

Abstract
Traumatic brain injury (TBI), which leads to disability, dysfunction, and even death, is a prominent health problem worldwide with no effective treatment. A brain-permeable flavonoid named (−)-epicatechin (EC) modulates redox/oxidative stress and has been shown to be beneficial for vascular and cognitive function in humans and for ischemic and hemorrhagic stroke in rodents. Here we examined whether EC is able to protect the brain against TBI-induced brain injury in mice and if so, whether it exerts neuroprotection by modulating the NF-E2-related factor (Nrf2) pathway. We used the controlled cortical impact model to mimic TBI. EC was administered orally at 3 h after TBI and then every 24 h for either 3 or 7 days. We evaluated lesion volume, brain edema, white matter injury, neurologic deficits, cognitive performance and emotion-like behaviors, neutrophil infiltration, reactive oxygen species (ROS), and a variety of injury-related protein markers. Nrf2 knockout mice were used to determine the role of the Nrf2 signaling pathway after EC treatment. In wild-type mice, EC significantly reduced lesion volume, edema, and cell death and improved neurologic function on days 3 and 28; cognitive performance and depression-like behaviors were also improved with EC administration. In addition, EC reduced white matter injury, heme oxygenase-1 expression, and ferric iron deposition after TBI. These changes were accompanied by attenuation of neutrophil infiltration and oxidative insults, reduced activity of matrix metalloproteinase 9, decreased Keap 1 expression, increased Nrf2 nuclear accumulation, and increased expression of superoxide dismutase 1 and quinone 1. However, EC did not significantly reduce lesion volume or improve neurologic deficits in Nrf2 knockout mice after TBI. Our results show that EC protects the TBI brain by activating the Nrf2 pathway, inhibiting heme oxygenase-1 protein expression, and reducing iron deposition. The latter two effects could represent an Nrf2-independent mechanism in this model of TBI.
Keywords: (−)-Epicatechin, NF-E2-related factor, Traumatic brain injury
Introduction
Traumatic brain injury (TBI) causes extensive neurologic disability and mortality for individuals worldwide, especially for those under 45 years old [1]. Despite progress made in diagnosis, neurosurgical care, and functional rehabilitation, no effective therapy is currently available for TBI. An understanding of the pathophysiology of TBI and effective treatments that can repair the damaged brain and improve functional recovery are urgently needed. After TBI, tissue loss and cell death cause the primary damage; subsequently, inflammatory damage from activated microglia and recruited immune cells (mainly neutrophils and macrophages) and reactive oxygen species (ROS) from blood metabolites contribute to secondary brain injury [2–4]. Theoretically, diminishing inflammation, iron toxicity, and ROS production would improve brain recovery in TBI patients by reducing secondary brain damage [5].
Natural flavanols have the potential to improve an individual’s oxidant defense systems by activating endogenous protective signaling pathways [6, 7]. Studies have shown that flavanols, such as epicatechins, catechins, and procyanidins, have great potential for neurovascular protection [8]. With anti-inflammatory, anti-oxidant, and iron-chelating properties [6, 7], (−)-epicatechin (EC) could be a potential drug candidate for treating TBI. We and others have shown previously that EC penetrates the blood-brain barrier (BBB) after intravenous or oral administration [9–12], produces a positive effect on cognition [13, 14], and reduces hemorrhagic/ischemic stroke injury [15, 16]. EC is thought to offer these benefits by activating the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway [16, 17]. By binding to antioxidant response elements (AREs), Nrf2 increases production of proteins involved in reduction of inflammatory responses, oxidative stress, and accumulation of toxic metabolites [15, 18, 19], as well as proteins that control cell redox state during oxidative stress [20, 21]. Kelch-like ECH-associated protein (Keap1) is recognized as a substrate linker protein involved in compartmentalization and degradation of Nrf2 [22]. In the cytoplasm, Keap1 anchors Nrf2, which is then degraded by the ubiquitin–26S proteasome pathway [20]. Our group has shown that heme oxygenase 1 (HO-1), a downstream target gene of the Nrf2-ARE pathway, may play an important role in iron deposition and may be a prooxidant in a mouse model of intracerebral hemorrhage [15, 23].
The bioavailability of pure EC in rats after oral administration is 34.2%, and its terminal elimination half-life is 71 min [24]. Its absorption in humans is relatively efficient [25]. EC is mainly distributed in the intestine and kidney in rats and is rapidly converted to conjugated (>90%) and nonconjugated (<10%) metabolites and excreted through both the bile and the urine [26, 27]. The primary metabolites of EC in plasma are glucuronide and sulfoglucuronide in nonmethylated forms, and sulfate in the 3′-O-methylated forms [9, 27–29]. A recent study in rats [10] showed that an intravenous dose of 20 mg/kg EC was able to rapidly (within 20 min) cross the BBB and enter the extracellular space. Additionally, its elimination half-life was 13.67 ± 4.33 min for blood and 41.67 ± 9.14 min for brain, the maximum brain concentration was observed at approximately 20 min after administration, and the mean maximum brain concentration was 0.83 ± 0.45 μg/mL [10].
Currently, it is unknown whether EC can activate Nrf2 and its downstream transcripts, such as HO-1, superoxide dismutase 1 (SOD1), and quinone 1 (NQO1), after TBI. Therefore, in this study, we examined whether EC can protect the brain from injury in mice subjected to the controlled cortical impact (CCI) model of TBI and investigated the underlying mechanisms. Further, we explored the relationship among the Nrf2-ARE pathway, HO-1 expression, and iron deposition in this model. We found that EC protects the brain after TBI by activating Nrf2 but that it also inhibits HO-1 expression and iron deposition through an Nrf-2-independent pathway.
Materials and Methods
Animals
Male C57BL/6 (wild-type, WT) mice (Charles River Laboratories) and Nrf2 gene knockout (KO) mice (on C57BL/6 background, originally generated by Dr. Masayuki Yamamoto) were used in this study at 8–12 weeks of age (20–26 g). The mice were housed with free access to food and water on a 12-h light/dark cycle in a pathogen-free environment. Nrf2 KO mice were genotyped by PCR amplification of genomic DNA extracted from tail snips. Experimental protocols were approved by the Johns Hopkins University Animal Care and Use Committee. Animal experiments were reported in accordance with the ARRIVE guidelines.
CCI model of TBI
The mice were anesthetized with isoflurane (4.0% for induction, 2.0% for maintenance) and ventilated with oxygen-enriched air (20%:80%) via a nose cone. We used ear bars and an incisor bar to hold the head in place. After the scalp was incised at the midline, a 5-mm-diameter craniotomy was created approximately midway between the bregma and the lambda on the left side, with the edge of the craniotomy 1 mm lateral to the midline. CCI was carried out with the Benchmark CCI Stereotaxic Impactor (Benchmark Deluxe; MyNeurolab, St Louis, MO). The impact tip (3 mm in diameter) was directed perpendicular to the brain surface with an impact velocity of 6 m/s, an impact duration of 100 ms, and a depth of 2 mm. Rectal temperature was monitored and maintained at 37.0 ± 0.5°C by an electronic thermostat-controlled warming blanket (Stoelting Co., Wood Dale, IL) throughout the experimental and recovery periods. The sham group received anesthesia and craniotomy, but no impact.
Experimental groups
The website Randomization.com (http://www.randomization.com) was used to assign mice randomly to study groups [15]. For short-term observation, mice were administered vehicle (sterilized water; Hospira, San Jose, CA) or 5, 15, or 45 mg/kg EC (Sigma, St. Louis, MO) by gavage at 3 h after TBI and once daily for 3 days. For long-term observation, mice received 15 mg/kg EC at 3 h after TBI and then once daily for 7 days. We chose the delivery route, dosing, and treatment regimen for EC based on previous work from our group [15] and others [9, 16, 30].
We used a total of 151 WT mice and 31 Nrf2 KO mice in this experiment. Five to six mice were randomly included in each group for histology, Western blot analysis, and brain water content measurement. Eight to 10 mice were used per group for behavior tests and lesion volume measurement. Investigators were blinded to treatment group during experimental tests and data analysis according to published guidelines [31].
Neurologic deficit score
On days 1, 2, 3, 7, 14, 21, and 28 post-TBI, we tested each mouse for neurologic deficits. Using our previously published protocol [32], we scored mice on six parameters, including body symmetry, gait, climbing, circling behavior, front limb symmetry, and compulsory circling. Each test was graded from 0 to 4, establishing a maximum deficit score of 24.
Forelimb placing test
Mice were trained in the forelimb placing test before surgery. We gently held the animal by the torso and brushed its vibrissae against the edge of a tabletop. Normal mice place their ipsilateral forelimb quickly onto the tabletop. Placing was quantified as the percentage of successful responses in 10 trials [15, 33].
Wire-hanging test
The wire-hanging test was performed at 1, 2, 3, 7, 14, 21, and 28 days after surgery. We used a previously published wire-hanging test with minor modification to evaluate grip strength, balance, and endurance [34, 35]. A metallic wire (1 mm × 55 cm) was stretched horizontally between two posts, 50 cm above the ground. The mice were placed on the wire, and latency to fall was recorded. We gently covered their hind limbs with adhesive tape to prevent the mice from using all four paws and put a pillow beneath the mice to prevent injury from falls.
Rotarod test
An accelerating rotarod was used to measure motor function and balance [36]. The rotarod treadmill (Accuscan, Inc., Columbus, OH, USA) provided a motor balance and coordination assessment. Each mouse was placed in a neutral position on a cylinder (4 cm in diameter), which then began to rotate with accelerating speed (1 rpm/5 s) linearly from 1 rpm. The time spent on the rotarod was recorded automatically. Mice were given three trials, and the individual times from these trials were averaged.
Novel object recognition (NOR) test
On day 28 post-TBI, we used an established protocol to test each mouse with the NOR test [34, 35]. The experiments were carried out in a black, open-field box measuring 50 × 25 × 50 cm. On day 27 post-TBI, the mouse was placed in the box and habituated for 10 min. The next day, two identical novel objects (black cubes, 4×4×4 cm) were placed in the arena, and the mouse was allowed to explore the area for 10 min. After 1 h, one novel object (blue ball, 4 cm in diameter) and one old object (black cube) were placed in the box, and the mouse was allowed to explore for 5 min while being recorded by camera. We compared the total time spent exploring the old and new objects. The exploration time included time in direct contact with the objects and time within the object area; a discrimination index (total time spent with new object/total time devoted to exploration of objects) was also calculated for each mouse.
Tail suspension test (TST)
The TST was performed with each mouse 28 days post-TBI. Briefly, according to methods described previously [34, 35], mice were suspended by the tail with a piece of adhesive tape (1 cm from the tip, 17 cm long) 55 cm above the desk. A camera recorded the movement of the mouse for 6 min. Mice were considered immobile when they hung passively and motionless. The immobility time was calculated by subtracting the total amount of mobility time from the 360 s of test time.
Forced swim test (FST)
On day 28 post-TBI, each mouse underwent the FST based on an established protocol with minor modifications [34, 35]. Mice were placed individually in clear cylindrical tanks (height = 25 cm; diameter = 22 cm) with 15 cm of water at 23 ± 1°C. The trial was conducted for 6 min, and the period of immobility during the last 4 min was measured manually. The immobility time was calculated by subtracting the total amount of mobility time from the 240 s of test time. The mouse was judged to be immobile when it remained floating in an upright position and made only small movements to keep its head above the water.
Sucrose preference test
The sucrose preference test was carried out at 21 days after TBI. The test was performed as described previously with slight modifications [34]. Before the test, we adapted the mice to the sucrose solution (1%, w/v) by placing two bottles of sucrose solution in each cage for a period of 3 days; then we replaced one bottle of sucrose with water. Each bottle contained 50 mL of liquid. After 4 days, the respective weights of the sucrose solution and water consumed were recorded, and the percent sucrose preference was calculated by using the following formula: % Sucrose preference = [sucrose consumption in g / (water + sucrose consumption in g)] × 100.
Tissue processing and histology
At 3 and 28 days, mice were deeply anesthetized with isoflurane and perfused intracardially with phosphate-buffered saline (PBS) followed by 4% paraformaldehyde. All solutions were maintained at pH 7.4 and 4°C. Brains were removed and stored in 4% paraformaldehyde overnight and then transferred to 30% sucrose in PBS for another 48 h. Frozen serial coronal brain sections were sliced at 30 μm on a cryostat. Sections were used for Luxol fast blue staining (day 28, n=5/group), Cresyl violet staining (days 3 and 28, n = 8/group), Fluoro-Jade B (FJB) staining (day 3, n=6/group), and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL; day 3, n=6/group) [32, 36]. Mice assigned to propidium iodide (PI) staining (day 3, n=6/group) and hydroethidine analysis (day 3, n=6/group) were perfused with PBS only.
Lesion volume
On days 3 and 28 after TBI, coronal brain sections were stained with Cresyl violet (for Nissl bodies) as described previously [37]. Image J software (NIH, Bethesda, MD, USA) was used to quantify lesion volumes. The volume of the lesion in cubic millimeters was calculated as the sum of the damaged areas of each section multiplied by the interslice distance [3].
In vivo PI staining
To detect cell death in the injured brain, we diluted PI (10 mg/mL; Sigma-Aldrich Corporation, St Louis, MO, USA) in 0.9% NaCl and administered it to mice by intraperitoneal injection at 0.4 mg/kg 1 h before they were killed [32]. The sections were stained with 4′, 6-diamidino-2-phenylindole (DAPI) to show total nuclei. Samples were observed and photographed under a fluorescence microscope (Eclipse TE2000-E; Nikon, Tokyo, Japan) at excitation and emission wavelengths of 535 and 617 nm, respectively.
FJB histochemistry
FJB staining was used to quantify degenerating neurons as previously described [37, 38] (n=6 mice/group). Stained brain sections were examined with a fluorescence microscope (Eclipse TE2000-E; Nikon) at an excitation wavelength of 450–490 nm.
In situ detection of ROS
We investigated ROS production by detecting oxidized hydroethidine (HEt, a cell-permeable oxidative fluorescent dye) in situ as previously described [32, 37]. At 3 days after TBI, mice were injected intraperitoneally with 200 μL of 1 mg/mL HEt and euthanized 1 h later. Sections with similar lesion areas were selected, visualized, and photographed under a fluorescence microscope (Eclipse TE2000-E; Nikon) at excitation and emission wavelengths of 518 and 605 nm, respectively. For measurement of fluorescence intensity, all images were captured at the same exposure times, contrast settings, and intensity.
Perls staining
Ferric iron accumulation was detected by DAB-enhanced Perls staining as previously described [38, 39]. Samples of brain tissue were washed with PBS and incubated in freshly prepared Perls’ solution (5% potassium ferrocyanide plus 10% hydrochloric acid) for 30 min, followed by PBS washes (3×5 min). After DAB incubation for 3 min and hematoxylin counterstaining, Image J software was used to analyze iron deposition.
Immunofluorescence staining
Immunofluorescence staining was carried out as described previously [39, 40]. After being blocked, brain sections (n=5 mice/group) were incubated with rabbit anti-myelin basic protein (MBP; 1:1000, Abcam, Cambridge, MA) or rabbit anti-myeloperoxidase (MPO; 1:500, Dako, Carpentaria, CA) at 4°C overnight and then with Alexa Fluor 488-conjugated secondary antibody (1:1000; Molecular Probes, Eugene, OR) for 1 h at room temperature. Stained sections were examined with a microscope (Eclipse TE2000-E; Nikon). MBP-positive area was measured around the lesion from nine randomly selected locations per mouse (three 400× fields per section, three sections per mouse) with Image J software.
Quantification of Luxol fast blue, MBP staining, MPO staining, Cresyl violet, PI, FJB, HEt, and Perls staining
Luxol fast blue- and MBP-positive areas were calculated as %Area per 400× field near the lesion in the striatum. For Cresyl violet staining, we analyzed the surviving neurons in the top half of the cortex in the injured hemisphere (between bregma −0.8 mm and bregma −2.8 mm) with a Nikon Eclipse 600 microscope and quantified them with stereology using StereoInvestigator software, version 10 (MicroBrightField, Colchester, VT). PI-, FJB-, and iron-positive cells were counted immediately adjacent to the injured region at 200× magnification in three randomly selected sections per animal. The region of interest was defined within one 200× or 100× field that corresponded to ~460 μm from the edge of the injury area. PI-, FJB-, and iron-positive cells were expressed as cells/200× field, and MPO-positive cells were expressed as cells/100× field. Fluorescence intensity of HEt staining was quantified near the injured area. Regions sampled for quantification of positive cells and areas and for fluorescence intensity in brain sections are shown in Supplementary Fig. 1.
Brain water content
The extent of brain edema after TBI was determined by brain water content measurement. Brain water content was measured on day 3 after TBI, as described previously [41]. Mice were euthanized by decapitation. The brains were removed and divided into ipsilateral and contralateral hemisphere and cerebellum (which served as an internal control). Samples were weighed immediately on an analytical balance to obtain the wet weight and then dried for 24 h at 100°C to obtain the dry weight. Water content was calculated as (wet weight – dry weight)/wet weight × 100%.
Spectrophotometric assay for hemoglobin
The hemoglobin content of brains was quantified with the Hemoglobin Assay Kit (Sigma) at 72 h post-TBI (n = 6/group). Mice were anesthetized and then transcardially perfused with PBS. The injured hemisphere was collected in a tube with 250 μL of distilled water. The tissue was homogenized for 30 s, sonicated for 1 min, and then centrifuged at high speed (16000g) for 15 min. Fifty microliters of supernatant were transferred to flat-bottom, 96-well plates. After a 5-min incubation, we measured the absorbance at 400 nm. Hemoglobin content was calculated by a standard curve.
Western blot analysis
Mice were anesthetized and decapitated at 72 h after TBI (n = 6/group). A 3-mm coronal section around the major area of injury was collected in ice-cold PBS with a mouse brain slicer matrix (Zivic Instruments, Pittsburgh, PA) [15, 41]. We used the Nuclear Extraction Kit (Origene, Rockville, MD) to isolate cytosolic and nuclear protein fractions. To detect protein levels of HO-1, SOD1, NQO1, and Keap-1, we homogenized tissues in ice-cold protein extraction reagent (T-PER reagent; Pierce, Rockford, IL) with a complete mini protease inhibitor cocktail (Roche Molecular Biochemicals, Mannheim, Germany). The Bradford protein assay (Bio-Rad, Hercules, CA) was used to quantify total protein. Western blotting was carried out as previously described [41]. Equal amounts of protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (Bio-Rad). Membranes were blocked with 5% nonfat milk in a solution of Tris-buffered saline (TBS) and 0.1% Tween 20 and probed with primary antibodies against Nrf2 (1:500; Santa Cruz Biotechnology, Dallas, TX), HO-1 (1:2000; Stressgen, San Diego, CA), Keap-1 (1:1000; Santa Cruz Biotechnology), SOD1 (1:1000; Abcam, Cambridge, MA), NQO1 (1:1000; Novus, Littleton, CO), β-actin (1:5000; Santa Cruz Biotechnology), and histone (1:100; Santa Cruz Biotechnology) at 4°C overnight. The membranes were washed and incubated with horseradish peroxidase–linked anti-rabbit, anti-mouse, or anti-goat secondary antibody (1:3000; Santa Cruz Biotechnology) for 1 h. The intensity of the resulting protein bands was quantified with Image J software [42].
Gelatin gel zymography
For gelatin gel zymography [37, 43], protein samples were prepared from mouse brains 72 h post-TBI (n=6/group). Equal amounts of protein were separated on a 10% Tris-glycine gel with 0.1% gelatin as substrate. After separation, the gel was incubated in renaturing buffer (2.5% Triton X-100) twice for 1 h each at room temperature with gentle agitation and then in developing buffer (50 mM Tris-HCl [pH 7.5], 200 mM NaCl, 5 mM CaCl2, 0.05% Brij-35, 0.02% NaN3) for 24 h. Then the gel was incubated with 0.5% Coomassie blue R-250 for 30 min and destained appropriately to be photographed and analyzed densitometrically by the Image Analyzer LAS-4000 system (GE Healthcare, Pittsburgh, PA). Gelatinolytic activity was determined by bands at the appropriate molecular weights (pro-MMP-9, ~98 kDa; pro-MMP-2, ~ 72 kDa; R&D Systems, Minneapolis, MN).
Statistical analyses
Data are presented as mean ± SEM. We used two-way ANOVA to analyze all behavioral tests between and among treatment groups. In anatomical and biochemical studies, we used one-way or two-way ANOVA to compare multiple groups. Bonferroni post hoc analysis was used to determine where those differences occurred. Differences between two groups were tested with the two-tailed Student’s t-test. The criterion for statistical significance was p < 0.05.
Results
Mortality
Ten WT mice (6 in the vehicle-treated group and 4 in the EC-treated group) and three Nrf2 KO mice (1 in the vehicle-treated group and 2 in the EC-treated group) died and were excluded from data analysis.
EC treatment reduces short-term lesion volume and neurologic deficit after TBI
To identify the optimal dosage of EC for TBI treatment, we administered three doses of EC (5, 15, or 45 mg/kg) after TBI. Cresyl violet staining at 3 days after TBI (Fig. 1A) showed that the lesions were significantly smaller in the 15 mg/kg EC group (7.33±0.33 mm3) and 45 mg/kg EC group (7.15±0.22 mm3) than in the vehicle group (8.54±0.32 mm3; n=6/group, p<0.05; Fig. 1B). There was no significant difference between the 5 mg/kg EC group (7.99±0.22 mm3) and the vehicle group (Fig. 1B).
Fig. 1.
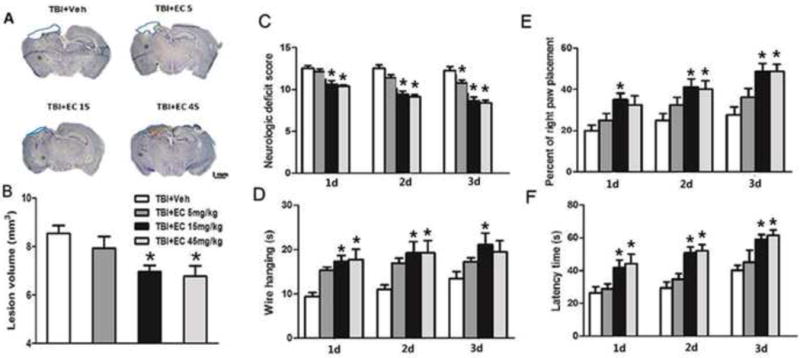
EC reduces brain lesion volume and improves neurologic function in mice in the short-term after TBI. (A) Cresyl violet-stained brain sections at 3 days post-TBI. Injured areas lack staining and are circled in blue (Scale bar: 1 mm). (B) Lesion volumes in the groups treated with 15 mg/kg or 45 mg/kg EC were significantly smaller than those in the vehicle-treated group at 3 days post-TBI. n=8/group, *p<0.05 (one-way ANOVA followed by Newman-Keuls test). (C) Neurologic deficit scores also were significantly lower in the groups treated with 15 mg/kg or 45 mg/kg EC than in the vehicle-treated group on days 1–3 after TBI. n=8/group, *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test). (D) EC treatment increased the latency to fall in the wire-hanging test compared with that in the vehicle-treated group beginning on day 1 after TBI. n=8/group, *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test). (E) In the forelimb placing test, 15 mg/kg EC treatment increased the percent of right paw placement on days 1–3, and 45 mg/kg EC increased the percent of right paw placement on days 2 and 3 after TBI, compared with that in the vehicle-treated group. n=8/group, *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test). (F) In the rotarod test, EC treatment increased the latency to fall on days 1–3 after TBI compared with that in the vehicle-treated group. n=8/group, *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test).
The neurologic deficit score was significantly lower in the 15 mg/kg group (day 1: 10.63±0.42; day 2: 9.38±0.42; day 3: 8.63±0.46) and in the 45 mg/kg EC group (10.38±0.18, 9.13±0.23, 8.38±0.32) than in the vehicle group (12.50±0.32, 12.50±0.42, 12.25 ±0.49; n=8/group, F=3.20, p<0.05; Fig. 1C) on days 1–3 after TBI. The 5 mg/kg EC dose promoted mouse locomotor function only at 3 days in this test (10.75±0.37; n=8/group, p<0.05; Fig. 1C). In addition, the wire hanging time was longer in the EC 15 mg/kg group (17.38±1.25 s, 19.25 ±2.52 s, 21.13± 2.69 s) than in the vehicle group (9.37±0.94 s, 11.00±0.98 s, 13.25±1.60 s; n=8/group, F=1.74, p<0.05; Fig. 1D) on days 1, 2, and 3 post-TBI. The group that received 45 mg/kg EC had longer wire-hanging time than did the vehicle group at 1 and 2 days (17.75±2.34 s, 19.25±2.81 s; n=8/group; p<0.05; Fig. 1D). In the forelimb placing test, 15 mg/kg EC increased the placing reflex on days 1 to 3 post-TBI (35.0±3.27%, 41.3 ±3.98%, 48.8±3.98%), and 45 mg/kg EC increased the placing reflex on days 2 and 3 post-TBI (40.0±4.22, 48.75± 3.50) compared to that in the vehicle group (20.0±2.67%, 25.0 ±3.27%, 27.5 ±4.12%, n=8/group, F=2.50, p<0.05; Fig. 1E). In the rotarod test, EC at 15 mg/kg (41.85±4.37 s, 50.79±3.58 s, and 59.00±2.89 s) and 45 mg/kg (44.25±5.88 s, 52.00±3.81 s, and 61.50±3.27 s) increased the latency to fall on days 1, 2, and 3 after TBI compared with that in the vehicle-treated group (26.26±3.84 s, 29.52±3.56 s, 40.19±3.04 s, n=8/group, F=1.66, p<0.05; Fig. 1F), but 5 mg/kg EC did not promote locomotor function in this test (p>0.05; Fig. 1F). Based on the results of these tests, we found that 15 mg/kg was the most efficacious dosage. Therefore, we used this dose in all subsequent studies.
EC decreases lesion volume and improves neurologic function in the long-term after TBI
EC (15 mg/kg) decreased lesion volume (8.23±0.68 mm3) compared to that in the vehicle group (10.94±0.90 mm3; n=8/group, p<0.05; Fig. 2A, B) at 28 days after TBI. EC also improved the neurologic deficit score and results of the forelimb-placing test, the wire-hanging test, and the rotarod test compared with those of the vehicle group (n=8/group, F=2.08, 1.77, 1.71, 3.65, p<0.05; Fig. 2C–F). In the NOR test for cognitive ability, the discrimination index decreased from 78.67±3.30% to 55.20±2.55% (n=8/group, F=13.62, p<0.05; Fig. 2G) after TBI, but EC significantly reversed this decrease to 64.43±2.08% (n=8/group, F=13.62, p<0.05; Fig. 2G). Similarly, TBI caused a decrease in the sucrose preference index from 79.23±3.40% in the sham group to 56.67±2.31% (n=8/group, F=9.904, p<0.05; Fig. 2H) in the vehicle-treated TBI group. However, EC significantly reversed the effect of TBI on sucrose preference to 67.02±2.17% (n=8/group, F=9.904, p<0.05; Fig. 2H). In the forced swim test and tail suspension test, the immobility time of the vehicle-treated group was significantly increased after TBI, whereas that of the EC group was decreased compared with that of the vehicle-treated group (n=8/group, F=13.72, 7.647, p<0.05; Fig. 2I, J) at 28 days after TBI.
Fig. 2.
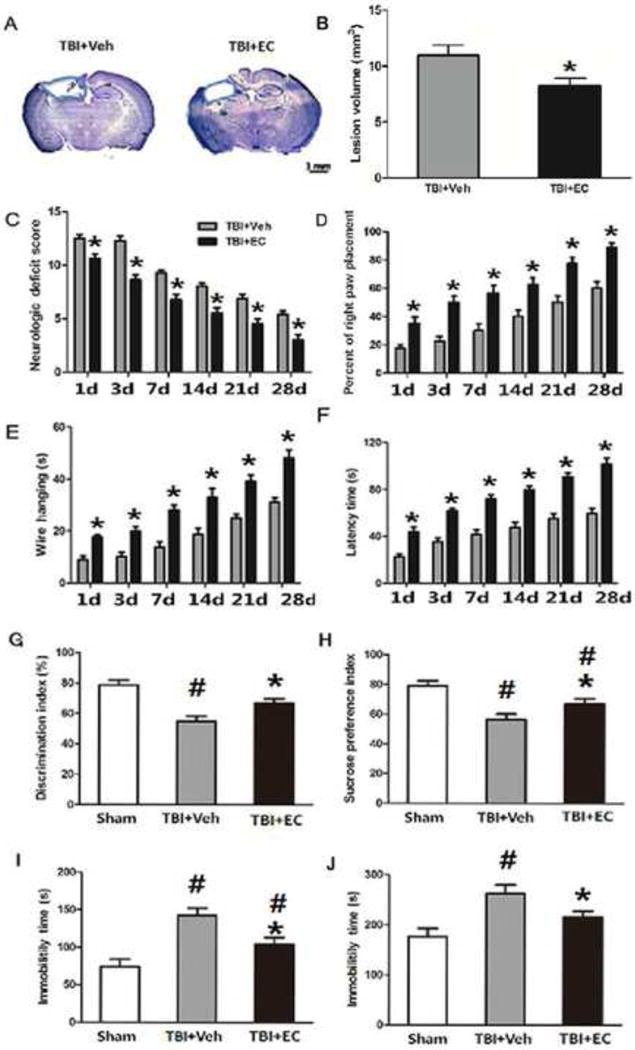
EC decreases lesion volume and improves long-term neurologic function in mice after TBI. Mice were treated for 7 days after TBI with 15 mg/kg EC or vehicle (n=8/group). (A) Cresyl violet-stained brain sections at 28 days post-TBI. Injured areas lack staining and are circled in blue (Scale bar: 1 mm). (B) Lesion volume was significantly smaller in the EC-treated group than in the vehicle-treated group on day 28 post-TBI. *p<0.05 (t-test). (C) Neurologic deficit scores were significantly lower in the EC-treated group than in the vehicle-treated group at all time points measured *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test). (D–F) EC treatment improved performance in the right paw placement test (D), the wire-hanging test (E), and the rotarod test (F) compared with that in the vehicle-treated group on days 1, 3, 7, 14, 21, and 28 after TBI. *p<0.05 (two-way ANOVA followed by Bonferroni post-hoc test). (G) In the novel object recognition test, TBI decreased the discrimination index compared with that of the sham group, but EC-treated TBI mice had a higher discrimination index than did vehicle-treated TBI mice.*p<0.05 versus the vehicle group; #p<0.05 versus the sham group (one-way ANOVA followed by Newman-Keuls test). (H) On day 28, TBI mice had a lower sucrose preference index than did sham mice. However, EC treatment increased the sucrose preference index compared with that in the vehicle-treated group. *p<0.05 versus the vehicle group; #p<0.05 versus the sham group (one-way ANOVA followed by Newman-Keuls test). Additionally, TBI increased immobility time in both the forced swim test (I) and the tail suspension test (J). EC treatment significantly decreased immobility time compared with that in the vehicle-treated group in both tests. n=8/group, *p<0.05 versus the vehicle group; #p<0.05 versus the sham group (one-way ANOVA followed by Newman-Keuls test).
EC decreases cell death and neuronal degeneration, increases cell survival, and decreases brain water content after TBI
As EC significantly attenuated brain injury and improved the results of the behavior tests, we further investigated whether this treatment rescued cell death. At 3 days after TBI, EC decreased the number of PI-positive cells (118.56±7.16 cells) compared with that in the vehicle group (164.58±9.64 cells; n=6/group, p<0.05; Fig. 3A, B). Additionally, the EC group had fewer FJB-positive degenerating neurons (56.56±6.24) in the peri-injury area than did the vehicle group (83.02±7.77 cells; n=6/group, p<0.05; Fig. 3C, D) and had a greater number of surviving neurons (1,126,000±123,700 cells vs. 758,100±111,200; n=7/group, p<0.05; Fig. 3E, F). At 3 days post-TBI, brain water content of the ipsilateral hemisphere was significantly lower in the EC-treated group than in the vehicle-treated group (78.86±0.17% vs. 79.87±0.40%; n=6/group, p<0.05; Fig. 3G). Brain water content of the contralateral hemisphere and cerebellum did not differ significantly between the two groups (p>0.05; Fig. 3G).
Fig. 3.
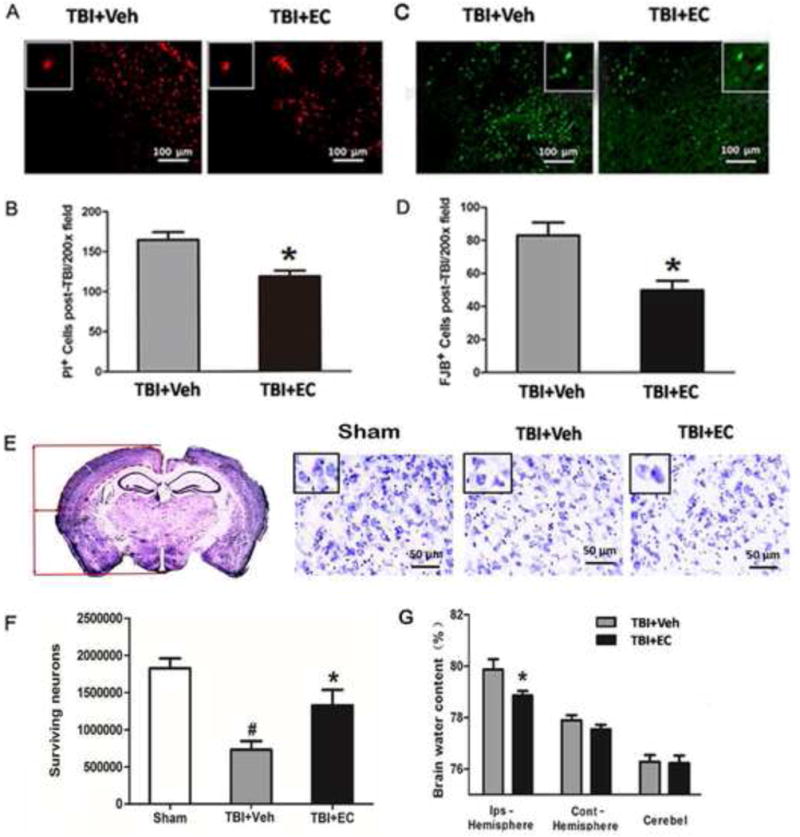
EC reduces cell death and neurodegeneration, increases cell survival, and decreases brain water content after TBI. (A) Representative propidium iodide (PI)-stained brain sections are shown for the vehicle-treated and the EC-treated groups. (B) EC decreased the number of PI-positive cells per 200× field. n=6/group, *p<0.05 (t-test). (C) Fluoro-Jade B (FJB)-stained brain sections. (D) EC-treated mice had significantly fewer FJB-positive cells per 200× field than did vehicle-treated mice. n=6/group, *p<0.05 (t-test). (E) Left: a representative Cresyl violet-stained brain section on day 28 after TBI. The location in the cortex used to determine the density of surviving neurons by stereology is delineated by a dashed red line. Right: three representative images of Cresyl violet-stained brain cortical sections from the sham group, the TBI+vehicle group, and the TBI+EC group. Insets represent higher magnification images of Cresyl violet-stained cells in the cortex. Scale bar, 50 μm. (F) Quantitative stereological analysis showed significantly more surviving neurons in the EC-treated mice than in the vehicle-treated mice on day 28 after TBI. n=7/group, *p<0.05 versus the vehicle-treated group; #p<0.05 versus the sham group (one-way ANOVA followed by Newman-Keuls test). (G) At 3 days post-TBI, brain water content of the ipsilateral hemisphere (but not contralateral hemisphere or cerebellum [cerebel]) was significantly lower in the EC-treated group than in the vehicle-treated group. n=8/group, *p<0.05 (t-test).
EC decreases white matter injury on day 28 post-TBI
On day 28 after TBI, we used Luxol fast blue (Fig. 4A) and MBP staining (Fig. 4C) to label normal myelin in brain sections. Both staining procedures showed that the percentage of area with normal myelin was higher in the EC-treated group than in the vehicle-treated group (n=6/group; p<0.05; Fig. 4B, D).
Fig. 4.
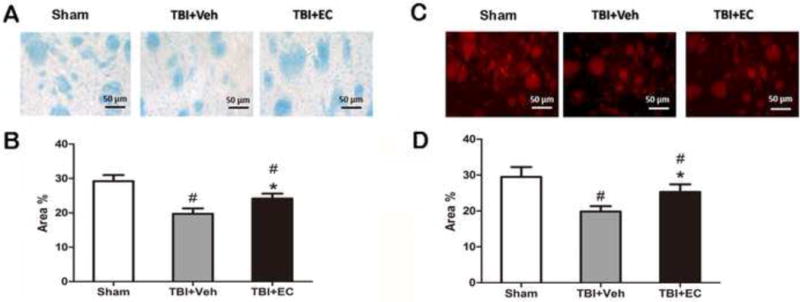
EC increases white matter recovery on day 28 post-TBI. On day 28 post-TBI, Luxol fast blue (A, B) and anti-myelin basic protein (MBP) antibody (C, D) were used to label normal myelin in brain sections. The percentage of area with normal myelin was higher in sections from EC-treated mice than in those from vehicle-treated mice by both methods (B, D). n=6/group; *p<0.05 (t-test).
EC attenuates neutrophil infiltration, alleviates reactive oxygen species (ROS) production, and decreases MMP-9 enzyme activity
On day 1 after TBI, we examined neutrophil infiltration by MPO staining. EC decreased the number of the MPO-positive cells compared with that in the vehicle-treated group (n=6/group, p<0.05; Fig. 5A, B). After mice were injected with HEt, ROS were observed as red fluorescence signal around the lesion at 1 day post-TBI (Fig. 5C). However, the EC-treated mice exhibited less ROS production than did the vehicle-treated mice (n=6/group, p<0.05; Fig. 5D).
Fig. 5.
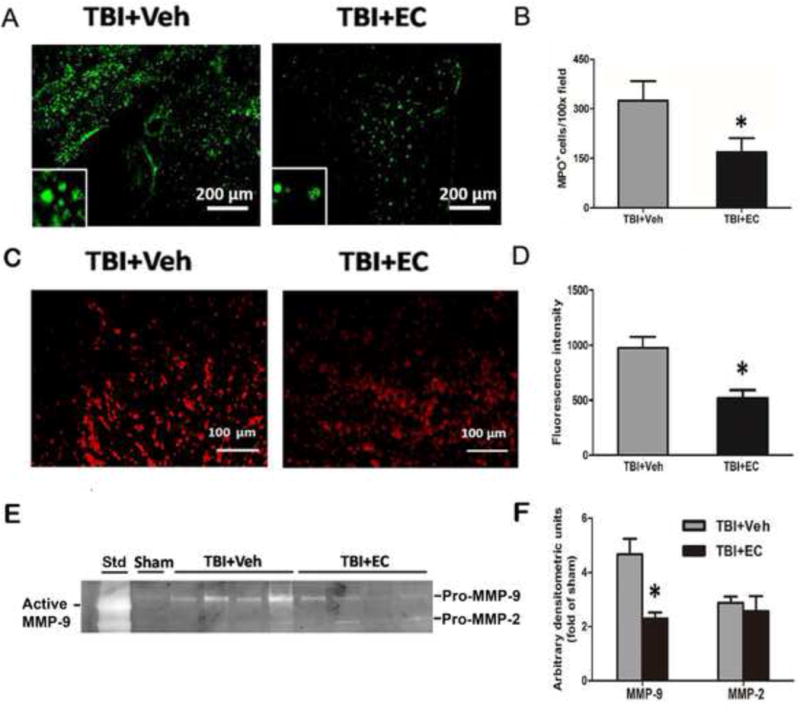
EC reduces TBI-induced neutrophil infiltration, reactive oxygen species (ROS) production, and MMP-9 enzyme activity. (A) Representative MPO immunoreactivity on brain sections from vehicle- and EC-treated groups on day 1 after TBI. (B) Quantification analysis showed that the EC-treated TBI group had less MPO-positive infiltrating neutrophils than did the vehicle-treated group. n=6/group, *p<0.05. (C) Mice were injected intraperitoneally with hydroethidine, which is oxidized to ethidium in the presence of superoxide. After TBI, ethidium fluorescent signal (seen as small red particles) was evident around the lesion in brain sections at 1 day. Scale bar: 100 μm. (D) Quantification analysis of fluorescence intensity indicated that ROS production was significantly less in the EC group than in the vehicle group at 1 day after TBI. n=6/group, *p<0.05 (t-test). (E) Representative gelatin gel zymogram of MMP-9 and MMP-2 activity at 3 days post-TBI. (F) Quantification analysis indicated that EC decreased MMP-9 activity, but not MMP-2 activity, compared with that in the vehicle-treated group. n=6/group, *p<0.05 (t-test).
We used gelatin gel zymography to detect MMP-2 and MMP-9 enzyme activity. The results showed that EC decreased MMP-9 activity by ~51% compared with that in the vehicle-treated group (n=6/group, p<0.05; Fig. 5E, F). However, we observed no difference in the MMP-2 gelatinolytic activity between the EC- and vehicle-treated groups (n=6/group, p>0.05; Fig. 5E, F).
EC reduces Keap 1 expression while increasing Nrf2 nuclear accumulation
Next we used Western blot analysis to determine whether EC reduces Keap 1 expression and promotes Nrf2 nuclear translocation. As shown in Figure 6A, Keap 1 expression was significantly lower in the vehicle-treated TBI group than in the sham group (n=6/group, F=49.07, p<0.05). EC further decreased Keap 1 protein expression compared with that in the vehicle group at 3 days post-TBI (n=6/group, F=49.07, p<0.05; Fig. 6A). EC did not alter Nrf2 expression in the cytosol (n=6/group, F=0.07899, p>0.05; Fig. 6B), but it did increase nuclear Nrf2 expression compared with that in the vehicle-treated group (n=6/group, F=4.699, p<0.05; Fig. 6C) at 3 days post-TBI.
Fig. 6.
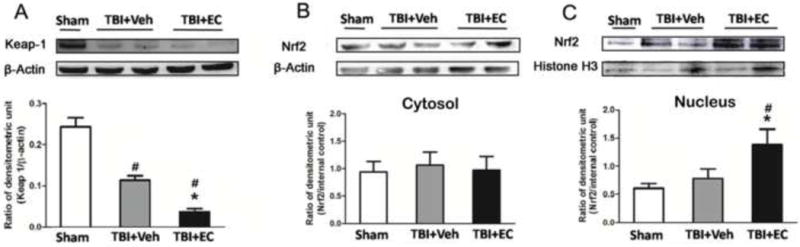
EC reduces cytosolic Keap 1 expression and increases Nrf2 nuclear accumulation at 3 days post-TBI. (A–C) The upper panels show immunoblots of ipsilateral hemispheres 3 days after sham surgery or TBI. The lower panels represent the quantitative analysis. (A) Keap 1 expression decreased significantly after TBI and was further decreased by EC treatment. Nrf2 expression was unchanged in the cytosol of mice treated with EC (B) but was significantly elevated in the nucleus (C). n=6/group, *p<0.05 versus vehicle group; #p<0.05 versus sham group (one-way ANOVA followed by Newman-Keuls test).
EC increases SOD1 and NQO1 expression and reduces HO-1 expression and iron deposition without reducing the brain hemoglobin level
At 3 days after TBI, we examined the expression of transcripts downstream of Nrf2. Expression levels of SOD1 (Fig. 7A) and NQO1 (Fig. 7C) were increased after TBI (n=6/group, F=2.819, 3.675, p<0.05; Fig. 7B, D) compared with those in the sham group and were further increased by EC (n=6/group, F=2.819, 3.675, p<0.05; Fig. 7B, D) compared with levels in the vehicle-treated group. In contrast, expression level of HO-1, one of the key downstream effectors of Nrf2, was upregulated after TBI but was reduced by approximately two-fold after EC treatment (n=6/group, F=17.72, p<0.05; Fig. 7E, F).
Fig. 7.
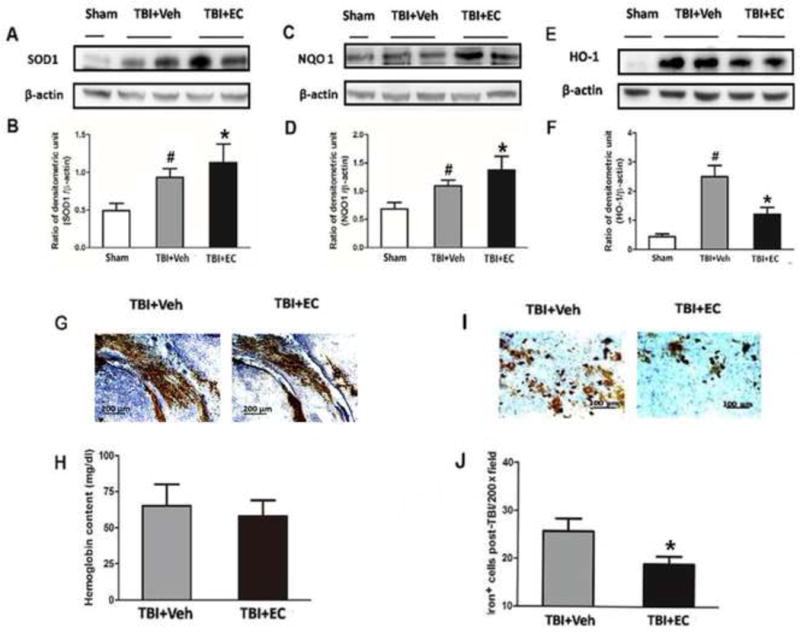
EC increases superoxide dismutase (SOD1) and quinone 1 (NQO1) expression and reduces heme oxygenase 1 (HO-1) protein expression and iron deposition on day 3 post-TBI. (A–F) The upper panels show immunoblots of ipsilateral hemispheres 3 days after sham surgery or TBI. The lower panels represent the quantitative analysis. SOD1 and NQO1 expression each increased after TBI and were further increased in mice treated with EC. In the absence of EC, TBI significantly increased HO-1 expression compared with that in the sham group. EC treatment significantly prevented this increase. *p<0.05 versus vehicle group; #p<0.05 versus sham group (one-way ANOVA followed by Newman-Keuls test). (G) Images of bleeding in the injury area on day 3 post-TBI. (H) EC did not reduce brain hemoglobin level. n=6/group, p>0.05 (t-test). (I) Representative Perls-stained brain sections on day 3 post-TBI. (J) Quantification analysis showed that TBI mice treated with EC had fewer iron-positive cells in the injury area than did vehicle-treated mice. *p<0.05 (t-test).
We found that all mice had various amounts of bleeding after TBI. Therefore, we measured brain hemoglobin at day 3 after TBI but observed no difference between the vehicle-treated and EC-treated TBI groups (65.08±14.85 vs 58.00±10.89 mg/dL, n=6/group, p>0.05; Fig. 7G, H). Because HO-1 is the main enzyme to metabolize heme (hemoglobin degradation products) to free iron, we examined ferric iron deposition at 3 days after TBI (Fig. 7I). EC significantly reduced the number of iron-positive cells compared with that in the vehicle-treated group (18.91±1.62 vs. 25.71±2.61, n=6/group, p<0.05; Fig. 7J).
EC does not decrease lesion volume or promote neurologic function in Nrf2 KO mice after TBI
To verify whether EC protects the brain from injury via the Nrf2 signaling pathway, we subjected Nrf2 KO mice to the TBI model. At 28 days post-TBI, lesion volume in the vehicle-treated and EC-treated Nrf2 KO mice did not differ significantly (n=8/group, p>0.05; Fig. 8A, B). However, EC decreased the neurologic deficit score at 3, 7, 14, and 28 days after TBI compared with that in the vehicle-treated group (n=8/group, F=2.07, p<0.05; Fig. 8C). In the forelimb placing test and the wire-hanging test, we observed no significant difference between the vehicle- and EC-treated groups at any time point (n=8/group, F=17.59, 14.89, p>0.05; Fig. 8D, E). In the rotarod test, the latency to fall was longer in the EC-treated group than in the vehicle treated group on day 3 post-TBI (n=8/group, F=0.50, p<0.05), but not at other time points (n=8/group, p>0.05; Fig. 8F). At 28 days post-TBI, there was no significant difference between the vehicle- and EC-treated groups in the discrimination index of the NOR test or in the sucrose preference index (n=6/group, F=56.09, 14.15, p>0.05), although these parameters decreased in the vehicle group compared with those in the sham group (n=6/group, F=56.09, 14.15, p<0.05; Fig. 8G, H). Furthermore, EC did not provide any benefit in the TST or FST. Immobility time in both the TST and FST increased after TBI in the vehicle group (n=6/group, F=6.25, 12.144, p<0.05) but was not significantly reduced by EC treatment (n=6/group, F=6.25, 12.144, p>0.05; Fig. 8I, J).
Fig. 8.
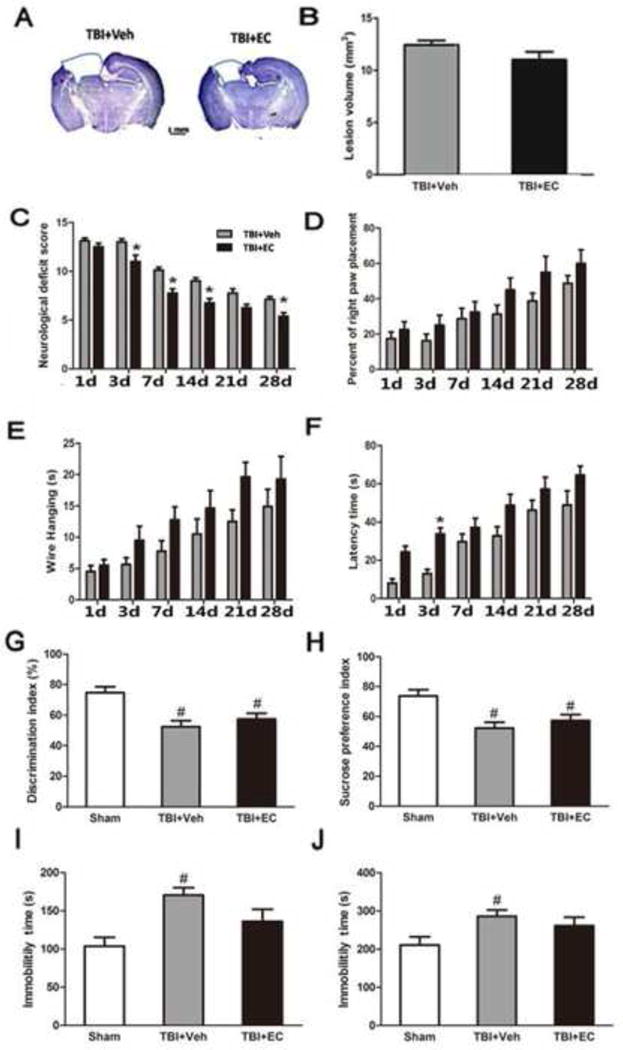
EC does not decrease lesion volume or promote neurologic function in Nrf2 knockout (KO) mice after TBI. (A) Cresyl violet-stained brain sections at 28 days post-TBI. Injured areas lack staining and are circled in blue (Scale bar: 1 mm). (B) Lesion volume did not differ significantly between the vehicle- and EC-treated groups. n=8/group, p>0.05 (t-test). (C) Neurologic deficit score was significantly lower in the EC-treated group than in the vehicle-treated group on days 3, 7, 14, and 28 after TBI. n=8/group, *p<0.05 (two-way ANOVA followed by Bonferroni test). Performance in the forelimb-placing test (D), wire-hanging test (E), and rotroard test (F) did not differ between the EC-treated Nrf2 KO mice and the vehicle-treated Nrf2 KO mice after TBI. n=8/group, all p>0.05. The discrimination index in the novel object recognition test (G) and the sucrose preference index (H) were both significantly decreased in Nrf2 KO mice after TBI but did not differ between vehicle-treated and EC-treated TBI groups. n=6/group, both p>0.05. Immobility time in the tail suspension test (I) and forced swim test (J) was significantly increased in Nrf2 KO mice after TBI but did not differ between vehicle-treated and EC-treated TBI groups. n=6/group, both p>0.05. *p<0.05 versus vehicle group; #p<0.05 versus sham group (one-way ANOVA followed by Newman-Keuls test).
Discussion
Fruit, dark chocolate, and green tea contain a large percentage of flavonoids that activate endogenous protective signaling pathways in the body and brain [13]. The flavonoid EC has been shown to be able to cross the BBB after oral or intravenous administration in rodents [9–12], and we have reported recently that EC can protect the brain via synergistic Nrf2 pathways after intracerebral hemorrhage [15]. In this study, we found that in the mouse CCI model, (1) EC reduces brain injury after TBI both in the short- and long-term; (2) EC reduces TBI-induced cell death and increases neuron survival in the peri-injury region; (3) EC reduces Keap 1 expression and promotes Nrf2 nuclear accumulation and the expression of SOD1 and NQO1 but reduces HO-1 expression and iron deposition; (4) EC reduces neutrophil infiltration, oxidative brain damage, and MMP-9 activity and has a protective effect on white matter; and 5) EC does not produce a clear protective effect in Nrf2 KO mice.
According to the World Health Organization, TBI is the leading cause of death in young adults [5]. We used 2- to 3-month-old male animals in this study to enhance clinical relevance, as TBI is most common in young male adults. Although a few studies [15, 16] have investigated the use of oral EC to treat stroke in rodents, to our knowledge, no study has investigated the use of EC for treatment of TBI. We used a moderate CCI model to mimic TBI because it is a well-researched model and is being used with increasing frequency to investigate TBI [44, 45]. In our study, we administered 15 mg/kg EC, which equates to a human dose of 1.22 mg/kg, as calculated by a body surface area conversion factor [46]. This dose decreased the lesion volume and improved neurologic function of WT mice at both short- and long-term time points. These results mirror those in an intracerebral hemorrhage model [15].
Because excess fluid increases brain tissue volume and intracranial pressure, brain edema contributes to morality after TBI [47]. Cell death and neurodegeneration after TBI occur not only because of the first impact, but also from secondary brain injury [45, 48]. Our data showed that EC reduced water content at 3 days post-TBI and lessened cell and neuronal death (as evidenced by a reduction in PI and FJB staining), while increasing neuronal survival (as shown by stereological analysis of Cresyl violet-stained neurons). These data suggest a direct protective effect of EC on neurons after TBI.
After brain injury, infiltrating neutrophils produce oxidative stress-causing ROS [49], which contribute to neurodegeneration and white matter injury. In this study, we found that EC attenuated neutrophil infiltration, decreased ROS production, and protected against white matter injury after TBI, as evidenced by an increase in MBP and Luxol Fast blue staining. An endogenous cytoprotective defense system protects the brain by balancing ROS and antioxidant/defense enzymes [50]. The pleiotropic transcription factor Nrf2 plays a key role in adjusting the inducible cytoprotective response by activating AREs [5, 51, 52]. Under physiologic conditions, Nrf2 binds with its negative regulator, Keap 1, in the cytoplasm [53]. However, under conditions of oxidative stress, Nrf2 is liberated from the Keap1-Nrf2 complex and translocates into the nucleus, where it activates the Nrf2-ARE pathway. We found that ROS and Keap 1 expression were decreased after EC treatment in the TBI model. Moreover, EC treatment increased nuclear accumulation of Nrf2 and cytoplasmic expression of phase II enzymes SOD1 and NQO1, which can combat oxidative stress. To verify whether EC protects against brain injury via this Nrf2-ARE pathway, we treated Nrf2 KO mice with EC after TBI. We found that EC did not decrease lesion volume or improve neurologic function in most of the behavior tests. These results indicate that EC likely produces neuroprotection mainly via the Nrf2-ARE pathway and that exogenous agents have the potential to enhance the endogenous Nrf2-ARE signaling pathway and thereby help the brain defend itself against oxidative damage after TBI.
HO-1, one of the cytoprotective enzymes activated by the Nrf2-ARE pathway [54], degrades heme into iron, carbon monoxide, and biliverdin. Studies show that increased iron toxicity may play a major role in BBB disruption, brain edema, oxidative stress, and neurologic deficits after TBI [55]. To our knowledge, there is no published evidence that EC can reduce iron deposition/toxicity after TBI. Nevertheless, in our study, HO-1 protein expression decreased in concert with a reduction in iron deposition after EC treatment in the TBI model. This finding is consistent with our recent study in wild-type and HO-1 knockout mice, which showed a causal relationship between HO-1 expression and iron deposition in the hemorrhagic brain [15]. Because EC treatment reduced HO-1 protein expression, we speculate that EC might activate an Nrf2-independent pathway that modulates HO-1 expression and iron deposition in the CCI model. This finding is in line with previous studies which showed that activator protein 1 (AP-1) regulates HO-1 gene expression in Nrf2-deficient fibroblasts [56] and normal brain astrocytes [57]. AP-1 is a stress-activated transcription factor and has been regarded as an inflammatory signaling molecule after TBI [58]. EC treatment reduced MMP-9 activity on day 3 post-TBI, an effect that could have been caused by reduced neutrophil infiltration. Because MMP-9 also lies downstream of AP-1 [59], we hypothesize that in addition to activating the Nrf2-ARE pathway, EC may also inhibit activation of the AP-1 pathway after TBI, which would lead to downregulation of HO-1 and MMP-9 expression/activity and a subsequent reduction in iron-induced brain damage and MMP-9-mediated neurovascular injury.
In conclusion, we demonstrated that EC protects the brain against injury after TBI via Nrf2-dependent and –independent pathways. In future research, we will investigate whether EC inhibits the AP-1 pathway to decrease HO-1/MMP-9 expression/activity and iron deposition, as we have shown in the hemorrhagic stroke model [15]. Studies also are needed to examine whether EC targets multiple signaling pathways that cause crosstalk. Overall, our results suggest that EC might be a promising drug candidate for treatment of TBI.
Supplementary Material
Highlights.
(−)-Epicatechin mitigated traumatic brain injury.
(−)-Epicatechin activated the Nrf2 pathway.
(−)-Epicatechin prevented HO-1 overexpression.
(−)-Epicatechin reduced iron deposition.
Protection by (−)-epicatechin involves Nrf2-depedent and –independent pathways.
Acknowledgments
We thank Dr. Zengjin Yang for help with the stereology protocol, Drs. Raymond Koehler and Zengjin Yang for helpful discussions, and Claire Levine for assistance with manuscript editing.
Funding
This study was supported by the Natural Science Foundation of China (81071008), the Excellent Youth Foundation of Henan Scientific Committee (114100510005), the Technology Foundation for Selected Overseas Chinese Scholars, American Heart Association grant 13GRNT15730001, and National Institutes of Health grants R01NS078026 and R01AT007317. T.C. is the recipient of the China Scholarship Council Joint PhD Training award.
Abbreviations
- BBB
blood-brain barrier
- CCI
controlled cortical impact
- EC
(−)-epicatechin
- FJB
Fluoro-Jade B
- FST
forced swim test
- NOR
novel object recognition
- Nrf2
NF-E2-related factor
- PBS
phosphate-buffered saline
- PI
propidium iodide
- ROS
reactive oxygen species
- TBI
traumatic brain injury
- TST
tail suspension test
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fernandez-Gajardo R, Matamala JM, Carrasco R, Gutierrez R, Melo R, Rodrigo R. Novel therapeutic strategies for traumatic brain injury: acute antioxidant reinforcement. CNS Drugs. 2014;28:229–248. doi: 10.1007/s40263-013-0138-y. [DOI] [PubMed] [Google Scholar]
- 2.Xu J, Wang H, Ding K, Zhang L, Wang C, Li T, Wei W, Lu X. Luteolin provides neuroprotection in models of traumatic brain injury via the Nrf2-ARE pathway. Free Radic Biol Med. 2014;71:186–195. doi: 10.1016/j.freeradbiomed.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 3.Wang W, Li H, Yu J, Hong M, Zhou J, Zhu L, Wang Y, Luo M, Xia Z, Yang ZJ, Tang T, Ren P, Huang X, Wang J. Protective effects of Chinese herbal medicine Rhizoma drynariae in rats after traumatic brain injury and identification of active compound. Mol Neurobiol. 2015 doi: 10.1007/s12035-12015-19385-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu J, Wang H, Ding K, Lu X, Li T, Wang J, Wang C, Wang J. Inhibition of cathepsin S produces neuroprotective effects after traumatic brain injury in mice. Mediators Inflamm. 2013;2013:187873. doi: 10.1155/2013/187873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mendes Arent A, de Souza LF, Walz R, Dafre AL. Perspectives on molecular biomarkers of oxidative stress and antioxidant strategies in traumatic brain injury. Biomed Res Int. 2014;2014:723060. doi: 10.1155/2014/723060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fraga CG, Oteiza PI. Dietary flavonoids: Role of (−)-epicatechin and related procyanidins in cell signaling. Free Radic Biol Med. 2011;51:813–823. doi: 10.1016/j.freeradbiomed.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 7.Mak JC. Potential role of green tea catechins in various disease therapies: progress and promise. Clin Exp Pharmacol Physiol. 2012;39:265–273. doi: 10.1111/j.1440-1681.2012.05673.x. [DOI] [PubMed] [Google Scholar]
- 8.Sokolov AN, Pavlova MA, Klosterhalfen S, Enck P. Chocolate and the brain: neurobiological impact of cocoa flavanols on cognition and behavior. Neurosci Biobehav Rev. 2013;37:2445–2453. doi: 10.1016/j.neubiorev.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 9.van Praag H, Lucero MJ, Yeo GW, Stecker K, Heivand N, Zhao C, Yip E, Afanador M, Schroeter H, Hammerstone J, Gage FH. Plant-derived flavanol (−)epicatechin enhances angiogenesis and retention of spatial memory in mice. J Neurosci. 2007;27:5869–5878. doi: 10.1523/JNEUROSCI.0914-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu L, Zhang QL, Zhang XY, Lv C, Li J, Yuan Y, Yin FX. Pharmacokinetics and blood-brain barrier penetration of (+)-catechin and (−)-epicatechin in rats by microdialysis sampling coupled to high-performance liquid chromatography with chemiluminescence detection. J Agric Food Chem. 2012;60:9377–9383. doi: 10.1021/jf301787f. [DOI] [PubMed] [Google Scholar]
- 11.Abd El, Mohsen MM, Kuhnle G, Rechner AR, Schroeter H, Rose S, Jenner P, Rice-Evans CA. Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Radic Biol Med. 2002;33:1693–1702. doi: 10.1016/s0891-5849(02)01137-1. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Ferruzzi MG, Ho L, Blount J, Janle EM, Gong B, Pan Y, Gowda GA, Raftery D, Arrieta-Cruz I, Sharma V, Cooper B, Lobo J, Simon JE, Zhang C, Cheng A, Qian X, Ono K, Teplow DB, Pavlides C, Dixon RA, Pasinetti GM. Brain-targeted proanthocyanidin metabolites for Alzheimer’s disease treatment. J Neurosci. 2012;32:5144–5150. doi: 10.1523/JNEUROSCI.6437-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nehlig A. The neuroprotective effects of cocoa flavanol and its influence on cognitive performance. Br J Clin Pharmacol. 2013;75:716–727. doi: 10.1111/j.1365-2125.2012.04378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cox CJ, Choudhry F, Peacey E, Perkinton MS, Richardson JC, Howlett DR, Lichtenthaler SF, Francis PT, Williams RJ. Dietary (−)-epicatechin as a potent inhibitor of betagamma-secretase amyloid precursor protein processing. Neurobiol Aging. 2015;36:178–187. doi: 10.1016/j.neurobiolaging.2014.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chang CF, Cho S, Wang J. (−)-Epicatechin protects hemorrhagic brain via synergistic Nrf2 pathways. Ann Clin Transl Neurol. 2014;1:258–271. doi: 10.1002/acn3.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shah ZA, Li RC, Ahmad AS, Kensler TW, Yamamoto M, Biswal S, Dore S. The flavanol (−)-epicatechin prevents stroke damage through the Nrf2/HO1 pathway. J Cereb Blood Flow Metab. 2010;30:1951–1961. doi: 10.1038/jcbfm.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruijters EJ, Weseler AR, Kicken C, Haenen GR, Bast A. The flavanol (−)-epicatechin and its metabolites protect against oxidative stress in primary endothelial cells via a direct antioxidant effect. Eur J Pharmacol. 2013;715:147–153. doi: 10.1016/j.ejphar.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 18.Yan W, Wang HD, Hu ZG, Wang QF, Yin HX. Activation of Nrf2-ARE pathway in brain after traumatic brain injury. Neurosci Lett. 2008;431:150–154. doi: 10.1016/j.neulet.2007.11.060. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Fields J, Zhao C, Langer J, Thimmulappa RK, Kensler TW, Yamamoto M, Biswal S, Dore S. Role of Nrf2 in protection against intracerebral hemorrhage injury in mice. Free Radic Biol Med. 2007;43:408–414. doi: 10.1016/j.freeradbiomed.2007.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ding K, Wang H, Xu J, Li T, Zhang L, Ding Y, Zhu L, He J, Zhou M. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2-ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1–11. doi: 10.1016/j.freeradbiomed.2014.04.031. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 22.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47:1304–1309. doi: 10.1016/j.freeradbiomed.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Dore S. Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain. 2007;130:1643–1652. doi: 10.1093/brain/awm095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang Q, Zuo Z, Ho WK, Chow MS. Comparison of the pharmacokinetics of hawthorn phenolics in extract versus individual pure compound. J Clin Pharmacol. 2005;45:106–112. doi: 10.1177/0091270004270500. [DOI] [PubMed] [Google Scholar]
- 25.Roura E, Andres-Lacueva C, Jauregui O, Badia E, Estruch R, Izquierdo-Pulido M, Lamuela-Raventos RM. Rapid liquid chromatography tandem mass spectrometry assay to quantify plasma (−)-epicatechin metabolites after ingestion of a standard portion of cocoa beverage in humans. J Agric Food Chem. 2005;53:6190–6194. doi: 10.1021/jf050377u. [DOI] [PubMed] [Google Scholar]
- 26.Chen L, Lee MJ, Li H, Yang CS. Absorption, distribution, elimination of tea polyphenols in rats. Drug Metab Dispos. 1997;25:1045–1050. [PubMed] [Google Scholar]
- 27.Rodriguez-Mateos A, Cifuentes-Gomez T, Gonzalez-Salvador I, Ottaviani JI, Schroeter H, Kelm M, Heiss C, Spencer JP. Influence of age on the absorption, metabolism, and excretion of cocoa flavanols in healthy subjects. Mol Nutr Food Res. 2015;59:1504–1512. doi: 10.1002/mnfr.201500091. [DOI] [PubMed] [Google Scholar]
- 28.Baba S, Osakabe N, Natsume M, Muto Y, Takizawa T, Terao J. In vivo comparison of the bioavailability of (+)-catechin, (−)-epicatechin and their mixture in orally administered rats. J Nutr. 2001;131:2885–2891. doi: 10.1093/jn/131.11.2885. [DOI] [PubMed] [Google Scholar]
- 29.Schroeter H, Heiss C, Balzer J, Kleinbongard P, Keen CL, Hollenberg NK, Sies H, Kwik-Uribe C, Schmitz HH, Kelm M. (−)-Epicatechin mediates beneficial effects of flavanol-rich cocoa on vascular function in humans. Proc Natl Acad Sci U S A. 2006;103:1024–1029. doi: 10.1073/pnas.0510168103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cuevas E, Limon D, Perez-Severiano F, Diaz A, Ortega L, Zenteno E, Guevara J. Antioxidant effects of epicatechin on the hippocampal toxicity caused by amyloid-beta 25–35 in rats. Eur J Pharmacol. 2009;616:122–127. doi: 10.1016/j.ejphar.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Landis SC, Amara SG, Asadullah K, Austin CP, Blumenstein R, Bradley EW, Crystal RG, Darnell RB, Ferrante RJ, Fillit H, Finkelstein R, Fisher M, Gendelman HE, Golub RM, Goudreau JL, Gross RA, Gubitz AK, Hesterlee SE, Howells DW, Huguenard J, Kelner K, Koroshetz W, Krainc D, Lazic SE, Levine MS, Macleod MR, McCall JM, Moxley RT, 3rd, Narasimhan K, Noble LJ, Perrin S, Porter JD, Steward O, Unger E, Utz U, Silberberg SD. A call for transparent reporting to optimize the predictive value of preclinical research. Nature. 2012;490:187–191. doi: 10.1038/nature11556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao X, Wu T, Chang CF, Wu H, Han X, Li Q, Gao Y, Li Q, Hou Z, Maruyama T, Zhang J, Wang J. Toxic role of prostaglandin E2 receptor EP1 after intracerebral hemorrhage in mice. Brain Behav Immun. 2015;46:293–310. doi: 10.1016/j.bbi.2015.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sugawara T, Jadhav V, Ayer R, Chen W, Suzuki H, Zhang JH. Thrombin inhibition by argatroban ameliorates early brain injury and improves neurological outcomes after experimental subarachnoid hemorrhage in rats. Stroke. 2009;40:1530–1532. doi: 10.1161/STROKEAHA.108.531699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu HT, Bian C, Yuan JC, Chu WH, Xiang X, Chen F, Wang CS, Feng H, Lin JK. Curcumin attenuates acute inflammatory injury by inhibiting the TLR4/MyD88/NF-kappaB signaling pathway in experimental traumatic brain injury. J Neuroinflammation. 2014;11:59. doi: 10.1186/1742-2094-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhu W, Gao Y, Chang CF, Wan JR, Zhu SS, Wang J. Mouse models of intracerebral hemorrhage in ventricle, cortex, and hippocampus by injections of autologous blood or collagenase. PLoS One. 2014;9:e97423. doi: 10.1371/journal.pone.0097423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han X, Lan X, Li Q, Gao Y, Zhu W, Cheng T, Maruyama T, W J. Inhibition of prostaglandin E2 receptor EP3 mitigates thrombin-induced brain injury. J Cereb Blood Flow Metab. 2015 doi: 10.1177/0271678X15606462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu H, Wu T, Hua W, Dong X, Gao Y, Zhao X, Chen W, Cao W, Yang Q, Qi J, Zhou J, Wang J. PGE2 receptor agonist misoprostol protects brain against intracerebral hemorrhage in mice. Neurobiol Aging. 2015;36:1439–1450. doi: 10.1016/j.neurobiolaging.2014.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu H, Wu T, Li M, Wang J. Efficacy of the lipid-soluble iron chelator 2,2′-dipyridyl against hemorrhagic brain injury. Neurobiol Dis. 2012;45:388–394. doi: 10.1016/j.nbd.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu H, Wu T, Xu X, Wang J, Wang J. Iron toxicity in mice with collagenase-induced intracerebral hemorrhage. J Cereb Blood Flow Metab. 2011;31:1243–1250. doi: 10.1038/jcbfm.2010.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu T, Wu H, Wang J, Wang J. Expression and cellular localization of cyclooxygenases and prostaglandin E synthases in the hemorrhagic brain. J Neuroinflammation. 2011;8:22. doi: 10.1186/1742-2094-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cheng T, Yang B, Li D, Ma S, Tian Y, Qu R, Zhang W, Zhang Y, Hu K, Guan F, Wang J. Wharton’s Jelly Transplantation Improves Neurologic Function in a Rat Model of Traumatic Brain Injury. Cell Mol Neurobiol. 2015;35:641–649. doi: 10.1007/s10571-015-0159-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zan L, Zhang X, Xi Y, Wu H, Song Y, Teng G, Li H, Qi J, Wang J. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience. 2014;262:118–128. doi: 10.1016/j.neuroscience.2013.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Tsirka SE. Neuroprotection by inhibition of matrix metalloproteinases in a mouse model of intracerebral haemorrhage. Brain. 2005;128:1622–1633. doi: 10.1093/brain/awh489. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Gao X, Zhao S, Hu W, Chen J. The small-molecule TrkB agonist 7, 8-dihydroxyflavone decreases hippocampal newborn neuron death after traumatic brain injury. J Neuropathol Exp Neurol. 2015;74:557–567. doi: 10.1097/NEN.0000000000000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang G, Zhang J, Hu X, Zhang L, Mao L, Jiang X, Liou AK, Leak RK, Gao Y, Chen J. Microglia/macrophage polarization dynamics in white matter after traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1864–1874. doi: 10.1038/jcbfm.2013.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Faseb J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 47.Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z, Xiao G. The beta-lactam antibiotic, ceftriaxone, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J Trauma Acute Care Surg. 2012;73:654–660. doi: 10.1097/TA.0b013e31825133c0. [DOI] [PubMed] [Google Scholar]
- 48.Jin W, Kong J, Wang H, Wu J, Lu T, Jiang J, Ni H, Liang W. Protective effect of tert-butylhydroquinone on cerebral inflammatory response following traumatic brain injury in mice. Injury. 2011;42:714–718. doi: 10.1016/j.injury.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 49.Bains M, Hall ED. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim Biophys Acta. 2012;1822:675–684. doi: 10.1016/j.bbadis.2011.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ramkumar KM, Sekar TV, Bhakkiyalakshmi E, Foygel K, Rajaguru P, Berger F, Paulmurugan R. The impact of oxidative stress on islet transplantation and monitoring the graft survival by non-invasive imaging. Curr Med Chem. 2013;20:1127–1146. doi: 10.2174/0929867311320090003. [DOI] [PubMed] [Google Scholar]
- 51.Miller DM, Wang JA, Buchanan AK, Hall ED. Temporal and spatial dynamics of nrf2-antioxidant response elements mediated gene targets in cortex and hippocampus after controlled cortical impact traumatic brain injury in mice. J Neurotrauma. 2014;31:1194–1201. doi: 10.1089/neu.2013.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miller DM, Singh IN, Wang JA, Hall ED. Nrf2-ARE activator carnosic acid decreases mitochondrial dysfunction, oxidative damage and neuronal cytoskeletal degradation following traumatic brain injury in mice. Exp Neurol. 2015;264:103–110. doi: 10.1016/j.expneurol.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hong Y, Yan W, Chen S, Sun CR, Zhang JM. The role of Nrf2 signaling in the regulation of antioxidants and detoxifying enzymes after traumatic brain injury in rats and mice. Acta Pharmacol Sin. 2010;31:1421–1430. doi: 10.1038/aps.2010.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog Neurobiol. 2010;92:463–477. doi: 10.1016/j.pneurobio.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhao J, Chen Z, Xi G, Keep RF, Hua Y. Deferoxamine attenuates acute hydrocephalus after traumatic brain injury in rats. Transl Stroke Res. 2014;5:586–594. doi: 10.1007/s12975-014-0353-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harada H, Sugimoto R, Watanabe A, Taketani S, Okada K, Warabi E, Siow R, Itoh K, Yamamoto M, Ishii T. Differential roles for Nrf2 and AP-1 in upregulation of HO-1 expression by arsenite in murine embryonic fibroblasts. Free Radic Res. 2008;42:297–304. doi: 10.1080/10715760801975735. [DOI] [PubMed] [Google Scholar]
- 57.Hsieh HL, Wang HH, Wu CY, Yang CM. Reactive Oxygen Species-Dependent c-Fos/Activator Protein 1 Induction Upregulates Heme Oxygenase-1 Expression by Bradykinin in Brain Astrocytes. Antioxid Redox Signal. 2010;13:1829–1844. doi: 10.1089/ars.2009.2957. [DOI] [PubMed] [Google Scholar]
- 58.Raivich G, Behrens A. Role of the AP-1 transcription factor c-Jun in developing, adult and injured brain. Prog Neurobiol. 2006;78:347–363. doi: 10.1016/j.pneurobio.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 59.Hsieh HL, Chi PL, Lin CC, Yang CC, Yang CM. Up-regulation of ROS-dependent matrix metalloproteinase-9 from high-glucose-challenged astrocytes contributes to the neuronal apoptosis. Mol Neurobiol. 2014;50:520–533. doi: 10.1007/s12035-013-8628-y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.