to regulate serum deprivation-induced cellular apoptosis.
To determine whether exogenous rCCN3 was able to promote the serum deprivation-induced apoptosis of CEP cells, cells cultured under serum deprivation conditions were treated with a dose-dependent concentration gradient of rCCN3. Flow cytometric analysis detected a dose-dependent increase in apoptosis in serum-deprived cells, whereas CCN3 did not exert a proapoptotic effect on normal serum-cultured cells. These results suggested that CCN3 can only promote cellular apoptosis induced by serum deprivation, and cannot trigger apoptosis, which is consistent with the findings on TNFα and FasL-induced fibroblast apoptosis (17,18). The present study demonstrated that CCN3 may influence CEP cell viability, and that serum deprivation-induced abnormal expression of CCN3 may promote excess cellular apoptosis and subsequent CEP degeneration.
The present study also detected the expression levels of Bax and Bcl-2 by western blot analysis following treatment with exogenous rCCN3. Bax expression was markedly increased, whereas Bcl-2 expression was decreased under serum deprivation conditions with or without rCCN3 treatment. These results indicated that the mitochondrial apoptotic pathway was involved in cellular apoptosis. Notably, the alterations in Bax and Bcl-2 expression following treatment with rCCN3 were more obvious, thus suggesting that CCN3 may engage in the cell apoptosis process by regulating mitochondrial apoptosis-associated proteins. Furthermore, Fas expression was detected at a high level under serum deprivation conditions, and CCN3 was shown to synergize with TNF family cytokines in order to promote cellular apoptosis by recruiting ROS to increase cytochrome c release via an imbalance in Bax and Bcl-2 levels (16). As a result, CCN3 may synergize with cytokines and their receptors to promote serum deprivation-induced apoptosis, as with Fas/FasL.
The effects of rCCN3 on the expression levels of critical ECM genes were also examined. Aggrecan and type II collagen are the most important components of endplate ECM, and a decrease in these factors will result in the compression or calcification of ECM, followed by accelerated disc degeneration (28). Following treatment with rCCN3, the expression levels of aggrecan and type II collagen were inhibited not only under normal conditions but also under serum deprivation conditions, and the decrease was more obvious under serum deprivation conditions. These findings suggested that CCN3 was a harmful factor that acted antagonistically to aggrecan and type II collagen expression, which is similar to previous in vitro and in vivo findings in NP cells (12). The reciprocal effects of CCN2 were also investigated following treatment with rCCN3. The decreases in CCN2 expression in the 1 and 10% FBS groups were consistent with the study results associated with other tissues (11,12). However, the decrease in CCN2 expression was not as apparent under serum deprivation, which may imply that CCN2 expression was also affected by serum deprivation; however, this mechanism requires further exploration.
In conclusion, the results of the present study suggested that CCN3 may be associated with serum deprivation-induced CEP cell apoptosis as a regulator, rather than an initiator, and that CCN3 has a catabolic effect on the modulation of ECM expression. Therefore, CCN3 may represent a novel therapeutic target for the prevention of CEP degeneration.
Acknowledgments
The authors thank American Journal Experts for English language editing. The present study was supported by the Jinshan District (and Shanghai Municipal) Health Bureau (grant nos. 2012-694 and 2012-341).
References
- 1.Hoy D, Brooks P, Woolf A, Blyth F, March L, Bain C, Baker P, Smith E, Buchbinder R. Assessing risk of bias in prevalence studies: Modification of an existing tool and evidence of interrater agreement. J Clin Epidemiol. 2012;65:934–939. doi: 10.1016/j.jclinepi.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 2.de Schepper EI, Damen J, van Meurs JB, Ginai AZ, Popham M, Hofman A, Koes BW, Bierma-Zeinstra SM. The association between lumbar disc degeneration and low back pain: The influence of age, gender, and individual radiographic features. Spine (Phila Pa 1976) 2010;35:531–536. doi: 10.1097/BRS.0b013e3181aa5b33. [DOI] [PubMed] [Google Scholar]
- 3.Magnier C, Boiron O, Wendling-Mansuy S, Chabrand P, Deplano V. Nutrient distribution and metabolism in the intervertebral disc in the unloaded state: A parametric study. J Biomech. 2009;42:100–108. doi: 10.1016/j.jbiomech.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 4.Raj PP. Intervertebral disc: Anatomy-physiology-pathophysiology-treatment. Pain Pract. 2008;8:18–44. doi: 10.1111/j.1533-2500.2007.00171.x. [DOI] [PubMed] [Google Scholar]
- 5.Grunhagen T, Shirazi-Adl A, Fairbank JC, Urban JP. Intervertebral disk nutrition: A review of factors influencing concentrations of nutrients and metabolites. Orthop Clin North Am. 2011;42:465–477. doi: 10.1016/j.ocl.2011.07.010. [DOI] [PubMed] [Google Scholar]
- 6.Shirazi-Adl A, Taheri M, Urban JP. Analysis of cell viability in intervertebral disc: Effect of endplate permeability on cell population. J Biomech. 2010;43:1330–1336. doi: 10.1016/j.jbiomech.2010.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Ding F, Shao ZW, Xiong LM. Cell death in intervertebral disc degeneration. Apoptosis. 2013;18:777–785. doi: 10.1007/s10495-013-0839-1. [DOI] [PubMed] [Google Scholar]
- 8.Kang R, Li H, Ringgaard S, Rickers K, Sun H, Chen M, Xie L, Bünger C. Interference in the endplate nutritional pathway causes intervertebral disc degeneration in an immature porcine model. Int Orthop. 2014;38:1011–1017. doi: 10.1007/s00264-014-2319-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubota S, Takigawa M. The CCN family acting throughout the body: Recent research developments. Biomol Concepts. 2013;4:477–494. doi: 10.1515/bmc-2013-0018. [DOI] [PubMed] [Google Scholar]
- 10.Chen CC, Lau LF. Functions and mechanisms of action of CCN matricellular proteins. Int J Biochem Cell Biol. 2009;41:771–783. doi: 10.1016/j.biocel.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jun JI, Lau LF. Taking aim at the extracellular matrix: CCN proteins as emerging therapeutic targets. Nat Rev Drug Discov. 2011;10:945–963. doi: 10.1038/nrd3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tran CM, Smith HE, Symes A, Rittié L, Perbal B, Shapiro IM, Risbud MV. Transforming growth factor β controls CCN3 expression in nucleus pulposus cells of the intervertebral disc. Arthritis Rheum. 2011;63:3022–3031. doi: 10.1002/art.30468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bedore J, Sha W, McCann MR, Liu S, Leask A, Séguin CA. Impaired intervertebral disc development and premature disc degeneration in mice with notochord-specific deletion of CCN2. Arthritis Rheum. 2013;65:2634–2644. doi: 10.1002/art.38075. [DOI] [PubMed] [Google Scholar]
- 14.Ren Z, Hou Y, Ma S, Tao Y, Li J, Cao H, Ji L. Effects of CCN3 on fibroblast proliferation, apoptosis and extracellular matrix production. Int J Mol Med. 2014;33:1607–1612. doi: 10.3892/ijmm.2014.1735. [DOI] [PubMed] [Google Scholar]
- 15.Janune D, Kubota S, Nishida T, Kawaki H, Perbal B, Iida S, Takigawa M. Novel effects of CCN3 that may direct the differentiation of chondrocytes. FEBS Lett. 2011;585:3033–3040. doi: 10.1016/j.febslet.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 16.Chen CC, Lau LF. Deadly liaisons: Fatal attraction between CCN matricellular proteins and the tumor necrosis factor family of cytokines. J Cell Commun Signal. 2010;4:63–69. doi: 10.1007/s12079-009-0080-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Juric V, Chen CC, Lau LF. Fas-mediated apoptosis is regulated by the extracellular matrix protein CCN1 (CYR61) in vitro and in vivo. Mol Cell Biol. 2009;29:3266–3279. doi: 10.1128/MCB.00064-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen CC, Young JL, Monzon RI, Chen N, Todorović V, Lau LF. Cytotoxicity of TNFalpha is regulated by integrin-mediated matrix signaling. EMBO J. 2007;26:1257–1267. doi: 10.1038/sj.emboj.7601596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tran CM, Schoepflin ZR, Markova DZ, Kepler CK, Anderson DG, Shapiro IM, Risbud MV. CCN2 suppresses catabolic effects of interleukin-1β through α5β1 and αVβ3 integrins in nucleus pulposus cells: Implications in intervertebral disc degeneration. J Biol Chem. 2014;289:7374–7387. doi: 10.1074/jbc.M113.526111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heath E, Tahri D, Andermarcher E, Schofield P, Fleming S, Boulter CA. Abnormal skeletal and cardiac development, cardiomyopathy, muscle atrophy and cataracts in mice with a targeted disruption of the Nov (Ccn3) gene. BMC Dev Biol. 2008;8:18. doi: 10.1186/1471-213X-8-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivkovic S, Yoon BS, Popoff SN, Safadi FF, Libuda DE, Stephenson RC, Daluiski A, Lyons KM. Connective tissue growth factor coordinates chondrogenesis and angiogenesis during skeletal development. Development. 2003;130:2779–2791. doi: 10.1242/dev.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ding L, Wu JP, Xu G, Zhu B, Zeng QM, Li DF, Lu W. Lentiviral-mediated RNAi targeting caspase-3 inhibits apoptosis induced by serum deprivation in rat endplate chondrocytes in vitro. Braz J Med Biol Res. 2014;47:445–451. doi: 10.1590/1414-431X20143198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran CM, Markova D, Smith HE, Susarla B, Ponnappan RK, Anderson DG, Symes A, Shapiro IM, Risbud MV. Regulation of CCN2/connective tissue growth factor expression in the nucleus pulposus of the intervertebral disc: Role of Smad and activator protein 1 signaling. Arthritis Rheum. 2010;62:1983–1992. doi: 10.1002/art.27445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tran CM, Shapiro IM, Risbud MV. Molecular regulation of CCN2 in the intervertebral disc: Lessons learned from other connective tissues. Matrix Biol. 2013;32:298–306. doi: 10.1016/j.matbio.2013.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bedore J, Leask A, Séguin CA. Targeting the extracellular matrix: Matricellular proteins regulate cell-extracellular matrix communication within distinct niches of the intervertebral disc. Matrix Biol. 2014;37:124–130. doi: 10.1016/j.matbio.2014.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Kondo S, Kubota S, Mukudai Y, Moritani N, Nishida T, Matsushita H, Matsumoto S, Sugahara T, Takigawa M. Hypoxic regulation of stability of connective tissue growth factor/CCN2 mRNA by 3′-untranslated region interacting with a cellular protein in human chondrosarcoma cells. Oncogene. 2006;25:1099–1110. doi: 10.1038/sj.onc.1209129. [DOI] [PubMed] [Google Scholar]
- 27.Kivelä R, Kyröläinen H, Selänne H, Komi PV, Kainulainen H, Vihko V. A single bout of exercise with high mechanical loading induces the expression of Cyr61/CCN1 and CTGF/CCN2 in human skeletal muscle. J Appl Physiol (1985) 2007;103:1395–1401. doi: 10.1152/japplphysiol.00531.2007. [DOI] [PubMed] [Google Scholar]
- 28.Hee HT, Chuah YJ, Tan BH, Setiobudi T, Wong HK. Vascularization and morphological changes of the endplate after axial compression and distraction of the intervertebral disc. Spine (Phila Pa 1976) 2011;36:505–511. doi: 10.1097/BRS.0b013e3181d32410. [DOI] [PubMed] [Google Scholar]