

Summary
The response to DNA double-strand breaks (DSBs) requires alterations in chromatin structure to promote the assembly of repair complexes on broken chromosomes. Non-homologous end-joining (NHEJ) is the dominant DSB repair pathway in human cells, but our understanding of how it operates in chromatin is limited. Here, we define a mechanism that plays a crucial role in regulating NHEJ in chromatin. This mechanism is initiated by DNA damage-associated poly(ADP-ribose) polymerase 1 (PARP1), which recruits the chromatin remodeler CHD2 through a poly(ADP-ribose)-binding domain. CHD2 in turn triggers rapid chromatin expansion and the deposition of histone variant H3.3 at sites of DNA damage. Importantly, we find that PARP1, CHD2, and H3.3 regulate the assembly of NHEJ complexes at broken chromosomes to promote efficient DNA repair. Together, these findings reveal a PARP1-dependent process that couples ATP-dependent chromatin remodeling with histone variant deposition at DSBs to facilitate NHEJ and safeguard genomic stability.
Graphical Abstract
Highlights
-
•
PARP1 recruits the chromatin remodeler CHD2 to DNA damage
-
•
CHD2 promotes chromatin expansion and H3.3 deposition at DNA breaks
-
•
CHD2 promotes the assembly of NHEJ repair complexes at DNA breaks
-
•
PARP1 drives CHD2- and H3.3-dependent DNA repair by NHEJ
Luijsterburg et al. define a PARP1-dependent mechanism that regulates NHEJ through localized chromatin expansion and deposition of the histone variant H3.3 by the nucleosome remodeler CHD2 at DNA breaks. Their data also show that these CHD2-mediated events promote DNA repair by facilitating the assembly of NHEJ complexes in chromatin.
Introduction
DNA double-strand breaks (DSBs) are a considerable threat to the integrity of the human genome and, if not properly dealt with, can cause genomic instability and cancer. The response to DSBs entails a coordinated series of events known as the DNA damage response (DDR), which integrates the regulation of cell cycle progression with DSB repair mechanisms through DNA damage signaling pathways (Polo and Jackson, 2011).
In eukaryotes, DSBs are primarily repaired by two pathways: homologous recombination (HR) and non-homologous end-joining (NHEJ). HR operates in the S and G2 stages of the cell cycle and requires extensive resection of DSBs to generate stretches of single-stranded DNA, which are acted upon by the single-stranded DNA-binding protein RPA and the recombinase RAD51. These and other factors subsequently facilitate the error-free repair of DSBs by using the sister chromatid as a template (Symington and Gautier, 2011). In contrast, NHEJ, which is the dominant DSB repair pathway in mammalian cells, requires minimal DNA-end processing. Initiation of NHEJ involves the binding of the KU70-KU80 complex to broken DNA ends followed by the assembly of the DNA-dependent protein kinase (DNA-PK) and the XRCC4-LigIV complex (Lieber, 2010).
DSB repair takes place on genomic DNA that is packaged together with histone proteins into an often-inaccessible structure called chromatin. Regulating the accessibility of damaged DNA requires a high degree of coordination between DSB repair machineries and chromatin-modifying enzymes (Luijsterburg and van Attikum, 2011, Smeenk and van Attikum, 2013).
Initial studies using photo-activatable GFP fused to the core histone H2B revealed that DNA damage triggers the localized expansion of chromatin in an ATP-dependent fashion (Kruhlak et al., 2006). Subsequent studies uncovered that this localized chromatin expansion requires the activity of poly(ADP-ribose) polymerase (PARP) enzymes and promotes DNA damage signaling by the RNF168 ubiquitin ligase (Smeenk et al., 2013). The initial rapid expansion of chromatin is followed by the localized compaction of chromatin (Burgess et al., 2014, Khurana et al., 2014), suggesting that specific chromatin configurations regulate different aspects of the DDR. In particular, localized chromatin compaction, which is regulated by the PRDM2 histone methyltransferase, regulates DNA-end resection and promotes DSB repair by HR (Khurana et al., 2014). In addition to chromatin compaction, a number of ATP-dependent chromatin remodelers (e.g., SMARCAD1, INO80, p400, and CHD4) that are usually associated with chromatin decondensation have also been linked to regulating end resection or other steps during HR (Smeenk and van Attikum, 2013). These findings suggest that HR is tightly regulated by dynamic changes in chromatin structure.
Despite these considerable insights into dynamic changes in chromatin structure during DNA damage signaling and HR, we know very little about alterations in chromatin structure that may play a role in NHEJ. To fill this gap, we sought to characterize chromatin changes that play a role in NHEJ in human cells and identify a previously uncharacterized pathway involved in this process.
Results
PARP1 Promotes Chromatin Expansion and Spreading of NHEJ Factor XRCC4
We sought to characterize changes in chromatin structure in response to DNA damage that may play a role in NHEJ. To this end, we revisited a method to locally inflict DNA damage and simultaneously activate histone H2A fused to a photo-activatable version of GFP (PA-GFP) using multiphoton micro-irradiation (Figure 1A) (Kruhlak et al., 2006, Smeenk et al., 2013). Local irradiation triggered the rapid expansion of PAGFP-H2A tracks in control cells, but not in cells treated with the PARP inhibitor (Figures 1B and 1C), suggesting that DNA damage-induced chromatin changes depend on the activity of PARP enzymes. To monitor possible chromatin changes involved in NHEJ, we generated U2OS cells stably expressing a GFP-tagged version of the core NHEJ protein XRCC4. Local irradiation triggered the accumulation of GFP-XRCC4 in laser tracks, which displayed considerable expansion over time (Figures 1D and 1E). Strikingly, treatment of cells with PARP inhibitor or knockdown of PARP1 significantly reduced the expansion of GFP-XRCC4 tracks, suggesting that PARP1-dependent changes in chromatin structure may play a role in NHEJ (Figures 1D and 1E; Figures S1A and S1B).
Figure 1.

Chromatin Changes in Response to DNA Damage Depend on PARP1
(A) Outline of the chromatin expansion approach.
(B) PAGFP-H2A expansion in U2OS cells treated with DMSO or 10 μM PARPi.
(C) Quantification of (B).
(D) GFP-XRCC4 expansion in U2OS cells treated with DMSO or 10 μM PARPi.
(E) Quantification of (D). 40–65 cells were analyzed from three independent experiments.
(F) Outline of the chromatin-tethering approach in U2OS 2-6-3 cells, which identified CHD2 as a PARP1 interactor.
(G) CoIP of endogenous PARP1 and CHD2 in HEK293 cells.
(H) Western blot on U2OS CHD2-GFP cells.
(I) SILAC of HEK293 cells expressing GFP (L) or CHD2-GFP (H).
(J) CoIP of CHD2-GFP and endogenous PARP1 in HEK293 cells.
(K) PARylation of CHD2-GFP in HEK293 cells.
(L) Recruitment of endogenous CHD2 to UV-A tracks in U2OS cells. CHD2 knockdown confirms antibody specificity.
(M) Recruitment of CHD2-GFP to multi-photon tracks in U2OS cells.
The Chromatin Remodeler CHD2 Is an Interactor of PARP1
We then set out to identify factors that regulate these chromatin changes by analyzing PARP1-associated chromatin-modifying proteins using a previously described chromatin-tethering approach (Luijsterburg et al., 2012a). To this end, we fused PARP1 to the lactose repressor protein (LacR) and expressed the fusion protein in U2OS cells harboring stably integrated LacO repeats. These cells were subsequently transfected with a representative collection of GFP-tagged SWI2/SNF2 ATPases from the four major chromatin remodeling families (SWI/SNF, ISWI, INO80, and CHD). This approach identified the chromodomain helicase DNA-binding protein 2 (CHD2) as a possible interactor of PARP1 (Figure 1F; Figure S1C). Co-immunoprecipitation (coIP) confirmed the interaction between endogenous PARP1 and CHD2, which was completely abolished by treatment with PARP inhibitor (Figure 1G). This suggests that the association between CHD2 and PARP1 is not mediated by protein-protein interactions but may involve the association of CHD2 with poly(ADP-ribose) (PAR) chains on PARP1 (Figure 1G).
To further study the interaction between CHD2 and PARP1, we stably expressed GFP-tagged CHD2 in U2OS cells at levels roughly similar to endogenous CHD2 (Figure 1H). Immunoprecipitation of CHD2-GFP followed by mass spectrometry (MS) after SILAC revealed 139 proteins that were at least 2-fold enriched compared with control cells (Table S1). This analysis not only revealed interactions with all core histones and the related chromatin remodeler CHD1, but it also confirmed PARP1 as a robust CHD2-interacting protein (Figure 1I). CoIP analysis of CHD2-GFP revealed endogenous PARP1 in the IP fraction, further establishing this interaction (Figure 1J). To test if CHD2 is a substrate of PARP1, we immunoprecipitated GFP or CHD2-GFP from cells under denaturing conditions and analyzed their PARylation status by western blotting. This revealed that CHD2 is indeed a substrate of poly(ADP-ribosyl)ation (Figure 1K). We conclude that CHD2 is an interactor of PARP1 and decided to study its role in DSB repair.
CHD2 Accumulates at Sites of DNA Damage
To determine if CHD2 acts locally at sites of DNA damage, we tested its recruitment to laser-inflicted DNA DSBs. Endogenous CHD2 was rapidly recruited to DNA damage tracks marked by γH2AX following UV-A micro-irradiation (Figure 1L) or multi-photon laser irradiation (Figure 2C). Control experiments showed that siRNA-mediated knockdown of CHD2 abolished CHD2 signals in laser tracks, demonstrating the specificity of the CHD2 antibody (Figure 1L). Moreover, GFP-tagged CHD2 also rapidly localized to sites of laser-induced DNA damage in both G1 and S/G2 cells (Figure 1M; Figures S1D and S1E). These findings show that CHD2 is recruited to sites of DNA damage.
Figure 2.

CHD2 Recruitment to DNA Damage Requires PARP1
(A) Recruitment of CHD2-GFP to multi-photon tracks in U2OS cells treated with DMSO or 1 μM PARPi.
(B) Quantification of (A).
(C) Recruitment of endogenous CHD2 in U2OS cells treated with DMSO or 10 μM PARPi.
(D) Recruitment of CHD2-GFP in cells transfected with the indicated siRNAs.
(E) Western blot showing PARP1/2 knockdown efficiency in cells from (D).
(F) Quantification of (D). 30–170 cells were analyzed from three independent experiments.
PARP1 Recruits CHD2 to Sites of DNA Damage
To assess a potential role of PARP1’s catalytic activity in CHD2 recruitment to sites of DNA damage, we measured the association kinetics of CHD2 using live-cell imaging. A time-course analysis revealed that the rapid (t1/2 = 5 s) accumulation of CHD2 reached a maximum around ∼1 min after which the steady-state bound levels gradually decreased within ∼5 min (Figures 2A and B). Treatment of cells with PARP inhibitor, which prevented PAR chain formation in laser tracks (Figure S1F), completely abolished the recruitment of CHD2-GFP (Figures 2A and 2B), as well as that of endogenous CHD2 into γH2AX-positive laser tracks (Figure 2C). This effect was phenocopied by siRNA-mediated depletion of PARP1, but not PARP2 (Figures 2D–2F), suggesting that CHD2 recruitment is strictly dependent on PARP1. Although PARP1 is responsible for ∼85% of the synthesized PAR chains in response to DNA damage, such chains are rapidly hydrolyzed by the activity of poly(ADP-ribose) glycohydrolase 1 (PARG), which explains the rapid turnover of PAR chains at sites of DNA damage (Pines et al., 2013). To prevent this rapid turnover, we increased the steady-state levels of chromatin-associated PAR chains by depletion of PARG (Luijsterburg et al., 2012b). Live-cell imaging showed that PARG knockdown dramatically increased the retention time of CHD2 on damaged chromatin (Figures 2D and 2F). To extend these findings, we overexpressed mCherry-tagged PARGWT or catalytically dead PARGE756D (Ismail et al., 2012), which completely suppressed CHD2-GFP recruitment (Figures S1G and S1H). Of note, catalytically dead PARG accumulated more strongly likely due to its inability to hydrolyze PAR chains, suggesting that this mutant protein may suppress CHD2 recruitment by direct competition for PAR binding. In conclusion, CHD2 recruitment to DSB-containing chromatin requires the PARP1-mediated synthesis of PAR chains.
A Conserved Region in CHD2 Mediates DNA Damage Accumulation and PAR Binding
To gain more insight into how CHD2 is recruited in a PAR-dependent manner, we generated a series of deletion mutants spanning the whole CHD2 protein (Figure 3A). Immunoblotting confirmed that the GFP-tagged deletion mutants were expressed at the correct molecular weight (Figure 3B). We then co-expressed these mutant proteins with NBS1-mCherry in cells depleted for endogenous CHD2 and subsequently assessed their ability to accumulate at DSBs. Regions of CHD2 spanning its chromodomains (CHD21–461), ATPase/helicase domains (CHD2462–951) or putative SANT-SLIDE motif (CHD2952–1391; Figure S2A) failed to accumulate in cells that did accumulate NBS1-mCherry (Figures 3C and 3D). Conversely, CHD21392–1828 robustly accumulated in laser tracks (Figures 3C and 3D). An in silico analysis of the minimal recruitment region identified a putative PAR-binding motif that almost matches the consensus (Figure S2B) (Gagné et al., 2008). Surprisingly, however, a region encompassing this putative PAR-binding region (CHD21392–1610) failed to accumulate (Figures 3C and 3D) even when fused to an NLS (Figure S2C), whereas the last C-terminal ∼200 amino acids (CHD21611–1828) were sufficient to mediate PAR-dependent accrual at DSBs (Figures 3C and 3D). Indeed, full-length CHD2 lacking this region (CHD21–1610) failed to accumulate at DSBs (Figures 3C and 3D), suggesting that the region spanning residues 1611–1828 regulates the PAR-dependent recruitment of CHD2. To further corroborate these findings, we carried out in vitro PAR-binding studies by incubating immunoprecipitated GFP-tagged wild-type and mutant CHD2 protein with 32P-radiolabeled PAR chains using southwestern blotting. This revealed that both CHD2WT and CHD21611–1828 indeed bind PAR chains with equal efficiency (Figures 3E and 3F). Although it did not support recruitment to laser tracks (Figures 3C and 3D), we could also detect PAR binding of CHD21392–1610, albeit almost 3-fold weaker compared to the minimal recruitment region (CHD21611–1828) (Figures 3E and 3F). Southwestern analysis with recombinant proteins confirmed the ∼3-fold stronger association of CHD21611–1828 with PAR chains in vitro compared to CHD21392–1610 (Figure S2D). Notably, the CHD21392–1610 fragment failed to support PAR binding in the context of the larger CHD2 protein because we did not detect appreciable PAR binding for the CHD21–1610 mutant containing this region (Figures 3E and 3F). Of note, coIPs showed that CHD21611–1828 interacted only weakly with PARP1, whereas CHD2WT and CHD21–1610 robustly pulled down PARP1 (Figure 3E), suggesting that the PAR-binding region is distinct from the PARP1-interacting region. Indeed, we found that fragment CHD21392–1610, which failed to support recruitment to sites of DNA damage, was sufficient to bind PARP1 (Figure 3E). In conclusion, we have mapped the PAR-binding region in CHD2 and show that this region is sufficient and required for CHD2 recruitment to DSBs.
Figure 3.

The C Terminus of CHD2 Is Required for DNA Damage Recruitment and PAR Binding
(A) Schematic representation of CHD2 and its deletion mutants.
(B) Western blot showing expression of the mutants from A in U2OS cells.
(C) Recruitment of CHD2-GFP deletion mutants in cells depleted for endogenous CHD2 by siCHD2-69 or siCHD2-17 in case of CHD2952–1391-GFP. CHD21–1611-GFP was rendered siCHD2-69-resistant. NBS1-mCherry was a DNA damage marker.
(D) Quantification of (C). 10–30 cells were analyzed from two independent experiments.
(E) IP of the indicated CHD2-GFP fragments from HEK293 cells followed by southwestern blotting to monitor association with radiolabeled PAR. Recombinant PARP1 was a control. CoIP with endogenous PARP1 is shown in the bottom panel.
(F) Quantification of (E) and two additional independent experiments. PAR binding levels of CHD2WT-GFP were set to 1.
CHD2 Protects Human Cells against IR
To test whether the recruitment of CHD2 bears any functional significance for DSB repair, we addressed whether this chromatin remodeler protects human cells against the deleterious consequences of ionizing radiation (IR)-induced DSBs. Depletion of CHD2 from SV40-immortalized VH10 human fibroblasts using three independent siRNAs reduced the clonogenic survival of these cells after IR to the same extent as knockdown of DSB repair factor XRCC4 (Figure 4A; Figure S3A). To corroborate these findings, we stably expressed three different shRNAs targeting CHD2 and one shRNA against the key DDR kinase ATM in hTERT-immortalized VH10 human fibroblasts. This approach confirmed that loss of CHD2 renders human cells highly sensitive to IR (Figure S3B) and raises the question how CHD2 is involved in DSB repair.
Figure 4.

CHD2 Promotes DSB Repair by NHEJ
(A) Clonogenic survival after IR exposure of VH10-SV40 cells transfected with the indicated siRNAs.
(B) Schematic representation of the EJ5-GFP reporter for NHEJ.
(C) Quantification of EJ5-GFP-positive HEK293 cells corrected for I-SceI transfection efficiency by co-transfection with mCherry. The average of four independent experiments is shown.
(D) Schematic representation of the plasmid integration assay.
(E) Quantification of the plasmid integration efficiency in U2OS cells from three independent experiments.
(F) TRF2ts MEFs were shifted to the non-permissive temperature to induce telomere uncapping and NHEJ-dependent chromosome fusions.
(G) Representative images of metaphases from TRF2ts MEFs transduced with the indicated shRNAs after 24 hr of telomere uncapping. Telomere-FISH shows the position of the telomeres (green), and chromosomes are stained by DAPI (blue).
(H) Western blot showing CHD2 and LigIV knockdown efficiency in TRF2ts MEFs.
(I) Quantification of interchromosomal fusions observed in cells transduced with the indicated shRNAs. Scrambled control shRNA (shScr) was normalized to 100%. 4500–8000 chromosomes were analyzed from three to eight independent experiments.
CHD2 Promotes DSB Repair by NHEJ
Chromosomal DSBs are repaired by HR or NHEJ. To test if CHD2 may be involved in these DSB repair pathways, we utilized GFP-based assays that rely on the repair of DSBs in the GFP gene generated by the I-SceI endonuclease. Flow cytometric analysis of DR-GFP reporter cells for HR (Figure S3C) showed a dramatic increase in GFP-positive cells following I-SceI expression, which was severely suppressed by siRNA-mediated depletion of the core HR factor BRCA2 (Figure S3D). However, three independent siRNAs against CHD2 did not substantially impair repair by HR and neither did depletion of the core NHEJ factor XRCC4 (Figures S3D and S3E). In addition, RAD51 foci formation after IR in S-phase cells was also not affected by depletion of CHD2, confirming that CHD2 does not promote HR (Figures S3F and S3G).
Conversely, analysis of EJ5-GFP reporter cells for NHEJ (Figure 4B) revealed a reproducible decrease in NHEJ efficiency upon depletion of CHD2 by six independent siRNAs, which was comparable to the defect observed after depletion of XRCC4 (Figure 4C; Figure S4A). As expected, depletion of the HR factor BRCA2 did not affect NHEJ (Figure 4C). The EJ5-GFP reporter provides a readout for total NHEJ activity, but it is not specific for canonical NHEJ (cNHEJ) or alternative NHEJ (aNHEJ), which comprise the two major pathways of NHEJ (Bennardo et al., 2008). To address if PARP1 and CHD2 play a role in cNHEJ, we used random plasmid integration into genomic DNA as a measure for cNHEJ (Figure 4D) (Galanty et al., 2009). Knockdown of the core NHEJ factors KU80 or DNA-PKcs nearly abrogated plasmid integration, confirming that this process largely depends on cNHEJ (Figure 4E; Figure S4B). Significantly reduced cNHEJ activity was also observed following depletion of either CHD2 or PARP1 with two independent siRNAs per gene (Figure 4E; Figure S4B), suggesting that both of these factors contribute to DSB repair by cNHEJ. Importantly, knockdown of CHD2 did not affect the steady-state levels of NHEJ proteins or PARP1 arguing against indirect effects due to transcriptional misregulation (Figure S4C).
As an alternative means to study NHEJ, we exploited the notion that loss of the shelterin complex at telomeres causes NHEJ-dependent chromosome-end fusions (Smogorzewska et al., 2002). To study the role of CHD2 in this process, we used mouse embryonic fibroblasts containing a temperature-sensitive allele of the shelterin protein TRF2 (TRF2ts) (Boersma et al., 2015). At the non-permissive temperature of 39°C, TRF2ts-mediated telomere protection is lost, subsequently triggering the NHEJ-dependent fusion of uncapped chromosome ends (Figure 4F). Strikingly, knockdown of CHD2 by three individual shRNAs significantly lowered the number of fused chromosomes in comparison to a control shRNA in TRF2ts cells grown at non-permissive temperatures (Figures 4G–4I; Figure S4D). A comparable decrease in chromosome-end fusions was observed in cells depleted for the core NHEJ factor LigIV (Figures 4G–4I). In line with previous findings (Sfeir and de Lange, 2012), we found that knockdown of PARP1, in contrast to CHD2 knockdown, did not reduce the formation of NHEJ-mediated chromosome fusions (Figures S4E and S4F), suggesting that at uncapped telomeres, CHD2 may act through a PARP1-independent mechanism. These findings suggest that CHD2 contributes to cNHEJ activity in mouse cells and, as such, promotes chromosome-end fusions at uncapped telomeres. Thus, although dispensable for repair by HR, our findings implicate CHD2 in DSB repair by NHEJ. We next set out to determine how CHD2 affects this DSB repair pathway.
CHD2 Promotes the Recruitment of Core NHEJ Factors
The repair of chromosomal DSBs by NHEJ depends on the binding of the KU70-KU80 dimer to broken DNA ends followed by the recruitment of the DNA-PKcs kinase (Mari et al., 2006). The XRCC4-LigIV complex is subsequently recruited to DSBs to seal the break. To monitor interactions between CHD2 and NHEJ proteins, we performed reciprocal coIP experiments, which revealed an interaction between endogenous CHD2 and endogenous KU70 (Figure 5A) or GFP-tagged KU70 (Figure S4G). Notably, this interaction was not affected by PARP inhibition or IR-induced DNA damage (Figure S4G). To subsequently test if CHD2 promotes the recruitment of NHEJ factors, we monitored their accumulation at DSBs by live-cell imaging using cells stably expressing GFP-tagged KU70 (Mari et al., 2006) or GFP-tagged XRCC4. Multiphoton micro-irradiation triggered rapid GFP-KU70 recruitment to DSBs within several seconds (t1/2 = 5 s; Figure 5B), whereas GFP-XRCC4 accrual was significantly slower (t1/2 = 25 s; Figure 5C). Importantly, the detection of KU70 in laser tracks required much high laser power than that of XRCC4, suggesting that these factors do not accumulate at the same stoichiometric amounts (Figure S4H). Importantly, the depletion of CHD2 by two independent siRNAs significantly suppressed both GFP-KU70 (Figure 5B) and GFP-XRCC4 (Figure 5C; Figure S4I) recruitment to laser-generated DSBs, whereas the recruitment of GFP-XRCC1 to single-stranded breaks was unaffected (Figures S4J–S4L). As expected, GFP-XRCC4 recruitment in these cells was fully dependent on functional KU80 (Figure 5C). Moreover, the recruitment of endogenous XRCC4 was also suppressed in CHD2-depleted cells after micro-irradiation (Figures S5A–S5D) and at nuclease-induced DSBs (Figure 5D). The levels of localized γH2AX in control or CHD2-depleted cells were similar, showing that CHD2 depletion did not affect DNA damage induction (Figure 5D; Figure S5C). At the single-cell level, we confirmed that cells with decreased GFP-XRCC4 accrual following depletion of CHD2 failed to accumulate endogenous CHD2 in laser tracks, whereas CHD2 did clearly accumulate in control cells (Figure S5E). Notably, overexpression of ATPase-dead CHD2 (K515R; Figure S5F), but not wild-type CHD2, also reduced the recruitment of XRCC4 (Figure 5E). Together, these findings suggest that the chromatin remodeling activity of CHD2 promotes the efficient assembly of NHEJ complexes at DSBs.
Figure 5.

CHD2 Promotes the Assembly of NHEJ Complexes at DSBs
(A) Reciprocal coIPs of endogenous CHD2 and KU70 in HEK293 cells.
(B) Recruitment of GFP-KU70 to multi-photon tracks in HeLa cells.
(C) As in (B), except for GFP-XRCC4 in U2OS cells.
(D) siRNA-transfected pTuner265 cells were induced for FokI-LacR expression and stained for γH2AX and XRCC4.
(E) U2OS cells transfected with the indicated constructs were UV-A micro-irradiated and stained for γH2AX and XRCC4.
(F and G) Recruitment kinetics of GFP-XRCC4 to multi-photon tracks in (F) U2OS cells treated with DMSO or 10 μM PARPi or in (G) U2OS cells transfected with the indicated siRNAs. 40–160 cells were analyzed from (B) three or (C–E) two independent experiments.
Given that CHD2 recruitment is PARP1 dependent, we next asked whether the CHD2-mediated recruitment of XRCC4 to damaged chromatin relies on PARP1. Micro-irradiation experiments showed that treatment of cells with PARP inhibitor (Figure 5F) or siRNAs against PARP1 (Figure 5G; Figure S5G) significantly suppressed GFP-XRCC4 accumulation at laser-generated DSBs. We conclude that PARP1-mediated recruitment of CHD2 is required for the efficient assembly of NHEJ factors.
CHD2 Promotes Chromatin Changes in Response to DNA Damage
The fact that the PARP1-mediated recruitment of CHD2 promotes the assembly of NHEJ complexes raises the question of whether this phenomenon requires CHD2-mediated chromatin remodeling. We first assessed whether CHD2 is capable of mediating large-scale chromatin unfolding by utilizing an in vivo chromatin-remodeling assay (Figure 6A). Human U2OS cells harboring a 4-Mbp heterochromatic region containing lactose operator (LacO) repeats were transfected with a plasmid encoding a LacR-tagged single-domain GFP antibody. The resulting fusion protein (αGFP-LacR) efficiently tethered co-expressed GFP-tagged proteins to the LacO array (Figure 6A). Tethering of wild-type CHD2-GFP elicited a 2-fold expansion of chromatin at the LacO array compared to tethering GFP only (Figure 6B). However, ATPase-dead CHD2 (K515R; Figure S5F) failed to unfold chromatin, even though it was recruited as efficiently as wild-type CHD2 (Figure S6A), whereas a mutant lacking its PAR-binding motif (Δ1611–1828; Figure 3) was proficient in mediating chromatin unfolding (Figures 6A and 6B). These findings confirm that CHD2 is an ATP-dependent chromatin remodeler and that its PAR-binding motif (1611–1828) is dispensable for its chromatin-remodeling function.
Figure 6.

CHD2 Promotes DNA Damage-Induced Chromatin Changes
(A) U2OS 2-6-3 cells containing a LacO array were co-transfected with αGFP-mCherry-LacR and GFP-tagged CHD2 variants or GFP only.
(B) Quantification of the array size upon tethering of the indicated proteins.
(C) Outline of the PAGFP-H2A expansion approach.
(D and E) PAGFP-H2A expansion in U2OS cells transfected with the indicated siRNAs. NBS1-mCherry was a DNA damage marker.
(F) Quantification of the PAGFP-H2A expansion experiments. Values were corrected for the expansion in fixed cells and normalized to 1 for siLuc at 5 s post irradiation.
(G) U2OS 2-6-3 cells were transfected with mCherry-LacR, whereas 2-6-3-derived pTuner265 cells were transfected with the indicated siRNAs and induced for FokI-LacR expression. 60–100 cells were analyzed from (B and F) three or (G) two independent experiments.
We next investigated if CHD2 would be required for DNA damage-induced chromatin changes. To this end, we co-expressed NBS1-mCherry together with histone H2A fused to PAGFP-H2A (Figures 1B and 6C). Laser micro-irradiation triggered robust recruitment of NBS1 and localized activation of PAGFP-H2A in tracks, which rapidly expanded within the first minutes after irradiation indicative of DNA damage-induced chromatin changes (Figures 6D and 6F). Expansion of PAGFP-H2A occurred in both G1 and S/G2 cells (Figure S6B) and was not observed in chemically fixed cells (Figures S6C and S6E), showing that it represents a cell cycle-independent biological phenomenon that is induced by DNA damage. Importantly, when we used laser conditions that efficiently activated PAGFP-H2A and led to recruitment of single-strand break repair factor GFP-XRCC1 but failed to recruit DSB repair factor XRCC4, we did not detect any expansion of H2A tracks (Figures S6D–S6I). These findings suggest that laser-induced chromatin changes are linked to the generation of DSBs. Having established conditions to detect DSB-induced chromatin changes, we next examined whether CHD2 is required for these events. Knockdown of CHD2 using two independent siRNAs substantially reduced expansion of PAGFP-H2A (Figures 6E and 6F; Figures S6E and S6J), whereas CHD4 or SNF2H depletion did not affect this process (Figures S6K and S6L). Similarly, knockdown of CHD2 also impaired unfolding of LacO arrays following DSB induction by the FokI nuclease (Figure 6G). Consistent with the PARP1-mediated recruitment of CHD2, we found that the depletion of PARP1 efficiently antagonized chromatin expansion (Figure 6F; Figure S6L). These findings are consistent with a model in which PARP1-mediated recruitment of CHD2 triggers localized chromatin remodeling that promotes the efficient assembly of NHEJ complexes.
PARP1 and CHD2 Link Histone H3.3 to NHEJ
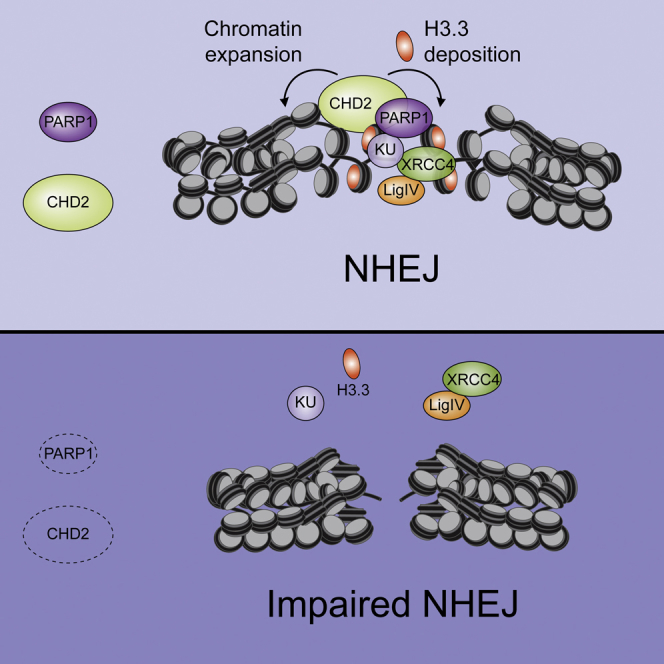
To further probe into possible mechanisms that underlie CHD2’s ability to promote NHEJ, we turned our attention to histone variant H3.3. Recent studies revealed that CHD2 associates with H3.3 and incorporates this variant at sites of transcription (Harada et al., 2012, Siggens et al., 2015). Moreover, H3.3 is also incorporated at sites of UV-C-induced DNA damage (Adam et al., 2013). To confirm the interaction between these proteins, we performed coIP experiments, which indeed showed a robust interaction between endogenous CHD2 and H3.3 (Figure 7A). Prompted by these findings, we sought to address whether CHD2 also cooperates with H3.3 in DSB repair. To study this, we generated cells stably expressing low levels of SNAP-tagged H3.3, which allows the fluorescent labeling and monitoring of newly synthesized histone H3.3 (Adam et al., 2013) (Figure 7B; Figure S7A). Local micro-irradiation revealed the rapid deposition of H3.3 at sites of DSBs within several minutes after damage induction, which was severely reduced in CHD2-depleted cells (Figures 7C and 7D). Similarly, overexpression of ATPase-dead CHD2 (K515R; Figure S5F), but not wild-type CHD2, significantly reduced H3.3 deposition (Figure 7E), suggesting that CHD2’s chromatin remodeling activity contributes to H3.3 assembly at DSBs. Incorporation of H3.1 could also be detected, albeit much weaker than the H3.3 assembly (Figure S7B). Importantly, deposition of H3.3 only occurred following micro-irradiation of cells that were pre-sensitized with BrdU, showing that not the UV-A irradiation itself, but rather the generation of laser-induced DSBs caused by BrdU sensitization triggers this response (Figure S7C) (Limoli and Ward, 1993). Considering that CHD2 recruitment is mediated by PARP1, we next addressed if PARP1 is involved in H3.3 deposition. CoIP experiments revealed an interaction between H3.3 and PARP1, which was not enhanced by DNA damage induction (Figure 7F). Importantly, treatment of cells with PARP inhibitor or siRNA-mediated depletion of PARP1 significantly reduced the de novo incorporation of H3.3 at DSBs (Figures 7G and 7H; Figures S7D–S7F). To analyze if H3.3 is required for DSB repair, we transfected siRNAs targeting both H3.3 genes in human cells, which efficiently depleted H3.3 (Figure 7I) without affecting cell cycle progression (Figure S7G) or the steady-state levels of PARP1 and core NHEJ proteins (Figure S7H). Knockdown of H3.3 caused a severe reduction in NHEJ activity as measured by the EJ5-GFP reporter (Figure 7J) and the plasmid integration assay (Figure 7K). In addition, it also moderately reduced aNHEJ (Figures S7I and S7J) and HR (Figures S7K and S7L), the latter of which agrees with a published report (Yang et al., 2013). Consistent with a defect in DSB repair, we found that loss of H3.3 rendered VH10-SV40 and U2OS cells highly sensitive to IR (Figure 7L; Figure S7M). Notably, knockdown of H3.3 did not affect CHD2 recruitment (Figures S7N–S7P), in agreement with the notion that PARP1 and CHD2 act upstream of H3.3. Depletion of H3.3 also did not affect PAGFP-H2A dynamics (Figures S6K and S7L), suggesting that CHD2-mediated chromatin expansion precedes H3.3 deposition. Similar to CHD2, coIP experiments revealed that H3.3 interacts with a member of the KU complex (KU80; Figure 7F). Moreover, just like depletion of PARP1 or CHD2, depletion of H3.3 severely reduced the KU-dependent assembly of XRCC4 in damaged chromatin (Figures 7M, and7N). To test if H3.3, similar to CHD2, affects NHEJ-dependent chromosome-end fusions (Figures 4F–4I), we simultaneously knocked down both H3.3 genes in mouse TRF2ts cells and induced telomere uncapping. Strikingly, knockdown of H3.3 by two combinations of shRNAs significantly reduced interchromosomal end fusions (Figure 7O). These findings suggest that both CHD2 and H3.3 promote NHEJ-driven chromosome fusion at uncapped telomeres. In summary, our findings suggest that the recruitment of CHD2 by PARP1 triggers the assembly of H3.3 at sites of DNA damage creating an accessible chromatin micro-environment that is amenable for DSB repair by NHEJ (Figure 7P).
Figure 7.

CHD2-Mediated H3.3 Deposition Regulates NHEJ
(A) CoIP of endogenous CHD2 and H3.3 in HEK293 cells.
(B) Outline of the H3.3 deposition approach.
(C) Deposition of H3.3 at UV-A tracks in U2OS cells transfected with the indicated siRNAs.
(D) Quantification of (C).
(E) U2OS H3.3-SNAP cells transfected with the indicated GFP constructs were UV-A micro-irradiated, SNAP-labeled, and stained for γH2AX.
(F) CoIP of GFP-H3.3 and KU80 or PARP1 in U2OS cells.
(G) Deposition of H3.3 at UV-A tracks in U2OS cells treated with DMSO or 10 μM PARPi.
(H) Quantification of (G) and of cells transfected with PARP1 siRNAs.
(I) Western blot showing H3.3 knockdown efficiency in U2OS cells.
(J) Quantification of GFP-positive EJ5-GFP HEK293 cells corrected for I-SceI transfection efficiency by co-transfection with mCherry. The average of two independent experiments is shown.
(K) Plasmid integration assay after H3.3 knockdown in U2OS cells. The average of two independent experiments is shown.
(L) Clonogenic survival after IR exposure of siRNA-transfected VH10-SV40 cells.
(M) XRCC4 recruitment to UV-A tracks in siRNA-transfected U2OS cells.
(N) Quantification of (M). 45–100 cells were analyzed from (E) two or (D and H) three independent experiments.
(O) Quantification of interchromosomal fusions observed in shRNA-transduced TRF2ts MEFs after telomere uncapping. 3,200–4,600 chromosomes were analyzed from two independent experiments. Western blot showing H3.3 knockdown efficiency.
(P) Model for how PARP1 links CHD2-mediated chromatin expansion and H3.3 assembly to DSB repair by NHEJ.
Discussion
In this study, we established that DSB-containing chromatin undergoes a rapid PARP1-dependent expansion, which coincides with the spreading and efficient recruitment of NHEJ factor XRCC4. The poorly characterized chromatin remodeler CHD2 is the effector of PARP1 in this process and is required for DNA damage-induced chromatin expansion, the DNA damage-induced deposition of histone variant H3.3, and the efficient recruitment and functioning of NHEJ repair complexes.
CHD2 Promotes cNHEJ
Initial studies have revealed that chromatin containing DSBs undergoes rapid ATP-dependent expansion (Kruhlak et al., 2006). We and others have recently shown that these chromatin changes rely on the activity of PARP enzymes (Khurana et al., 2014, Smeenk et al., 2013). Our current findings shed light on the underlying molecular mechanism by showing that CHD2 associates with PARP1-generated PAR chains through its C terminus and subsequently promotes DNA damage-induced chromatin expansion. Knockdown of PARP1 or CHD2 significantly reduces, but does not completely abolish chromatin expansion, suggesting that other mechanisms may also contribute to this phenomenon. Although local irradiation of cells using laser micro-irradiation produces different types of DNA lesions, including DSBs and single-stranded breaks, our results suggest that the CHD2-dependent chromatin response requires the presence of DSBs. First, we show that significant chromatin expansion is only observed when using laser conditions that trigger accrual of the DSB repair factor XRCC4. Second, we show that CHD2 promotes the recruitment of the KU complex and XRCC4, both of which respond specifically to DSBs. Third, we find that CHD2 is required for NHEJ-dependent repair of the EJ5-GFP reporter and genomic integration of linearized plasmid DNA. Both of these substrates contain bona fide DSBs generated by endonucleases, and we show that processing of the latter substrate is strictly dependent on canonical NHEJ. Fourth, we find that CHD2 promotes XRCC4 recruitment and chromatin expansion in chromatin surrounding nuclease-induced DSBs. Fifth, and finally, we show in mouse cells that the cNHEJ-dependent fusion of chromosomes containing uncapped telomeres requires the activity of CHD2. Together, these findings argue that the chromatin response mediated by CHD2 is triggered by the presence of DSBs and promotes repair of these lesions by the canonical KU-dependent NHEJ pathway.
PARP1 Contributes to cNHEJ
Our findings reveal that CHD2 is recruited to sites of DNA damage by PARP1 and promotes NHEJ. This raises the question: Is the PARP1-dependent recruitment of CHD2 required for NHEJ? The current view on NHEJ mechanisms distinguishes a fast KU-dependent cNHEJ pathway from a slower PARP1-dependent aNHEJ mechanism (Audebert et al., 2004, Wang et al., 2006). However, the demonstrated role of PARP1 in aNHEJ does not exclude a role for PARP1 in cNHEJ. Indeed, several studies have implicated PARP1 in cNHEJ, but its contribution is less clear due to conflicting results (reviewed in Pines et al., 2013). We provide evidence for a role of PARP1 in modulating the efficiency of cNHEJ in human cells. First, we show that both the inhibition and knockdown of PARP1 in human cells significantly reduces the assembly of XRCC4 to laser-induced DSBs. Second, we find that knockdown of PARP1 in human cells reduces the cNHEJ-dependent integration of linearized plasmid DNA. These findings suggest that PARP1 is not essential for cNHEJ, but together with CHD2 it promotes the efficiency of this DSB repair pathway in a chromatin environment.
PARP1 Activation in the Presence of KU
Biochemical evidence suggests that KU and PARP1 compete for the binding to broken DNA ends (Cheng et al., 2011, Wang et al., 2006), raising the question: How does PARP1 stimulate cNHEJ in the presence of functional KU? Interestingly, KU limits but does not prevent PARP1 recruitment to DSBs (Cheng et al., 2011). Moreover, live-cell imaging revealed that KU dimers exchange rapidly from DNA ends with a t1/2 of 2 min (Mari et al., 2006). This suggests that PARP1 could associate with DSBs recently vacated by KU. Another possibility is that PARP1 associates with DSBs that are not bound by KU and, through CHD2 recruitment, creates a local chromatin environment that promotes the assembly of NHEJ complexes in trans. Either way, our findings support a model in which PARP1 acts upon DSBs in the presence of KU and contributes to DSB repair by cNHEJ. Recent findings have shown that PARP3 also promotes cNHEJ, suggesting that PARP3 can also associate with DSBs in the presence of functional KU (Rulten et al., 2011). How PARP1 and PARP3 cooperate at DSBs is not clear, but the finding that the combined loss of PARP1 and PARP3 renders mice sensitive to IR beyond the impact of the single disruption of either gene, suggests a functional synergy between these PARP enzymes in the DSBs response (Boehler et al., 2011).
CHD2 Contains a PAR-Binding Domain
Our findings link PARP1-mediated PAR synthesis to the recruitment of CHD2. Surprisingly, although CHD2 contains a putative PAR-binding domain (Gagné et al., 2008), we found this domain to be dispensable for DNA damage recruitment. Instead, we identified a C-terminal region located between amino acids 1611 and 1828 to be essential for in vitro PAR binding and the localization of CHD2 at laser-induced DSBs. Interestingly, a heterozygous CHD2 mutant mouse was generated by gene trapping, which expresses a truncated CHD2-β-gal-neomycin fusion protein that contains the first 1198 amino acids of the wild-type protein. Notably, CHD2 gene-trap mice develop lymphomas, and cells from these mice display signs of defective DSB repair (Nagarajan et al., 2009). However, it is unclear whether these phenotypes are caused by a gain-of-function feature of the CHD2 fusion protein or through a loss-of-function feature of CHD2. Using siRNA- and shRNA-mediated knockdown of CHD2 in human and mouse cells, our results provide direct evidence for a role of CHD2 in DSB repair by KU-dependent NHEJ. It is tempting to speculate that the gene-trap allele, which produces a truncated CHD2 protein (CHD21–1198) lacking the C-terminal PAR-binding region, is not functional due to its inability to associate with PAR chains. This region of CHD2 bears no similarity to other known PAR-binding domains, and it would be interesting to biochemically define how it interacts with PAR chains and whether interactions with PAR stimulate the ATPase activity of CHD2.
A Pathway that Regulates NHEJ in Chromatin
How does the PARP1-dependent recruitment of CHD2 promote NHEJ? Our data reveal that, similar to transcription sites (Harada et al., 2012, Siggens et al., 2015), the deposition of H3.3 at DSBs is regulated by CHD2’s chromatin remodeling activity. However, whether CHD2 directly deposits H3.3, as was demonstrated in vitro for the p400 ATPase (Pradhan et al., 2016), or if it cooperates with known H3.3 chaperones remains to be investigated (Adam et al., 2013). Knockdown of H3.3, like knockdown of CHD2, results in a profound defect in DSB repair by NHEJ, suggesting that CHD2 promotes NHEJ, at least in part, through the DNA damage-dependent assembly of H3.3. The deposition of H3.3 may create a chromatin environment that facilitates the assembly of functional NHEJ complexes, and this process may be aided by interactions between H3.3 and the KU complex. Interestingly, somatic mutations in CHD2 and H3.3 genes have been found to drive tumorigenesis (Rodríguez et al., 2015, Yuen and Knoepfler, 2013), suggesting that the uncovered pathway may contribute to tumor suppression by maintaining genetic stability. In summary, we define a pathway involved in facilitating NHEJ in a chromatin context. Our findings support a model in which PARP1-associated PAR chains attract the chromatin-remodeling activity of CHD2 to deposit histone variant H3.3 and generate an accessible chromatin environment that promotes the efficient assembly of NHEJ complexes at DSBs. In addition to its described role in aNHEJ, PARP1 also contributes to efficient KU-dependent cNHEJ in human cells through its effectors CHD2 and H3.3. The strong link between these factors raises the question whether CHD2 and H3.3 play a role in other genome maintenance pathways that are modulated by PARP1 (Pines et al., 2013).
Experimental Procedures
Cell Lines, Chemicals, Plasmids, and Transfections
Cells (see the Supplemental Information) were cultured in DMEM, supplemented with antibiotics and 10% fetal calf serum. PARP inhibitor (KU-0058948) was used at a concentration of 1–10 μM. The CHD2 cDNA was inserted into pEGFP-N1 (Addgene). All indicated deletion mutants of CHD2 were generated by PCR. Plasmid DNA or siRNAs were transfected using Lipofectamine 2000 or RNAiMAX (Invitrogen). See the Supplemental Information for details on siRNAs, shRNAs, and primers.
Immunoprecipitation for Mass Spectrometry and PAR-Binding Assay
GFP-tagged PARP1 and CHD2 were immunoprecipitated, trypsinized, desalted, and analyzed on a Q-Exactive Orbitrap mass spectrometer (Thermo Scientific, Germany) coupled to an EASY-nanoLC 1000 system (Proxeon, Odense, Denmark). CHD2-GFP fragments were immunoprecipitated, separated by SDS-PAGE, and incubated with radioactive PAR. Radioactivity was detected by a phosphor-imager screen.
Microscopic Analysis
Laser micro-irradiation was performed by UV-A micro-irradiation of BrdU-sensitized cells or by multi-photon (MP) irradiation using a titanium-sapphire laser. PAGFP-H2A was photoactivated using the same MP laser and settings as those used to inflict localized DNA damage (see the Supplemental Information for details). Immunostaining was performed as described (Luijsterburg et al., 2012a). Primary antibodies are listed in the Supplemental Information.
H3.3-SNAP Labeling
U2OS H3.3-SNAP cells were blocked with SNAP-Cell Block (New England Biolabs), subjected to UV-A micro-irradiation after which newly synthesized histones were labeled with SNAP-cell TMR star (New England Biolabs).
DSB Repair Assays
EJ5-GFP, EJ2-GFP, and DR-GFP reporter assays were carried out as described previously (Smeenk et al., 2013). Gel-purified BamHI-EcoRI-linearized pEGFP-C1 plasmid was transfected into siRNA-depleted cells to measure random plasmid integration events (see the Supplemental Information) (Galanty et al., 2009).
Chromosome Fusion Assays
TRF2ts MEFs were infected with shRNAs constructs and shifted to the non-permissive temperature (39°C) for 24 hr to induce telomere uncapping followed by telomere-FISH, as described (Boersma et al., 2015).
Statistical Analysis
Statistical significance was assessed by a two-tailed, unpaired t test and is indicated as follows: ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, and ∗p < 0.05. ns, not significant. All error bars represent the SEM.
Author Contributions
M.S.L. generated constructs and stable cell lines; performed micro-irradiation and photo-activation experiments, LacR-based tethering assays, H3.3 deposition assays, and IPs for mass spectrometry; and wrote the paper. W.W.W. performed multi-photon and photo-activation experiments, clonogenic survivals, plasmid integration assays, coIPs, and DR-GFP, EJ5-GFP, and EJ2-GFP reporter assays. G.S. performed clonogenic survivals using shRNAs and siRNA as well as the EJ5-GFP reporter assay. A.J.L.d.G. performed CHD2 coIPs. A.J.L.d.G. and A.P. performed coIPs for mass spectrometry. A.P. and A.C.O.V. analyzed the mass spectrometry samples. R.G.S. and G.M.S. performed PARP1 coIPs and in vitro PAR-binding assays. I.d.K. and J.J.L.J. performed the telomere uncapping and chromosome fusion analysis. H.v.A. supervised the project and wrote the paper.
Acknowledgments
The authors acknowledge Jer-Gung Chang, Remco Derr, Jiabo Di, and Pierre Caron for valuable support and Penny Jeggo, Heinrich Leonardt, and Michael Hendzel for providing cDNA constructs. Dik van Gent, Mauro Modesti, Maria Jasin, and Jeremy Stark generously provided cell lines and reagents. We acknowledge Natalia Lukashchuk and Stephen Jackson for advice regarding the plasmid integration assay. This work was funded by a FEBS fellowship, LUMC research fellowship, and an NWO-VENI grant to M.S.L., an ERC Starting grant to A.C.O.V., an ERC Starting grant and an EMBO Young Investigator Award to J.J.L.J., a Discovery Grant from the Natural Sciences and Engineering Research Council of Canada to G.M.S., and an ERC Consolidator grant to H.v.A. M.S.L. and H.v.A. also acknowledge the Bontius Stichting for financial support.
Published: February 18, 2016
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and one table and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2016.01.019.
Contributor Information
Martijn S. Luijsterburg, Email: m.luijsterburg@lumc.nl.
Haico van Attikum, Email: h.van.attikum@lumc.nl.
Supplemental Information
References
- Adam S., Polo S.E., Almouzni G. Transcription recovery after DNA damage requires chromatin priming by the H3.3 histone chaperone HIRA. Cell. 2013;155:94–106. doi: 10.1016/j.cell.2013.08.029. [DOI] [PubMed] [Google Scholar]
- Audebert M., Salles B., Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- Bennardo N., Cheng A., Huang N., Stark J.M. Alternative-NHEJ is a mechanistically distinct pathway of mammalian chromosome break repair. PLoS Genet. 2008;4:e1000110. doi: 10.1371/journal.pgen.1000110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehler C., Gauthier L.R., Mortusewicz O., Biard D.S., Saliou J.M., Bresson A., Sanglier-Cianferani S., Smith S., Schreiber V., Boussin F., Dantzer F. Poly(ADP-ribose) polymerase 3 (PARP3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA. 2011;108:2783–2788. doi: 10.1073/pnas.1016574108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boersma V., Moatti N., Segura-Bayona S., Peuscher M.H., van der Torre J., Wevers B.A., Orthwein A., Durocher D., Jacobs J.J. MAD2L2 controls DNA repair at telomeres and DNA breaks by inhibiting 5′ end resection. Nature. 2015;521:537–540. doi: 10.1038/nature14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess R.C., Burman B., Kruhlak M.J., Misteli T. Activation of DNA damage response signaling by condensed chromatin. Cell Rep. 2014;9:1703–1717. doi: 10.1016/j.celrep.2014.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q., Barboule N., Frit P., Gomez D., Bombarde O., Couderc B., Ren G.S., Salles B., Calsou P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011;39:9605–9619. doi: 10.1093/nar/gkr656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagné J.P., Isabelle M., Lo K.S., Bourassa S., Hendzel M.J., Dawson V.L., Dawson T.M., Poirier G.G. Proteome-wide identification of poly(ADP-ribose) binding proteins and poly(ADP-ribose)-associated protein complexes. Nucleic Acids Res. 2008;36:6959–6976. doi: 10.1093/nar/gkn771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanty Y., Belotserkovskaya R., Coates J., Polo S., Miller K.M., Jackson S.P. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009;462:935–939. doi: 10.1038/nature08657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A., Okada S., Konno D., Odawara J., Yoshimi T., Yoshimura S., Kumamaru H., Saiwai H., Tsubota T., Kurumizaka H. Chd2 interacts with H3.3 to determine myogenic cell fate. EMBO J. 2012;31:2994–3007. doi: 10.1038/emboj.2012.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ismail I.H., Gagné J.P., Caron M.C., McDonald D., Xu Z., Masson J.Y., Poirier G.G., Hendzel M.J. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res. 2012;40:5497–5510. doi: 10.1093/nar/gks222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S., Kruhlak M.J., Kim J., Tran A.D., Liu J., Nyswaner K., Shi L., Jailwala P., Sung M.H., Hakim O., Oberdoerffer P. A macrohistone variant links dynamic chromatin compaction to BRCA1-dependent genome maintenance. Cell Rep. 2014;8:1049–1062. doi: 10.1016/j.celrep.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruhlak M.J., Celeste A., Dellaire G., Fernandez-Capetillo O., Müller W.G., McNally J.G., Bazett-Jones D.P., Nussenzweig A. Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J. Cell Biol. 2006;172:823–834. doi: 10.1083/jcb.200510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010;79:181–211. doi: 10.1146/annurev.biochem.052308.093131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limoli C.L., Ward J.F. A new method for introducing double-strand breaks into cellular DNA. Radiat. Res. 1993;134:160–169. [PubMed] [Google Scholar]
- Luijsterburg M.S., van Attikum H. Chromatin and the DNA damage response: the cancer connection. Mol. Oncol. 2011;5:349–367. doi: 10.1016/j.molonc.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M.S., Acs K., Ackermann L., Wiegant W.W., Bekker-Jensen S., Larsen D.H., Khanna K.K., van Attikum H., Mailand N., Dantuma N.P. A new non-catalytic role for ubiquitin ligase RNF8 in unfolding higher-order chromatin structure. EMBO J. 2012;31:2511–2527. doi: 10.1038/emboj.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg M.S., Lindh M., Acs K., Vrouwe M.G., Pines A., van Attikum H., Mullenders L.H., Dantuma N.P. DDB2 promotes chromatin decondensation at UV-induced DNA damage. J. Cell Biol. 2012;197:267–281. doi: 10.1083/jcb.201106074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mari P.O., Florea B.I., Persengiev S.P., Verkaik N.S., Brüggenwirth H.T., Modesti M., Giglia-Mari G., Bezstarosti K., Demmers J.A., Luider T.M. Dynamic assembly of end-joining complexes requires interaction between Ku70/80 and XRCC4. Proc. Natl. Acad. Sci. USA. 2006;103:18597–18602. doi: 10.1073/pnas.0609061103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan P., Onami T.M., Rajagopalan S., Kania S., Donnell R., Venkatachalam S. Role of chromodomain helicase DNA-binding protein 2 in DNA damage response signaling and tumorigenesis. Oncogene. 2009;28:1053–1062. doi: 10.1038/onc.2008.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines A., Mullenders L.H., van Attikum H., Luijsterburg M.S. Touching base with PARPs: moonlighting in the repair of UV lesions and double-strand breaks. Trends Biochem. Sci. 2013;38:321–330. doi: 10.1016/j.tibs.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Polo S.E., Jackson S.P. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 2011;25:409–433. doi: 10.1101/gad.2021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan S.K., Su T., Yen L., Jacquet K., Huang C., Cote J., Kurdistani S.K., Carey M.F. EP400 deposits H3.3 into promoters and enhancers during gene activation. Mol. Cell. 2016;61:27–38. doi: 10.1016/j.molcel.2015.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez D., Bretones G., Quesada V., Villamor N., Arango J.R., López-Guillermo A., Ramsay A.J., Baumann T., Quirós P.M., Navarro A. Mutations in CHD2 cause defective association with active chromatin in chronic lymphocytic leukemia. Blood. 2015;126:195–202. doi: 10.1182/blood-2014-10-604959. [DOI] [PubMed] [Google Scholar]
- Rulten S.L., Fisher A.E., Robert I., Zuma M.C., Rouleau M., Ju L., Poirier G., Reina-San-Martin B., Caldecott K.W. PARP-3 and APLF function together to accelerate nonhomologous end-joining. Mol. Cell. 2011;41:33–45. doi: 10.1016/j.molcel.2010.12.006. [DOI] [PubMed] [Google Scholar]
- Sfeir A., de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siggens L., Cordeddu L., Rönnerblad M., Lennartsson A., Ekwall K. Transcription-coupled recruitment of human CHD1 and CHD2 influences chromatin accessibility and histone H3 and H3.3 occupancy at active chromatin regions. Epigenetics Chromatin. 2015;8:4. doi: 10.1186/1756-8935-8-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeenk G., van Attikum H. The chromatin response to DNA breaks: leaving a mark on genome integrity. Annu. Rev. Biochem. 2013;82:55–80. doi: 10.1146/annurev-biochem-061809-174504. [DOI] [PubMed] [Google Scholar]
- Smeenk G., Wiegant W.W., Marteijn J.A., Luijsterburg M.S., Sroczynski N., Costelloe T., Romeijn R.J., Pastink A., Mailand N., Vermeulen W., van Attikum H. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J. Cell Sci. 2013;126:889–903. doi: 10.1242/jcs.109413. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A., Karlseder J., Holtgreve-Grez H., Jauch A., de Lange T. DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr. Biol. 2002;12:1635–1644. doi: 10.1016/s0960-9822(02)01179-x. [DOI] [PubMed] [Google Scholar]
- Symington L.S., Gautier J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Li L., Liang J., Shi L., Yang J., Yi X., Zhang D., Han X., Yu N., Shang Y. Histone acetyltransferase 1 promotes homologous recombination in DNA repair by facilitating histone turnover. J. Biol. Chem. 2013;288:18271–18282. doi: 10.1074/jbc.M113.473199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen B.T., Knoepfler P.S. Histone H3.3 mutations: a variant path to cancer. Cancer Cell. 2013;24:567–574. doi: 10.1016/j.ccr.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.