

Abstract
When activated by an acylating agent, pyridine boronic esters react with organometallic reagents to form a dihydropyridine boronic ester. This intermediate allows access to a number of valuable substituted pyridine, dihydropyridine and piperidine products.
Keywords: Pyridine, Piperidine, Boronate ester, Allylboration, Samarium iodide
Pyridines and their derivatives are common motifs found in drugs and natural products. Current FDA-approved drugs include 62 pyridines, 10 dihydropyridines and 72 piperidines.[1] For this reason, methods for their preparation are valuable for drug discovery and natural products synthesis. Traditional Chichibabin reactions involve addition of nucleophiles to activated pyridines, although they generally show poor regioselectivity (Scheme 1a). [2,3,4] Enhanced regioselectivity has been observed by Knochel and coworkers using substrates with electron-withdrawing groups at C3.[5] Alternatively, Minsci reactions feature addition of alkyl or aryl radicals to the heteroarene. Poor regioselectivity is usually observed, and the 2-substituted products generally predominate (Scheme 1b).[6] Transition metal-catalyzed coupling reactions can also generate substituted pyridines, but they do not provide dearomatized products.[7] Recently, C-H functionalization strategies have emerged as attractive routes to the pyridine scaffold.[8]
Scheme 1.
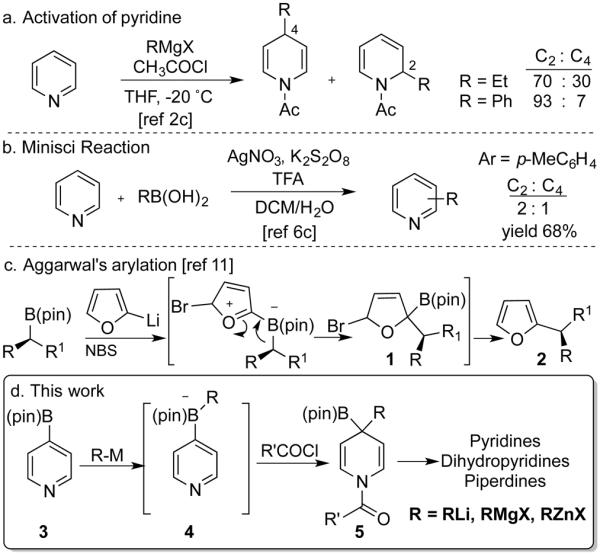
Selected functionalization of pyridines.
We became interested in exploring the utility of boronic esters in the synthesis of nitrogen heterocycles.[9] By way of background, Matteson demonstrated the ability of 2-chloro boronic esters to undergo homologation upon treatment with lithiated reagents.[10] In these reactions, an alkyl group migrates from boron to an adjacent electrophile. Building on this mechanistic platform, the Aggarwal group developed an ingenious coupling of aryl lithium reagents with boronic esters (Scheme 1c). Thus, addition of an aryl lithium to the boronic ester followed by a halogenation-induced 1,2-alkyl migration forms the new C-C bond (1). Loss of (pin)BBr formed the aromatic product (2).[11] We recognized that a similar mechanism could be accessible with pyridines, but that the dearomatized intermediates might be isolable. Thus, acylation of boronate 4 could prompt a 1,2-migration to form a new C-C bond (Scheme 1d). Importantly, this sequence would retain the boron moiety (5) and allow subsequent functionalization. Unlike prior work, the transformation would allow access to both the aromatic and dearomatized products. As we were preparing this manuscript, the Aggarwal group described a synthesis of substituted heteroarenes from boronic esters and heteroaryl lithium reagents using a related strategy.[12] Prompted by their disclosure, here we describe a method for the preparation of pyridines, dihydropyridines and piperidines that shows wide scope with regard to the organometallic nucleophile and leads to complex dearomatized products.
We began our studies with the addition of tert-BuLi to 4-pyridineboronic acid pinacol ester (Scheme 2). Upon activation of the pyridine, the desired dihydropyridines 6–12 were formed in good yield. The reaction worked well to yield amides, carbamates and sulfonamides. Likewise, both anhydrides and acid chlorides generated amides with equal facility. As shown below, the ability to incorporate diverse N-activating groups allows for subsequent derivatization of the dihydropyridine. The structure of amide 11 was confirmed by X-ray analysis.
Scheme 2.
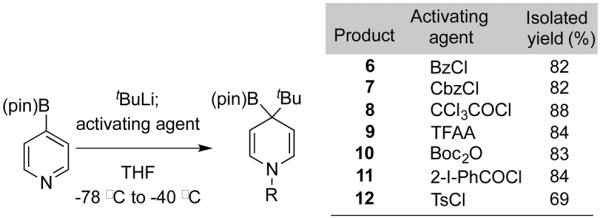
Generation of dihydropyridines.
Upon treatment of the dihydropyridine with oxygen and base, rapid formation of the substituted pyridine was observed.[13] A broad range of substituted pyridines and quinolones were synthesized as highlighted in Table 1. Primary, secondary and tertiary alkyl lithium reagents were successfully used, as were aryl and alkynyl lithium species. Optically active pyrrolidine was an excellent substrate, leading to the pyridines 26–28 in high yield and high enatiomeric ratios.[14] To introduce functionalized nucleophiles, we explored the use of aryl and alkyl zinc reagents. Ester, bromo and cyano groups were all accommodated (29–31). Although the reaction worked well with aryl zinc halides, the analogous reaction using alkyl zinc reagents proved more difficult. Accordingly, we used a mixed zinc reagent derived from the zinc bromide and tert-butyl magnesium chloride and achieved good yields of the ester 32.[5] Additionally, alkyl, aryl and vinyl Grignard reagents provided the pyridines in high yields. Two cyclopropyl Grignards were incorporated without any evidence of ring-opening (41, 42). Overall, the reaction is notable for its ability to 1) incorporate Csp3, Csp2 and Csp nucleophiles, 2) accommodate organo-lithium, -zinc and -magnesium reagents, and 3) introduce electrophilic functional groups including nitriles and esters.
Table 1.
Substrate scope for preparation of substituted pyridines[a]

|
Typical conditions: het-B(pin) (1 equiv., 0.3 mmol), R-M (1.1 equiv.), −78 °C, CCI3COCI (2 equiv.), −78 to −40 °C; 10% NaOH aq, rt, O2 balloon.
Isolated yield.
Typical conditions: (−)sparteine (1.3 equiv., 0.39 mmol), sBuLi (1.3 equiv., 0.39 mmol), N-Boc pyrrolidine (1.3 equiv., 0.39 mmol); het-B(pin) (1 equiv., 0.3 mmol), −78 °C, CCI3COCI (3.5 equiv.), −78 °C; saturated aq. Na2CO3 rt, O2 balloon. het-B(pin) = heteroaryl pinacol bononic ester.
This coupling showed little influence from substitution on the pyridine ring itself. Electron donating and withdrawing groups were tolerated at the 2 and 3 positions (17–20, 27–28, 34–35, 43–46). The observation that the 2-chloro-pyridine was a suitable substrate is particularly noteworthy since these reagents readily participate in cross-coupling reactions. Thus their successful application highlights the mildness of the method while retaining the 2-chloro group for subsequent chemistry. This reaction was also applied to quinoline 4-boronic esters, giving good yields for these substrates (21, 36, 48). Expansion of this methodology to 5- and 7-borono quinolines and 6-borono-isoquinoline was only modestly successful (49–51). Additionally, the 2-pyridyl boronic esters were too unstable for the current reaction conditions,[7b] providing large amounts of protodeborylated pyridines.
Typical oxidations of boronic esters require peroxides, so the facile oxidation shown in Table 1 was surprising.[12] The rate of this oxidation is insensitive to the nature of activating group (both steric and electronic) but qualitatively dependent on the substituent on the 4-position (Ph > 1° alkyl > 3° alkyl). These observations suggest a radical mechanism; indeed, trialkyl boranes are known to react with oxygen to generate alkyl radicals.[15] However, we observed no ring-opening of the cyclopropyl ring in 42, indicating that the lifetime of any radical must be short.
Hall and coworkers previously prepared tetrahydropyridines featuring an allylic boronic ester, and they showed that these participated in allylation reactions.[16] By contrast, the 4-boryl-4-dihydropyridine products described here are unknown reagents. Therefore we next explored their synthetic utility For example, addition of the allyl boronate to benzaldehyde generated the desired homoallylic alcohol 52 with good yield and diastereoselectivity (Scheme 3a).[17] Hydrogenation of this alcohol gave the trisubstituted piperidine 53 while oxidation provided the disubstituted pyridine 54, in which the benzoyl group migrated from N to O. In this way the (pin)B moiety was used to functionalize both C2 and C4 of the pyridine ring – a distinguishing characteristic of the current method relative to Suzuki couplings and related reactions.[7, 12]
Scheme 3.

Functionalization of Dihydropyridine Intermediates. [a]nBuLi (1 equiv.), BzCI (1.1 equiv); then PhCHO (4 equiv.)
In addition to trapping the allyl boronic ester, we also investigated reduction of the crude dihydropyridine (Scheme 3b). Surprisingly, upon treatment with Pd/C in methanol under a hydrogen atmosphere, the deborylated 4-substituted piperidines were the predominant products (56, 57). However, when the crude dihydropyridine was treated with an acidic resin prior to hydrogenation, we obtained the tetrahydropyridines 58 and 59. Longer reaction times yielded the piperidine boronic esters 63 and 64. We hypothesized that Amberlyst 15 could be quenching some base present in the reaction medium that was promoting protodeborylation.[18] Indeed, addition of LiOH in the reduction of tetrahydropyridine 58 generated the deborylated product 56.[13] Finally, both the tetrahydropyridines and the piperidines could be oxidized with peroxide to form tertiary alcohols (60, 61 and 65, 66). Alternatively allyl boronic ester 58 could add to benzaldehyde to furnish alcohol 62.
Our initial studies demonstrated that a variety of activating groups could be used in the dearomatization (Scheme 2). Building on this observation, we considered whether the acylating group could incorporate functionality to initiate a radical cyclization (Scheme 4a). In this context, we found that SmI2 affected the desired ring-closing reaction of aryl iodide 67.[19] Unexpectedly, C-C bond formation was accompanied by a 1,2-boron shift and olefin transposition to give the allyl boronic esters 68-73. Various additives were screened to improve the yield the reaction including HMPA, water, methanol, and trimethylamine. The addition of water (2–3 equiv. relative to Sm) proved essential to obtain good yields.[20] This reaction incorporated aryl, tertiary, secondary and primary alkyl groups derived from organo-lithium, magnesium and zinc reagents. Additionally DIBALH transferred hydride to provide 73, which was unstable but could be isolated after oxidation (84). While only modest diastereoselectivity was observed for the cyclization, the diastereomers could readily be separated by silica gel chromatography in high yield.
Scheme 4.

Cyclization of dihydropyridines with SmI2. DIBAL = iBu2AIH.
The tricyclic allyl boronic esters 68-73 emerged as versatile synthetic intermediates (Scheme 4b). Thus, addition to benzaldehyde afforded the desired homoallylic alcohols 74-78 with high stereoselectively. Alternatively, oxidation of the boronic esters with sodium perborate gave the corresponding secondary alcohols 79-84 in high yield and complete stereospecificity. Amination of the allyl boronic ester 69 with nitrosobenzene followed by reduction with Mo(CO)6/NaBH4 generated enamide 85 with very high diastereoselectivity.[21] X-ray crystallography confirmed the surprising observation that Mo(CO)6/NaBH4 isomerized the olefin to form the conjugated enamide 85.[22] Alternatively, hydrogenation with Pd/C yielded tetrasubstituted piperidine 86 with d.r. of 12:1. Interestingly, addition of allylboronates to nitrosobenzenes are generally O-selective.19b By contrast, we observed complete N-selectivity to afford the indicated amine products instead of alcohols.
The dearomatization/SmI2-mediated cyclization of 2-substituted pyridine 87 led to two noteworthy observations. First, cyclization favored the more hindered product possessing a quaternary stereocenter (5:1 88:89). Second, unlike the 2-unsubstitued pyridines, this substrate cyclized with high diastereoselectivity (10:1). The relative stereochemistry was initially assigned via comparison of experimental 1H and 13C chemical shifts with those computed for possible diastereomers using quantum chemical methods [mPW1PW91/6-311+G(2d,p)//M06-2X/6-31+G(d,p), scaled].23 The structure was later confirmed by X-ray crystallography indicating that DFT methods can be used for other products for which crystallography is not feasible.
The SmI2-mediated cyclization iodo-amide 67 presumably involves reductive generation of an aryl radical. Next, 5-exo-trig cyclization might lead to migration of the B(pin) moiety. We are unaware of previous examples of radical migrations of boronic esters. Current efforts aim to characterize this process more completely and determine its generality. Of note, performing the cyclization in the presence of D2O formed cyclized product 68 with deuterium incorporation at C6. Finally, we remain uncertain why the 2-substituted pyridine 87 cyclized with high regio- and diastereoselectivity. The ground state conformation of this substrate orients the large aryl ring towards the unsubstituted C6 carbon; clearly, cyclization occurs from an alternative conformation. Understanding these provocative observations and expanding the synthetic applications of the dihydropyridine boronic esters are the subjects of ongoing studies.
Supplementary Material
Footnotes
Financial support provided by the NIH (R01GM102403) and the Welch Foundation (I-1612). X-ray crystallography performed by Dr. M. F. Yousufuddin (UT Arlington) and Dr. V. Lynch (UT Austin). NSF CHE030089 supported computational resources. Supporting information for this article is given via a link at the end of the document.
References
- [1].a) Vitaku E, Smith DT, Njardarson JT. J. Med. Chem. 2014;57:10257–10274. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]; b) Walsh CT. Tetrahedron Lett. 2015;56:3075–3081. [Google Scholar]; c) Michael JP. Nat. Prod. Rep. 2008;25:166–187. doi: 10.1039/b612168n. [DOI] [PubMed] [Google Scholar]
- [2].Reviews on addition to activated pyridines: Bull JA, Mousseau JJ, Pelletier G, Charette AB. Chem. Rev. 2012;112:2642–2713. doi: 10.1021/cr200251d. Andersson H, Olsson R, Almqvist F. Org. Biomol. Chem. 2011;9:337–346. doi: 10.1039/c0ob00336k. Comins DL, O'Connor S. In: Advances in Heterocyclic Chemistry. Alan RK, editor. Volume 44. Academic Press; 1988. pp. 199–267. Stout DM, Meyers AI. Chem. Rev. 1982;82:223–243.
- [3].For examples of Lewis acid-mediated addition to pyridines see Chen Q, León T, Knochel P. Angew. Chem. 2014;126:8891–8895. doi: 10.1002/anie.201400750. Angew. Chem. Int. Ed. 2014;53:8746–8750. doi: 10.1002/anie.201400750. Jeffrey JL, Sarpong R. Org. Lett. 2012;14:5400–5403. doi: 10.1021/ol3024117.
- [4].For current examples of addition to activated pyridines see: Zhang P, Cook AM, Liu Y, Wolf CJ. Org. Chem. 2014;79:4167–4173. doi: 10.1021/jo500365h. Tsukanov SV, Comins DL. Angew. Chem. 2011;123:8785–8787. Angew. Chem. Int. Ed. 2011;50:8626–8628. doi: 10.1002/anie.201103596.
- [5].Chen Q, du Jourdin XM, Knochel P. J. Am. Chem. Soc. 2013;135:4958–4961. doi: 10.1021/ja401146v. [DOI] [PubMed] [Google Scholar]
- [6].For reviews and current example of Minsci reaction: Minisci F, Vismara E, Fontana F. Heterocycles. 1989;28:489–519. Harrowven DC, Sutton BJ. Prog. Heterocycl. Chem. 2004;16:27–53. Seiple IB, Su S, Rodriguez RA, Gianatassio R, Fujiwara Y, Sobel AL, Baran PS. J. Am. Chem. Soc. 2010;132:13194–13196. doi: 10.1021/ja1066459. Duncton MAJ. Med. Chem. Commun. 2011;2:1135–1161. O'Hara F, Blackmond DG, Baran PS. J. Am. Chem. Soc. 2013;135:12122–12134. doi: 10.1021/ja406223k.
- [7].For recent examples of Suzuki-Miyaura metal couplings see: Molander GA, Canturk B, Kennedy LE. J. Org. Chem. 2009;74:973–980. doi: 10.1021/jo802590b. Knapp DM, Gillis EP, Burke MD. J. Am. Chem. Soc. 2009;131:6961–6963. doi: 10.1021/ja901416p. Billingsley K, Buchwald SL. J. Am. Chem. Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. Billingsley KL, Anderson KW, Buchwald SL. Angew. Chem. 2006;118:3564–3568. doi: 10.1002/anie.200600493. Angew. Chem. Int. Ed. 2006;45:3484–3488. doi: 10.1002/anie.200600493. Kudo N, Perseghini M, Fu GC. Angew. Chem. 2006;118:1304–1306. doi: 10.1002/anie.200503479. Angew. Chem. Int. Ed. 2006;45:1282–1284. doi: 10.1002/anie.200503479.
- [8].a) Romanov-Michailidis F, Sedillo KF, Neely JM, Rovis T. J. Am. Chem. Soc. 2015;137:8892–8895. doi: 10.1021/jacs.5b04946. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Duttwyler S, Chen S, Takase MK, Wiberg KB, Bergman RG, Ellman JA. Science. 2013;339:678–682. doi: 10.1126/science.1230704. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Guo P, Joo JM, Rakshit S, Sames D. J. Am. Chem. Soc. 2011;133:16338–16341. doi: 10.1021/ja206022p. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wasa M, Worrell BT, Yu J-Q. Angew. Chem. 2010;122:1297–1299. doi: 10.1002/anie.200906104. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2010;49:1275–1277. doi: 10.1002/anie.200906104. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Nakao Y, Yamada Y, Kashihara N, Hiyama T. J. Am. Chem. Soc. 2010;132:13666–13668. doi: 10.1021/ja106514b. [DOI] [PubMed] [Google Scholar]; f) Larivée A, Mousseau JJ, Charette AB. J. Am. Chem. Soc. 2008;130:52–54. doi: 10.1021/ja710073n. [DOI] [PubMed] [Google Scholar]; g) Campeau L-C, Rousseaux S, Fagnou K. J. Am. Chem. Soc. 2005;127:18020–18021. doi: 10.1021/ja056800x. [DOI] [PubMed] [Google Scholar]
- [9].a) Burns M, Essafi S, Bame JR, Bull SP, Webster MP, Balieu S, Dale LW, Butts CP, Harvey JN, Aggarwal VK. Nature. 2014;513:183–188. doi: 10.1038/nature13711. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Suzuki A, Nozawa S, Miyaura N, Itoh M, Brown HC. Tetrahedron Lett. 1969;10:2955–2958. [Google Scholar]; c) Brown HC, Zweifel G. J. Am. Chem. Soc. 1961;83:486–487. [Google Scholar]
- [10].Matteson DS. J. Org. Chem. 2013;78:10009–10023. doi: 10.1021/jo4013942. [DOI] [PubMed] [Google Scholar]
- [11].a) Bonet A, Odachowski M, Leonori D, Essafi S, Aggarwal VK. Nat. Chem. 2014;6:584–589. doi: 10.1038/nchem.1971. [DOI] [PubMed] [Google Scholar]; b) Stymiest JL, Bagutski V, French RM, Aggarwal VK. Nature. 2008;456:778–782. doi: 10.1038/nature07592. [DOI] [PubMed] [Google Scholar]
- [12].Llaveria J, Leonori D, Aggarwal VK. J. Am. Chem. Soc. 2015;137:10958–10961. doi: 10.1021/jacs.5b07842. [DOI] [PubMed] [Google Scholar]
- [13].See supporting information for details.
- [14].Barker G, McGrath JL, Klapars A, Stead D, Zhou G, Campos KR, O'Brien P. J. Org. Chem. 2011;76:5936–5953. doi: 10.1021/jo2011347. [DOI] [PubMed] [Google Scholar]
- [15].a) Brown HC, Midland MM. Angew. Chem. 1972;84:702–710. [Google Scholar]; Angew. Chem. Int. Ed. 1972;11:692–700. [Google Scholar]; b) Brown HC, Midland MM. J. Am. Chem. Soc. 1971;93:3291–3293. [Google Scholar]
- [16].Lessard S, Peng F, Hall DG. J. Am. Chem. Soc. 2009;131:9612–9613. doi: 10.1021/ja903946f. [DOI] [PubMed] [Google Scholar]
- [17].Hesse MJ, Essafi S, Watson CG, Harvey JN, Hirst D, Willis CL, Aggarwal VK. Angew. Chem. 2014;126:6259–6263. doi: 10.1002/anie.201402995. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2014;53:6145–6149. doi: 10.1002/anie.201402995. [DOI] [PubMed] [Google Scholar]
- [18].Hesse MJ, Butts CP, Willis CL, Aggarwal VK. Angew. Chem. 2012;124:12612–12616. doi: 10.1002/anie.201207312. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:12444–12448. doi: 10.1002/anie.201207312. [DOI] [PubMed] [Google Scholar]
- [19].a) Szostak M, Fazakerley NJ, Parmar D, Procter DJ. Chem. Rev. 2014;114:5959–6039. doi: 10.1021/cr400685r. [DOI] [PubMed] [Google Scholar]; b) Edmonds DJ, Johnston D, Procter DJ. Chem. Rev. 2004;104:3371–3404. doi: 10.1021/cr030017a. [DOI] [PubMed] [Google Scholar]; c) Curran DP, Totleben MJ. J. Am. Chem. Soc. 1992;114:6050–6058. [Google Scholar]; d) Curran DP, Fevig TL, Jasperse CP, Totleben MJ. Synlett. 1992;1992:943–961. [Google Scholar]
- [20].a) Parmar D, Price K, Spain M, Matsubara H, Bradley PA, Procter DJ. J. Am. Chem. Soc. 2011;133:2418–2420. doi: 10.1021/ja1114908. [DOI] [PubMed] [Google Scholar]; b) Szostak M, Spain M, Procter DJ. Nat. Protocols. 2012;7:970–977. doi: 10.1038/nprot.2012.034. [DOI] [PubMed] [Google Scholar]; b) Prasad E, Flowers RA. J. Am. Chem. Soc. 2005;127:18093–18099. doi: 10.1021/ja056352t. [DOI] [PubMed] [Google Scholar]
- [21].a) Li Y, Chakrabarty S, Studer A. Angew. Chem. 2015;127:3657–3661. [Google Scholar]; Angew. Chem. Int. Ed. 2015;54:3587–3591. doi: 10.1002/anie.201410188. [DOI] [PubMed] [Google Scholar]; b) Kyne RE, Ryan MC, Kliman LT, Morken JP. Org. Lett. 2010;12:3796–3799. doi: 10.1021/ol101472k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Frauenrath H, Philipps T. Angew. Chem. 1986;98:261. [Google Scholar]; Angew. Chem. Int. Ed. 1986;25:274. [Google Scholar]
- [23].a) Lodewyk MW, Siebert MR, Tantillo DJ. Chem. Rev. 2012;112:1839–1862. doi: 10.1021/cr200106v. [DOI] [PubMed] [Google Scholar]; b) Nguyen QNN, Tantillo DJ. Chem. Asian. J. 2014:674–680. doi: 10.1002/asia.201301452. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.