

Abstract
The use of transgenic models for the study of neurodegenerative diseases has made valuable contributions to the field. However, some important limitations, including protein overexpression and general systemic compensation for the missing genes, has caused researchers to seek natural models that show the main biomarkers of neurodegenerative diseases during aging. Here we review some of these models—most of them rodents, focusing especially on the genetic variations in biomarkers for Alzheimer diseases, in order to explain their relationships with variants associated with the occurrence of the disease in humans.
Keywords: Degus, Genome, APP, APOE
Background
A valuably strategy for the study of neurodegenerative diseases like Alzheimer’s disease (AD), has been the use of transgenic mice bearing a particular human allele, to evaluate its pathogenic potential [43]. Unfortunately, most transgenic models don’t recapitulate the full spectrum of a particular disease and require protein overexpression. Although new knock-in mouse models promise to show a more realistic and faithful progress of human diseases [85], nevertheless the short life of mice still prevents an accurate association between age and sporadic diseases. Therefore, a promising alternative approach is the search for non-transgenic models (NTM), in which the main hallmarks of a pathological phenotype appear naturally during aging [15]. More recently, the extraordinary advent and growth in genomic information has lead to the availability of complete genomes from a large number of different species. The latter offer a unique opportunity to investigate the involvement of particular genes in different diseases, in NTM. So it is possible today to ask, What might be the genetic basis of a neuropathology? What would be the importance of inherited or risk genes for the start and/or progress of AD pathology?
Here we review NTM of neurodegeneration and use published genomes to compare the sequences of specific gene variants in relation to idiopathic or sporadic form of AD (SAD). The latter in order to identify in NTM of AD gene sequences (tau, APOE, APP, PSEN, Aβ) corresponding to gene variants for causing AD in human.
Genes implicated in familial Alzheimer´s disease
AD in its familial (early onset) or sporadic (late onset) form is characterized by the occurrence of a series of critical biomarkers. Among these the main indicators of neural degeneration, including synaptic failure and cognitive decline [88] are the accumulation of phosphorylated tau protein, which form neurofibrillary tangles (NFT), and the overexpression of amyloid precursor protein (APP), which leads to the accumulation of Amiloid-β (Aβ) peptide in senile plaques,
APP is an integral membrane protein present in the brain [50] and has been related to diverse functions including cell adhesion, growth factor and signaling associated with synaptogenesis and synaptic plasticity [98]. The proteolytic processing of APP releases potentially neurotoxic species, e.g., the Aβ peptide, which is considered one of the key pathogenic events in AD. The deregulation of both APP and Aβ has been linked to the hereditary or familial form of AD (FAD). For example, the Aβ peptide is present in meningovascular brain deposits in AD and in Down syndrome patients [46, 47], and is also a main component of senile plaques [49, 71, 89]. The cloning of the APP gene [58] showed that more than 20 mutations are associated to FAD. APP belongs to a conserved superfamily, which in mammals includes APP and the APP-like proteins, APLP1 and APLP2 [25]. APP isoforms 770, 751, and 695 residues in length, are produced by alternative splicing of exons 7 and 8 [90], the latter being the most abundant form in neurons [51]. A comparative analysis of APP revealed the existence of an important number of conserved amino acids [57]; Fig. 1). The Aβ region differs by 3 amino acids in rodents vs. humans (R5G, Y10F and H13R). In fact, the presence of these residues affects APP processing through its aminoacid oxidation induced by free radical generation systems and the subsequent generation of cross-linking protein interactions [37]. Interestingly, it has been suggested that the absence of these residues in Aβ rodent makes it less prone to forming amyloid aggregates [41, 77]). These differences between species could also provide protection against β-secretase processing [28]. This may also be the reason why the transgenic mouse model of AD requires human APP to be over-expressed in order to cause the AD pathogenesis, a feature that limits the utility of such models [32, 45].
Fig. 1.
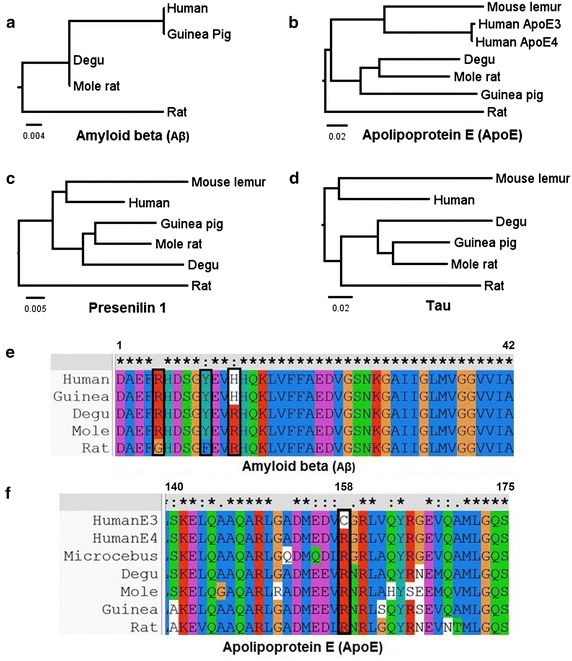
Phylogenetic analysis of 4 proteins involved in Alzheimer disease. a Phylogenetic tree of Aβ. The tree shows that the sequence for the degu is identical to that of the mole rat and is more similar to that of human and guinea pig than it is to that of the rat. b Phylogenetic tree of ApoE, showing that degu ApoE is more similar to the human protein than it is to rat ApoE. c Phylogenetic tree of Presenilin 1. The tree shows that the sequence for the degu is grouped with that of the mole rat and the guinea pig and is separate from the human and mouse lemur grouping. As occurs for Aβ and ApoE proteins, the Presenilin 1 sequence for the rat is outside of both groupings. d Phylogenetic tree of tau. In this case, the sequence for the rat is grouped with that of other rodents. e Multiple alignment of Aβ shows the three aminoacids (boxed) that differ between the rat and the other species; by contrast there is only one difference between degu and human APP sequence (H684R). f Multiple alignment of ApoE. There is a single difference between human ApoE3 and ApoE4 alleles (C112R, boxed); interestingly, this ApoE4 variant is the one that is present in all the other species. Aβ mouse lemur sequence is absent in the phylogram showed in a, because it was not available at the time of analysis
Other proteins involved in FAD are presenilins [87]. Mutations in the presenilin 1 gene (PSEN1) are present in certain families with clinical early onset AD [93], whereas mutations in the presenilin 2 gene (PSEN2) appear to lead to AD at a more advanced age [23, 64, 83]. Presenilins are catalytic proteins of γ-secretase complexes responsible for producing Aβ from APP [52]; thus, mutations in PSEN1 and PSEN2 affect the generation of Aβ peptide [13, 22, 27, 35, 40, 95]. To date, over 180 mutations in PSEN1 and 15 mutations in PSEN2 have been reported with potential pathogenicity related to AD. Remarkably, the incorporation of transgenes bearing missense mutations in presenilins in APP transgenic mice seems to accelerate the processing and production of Aβ [35, 53].
The search in human cerebrospinal fluid for proteins that could bind Aβ peptides led to the identification of apolipoprotein E (ApoE), which was classified as a genetic risk factor in SAD [97]. There are three alleles of ApoE: ApoE2, ApoE3, and ApoE4. The age of onset of AD decreases with increasing copies of the ApoE4 allele [24, 84] such that, for example, homozygosity for this allele increases the risk of AD 12-fold [24, 39]. Nevertheless, the inheritance of ApoE4 is not sufficient for the development of AD. Interestingly, the deletion of the two copies of mouse ApoE in APP transgenic mice causes a reduction in amyloid accumulation [4] and APP mice expressing human ApoE3 or ApoE4 develop less Aβ deposits than do animals without ApoE [54].
Another key factor for AD is the microtubule binding protein, tau, for which its hyperphosphorylated form is a component of neurofibrillary tangles (NFT) [60, 73, 102]. NFT are present in both familiar and sporadic forms of AD; however, to date specific mutations in the MAPT gene haven’t been associated with AD, suggesting that the tau pathology occurs downstream of the detrimental cascades caused by Aβ. Tau depositions are also associated with a number of other tauopathies, including frontotemporal dementia (FTD), Pick disease, and corticobasal degeneration [91]. Several mutations have been identified in tauopathies [100], which have then been used to generate transgenic mice for the study of this class of diseases [6]. Furthermore, a transgenic tau mouse related to FTD and bearing mutations in APP and PSEN1, have provided evidence that amyloid deposition develops prior to tangle pathologies [74] and that memory deficits are related to increases in Aβ [82].
Natural models of AD
During the last decades, and prompted by an understanding of the genetic basis of AD, several transgenic mice models have been developed that attempt to recreate sequence variants associated with FAD [36]. Moreover, newer knock-in mice models show a more realistic development of AD avoiding the overexpression of APP and of APP fragments such as Aβ, by showing a more precise pattern of expression [21, 65, 85]. However, despite these advantages the limited lifespan of mice precludes the appropriate analysis of these models in the context of normal aging. Nevertheless, the advent of modern genetics and bioinformatics has led to a more comprehensive characterization of the genomes of animal models and has facilitated the search of NTM for which aging is associated with neurodegeneration [68]. Below, we describe some NTM that are relevant for the study of AD (Table 1).
Table 1.
Natural animal models of AD
| Model | Lifespan | Neuropathology | Reference |
|---|---|---|---|
| Naked mole-rat | 28–32 years | Decreased Tau phosphorylation and amyloid-β accumulation starting at 3 months of age | [38, 76] |
| Mouse lemur | 10–15 years | Senile plaques and neurofibrillary tangles present in cortex at 9 years of age, and hippocampus at 10 years. Procedural memory is normal, but executive function is altered at 7–11 years | [79, 94] |
| Octodon degus | 10–12 years | Amyloid-β oligomers are present at 6 months, amyloid plaques and neurofibrillary tangles starting at 6 years of age. Synaptic dysfunction occurs prior to the detection of plaques and tangles | [2, 3, 55] |
| Guinea pig | 5–8 years | Amyloid-β deposition and Tau phosphorylation present in frontal cortex at 4 years of age | [7] |
Microcebus murinus (gray mouse lemur) is a small nocturnal primate native to Madagascar that reaches the age of 8–14 years in captivity. Interestingly, at 5 years of age, 20 % of M. murinus show morphological signs of brain neurodegeneration [61] and cognitive decline [78, 79] that are similar to those of humans with AD [12, 31]. These signs include the accumulation of Aβ plaques in 15 % of the adult population, the formation of NFT [44, 63], and the loss of cholinergic neurons [72]. In M. murinus the gene responsible for the formation of Aβ plaques shows high sequence similarity with the human allele [12], and 92,7 % similarity with human ApoE4 allele [18] (Fig. 1). However, Aβ deposits and plaques have a different distribution in humans: they occur first in hippocampus, whereas in M. murinus they are first detected in other cortical areas [44]. These signs have allowed the population of old M. murinus to be classified into 4 groups: (i) animals presenting amyloid plaques without (5–10 %) or (ii) with a tauopathy (1 %); (iii) animals presenting tauopathy in the absence of amyloid plaques (1 %) and (iv) animals with no lesions (80–90 %). These data suggest that most of the mouse lemurs undergo a normal aging, whereas some of them show this age-associated pathology [63].
Another interesting species is Cavia porcellus (Guinea pig), a rodent native to the Andes Mountains. The aged guinea pig presents well-described, diffuse, amyloid-β deposits in the region of the hippocampus [7]. Interestingly its APP is highly homologous to the human protein (97 %) [10], and is processed in a similar way in vitro [9]. The APP695 variant is the most abundant form in the brain, whereas the longest isoforms are primarily expressed in peripheral organs [10]. High sequence similarity between human and guinea pig PSEN1 has also been documented, highlighting guinea pig as a good model for SAD [92]. However, the presence of senile plaques and neurofibrillary tangles and their correlation with cognitive and synaptic impairments has not yet been reported for the aged guinea pig brain.
The naked mole-rat (Heterocephalus glaber) (NMR) is a small rodent with a lifespan of over 30 years in captivity [16]. It presents early signs of neurodegeneration related to vitamin D deficiencies [17] and high levels of oxidative stress [1]. Moreover, appreciable levels of Aβ can be detected, and its sequence differs in only one amino acid with that of the human protein. Remarkably the mole rat brain acquires and tolerates high levels Aβ, but does not form plaques [38]. Moreover, tau phosphorylation levels rise during the early stages of life, resulting in an increase in molecular weight, reaching 88 kDa in the first year of life. This increase in phosphorylation declines at the end of development but occurs throughout life. Unlike other transgenic models such as 3xTgAD the phosphorylation in NMR is specifically localized in the axonal region but not in the somatodendritic compartment, where synaptic alterations could be generated [76]. Although phosphorylation is related to the loss of stability of microtubules, in NMR it is a tightly regulated kinase mechanism and it is essential for maintaining the dynamics of the cytoskeleton [76]. Interestingly, NMR tau protein is very similar to human tau (95 % similarity) and shows 100 % identity in the microtubule-binding domain, suggesting the inmportance of the location and regulation of the phosphorylation processes and not the sequence per se.
Octodon degus (degu) is a rodent native of South America, belonging to the Octodontidae family [96]. The Octodon genus includes three species: O.degu, O. lunatus, and O. bridgesi and is related to the Chinchilloidea and Cavioidea (e.g. guinea pig) families [75]. During aging degus present intracellular and extracellular deposits of Aβ, intracellular accumulations of tau-protein, and strong astrocytic responses, suggesting that they represent a natural model for sporadic AD [55, 56]. More recently, Van Groen et al. [99] has shown that 6 year-old degus have Aβ and tau deposits in the hippocampus, and in the blood vessel walls. We have demonstrated that degu develops synaptic changes related to AD, which explains the early impairments in cognitive and neural plasticity observed before the appearance of fibrillar deposition [3]. We have also shown that the memory of degu declines during aging, correlated with an increase in the levels of soluble Aβ, in particular the Ab*56 oligomer. In a small number of cases, very old degus (7–9 years old) also seems to develop Aβ plaques, similar to what occurs in naked mole rats. Interesting, more recently we have shown that the retina (which is also part of the nervous system) of aged degus presents, as does the brain, the main hallmarks of AD [34].
The draft genome of the degu has recently been completed at the Broad Institute (Boston) and some important questions can now be addressed, such as the presence and homology of genes involved in the familial, or risk forms of human AD. Here we used this preliminary information to carry out bioinformatics analysis comparing genomic sequences of NTM.
Methods
Genomes
The species and protein sequence IDs used for this analysis were: Aβ (from APP sequence): Homo sapiens P05067.3, Cavia porcellus Q60495.2, Octodon degus XP_004627753.1, Heterocephalus glaber XP_004842285.1, Rattus norvegicus P08592.2; ApoE:Homo Sapiens P02649.1, Cavia porcellus P23529.1, Octodon degus XP_004644379.1, Heterocephalus glaber XP_004910131.1, Microcebus murinus ENSMICP00000012801, Rattus norvegicus NP_001257613.1; Tau:Homo SapiensP10636.5, Cavia porcellus XP_003465958.1, Octodon degus XP_004630049, Heterocephalus glaber EHB10652.1, Microcebus murinus, ENSMICP00000004446, Rattus norvegicus P19332.3; Presenilin 1:Homo Sapiens P49768.1, Cavia porcellus XP_003472446.1, Octodon degus XP_004624901.1, Heterocephalus glaber XP_004837306.1, Microcebus murinus CAA95930.1, Rattus norvegicus NP_062036.2.
Results and discussion
Phylogenetic trees
Multiple alignments followed by neighbor-joining analysis in order to generate the phylogenetic trees were performed using the Clustal X2 program as shown in Fig. 1.
AD is a neurodegenerative condition and is the main cause of dementia that affects over 24 millions of people worldwide [11]. AD occurs in two forms, the rare early-onset genetic or familial Alzheimer’s disease (FAD) which represents <1 % of diagnosed cases, and the frequent late-onset form or sporadic Alzheimer’s disease (SAD) [11]. Transgenes bearing mutations in genes linked to human FAD have been incorporated into mice to model the disease, and have provided valuable information about the neuropathology of the disease [36]. However, one difficulty associated with the use of this short-lived model is that aging is a main risk factor for AD. For this reason, it is imperative to search for long-lived animal models that can be used to study the neuropathology of AD.
Here we analyzed, for several natural models (naked mole, guinea pig, degus) as explained in the introduction, the sequences of proteins that have traditionally been associated with AD. These include ApoE, tau, Presenilins and Aβ (APP). In the resulting phylogenetic trees or phylograms, the length of the branches represents the amount of change in a sequence with respect to a common ancestor [8]. Our results show different relationships between species for some important markers of AD. Thus, we found that the degu is grouped with the mole rat and the guinea pig in all analyses; interestingly, the Aβ sequence for the guinea pig is identical to that of the human, because it includes a histidine at position 13 instead of the arginine, as occurs in other rodents. However, the most important change is likely to be H13R that is a critical residue for the aggregation of the Aβ [66]. Degu Aβ presents high homology with the human protein, differing in only one amino acid; this supports the hypothesis that the occurrence of amyloidogenic cascade is associated with the presence of Aβ oligomers [3] and amyloid deposits in older animals [55]. However, we also note that the mole rate shows 100 % homology (Fig. 1), presents high levels of Aβ [38], but does not show amyloid deposits. It will be important to determine if this rodent shows alteration in memory processes, which would establish it as a model of neurodegeneration and memory impairments in the absence of amyloid plaque. It will also be important to understand the markers present in the degu, as this rodent shows memory and synaptic impairments, and an accumulation of soluble amyloid oligomer but appears not to develop amyloid plaques.
Although our results for Presenilin 1 show a greater relatedness between the isoforms of human and, degu, guinea pig, and mole rat, compared to those of others rodents such the rat (Fig. 1c), there are more than 150 mutations in these proteins associated with Familial AD [26]; for this reason we need to perform more detailed analysis for each mutation to establish differences in the presence of potentially mutagenic variants in each of these species. Nevertheless, it should be noted that D257 and D385, two essential aminoacids of the catalytic site [101], are conserved in all species. In addition the aminoacid alanine 246, for which the substitution for a glutamate is associated with increased deposition of beta amyloid in transgenic models of AD [14], is also conserved in all species of this study. Interestingly, Sharman et al. reported that the S212Y mutation occurring in transmembrane domain 4 of PSEN1, and which was previously identified in a family with FAD [81], is conserved in guinea pig [92]. Additionally, a normal truncated PSEN2 isoform termed PS2V, which in humans has been implicated in AD [86], is also present in guinea pig but is absent in mice and rats [92], further confirming the close similarity between guinea pig and human presenilins.
In humans the ApoE gene has different isoforms that can be related to the occurrence of sporadic AD. These isoforms differ only in the variation of two amino acids and are designated as ApoE 2 (Cys112, Cys158), ApoE 3 (Cys112, Arg158), and ApoE4 (Arg112, Arg158). Within these isoforms ApoE 4 has been described as the major risk factor in sporadic late onset Alzheimer’s disease (LOAD) [67], where homozygosity for the E4 allele may be sufficient to develop AD at the age of 80 years [24]. This allele has a single amino acid change with respect to the E3 allele (C112R) [24]. ApoE is fundamental for the regulation of clearance and deposition of Aβ in the brain, which may explain its association with sporadic cases of AD [67]. Studies in HEK-293 cells demonstrate that the binding between ApoE and Aβ is less effective for ApoE4 than ApoE3 [62]. On the other hand ApoE can disrupt the clearance of Aβ from brain mouse in an isoform specific manner [30]. These findings suggest that the differences in the amino acids between the three isoforms of ApoE can fundamentally affect its role in the regulation of Aβ.
Surprisingly, we found that degu have the arginine substitution present in the ApoE4 pathogenic human allele. In addition, our results show that, in the sequence of human of ApoE 3, the amino acid at position 112 is a cysteine, unlike degu and other rodents, which have an arginine at this position as is found in the human ApoE4 form. These data would suggest that this protein could potentially be pathological for the rodents. However all rodents have this amino acid, not only those that show sporadic AD such as the degu. Thus, the functional consequences of these substitutions await further sequence and protein structure analyses.
One possible explanation for this anomaly arises from the fact that ApoE has a region that is essential for binding lipids. In humans, this region has a glutamate at position 255 that is critical for the structure of the “toxic” E4 allele because it generates an ionic interaction with Arg61 that changes the position of the lipid binding region and the protein structure. On the other hand, the presence of Cys112 in the E3 allele leads to an interaction between this amino acid and Arg61, causing Glu255 and Arg61 not to form the interaction that occurs in E4 [69]. Studies in cell culture show that the substitution of an Arg at position 61 for a Thr generates a disruption in the interaction with Glu255, generating an E3 like structure [103]. In the case of rodents such as degu this amino acid is a Thr so an interaction with the lipid binding region would not possible despite the presence of Arg112. Thus, the presence of Arg112 may not be a “toxic” substitution in rodents. However, it is clear that amino acids Thr61, Arg112, and Glu255 are important for maintaining the function of ApoE. More research is needed to determine the presence of polymorphisms in these and other amino acids that can account for potential pathogenic substitutions that are present in models of sporadic AD such as degu.
On the other hand, it has been speculated with the possibility of certain “protective” polymorphisms in the case of human ApoE, since that inheritance of ApoE2 has been associated with a decrease in the AD risk [39]. This allele has a lower affinity to LDL compare to other alleles [42], leading to a decrease in their clearance resulting in an increase in plasma, cerebrospinal fluid and brain levels [70]. That increase in ApoE2 availability in the brain could explain the greater Aβ clearance observed in human and mice models. [5, 20]. In the E2 allele, Cys158 causes Asp154 to now interact with Arg150, thus modifying the LDL binding domain [33]. By contrast, in the E3 allele the amino acid Arg158 forms a salt bridge interaction with Asp154, which does not affect the LDL binding domain (amino acids 134–150).
In degu, the amino acids at these positions are those present in the E3 allele (Arg150, Asp 154, Arg158). Thus, it will also be necessary to determine the presence of different protective polymorphisms in this rodent. Consistent with this, brain levels of ApoE are altered (in the frontal cortex and hippocampus) depending on ApoE genotype, where animals homozygous for the E4 allele have lower protein levels than those with the E3 allele, which in turn are lower than those with the E2 allele [80]. Genetic and crystallographic research is needed to understand the consequences of each of the various polymorphisms on ApoE protein structure and function. It will also be important to determine how hetero- and homozygosity for the different ApoE alleles might affect the onset of sporadic of AD in the degu.
Interestingly, a recent report by Deacon et al. [29] shows that poor-burrower degus have high levels of Aβ1–42, APOE, and APP, cytokine TNF-α and oxidative stress marker NFE2L2 compared to good-burrowers degus. This result, although preliminary, suggest that degus present increased inflammation during aging and AD like diseases, adding a new group of protein targets to be considered in this type of bioinformatics analysis.
Regarding the tau protein, we found that for all rodents its sequences are grouped in a branch of the tree that is separate from that for lemurs and humans (Fig. 1), which show the highest similarity [76]. Since there is no evidence for a link between polymorphisms in tau and risk of AD, and the SNPs identified in the MAPT gene do not influence the risk of AD [59], we cannot extrapolate much from these data. However, the regulation of MAPT transcriptional splicing is known to be critical for normal tau function [48]. It has also been shown that an imbalance in the ratio of tau isoforms with 3 (3R) vs. 4 (4R) repeats, which are derived from the alternative splicing of Exon10 of the human MAPT, is associated with AD and Tauopathies [19]. In this regard, guinea pig show both 3R and 4R tau isoforms and, interestingly, under cholesterol intake, guinea pig shows an increase in 3R isoforms suggesting a relationship between AD risks factors and AD in guinea pig [92]. It would be interesting to determine if the 3R/4R ratio also is disturbed in the other NTM’s.
Overall, our analyses reveal that the sequences of genes associated with AD risk are more similar to human in NTM than in rat or mouse, explaining why the biomarkers encoded by these genes appear during aging in NTM. The fact that the temporary or regional expression of these markers differ between the different models discussed here provide an opportunity to explore different factors that can accelerate the progress of AD-related pathological events. The understanding of these processes also provides a unique opportunity to explore new drugs or therapeutic strategies against this devastating disease.
Authors’ contributions
AGP conceived of the study and wrote the paper. CZ and AA participated to draft the literature and the manuscript. GV and JE contribute with the sequence alignment and discussion. All authors read and approved the final manuscript.
Acknowledgements
This work was partially supported by FONDECYT #1150638; Millennium Institute ICM-P09-022-F. GV is an undergraduate student from the Biochemistry program, Instituto de Química, Pontificia Universidad Católica de Valparaíso, Chile.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Claudia Salazar, Email: csalazars87@gmail.com.
Gonzalo Valdivia, Email: gonzalo.vu@gmail.com.
Álvaro O. Ardiles, Email: alvaro.ardiles@cinv.cl
John Ewer, Email: john.ewer@uv.cl.
Adrián G. Palacios, Phone: +56 32 2508048, Email: adrian.palacios@uv.cl
References
- 1.Andziak B, O’Connor TP, Qi W, DeWaal EM, Pierce A, Chaudhuri AR, Van Remmen H, Buffenstein R. High oxidative damage levels in the longest- living rodent, the naked mole-rat. Aging Cell. 2006;5:463–471. doi: 10.1111/j.1474-9726.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 2.Ardiles AO, Ewer J, Acosta ML, Kirkwood A, Martinez AD, Ebensperger LA, Bozinovic F, Lee TM, Palacios AG. Octodon degus (Molina 1782): a model in comparative biology and biomedicine. Cold Spring Harb Protoc. 2013;2013(4):312–318. doi: 10.1101/pdb.emo071357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ardiles AO, Tapia-Rojas CC, Mandal M, Alexandre F, Kirkwood A, Inestrosa NC, Palacios AG. Postsynaptic dysfunction is associated with spatial and object recognition memory loss in a natural model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2012;109(34):13835–13840. doi: 10.1073/pnas.1201209109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM. Lack of apolipprotein E dramatically reduces amyloid β-peptide deposition. Nature Genet. 1997;17:263–264. doi: 10.1038/ng1197-263. [DOI] [PubMed] [Google Scholar]
- 5.Bales KR, Liu F, Wu S, Lin S, Koger D, DeLong C, Hansen JC, Sullivan PM, Paul SM. Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J Neurosci. 2009;29(21):6771–6779. doi: 10.1523/JNEUROSCI.0887-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballatore C, Lee VM, Trojanowski JQ. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat Rev Neurosci. 2007;8:663–672. doi: 10.1038/nrn2194. [DOI] [PubMed] [Google Scholar]
- 7.Bates K, Vink R, Martins R, Harvey A. Aging, cortical injury and Alzheimer’s disease-like pathology in the guinea pig brain. Neurobiol Aging. 2014;35(6):1345–1351. doi: 10.1016/j.neurobiolaging.2013.11.020. [DOI] [PubMed] [Google Scholar]
- 8.Baum D. Reading a phylogenetic tree: the meaning of monophyletic groups. Nature Educ. 2008;1(1):190. [Google Scholar]
- 9.Beck M, Bigl V, Roßner S. Guinea pigs as a nontransgenic model for APP processing in Vitro and in Vivo. Neurochem Res. 2003;28(3–4):637–644. doi: 10.1023/A:1022850113083. [DOI] [PubMed] [Google Scholar]
- 10.Beck M, Müller D, Bigl V. Amyloid precursor protein in guinea pigs–complete cDNA sequence and alternative splicing. Biochim Biophys Acta. 1997;1351(1–2):17–21. doi: 10.1016/S0167-4781(96)00232-1. [DOI] [PubMed] [Google Scholar]
- 11.Bekris LM, Yu CE, Bird TD, Tsuang DW. Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol. 2010;23(4):213–227. doi: 10.1177/0891988710383571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bons N, Rieger F, Prudhomme D, Fisher A, Krause KH. Microcebus murinus: a useful primate model for human cerebral aging and Alzheimer’s disease? Genes, Brain and Behavior. 2006;5(2):120–130. doi: 10.1111/j.1601-183X.2005.00149.x. [DOI] [PubMed] [Google Scholar]
- 13.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17(5):1005–1013. doi: 10.1016/S0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 14.Borchelt D, Ratovitski T, Van Lare J, Lee M, Gonzales V, Jenkins N, Copeland N, Price D, Sisodia S. Accelerated Amyloid Deposition in the Brains of Transgenic Mice Coexpressing Mutant Presenilin 1 and Amyloid Precursor Proteins. Neuron. 1997;19:939–945. doi: 10.1016/S0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 15.Braidy N, Munoz P, Palacios AG, Castellano-Gonzalez G, Inestrosa NC, Chung RS, Sachdev P, Guillemin GJ. Recent rodent models for Alzheimer’s disease: clinical implications and basic research. J Neural Transm. 2012;119(2):173–195. doi: 10.1007/s00702-011-0731-5. [DOI] [PubMed] [Google Scholar]
- 16.Buffenstein R. The naked mole-rat: a new long-living model for human aging research. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 2005;60(11):1369–1377. doi: 10.1093/gerona/60.11.1369. [DOI] [PubMed] [Google Scholar]
- 17.Buffenstein R, Yahav S. Cholecalciferol has no effect on calcium and inor- ganic phosphorus balance in a naturally cholecalciferol-deplete subterranean mammal, the naked mole rat (Heterocephalus glaber) J Endocrinol. 1991;129:21–26. doi: 10.1677/joe.0.1290021. [DOI] [PubMed] [Google Scholar]
- 18.Calenda A, Jallageas V, Silhol S, Bellis M, Bons N. Identification of a unique apolipoprotein E allele in Microcebus murinus; ApoE brain distribution and co-localization with beta-amyloid and tau proteins. Neurobiol Dis. 1995;2(3):169–76. doi: 10.1006/nbdi.1995.0018. [DOI] [PubMed] [Google Scholar]
- 19.Cárdenas AM, Ardiles AO, Barraza N, Baéz-Matus X, Caviedes P. Role of tau protein in neuronal damage in Alzheimer’s disease and Down syndrome. Arch Med Res. 2012;43(8):645–654. doi: 10.1016/j.arcmed.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 20.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011;3(89):89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer’s disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103(9):3410–3415. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, Fraser P, St George Hyslop P, Selkoe DJ. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat Med. 1997;3(1):67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- 23.Clarke RF, Hutton M. and the Alzheimer’s Collaborative Group The structure of the presenilin-1 (S182) gene and identification of six novel mutations in early onset AD families. Nature Genet. 1995;11:219–222. doi: 10.1038/ng1095-219. [DOI] [PubMed] [Google Scholar]
- 24.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261(5123):921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 25.Coulson EJ, Paliga K, Beyreuther K, Masters CL. What the evolution of the amyloid protein precursor supergene family tells us about its function. Neurochem Int. 2000;36:175–184. doi: 10.1016/S0197-0186(99)00125-4. [DOI] [PubMed] [Google Scholar]
- 26.De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8(2):141–6. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391(6665):387–90. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 28.De Strooper B, Simons M, Multhaup G, van Leuven F, Beyreuther K, Dotti CG. Production of intracellular amyloid-containing fragments in hippocampal neurons expressing human amyloid precursor protein and protection against amyloidogenesis by subtle amino acid substitutions in the rodent sequence. EMBO J. 1995;14:4932–8. doi: 10.1002/j.1460-2075.1995.tb00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deacon RM, Altimiras FJ, Bazan-Leon EA, Pyarasani RD, Nachtigall FM, Santos LS, Tsolaki AG, Pednekar L, Kishore U, Biekofsky RR, Vasquez RA, Cogram P. Natural AD-Like neuropathology in Octodon degus: impaired burrowing and neuroinflammation. Curr Alzheimer Res. 2015;12(4):314–322. doi: 10.2174/1567205012666150324181652. [DOI] [PubMed] [Google Scholar]
- 30.Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. ApoE isoform – specific disruption of amyloid β peptide clearance from mouse brain. J Clin Investig. 2008;118(12):4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhenain M, Chenu E, Hisley CK, Aujard F, Volk A. Regional Atrophy in the Brain of Lissencephalic Mouse Lemur Primates: measurement by automatic histogram-based segmentation of MR images. Magn Reson Med. 2003;50(5):984–992. doi: 10.1002/mrm.10612. [DOI] [PubMed] [Google Scholar]
- 32.Dodart JC, Mathis C, Bales KR, Paul SM. Does my mouse have Alzheimer’s disease? Genes Brain Behav. 2002;1(3):142–155. doi: 10.1034/j.1601-183X.2002.10302.x. [DOI] [PubMed] [Google Scholar]
- 33.Dong LM, Parkin S, Trakhanov SD, Rupp B, Simmons T, Arnold KS, Newhouse YM, Innerarity TL, Weisgraber KH. Novel mechanism for defective receptor binding of apolipoprotein E2 in type III hyperlipoproteinemia. Nat Struct Biol. 1996;3(8):718–722. doi: 10.1038/nsb0896-718. [DOI] [PubMed] [Google Scholar]
- 34.Du LY, Chang LY, Ardiles AO, Tapia-Rojas C, Araya J, Inestrosa NC, Palacios AG, Acosta ML. Alzheimer’s disease-related protein expression in the retina of Octodon degus. PLoS One. 2015;10(8):e0135499. doi: 10.1371/journal.pone.0135499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Duff K, Eckman C, Zehr C, Yu X, Prada C-M, Perez-Tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42 (43) in brains of mice expressing mutant presenilin1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- 36.Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dyrks T, Dyrks E, Masters CL, Beyreuther K. Amyloidogenicity of rodent and human beta A4 sequences. FEBS Lett. 1993;324(2):231–236. doi: 10.1016/0014-5793(93)81399-K. [DOI] [PubMed] [Google Scholar]
- 38.Edrey YH, Medina DX, Gaczynska M, Osmulski PA, Oddo S, Caccamo A, Buffenstein R. Amyloid beta and the longest-lived rodent: the naked mole-rat as a model for natural protection from alzheimer’s disease. Neurobiol Aging. 2013;34(10):2352–2360. doi: 10.1016/j.neurobiolaging.2013.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278(16):1349–1356. doi: 10.1001/jama.1997.03550160069041. [DOI] [PubMed] [Google Scholar]
- 40.Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23(3):335–348. doi: 10.1016/S0197-4580(01)00330-X. [DOI] [PubMed] [Google Scholar]
- 41.Fraser PE, Nguyen JT, Inouye H, Surewicz WK, Selkoe DJ, Podlisny MB, Kirschner DA. Fibril formation by primate, rodent, and Dutch-hemorrhagic analogues of Alzheimer amyloid β-protein. Biochemistry. 1992;31:10716–10723. doi: 10.1021/bi00159a011. [DOI] [PubMed] [Google Scholar]
- 42.Fryer JD, Simmons K, Parsadanian M, Bales KR, Paul SM, Sullivan PM, Holtzman DM. Human apolipoprotein E4 alters the amyloidbeta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005;25:2803–2810. doi: 10.1523/JNEUROSCI.5170-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gama Sosa MA, De Gasperi R, Elder GA. Modeling human neurodegenerative diseases in transgenic systems. Hum Genet. 2012;4:535–563. doi: 10.1007/s00439-011-1119-1. [DOI] [PubMed] [Google Scholar]
- 44.Giannakopoulos P, Silhol S, Jallageas V, Mallet J, Bons N, Bouras C, Dela P. Quantitative analysis of tau protein-immunoreactive accumulations and β amyloid protein deposits in the cerebral cortex of the mouse lemur. Microcebus murinus. Acta Neuropathol. 1997;94:131–139. doi: 10.1007/s004010050684. [DOI] [PubMed] [Google Scholar]
- 45.Glazner KA, Odero GL, Anema E, Motnenko A, Schapansky J, Grossman D, Oliver DR, Glazner GW, Albensi BC. Strain specific differences in memory and neuropathology in a mouse model of Alzheimer’s disease. Life Sci. 2010;86(25–26):942–950. doi: 10.1016/j.lfs.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 46.Glenner GG, Wong CW. Alzheimer’s disease and Down’s syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291X(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 47.Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 48.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1999;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 49.Gorevic P, Goni F, Pons-Estel B, Alvarez F, Peress R, Frangione B. Isolation and partial characterization of neurofibrillary tangles and amyloid plaque cores in Alzheimer’s disease: immunohistological studies. J Neuropathol Exp Neurol. 1986;45:647–664. doi: 10.1097/00005072-198611000-00004. [DOI] [PubMed] [Google Scholar]
- 50.Gralle M, Ferreira ST. Structure and functions of the human amyloid precursor protein: the whole is more than the sum of its parts. Prog neurobiol. 2007;82(1):11–32. doi: 10.1016/j.pneurobio.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 51.Haass C, Hung Y, Selkoe DJ. Processing of beta-amyloid precursor protein in microglia and astrocytes favors an internal localization over constitutive secretion. J Neurosci. 1991;11(12):3783–3793. doi: 10.1523/JNEUROSCI.11-12-03783.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haass C, Kaether C, Thinakaran G, Sisodia S. Trafficking and proteolytic processing of APP. S Cold Spring Harb Perspect Med. 2012;2(5):a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O’Campo Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin1 transgenes. Nature Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- 54.Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM. Expression of human apolipoprotein E reduces amyloid-beta deposition in a mouse model of Alzheimer’s disease. J Clin Invest. 1999;103:R15–R21. doi: 10.1172/JCI6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inestrosa NC, Reyes AE, Chacon MA, Cerpa W, Villalon A, Montiel J, Merabachvili G, Aldunate R, Bozinovic F, Aboitiz F. Human-like rodent amyloid-beta-peptide determines Alzheimer pathology in aged wild-type Octodon degu. Neurobiol Aging. 2005;26(7):1023–1028. doi: 10.1016/j.neurobiolaging.2004.09.016. [DOI] [PubMed] [Google Scholar]
- 56.Inestrosa NC, Ríos JA, Cisternas P, Tapia-Rojas C, Rivera DS, Braidy N, Zolezzi JM, Godoy JA, Carvajal FJ, Ardiles AO, Bozinovic F, Palacios AG, Sachdev PS. Age Progression of Neuropathological Markers in the Brain of the Chilean Rodent Octodon degus, a Natural Model of Alzheimer’s Disease. Brain Pathol. 2015 doi: 10.1111/bpa.12226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnstone EM, Chaney MO, Norris FH, Pascual R, Little SP. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10:299–305. doi: 10.1016/0169-328X(91)90088-F. [DOI] [PubMed] [Google Scholar]
- 58.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325(6106):733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 59.Kauwe JS, Cruchaga C, Mayo K, Fenoglio C, Bertelsen S, Nowotny P, Galimberti D, Scarpini E, Morris JC, Fagan AM, Holtzman DM, Goate AM. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci U S A. 2008;105(23):8050–8054. doi: 10.1073/pnas.0801227105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kosik KS, Joachim CL, Selkoe DJ. Microtubule-associated protein, tau, is a major antigenic component of paired helical filaments in Alzheimer’s disease. Proc Natl Acad Sci USA. 1986;83:4044–4048. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kraska A, Dorieux O, Picq JL, Petit F, Bourrin E, Chenu E, Volk A, Perret M, Hantraye P, Mestre-Frances N, Aujard F, Dhenain M. Age-associated cerebral atrophy in mouse lemur primates. Neurobiol Aging. 2011;32:894–906. doi: 10.1016/j.neurobiolaging.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE. Isoform-specific binding of apolipoprotein E to B-amyloid. J Biol Chem. 1994;269(38):23403–23406. [PubMed] [Google Scholar]
- 63.Languille S, Blanc S, Blin O, Canale CI, Dal-Pan A, Devau G, Aujard F. The grey mouse lemur: a non-human primate model for ageing studies. Ageing Res Rev. 2012;11(1):150–162. doi: 10.1016/j.arr.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 64.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell H, Yu C, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu Y-H, Guentette SY, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 65.Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, Pautler RG, Taffet GE, Zheng H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: interplay with cerebral blood flow. Mol Neurodegener. 2014;9:28. doi: 10.1186/1750-1326-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu ST, Howlett G, Barrow CJ. Histidine-13 is a crucial residue in the Zinc ion-induced aggregation of the Aβ peptide of Alzheimer disease. Biochemistry. 1999;38(29):9373–9378. doi: 10.1021/bi990205o. [DOI] [PubMed] [Google Scholar]
- 67.Liu Chia-Chen, Liu Chia-Chan, Kanekiyo Takahisa, Huaxi Xu, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–118. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Magalhães JP, Costa JJ. A database of vertebrate longevity records and their relation to other life-history traits. Evol Biol. 2009;2009(8):1770–1774. doi: 10.1111/j.1420-9101.2009.01783.x. [DOI] [PubMed] [Google Scholar]
- 69.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer’s disease. Proc Natl Acad Sci USA. 2006;103(15):5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mahley RW, Weisgraber KH, Huang Y. Apolipoprotein E: structure determines function, from atherosclerosis to Alzheimer’s disease to AIDS. J Lipid Res. 2009;50:S183–S188. doi: 10.1194/jlr.R800069-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mestre N, Bons N. Age-related cytological changes and neuronal loss in basal forebrain cholinergic neurons in Microcebus murinus (Lemurian, Primate) Neurodegeneration. 1993;2:25–32. [Google Scholar]
- 73.Nukina N, Ihara Y. One of the antigenic determinants of paired helical filaments is related to tau protein. J Biochem. 1986;99:1541–1544. doi: 10.1093/oxfordjournals.jbchem.a135625. [DOI] [PubMed] [Google Scholar]
- 74.Oddo S, Caccamo A, Shepherd J, Murphy P, Golde T, Kayed R, Metherate R, Mattson M, Akbari Y, LaFerla F. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/S0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 75.Opazo JC. A molecular timescale for caviomorph rodents (Mammalia, Hystricognathi) Mol Phylogenet Evol. 2005;37:932–937. doi: 10.1016/j.ympev.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 76.Orr ME, Garbarino VR, Salinas A, Buffenstein R. Sustained high levels of neuroprotective, high molecular weight, phosphorylated tau in the longest-lived rodent. Neurobiol Aging. 2015;36(3):1496–1504. doi: 10.1016/j.neurobiolaging.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Otvos L, Jr, Szendrei GI, Lee VM, Mantsch HH. Human and rodent Alzheimer beta-amyloid peptides acquire distinct conformations in membrane-mimicking solvents. Eur J Biochem. 1993;211(1–2):249–257. doi: 10.1111/j.1432-1033.1993.tb19893.x. [DOI] [PubMed] [Google Scholar]
- 78.Picq JL, Aujardb F, Volkc A, Dhenain M. Age-related cerebral atrophy in nonhuman primates predicts cognitive impairments. Neurobiol Aging. 2012;33:1096–1109. doi: 10.1016/j.neurobiolaging.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Picq JL. Aging affects executive functions and memory in mouse lemur primates. Exp Gerontol. 2007;42(3):223–232. doi: 10.1016/j.exger.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 80.Riddell DR, Zhou H, Atchison K, Warwick HK, Atkinson PJ, Jefferson J, Xu L, Aschmies S, Kirksey Y, Hu Y, Wagner E, Parratt A, Xu J, Li Z, Zaleska MM, Jacobsen JS, Pangalos MN, Reinhart PH. Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J Neurosci. 2008;28(45):11445–11453. doi: 10.1523/JNEUROSCI.1972-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ringman JM, Gylys KH, Medina LD, Fox M, Kepe V, Flores DL, Apostolova LG, Barrio JR, Small G, Silverman DH, Siu E, Cederbaum S, Hecimovic S, Malnar M, Chakraverty S, Goate AM, Bird TD, Leverenz JB. Biochemical, neuropathological, and neuroimaging characteristics of early-onset Alzheimer’s disease due to a novel PSEN1 mutation. Neurosci Lett. 2011;487:287–292. doi: 10.1016/j.neulet.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ, Mucke L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 83.Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T, Mar T, Sorbi L, Nacmias S, Piacentini S, Amaducci L, Chumakov I, Cohen D, Lannfelt L, Fraser PE, Rommens JM, George-Hyslop PH. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- 84.Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-Machlachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993;43:1467–1472. doi: 10.1212/WNL.43.8.1467. [DOI] [PubMed] [Google Scholar]
- 85.Saito T, Matsuba Y, Mihira N, Takano J, Nilsson P, Itohara S, Iwata N, Saido TC. Single App knock-in mouse models of Alzheimer’s disease. Nat Neurosci. 2014;17(5):661–663. doi: 10.1038/nn.3697. [DOI] [PubMed] [Google Scholar]
- 86.Sato N, Hori O, Yamaguchi A, Lambert JC, Chartier-Harlin MC, Robinson PA, Delacourte A, Schmidt AM, Furuyama T, imaizumi K, Tohyama M, Takagi T. A novel presenilin-2 splice variant in human Alzheimer’s disease brain tissue. J Neurochem. 1999;72:2498–2505. doi: 10.1046/j.1471-4159.1999.0722498.x. [DOI] [PubMed] [Google Scholar]
- 87.Schellenberg GD, Bird TD, Wijsman EM, Orr HT, Anderson L, Nemens E, White JA, Bonnycastle L, Weber JL, Alonso ME, Potter H, Heston LH, Martin GM. Genetic linkage evidence for a familial Alzheimer’s disease locus on chromosome 14. Science. 1992;258:668–671. doi: 10.1126/science.1411576. [DOI] [PubMed] [Google Scholar]
- 88.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 89.Selkoe DJ, Abraham CR, Podlisny MB, Duffy LK. Isolation of low-molecular-weight proteins from amyloid plaque fibers in Alzheimer’s disease. J Neurochem. 1986;146:1820–1834. doi: 10.1111/j.1471-4159.1986.tb08501.x. [DOI] [PubMed] [Google Scholar]
- 90.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. β-Amyloid precursor protein of Alzheimer disease occurs as 110–135 kilodalton membrane-associated proteins in neural and non-neural tissues. Proc Natl Acad Sci USA. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sergeant N, Delacourte A, Buée L. Tau protein as a differential biomarker of tauopathies. Biochim Biophys Acta. 2005;1739(2):179–197. doi: 10.1016/j.bbadis.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 92.Sharman MJ, Moussavi Nik SH, Chen MM, Ong D, Wijaya L, Laws SM, Taddei K, Newman M, Larddelli M, Martins RN, Verdile G. The guinea pig as a model for sporadic Alzheimer’s disease (AD): the impact of cholesterol intake on expression of AD-related genes. PLoS One. 2013;8(6):e66235. doi: 10.1371/journal.pone.0066235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin J-F, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen DA, Roses AD, Fraser PE, Rommens JM, St. George-Hyslop PH. Cloning of a novel gene bearing missense mutations in early onset familial Alzheimer disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 94.Silhol S, Calenda A, Jallageas V, Mestre-Frances N, Bellis M, Bons N. β-amyloid protein precursor in microcebus murinus: genotyping and brain localization. Neurobiol Dis. 1996;3(3):169–182. doi: 10.1006/nbdi.1996.0017. [DOI] [PubMed] [Google Scholar]
- 95.Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20(23):8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Spotorno AE, Walter L, Contreras LC, Torres-Mura JC, Fernández-Donoso R, Berríos MS, Pincheira J. Chromosome divergence of Octodon lunatus and the origins of Octodontoidea (Rodentia, Hystricognathi) Rev Chilena Hist Nat. 1995;68:227–239. [Google Scholar]
- 97.Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283(44):29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Van Groen T, Kadish I, Popovic N, Popovic M, Caballero-Bleda M, Bano-Otalora B, Vivanco P, Rol MA, Madrid JA. Age-related brain pathology in Octodon degu: blood vessel, white matter and Alzheimer-like pathology. Neurobiol Aging. 2011;32:1651–1661. doi: 10.1016/j.neurobiolaging.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 100.Wolfe MS. Tau mutations in neurodegenerative diseases. J Biol Chem. 2009;284:6021–6025. doi: 10.1074/jbc.R800013200. [DOI] [PubMed] [Google Scholar]
- 101.Wolfe M, Xia W, Ostaszewski B, Diehl T, Kimberly W, Selkoe D. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and big gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 102.Wood JG, Mirra SS, Pollock NL, Binder LI. Neurofibrillary tangles of Alzheimer’s disease share antigenic determinants with the axonal microtubule-associated protein tau. Proc Natl Acad Sci USA. 1986;83:4040–4043. doi: 10.1073/pnas.83.11.4040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Xu Q, Brecht WJ, Weisgraber KH, Mahley RW, Huang Y. Apolipoprotein E4 domain interaction occurs in living neuronal cells as determined by fluorescence resonance energy transfer. J Biol Chem. 2004;279(24):25511–25516. doi: 10.1074/jbc.M311256200. [DOI] [PubMed] [Google Scholar]