

Abstract
We report a case of a female infant with an elevated 17-hydroxyprogesterone level detected in the newborn screening for 21-hydroxylase deficiency, the most common cause of congenital adrenal hyperplasia. The physical examination was unremarkable including no dysmorphism and no signs of virilisation. In the absence of clinical evidence of androgen excess, as would be expected in a female infant with 21-hydroxylase deficiency, further evaluation was performed and led to the diagnosis of the extremely rare disorder, 3β-hydroxysteroid dehydrogenase deficiency. This case highlights the differential diagnosis of elevated 17-hydroxyprogesterone levels in newborn screening and the importance of correct diagnosis for improving patient care.
Background
Congenital adrenal hyperplasia (CAH) refers to a group of autosomal-recessive disorders of cortisol biosynthesis.1 The incidence of CAH is about 1:10 000–1:15 000 in most populations.2 More than 90% of cases result from steroid 21-hydoxylase deficiency caused by mutations in CYP21A2. Newborn screening for CAH became practical in the late 1970s with the use of heel stick capillary blood specimens spotted onto filter paper for measuring 17-hydroxyprogesterone (17-OHP) level, the steroid precursor just proximal to the defect (figure 1). CAH screening is justified because it prevents a potentially deadly treatable disease and in rare instances avoids incorrect male sex assignment of severely virilized female newborns.3
Figure 1.

Schematic diagram of steroidogenesis in the placenta, adrenal and gonads, and peripheral tissues. (1) Fetal progesterone derived from the placenta serves as a precursor to the synthesis of aldosterone and cortisol. (2) Loss of 21-hydroxylase activity results in deficiencies of cortisol and aldosterone, as well as elevated levels of 17-hydroxyprogesterone. (3) Loss of 11β-hydroxylase deficiency results in deficiencies of cortisol and aldosterone. (4) In 3βHSD2 deficiency, large amounts of pregnenolone, 17-hydroxypregnenolone and DHEA are secreted. These hormones are converted in the peripheral tissues by the action of 3βHSD-I, resulting in elevated levels of 17-hydroxyprogesterone. 3βHSD-I, 3β-hydroxysteroid dehydrogenase type1; 3βHSD-II, 3β-hydroxysteroid dehydrogenase type 2; 17OH, 17-hydroxy; DHEA, dehydroepiandrosterone.
Currently, the 17-OHP level is usually measured rapidly by automated time-resolved dissociation-enhanced lanthanide fluorescence immunoassay (DELFIA). Since the screening test produced a high number of false-positive results due to cross-reactions of the antibody with other steroids, the assay was reformulated in 2009 to make it less sensitive to cross-reacting compounds.3 Independent of the specificity of the immunoassay, other aspects limit accuracy of diagnosis. Clear normal ranges that distinguish disease from normal state are difficult to establish because the concentrations of 17-OHP are normally high at birth and then decrease rapidly over the first few postnatal days. Thus, diagnostic accuracy is relatively low during the first 2 days. In addition, preterm and stressed infants typically have higher concentrations than do healthy full-term infants. Accordingly, an isolated 17-OHP level must be interpreted in the context of key additional information such as pregnancy history, gestational age and birth weight, stress level and the age of the newborn on the day of the test.4
The aim of this case report is to present a female infant with an unexpected positive screening test.
Case presentation
Newborn screening results obtained at day 2 after birth from a phenotypic female infant revealed an elevated level of 17-OHP 153 nmol/L (5095 ng/dL), normal range <40 nmol/L. The patient was the product of a full term, uneventful pregnancy and delivery. Birth weight was 3270 g. The parents were second-degree cousins of Jewish ethnicity from the Caucasus. The physical examination was unremarkable including no dysmorphism and no signs of virilization.
In the absence of clinical evidence of androgen excess, the screening test result could be interpreted as false positive. However, a repeated screening test from spotted blood at age 6 days revealed an unequivocally high 17-OHP level of 500 nmol/L (16 666 ng/dL). The results of a simultaneous venous sample are presented in table 1. In addition to elevated 17-OHP levels, 11-desoxycortisol, androstenedione and testosterone levels were elevated. Potassium and sodium levels were within the normal range but plasma renin was elevated, suggesting salt wasting. Therefore, vital signs and electrolyte levels were followed closely.
Table 1.
Laboratory data
| Results of venous sample at age 6 days | Normal range for female newborns | |
|---|---|---|
| 17-OH-progesterone | 181 nmol/L (6033 ng/dL) | <7.5 nmol/L (<250 ng/dL) |
| 11-deoxycortisol | 143.2 nmol/L (5114 ng/dL) | <23 nmol/L (<821 ng/dL) |
| Testosterone | >55 nmol/L (>1571 ng/dL) | 0.7–2.2 nmol/L (20–62.8 ng/dL) |
| Androstenedione | >34.5 nmol/L (>985 ng/dL) | 0.7–10.5 nmol/L (20–300 ng/dL) |
| Cortisol | 292 nmol/L (10.5 µg/dL) | 138–690 nmol/L (5–25 µg/dL) |
| Plasma renin activity | >50 ng/mL/h | 8–17 ng/mL/h |
At age 7 days, the serum potassium levels increased to 7.3 mmol/L and sodium level decreased to 132 mmol/L. Blood pressure was normal, 86/51 mm Hg. Glucose levels were in the normal range for this age (68–108 mg/dL).
Investigations
Abdominal and pelvic ultrasound revealed a normal uterus, the adrenal glands were enlarged with a cerebriform pattern characteristic of CAH in neonates (figure 2). The karyotype was 46XX.
Figure 2.
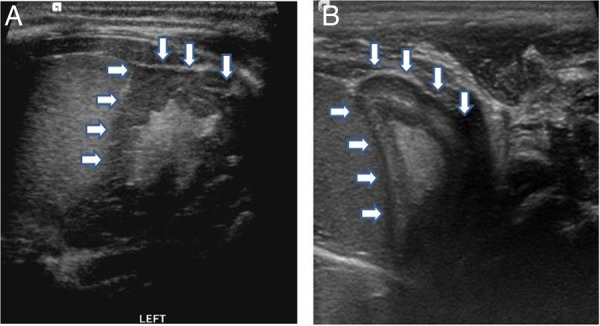
Ultrasound of the adrenal gland obtained on the 10th day, compared with a normal scan. (A) Transverse scan shows enlargement of the adrenal gland with ‘cerebriform’ appearance, the wrinkled surface resembles the appearance of brain gyri. (B) Normal adrenal gland of a healthy infant at the same age. The gland is relatively large and easy to see, the cortex is thick and hypoechoic, whereas the medulla is relatively thin and hyperechoic.
Genetic evaluations of the patient and her parents for mutations in the CYP21A2 and CYP11B1 genes, encoding the enzymes 21-hydroxylase and 11β-hydroxylase, respectively, were negative. Sequencing of HSD3B2, encoding the enzyme 3β-hydroxysteroid dehydrogenase (3βHSD), revealed a homozygous missense mutation in exon 4 of the HSD3B2 gene. This C>A mutation results in the substitution of proline for threonine in codon 222 (P222T) (figure 3). This mutation has been described as causing classic salt-wasting CAH with in vitro undetectable enzyme activity.5
Figure 3.

DNA sequencing of the patient's HSD3B2 gene. Partial nucleotide sequence of the sense strand of axon 4 of the HSD3B2 gene, depicting a homozygous missense mutation. A nucleotide substitution is indicated by an arrow from the WT sequence CCA (Pro) to ACA (Thr)—P222T.
Differential diagnosis
The abdominal ultrasound confirmed the diagnosis of CAH. Although CAH screening programmes are aimed to detect 21-hydroxylase deficiency, the most common cause of CAH, they appear to enable the detection of other rare causes of CAH: 11β-hydroxylase and 3βHSD deficiency as well. The possible differential diagnoses are detailed below.
21-hydroxylase deficiency: The vast majority of CAH cases are due to 21-hydroxylase deficiency.1 6 7 Steroid 21-hydroxylase converts 17-OHP to 11-deoxycortisol, and progesterone to 11-deoxycorticosterone (figure 1). As 11-deoxycortisol and 11-deoxycorticosterone are precursors to cortisol and aldosterone, respectively, moderate-to-complete loss of 21-hydroxylase activity results in deficiencies of both of these vital corticosteroids, as well as high levels of 17-OHP (figure 1 (2)). Depending on the severity of the genetic defect, the accumulating steroid precursors may be variably shunted to androgen pathways not impaired by the block; this can lead to prenatal virilization in affected girls. Our patient had elevated levels of 17-OHP on the screening test but no signs of androgen excess, suggesting a false-positive 17-OHP assay or a mild defect. The lack of virilization in the presence of 21-hydroxylase deficiency may also be explained by an additional disorder such as androgen insensitivity. Indeed, Giwercman et al8 described Swedish sisters who showed minimal signs of virilization due to both 21-hydroxylase deficiency and androgen receptor gene mutation.
In our case, a repeated screening test from spotted blood at age 6 days confirmed a very high level of 17-OHP. A simultaneous venous sample also revealed elevated levels of 11-desoxycortisol, androstenedione and testosterone. The cortisol level was within the normal range; however, normal levels have been measured in the postnatal days in newborns with enzyme deficiencies. These findings suggested a deficiency in other adrenal cortical enzymes that are more rarely observed as a cause of CAH.
11β-hydroxylase deficiency: The next most common cause of CAH, 11β-hydroxylase deficiency, has an incidence rate of about 1:100 000 persons and, depending on the population, may comprise as high as 5–10% of CAH cases.9 10 11β-Hydroxylase enzyme converts 11-deoxycortisol to cortisol and 11-doxycorticosterone (DOC) to corticosterone (figure 1(3)). In 11β-hydroxylase deficiency, in addition to elevated levels of 11-deoxycortisol, DOC and androgens, the chronic elevation of adrenocorticotropic hormone (ACTH) in response to low-serum cortisol, results in increased synthesis and secretion of upstream precursors, such as 17-OHP.11 Thus, it is reasonable to expect positive results in the screening test for 17-OHP.12 Virilisation and hypertension are the prominent clinical features of 11β-hydroxylase deficiency. Our patient had a high level of androgen but normal female phenotype.
3βHSD deficiency: Less than 1% of all patients with CAH have 3βHSD deficiency.5 13 The enzyme 3βHSD catalyses the conversion of Δ5–3 β-hydroxysteroid to Δ4–3 β-ketosteroid in the adrenal and gonads, and is essential for the formation of progesterone, 17-OHP and androstenedione, the precursor hormones for aldosterone, cortisol and testosterone, respectively (figure 1(1)). Salt wasting may occur in both males and females. In males, 3βHSD deficiency is characterised by incomplete virilisation of the external genitalia, due to impairment of androgen biosynthesis in the testis, whereas females exhibit mild virilisation or normal external genitalia, as in our patient.
Treatment
A combined therapy of glucocorticoid (hydrocortisone 20 mg/m²/day), mineralocorticoid (fludrocortisones 0.2 mg/day), and sodium chloride supplements (1 g NaCl/day) was initiated at the age of 7 days. Instructions to raise the hydrocortisone dose during stress were given to the parents.
Outcome and follow-up
The girl is currently 5 years old. She is treated with hydrocortisone (9 mg/m²/day) and fludrocortisone (0.1 mg/day). Her physical examination is unremarkable.
Discussion
Our case demonstrates an inherent challenge of screening programmes: what to do with laboratory results when the data do not fit the physical examination? In this current case, screening for relatively frequent 21-hydroxylase deficiency led ultimately to diagnosis of the extremely rare syndrome, 3βHSD deficiency.
In humans, there are two 3βHSD isoenzymes, types I and II, which share 93.5% homology. Type II isoenzyme (HSD3B2) is expressed in the adrenals and gonads, whereas type I (HSD3B1) is expressed in the placenta and peripheral tissues including the liver, mammary gland, prostate, skin, adipose tissue and brain.14 The adrenals of a patient with 3βHSD2 deficiency secrete very large amounts of three principal Δ5 steroids: pregnenolone, 17-hydroxypregnenolone, and dehydroepiandrosterone (DHEA). These may serve as precursor substrates for 3βHSD1. Some of the secreted 17-hydroxypregnenolone is converted by the extra-adrenal action of 3βHSD1, resulting in elevated levels of 17-OHP, confusing the diagnosis as 21-hydroxylase deficiency.15
Why were there no signs of virilization in the presence of elevated levels of androgens? Activity of fetal 3βHSD2 is greatest during urethral-fold fusion, before the 12th week of gestation. In contrast, peripheral 3βHSD1 activity occurs after the development of the external genitalia, during the second or third trimester. Consequently, males are born with incomplete virilisation, due to the lack of 3βHSD2 activity during this critical time. The relatively late appearance of 3βHSD1 activity results in elevated levels of androgens but does not induce urethral-fold fusion. Females are born with normal genitalia or only mild clitormegaly because they are exposed to high levels of androgens only during the last weeks of pregnancy.
Learning points.
A newborn screening test originally designed to identify a 21-hydroxylase deficiency enabled the discovery of 3β-hydroxysteroid dehydrogenase (3βHSD) deficiency.
3βHSD deficiency is a life-threatening condition that could be easily missed in female newborns in the absence of clinical signs of virilisation.
Footnotes
Contributors: YL-S treated the patient, carried out data collection and drafted the initial manuscript. OP-H designed the treatment plan for the patient, supervised the treatment and follow-up and edited the manuscript. Both the authors read and approved the final manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol 2009;5:490–8. 10.1038/nrendo.2009.148 [DOI] [PubMed] [Google Scholar]
- 2.Webb EA, Krone N. Current and novel approaches to children and young people with congenital adrenal hyperplasia and adrenal insufficiency. Best Pract Res Clin Endocrinol Metab 2015;29:449–68. 10.1016/j.beem.2015.04.002 [DOI] [PubMed] [Google Scholar]
- 3.White PC. Optimizing newborn screening for congenital adrenal hyperplasia. J Pediatr 2013;163:10–12. 10.1016/j.jpeds.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 4.Speiser PW. Congenital adrenal hyperplasia. F1000Res 2015;4:601 10.12688/f1000research.6543.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pang S, Wang W, Rich B et al. A novel nonstop mutation in the stop codon and a novel missense mutation in the type II 3beta-hydroxysteroid dehydrogenase (3beta-HSD) gene causing, respectively, nonclassic and classic 3beta-HSD deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab 2002;87: 2556–63. 10.1210/jcem.87.6.8559 [DOI] [PubMed] [Google Scholar]
- 6.Speiser PW, Azziz R, Baskin LS et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010;95:4133–60. 10.1210/jc.2009-2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Joint LWPES/ESPE CAH Working Group. Consensus statement on 21-hydroxylase deficiency from the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. J Clin Endocrinol Metab 2002;87:4048–53. 10.1210/jc.2002-020611 [DOI] [PubMed] [Google Scholar]
- 8.Giwercman YL, Nordenskjold A, Ritzen EM et al. An androgen receptor gene mutation (E653K) in a family with a congenital adrenal hyperplasia due to steroid 21-hyroxylase deficiency as well as in partial androgen insensitivity. J Clin Endocrinol Metab 2002;87:2623–8. 10.1210/jcem.87.6.8518 [DOI] [PubMed] [Google Scholar]
- 9.Zachmann M, Tassinari D, Prader A. Clinical and biochemical variability of congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. A study of 25 patients. J Clin Endocrinol Metab 1983;56:222–9. 10.1210/jcem-56-2-222 [DOI] [PubMed] [Google Scholar]
- 10.Holcombe JH, Keenan BS, Nichols BL et al. Neonatal salt loss in the hypertensive form of congenital adrenal hyperplasia. Pediatrics 1980;65: 777–81. [PubMed] [Google Scholar]
- 11.Nimkarn S, New MI. Steroid 11beta-hydroxylase deficiency congenital adrenal hyperplasia. Trends Endocrinol Metab 2008;19:96–9. 10.1016/j.tem.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 12.Janzen N, Riepe FG, Peter M et al. Neonatal screening: identification of children with 11beta-hydroxylase deficiency by second-tier testing. Horm Res Paediatr 2012;77:195–9. 10.1159/000337974 [DOI] [PubMed] [Google Scholar]
- 13.Pang S. Congenital adrenal hyperplasia owing to 3 beta-hydroxysteroid dehydrogenase deficiency. Endocrinol Metab Clin North Am 2001;30: 81–99, vi-vii 10.1016/S0889-8529(08)70020-3 [DOI] [PubMed] [Google Scholar]
- 14.Sahakitrungruang T. Clinical and molecular review of atypical congenital adrenal hyperplasia. Ann Pediatr Endocrinol Metab 2015;20:1–7. 10.6065/apem.2015.20.1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simard J, Ricketts ML, Gingras S et al. Molecular biology of the 3beta-hydroxysteroid dehydrogenase/delta5-delta4 isomerase gene family. Endocr Rev 2005;26: 525–82. 10.1210/er.2002-0050 [DOI] [PubMed] [Google Scholar]