

Abstract
Maintenance of protein homeostasis, also referred to as “Proteostasis”, integrates multiple pathways that regulate protein synthesis, folding, translocation, and degradation. Failure in proteostasis may be one of the underlying mechanisms responsible for the cascade of events leading to age-related macular degeneration (AMD). This review covers the major degradative pathways (ubiquitin-proteasome and lysosomal involvement in phagocytosis and autophagy) in the retinal pigment epithelium (RPE) and summarizes evidence of their involvement in AMD. Degradation of damaged and misfolded proteins via the proteasome occurs in coordination with heat shock proteins. Evidence of increased content of proteasome and heat shock proteins in retinas from human donors with AMD is consistent with increased oxidative stress and extensive protein damage with AMD. Phagocytosis and autophagy share key molecules in phagosome maturation as well as degradation of their cargo following fusion with lysosomes. Phagocytosis and degradation of photoreceptor outer segments ensures functional integrity of the neural retina. Autophagy rids the cell of toxic protein aggregates and defective mitochondria. Evidence suggesting a decline in autophagic flux includes the accumulation of autophagic substrates and damaged mitochondria in RPE from AMD donors. An age-related decrease in lysosomal enzymatic activity inhibits autophagic clearance of outer segments, mitochondria, and protein aggregates, thereby accelerating the accumulation of lipofuscin. This cumulative damage over a person’s lifetime tips the balance in RPE from a state of para-inflammation, which strives to restore cell homeostasis, to the chronic inflammation associated with AMD.
Keywords: age-related macular degeneration, autophagy, proteasome, proteostasis, lysosome, retinal pigment epithelium
1.0 Introduction to proteostasis and proteolysis
Maintenance of protein homeostasis, recently coined “Proteostasis”, involves multiple integrated pathways that regulate protein synthesis, folding, translocation, and degradation. Proteostasis is essential for preserving cellular function under both normal conditions and following conditions that upset the balance, for instance, exposure to environmental stressors or biological aging. A deficiency in cellular proteostasis has been associated with multiple age-related degenerative disorders (Marques et al., 2015) and has particularly devastating consequences in post-mitotic cells, such as neurons and the retinal pigment epithelium (RPE), where cell replacement to maintain tissue integrity and function is very limited. This review will examine pathways of degradation in the RPE and discuss the implications to age-related macular degeneration (AMD) when there is failure in this key part of proteostasis.
There are two main proteolytic machineries in eukaryotic cells that are an essential component of quality control: 1) the ubiquitin-proteasome system (UPS), and 2) the lysosomal/autophagosomal degradation system (Ciechanover, 2012; Ciechanover and Stanhill 2014). These are separate pathways with their own unique characteristics, but they also share similar major steps. Both systems recognize and actively select the material for degradation, and when the proteins have been degraded, the end products, i.e. amino acids, are recycled (Wong and Cuervo, 2010). In both systems, molecular chaperones play important roles in the recognition and selection of the proteins or organelles to be degraded (Kaarniranta et al., 2009b; Kaarniranta et al., 2010; Reggiori et al., 2012). An auxiliary degradation system that is present in the RPE involves the daily phagocytosis and degradation of the outer tips of photoreceptors, which is a fundamental process in disc renewal and maintenance of visual function. As will be explained in detail, there is some overlap between the molecules used for autophagy and those involved in phagocytic degradation of photoreceptor outer segments.
The basic molecular details of the UPS and lysosomal/autophagy degradation system have been developed from studies in cultured cells and by comparing WT and transgenic mice lacking specific proteins that are involved in proteolysis. Manipulation of these model systems to mimic conditions associated with AMD, such as stressors associated with oxidation or cytokines, provided a rational basis for inferring the contribution of proteolytic dysfunction in AMD. More recent work in tissue from human donors with and without AMD have provided key evidence either refuting or confirming results from model systems. Continued work in model systems, coupled with validating specific hypotheses in tissue from human donors with AMD, will provide a more accurate and detailed picture of how failure in degradative pathways contribute to AMD pathology.
Failure in proteostasis, which can be initiated when the degradative system is overwhelmed, may be one of the underlying mechanisms responsible for the cascade of events leading to the AMD phenotype. This review will cover the major players in each proteolytic system of the RPE, summarize evidence of their involvement in AMD, and highlight areas requiring more detailed study. Finally, we propose a model that includes defects in RPE proteolysis and the consequent cumulative damage as a potential mechanism underlying AMD pathology.
2.0 Phagocytosis in the retinal pigment epithelial cells
The RPE serves many physiological roles that are crucial to maintaining homeostasis of the retina. Phagocytosis is one of its most important functions; however, gaps remain in our understanding of the molecular details of this process. The RPE is one of the most active phagocytic cell types in the body, phagocytosing and degrading 10% of total photoreceptor volume daily (Kevany and Palczewski, 2010).
The process of photoreceptor disc shedding and renewal and the role of RPE in phagocytizing the shed discs have been known since the work of Young and colleagues (Young, 1967; Young and Droz, 1968; Young and Bok, 1969). Phagocytosis of the shed discs is essential to the survival of photoreceptor cells; several lines of evidence link abnormal RPE phagocytic function to degenerative diseases of the retina (Sparrow et al., 2010; Mustafi et al., 2011). A close proximity of photoreceptor outer segments (OS) and the RPE is required for the phagocytic process to take place (Williams and Fisher, 1987).
The process of phagocytosis exhibits a circadian rhythm (Lavail, 1976; Besharse et al., 1977; Young, 1977 & 1978; O’Day and Young, 1978; Fisher et al., 1983; Immel and Fisher, 1985; Bobu and Hicks, 2009) and can be divided into five distinct stages: 1) recognition and attachment of the OS; 2) ingestion of the OS; 3) formation of the phagosome, (4) fusion with lysosomes; and finally, 5) digestion (Figure 1). It is now recognized that abnormalities at any stage of this process or to the phagocytosis machinery can lead to disease, ranging from early-onset retinal dystrophies, such as retinitis pigmentosa or Usher’s syndrome to aging diseases affecting the central retina, such as AMD (Nandrot, 2014).
Figure 1. Schematic depiction of phagocytosis in the RPE.

The photoreceptor outer segment phagocytosis process includes outer segment recognition (1), engulfment (2), internalization (3), maturation and fusion with lysosomes (4) and final degradation within the phagolysosome (5). Please refer to the text for detailed information on the different stages of phagocytosis process.
A number of molecules that are essential in recognition, binding and internalization of OS have been identified (Kevany and Palczewski, 2010; Caberoy et al., 2010). The RPE cells can recognize phosphatidylserine “eat me” signals that trigger phagocytic uptake (Wu et al., 2006; Ruggiero and Finnemann, 2014). In various phagocytic cell types, phosphatidylserine can be detected via several membrane receptors, such as brain-specific angiogenesis inhibitor 1 (BAI1), which helps in recognition and engulfment (Park et al., 2007). Members of the T cell immunoglobulin mucin domain (TIM) family of proteins, such as TIM1, TIM 3 and TIM4 mediate phosphatidylserine binding (Freeman et al., 2010). Following phosphatidylserine binding, stabilin 2 also helps to promote engulfment by interacting with PTB domain-containing engulfment adaptor protein 1 (GULP) and thymosin β4 (Park et al., 2008). In addition, several ligands that are bridging molecules, such as milk fat globule EGF factor 8 (MFGE8), protein S and GAS6 also participate in the clearance process (Fricker et al., 2012; Li, 2013). There are a growing number of recognition molecules (Poon et al., 2014), for which a function in RPE phagocytosis remains to be determined. A defect in the initial stages of RPE phagocytosis can lead to photoreceptor death, as seen in the Royal College of Surgeons rat. This rat carries a mutation in the Mertk (a receptor tyrosine kinase) gene that impairs the phagocytic ability of the RPE (D’Cruz et al., 2000; Gal et al., 2000). In humans, mutations in the Mertk gene show rod-cone dystrophies (Tschernutter et al., 2006) with macular defects, as well as cone-rod dystrophies (Ebermann et al., 2007).
Once the photoreceptor OS are ingested by RPE cells, they are packaged into membrane-bound phagosomes. Cytoskeletal elements, particularly actin filaments and microtubule-dependent motor proteins, are known to play critical roles in the internalization of phagosomes by the RPE (Burnside and Bost-Usinger, 1998), and an abnormality in cytoskeletal reorganization that affects OS internalization leads to one type of Usher’s syndrome (Gibbs et al., 2003). Thus, some of the molecular events involved in the initial stages of the phagocytosis process are known. However, the processes of phagosome maturation and final degradation of photoreceptor OS remain obscure. Proteins that are involved in the phagosome maturation process in macrophages, cells where phagocytosis has been much more extensively studied, have been identified (Kinchen and Ravichandran, 2008). Cytoskeletal rearrangements occur through the evolutionarily conserved ELMO1 (engulfment and cell motility protein 1)-DOCK180 (dedicator of cytokinesis)-RAC complex (Elliott and Ravichandran, 2010). We have reported that some of these proteins, such as ELMO1, DOCK 180 and RAC1 are also expressed in the RPE cells of the rat, indicating that a similar signaling pathway may be involved in phagosome maturation in RPE (Zigler et al., 2011). We have also shown that Rab5 GTPase, which in macrophages is known to be crucial in the final maturation process before the degradation by lysosomal enzymes occurs, is also important in the maturation process in RPE (Zhou and Yu, 2008; Zigler et al., 2011). Information regarding the role of the cytoskeleton during the RPE degradation process is relatively sparse.
2.1 Lysosomal degradation of outer segments
RPE cells are polarized, with engulfment of photoreceptor OS occurring at the apical surface and the ingested OS (phagosome) being degraded by lysosomes in the basal region of the cell. Previous studies have suggested that lysosomal-mediated degradation of photoreceptor OS occurs in two steps (Bosch et al., 1993). In the first step, phagolysosomes are formed when small lysosomes fuse with phagosomes containing photoreceptor OS. When phagosomes fuse with lysosomes to form phagolysosomes, the cargo is degraded by hydrolytic enzymes. We have recently identified a protein, βA3/A1-crystallin that is expressed in the lysosomes of RPE cells (Zigler et al., 2011; Valapala et al., 2014b). Interestingly, while loss of this protein does not affect the engulfment or internalization of the photoreceptor OS or fusion of the phagosomes with lysosomes, the degradation phase of the process is impaired (Zigler et al., 2011; Valapala et al., 2014b). In Nuc1 rats that have a spontaneous mutation in this protein (Zigler et al., 2011) and in mice where Cryba1 (gene encoding for bA3/A1-crystallin) is conditionally knocked out (Cryba1 cKO) from RPE (Valapala et al., 2014b), electron microscopy clearly shows photoreceptor OS being retained at the basal side of RPE cells (Figures 2 and 3). Our studies indicate that βA3/A1-crystallin is required by RPE cells for the proper degradation of photoreceptor outer segments. The primary enzyme involved in the proteolysis of rhodopsin in the RPE phagosomes is Cathepsin D, and a decrease in the activity of Cathepsin D, as is seen in the βA3/A1-crystallin deficient genetic animal models, leads to the accumulation of photoreceptor OS-derived debris in RPE cells (Bosch et al., 1993; Deguchi et al., 1994; Rakoczy et al., 1997; Lai et al., 2000; Hoppe et al., 2004). The degradation of photoreceptor OS is also delayed in Mreg−/− mice, suggesting a role for melanoregulin in the process (Damek-Poprawa et al., 2009).
Figure 2. Disrupted phagocytosis with Nuc1 depletion.

(A, B) Transmission electron microscopy of RPE from 1-yr old wild type rats shows normal accumulation of lipofuscin-like particles (arrows) in the apical cytoplasm of RPE cells. (C) At higher magnification, RPE cells from 1 year old Nuc1 animals show abundant lipofuscin-like aggregates in the cytoplasm (arrows) and a large phagosome containing undigested outer segment discs (arrowhead). Scale bar = 500 nm. Reprinted with permission of J Cell Science.
Figure 3. Age-dependent abnormalities in the RPE cells of Cryba1 cKO mice.

Transmission electron microscopy of 2-month old cKO RPE shows many vacuole-like structures with degenerated membrane-bound cellular organelles (arrows, center), undigested photoreceptor outer segments (arrow, right) and loss and truncation of basal infoldings (asterisks), as compared to Cryba1fl/fl (left). Scale bar= 500 nm. Reprinted with permission of Autophagy.
Defects in clearance have been linked with various inflammatory diseases. In contrast, clearance during normal development, or in processes where cellular homeostasis is maintained, does not elicit an immune response. We have recently shown that defective lysosomal clearance of OS in the RPE may contribute to the induction of a chronic inflammatory response (Valapala et al., 2014a). Chronic inflammation is believed to contribute to the pathology of AMD (Ambati et al., 2013; Whitcup et al., 2013) and will be discussed in more detail later in Section 5.0.
2.2 Crosstalk between phagocytosis and autophagy
It has been proposed earlier that phagocytosis and autophagy might have a common mechanism because they share key molecular regulators of the phagosome maturation process, and because fusion with lysosomes is required for ultimate degradation in both processes (Kinchen and Ravichandran, 2008). An elegant study reported by the Ferguson laboratory has shown that phagocytosis and the visual cycle are linked through a non-canonical autophagy process, termed LC3-associated phagocytosis or LAP (Kim et al., 2013; Ferguson and Green, 2014). This study showed that degradation of photoreceptor OS is also dependent on LAP. The study demonstrated for the first time that a convergence of the phagocytosis process in RPE and the visual cycle is essential for normal vision. Therefore, defects in LAP could potentially be a contributing factor in AMD. However, additional studies are needed to gain a greater understanding of the interplay between the two lysosomal-mediated degradative pathways of phagocytosis and autophagy. To ensure the functional integrity of the neural retina, both phagocytosis and autophagy need to be in balance.
3.0 Autophagy and the lysosomal clearance system
During the past few years autophagy research has become one of the most exciting areas of science, with indications emerging of its pathologic role in cancer, cardiovascular, muscular dystrophy, lipid storage disorders and neurodegenerative diseases, as well as a non-pathologic role in development, ageing and innate immunity processes (Boya et al., 2013). Autophagy is an intracellular “self-eating” process involved in protein and organelle degradation via the lysosomal degradation system, by which it is able to recycle cellular material (Mizushima and Komatsu, 2011). Three different types of autophagic pathways have been documented: microautophagy, chaperone-mediated autophagy, and macroautophagy. In microautophagy, a portion of cytoplasm substrates are trapped inside vesicles that form via invagination of lysosomal membranes; lysosomal degradation of these endosomes occurs within the lumen of the lysosome. Microautophagy is classified as non-selective bulk degradation, which occurs under conditions of starvation and provides essential nutrients for cell survival (Boya et al., 2013). In chaperone-mediated autophagy (CMA), cellular chaperones and heat shock proteins assist in the targeting of proteins containing a specific KFERQ recognition signal to lysosomes for degradation (Eskelinen, 2008; Reggiori et al., 2012; Cuervo and Wong, 2014). This review will focus on macroautophagy, where cellular organelles and protein aggregates are encapsulated from the cytoplasm by double-membrane autophagosomes, which then fuse with lysosomal vesicles, delivering the engulfed cytoplasmic cargo for degradation. Macroautophagy and CMA are both classified as selective degradative processes.
3.1 Macroautophagy: Selective degradation involving lysosomes
Protein aggregates, cellular organelles, and protein complexes in signaling cascades are usually degraded via selective autophagy. Selective autophagy utilizes autophagy receptors, which bind to specific structural elements on the protein or organelle that needs to be eliminated (Boya et al., 2013). Ubiquitin modification is also part of the signal marking cellular components targeted for destruction. Autophagy receptors, such as SQSTM1/p62, NBR1 and optineurin, all contain domains that recognize ubiquitin and structural elements of an unfolded protein in the cargo (Birgisdottir et al., 2013; Rogov et al., 2014, Svenning and Johansen, 2013). Following receptor binding, the cargo is chaperoned to the autophagosome (explained in detail below) where it is degraded. The autophagosome is decorated with ATG8 (autophagy-related protein 8)/LC3 (microtubule-associated protein 1A/1B-light chain 3) proteins, which are recognized by the LIR (LC3-Interacting Regions) motif that is present on all autophagy receptors.
Recognition and chaperoning of cargo by autophagy receptors to the autophagosome is only the initial step in macroautophagy. More than 30 autophagy-related genes (ATG) regulate autophagy steps, which can be divided into induction, nucleation, elongation, fusion and degradation (Figure 4). Autophagosome formation starts at phagophore assembly sites (Suzuki et al., 2001; Rubinsztein et al., 2012). Phagophores are elongated by the class III phosphoinositide 3-kinase Vps34, which acts in a large macromolecular complex, along with Beclin-1 (mammalian Atg6), Atg14 and Vps15/p150 to form PI 3-phosphate (PI(3)P). The activity of this complex is dependent on upstream autophagy regulators, including ubiquitin like kinases-Atg13-FIP200 complex, which plays a crucial role in the formation of double-membrane autophagic vacuoles—the autophagosomes. The autophagosomes engulf cytoplasmic components in a process that involves the cytosolic form of LC3-I conjugating to phosphatidylethanolamine (PE) to form the LC3-PE conjugate (LC3-II), which is subsequently recruited to phagophore membranes (Figure 4A). An increase in LC3-II is a commonly used marker of autophagosome formation that can be monitored on Western immunoblots where the lipidated and non-lipidated proteins can be clearly resolved (Figure 4B). Formed autophagosomes fuse with lysosomes to create autolysosomes, and the cargo is degraded by lysosomal acidic enzymes, including Cathepsin D, B, and L (Hyttinen et al., 2014). At the same time, LC3-II in the autolysosomal lumen is degraded. Therefore, lysosomal turnover of the autophagosomal marker LC3-II reflects autophagic activity and has also been used as a marker of autophagic flux. Figure 4B shows examples of the changes in the LC3 protein that occur following autophagy induced by both rapamycin and serum starvation in primary cultures of human RPE. The formation of double membrane autophagosomes is an even more reliable marker of autophagy induction and can be monitored by electron microscopy, as shown in the micrograph of ARPE-19 cells treated with bafilomycin (Figure 4C).
Fig. 4.

Time course of autophagy induction.
(A) Steps in the autophagic degradation of cell contents include the induction of the isolation membrane followed by elongation of the double membrane to form the phagophore, which begins surrounding damaged organelles (eg., mitochondria) and protein aggregates. The lipidation of LC3-I by phosphatidylethanolamine (PE) to form LC3-II, which binds to the forming double membrane structure, is a crucial step in the maturation of the autophagosome. A commonly used indicator of autophagosome formation is the increase in LC3II. The autolysosome occurs when the autophagosome merges with a lysosome, after which lysosomal enzymes (LE) degrade the cargo. (B) Autophagic flux can be induced by inhibiting the mTOR pathway with rapamycin or under conditions of starvation (no serum). On Western blots, the lipidation by PE alters the migration of LC3 and permits quantification of both unlipidated (LC3-I) and lipidated (LC3-II) forms of the protein. Numbers below the figure are the ratio from densitometry of individual protein bands. Two examples of autophagy induction in primary cultures of human RPE show an increase in both total LC3 content and the lipidated form of the protein. At 24 hrs post-rapamycin treatment, hRPE show a dose-dependent increase in the LC3 II/I ratio. Serum deprivation for 2 hrs also dramatically increased the LC3 II/I ratio. Unpublished data. (C) The presence of the double membrane (arrows) verifies autophagosome formation in ARPE-19 cells 24 hrs after treatment with 50 nM bafilomycin. Scale bar =500 nm. Unpublished data.
Fusion of autophagosome and lysosome is regulated by multiple proteins, including Rab7, Lysosome membrane associated protein 2 (LAMP-2), and a large superfamily of small proteins, SNAREs (SNAP (soluble N-ethyl-maleimide-sensitive-fusion-protein attachment protein) receptors). Rab7 belongs to the small GTPase family (Hyttinen et al., 2013). It controls the maturation of endosomes and autophagosomes, directs the trafficking of proteins along microtubules, and participates in the fusion step with lysosomes (Hyttinen et al., 2014, Ryhänen et al., 2011). In connecting autophagy receptor SQSTM1/p62 to the fusion process, we have observed that silencing of SQSTM1/p62 during MG-132 exposed proteasome inhibition increases Rab7 expression in ARPE-19 cells (Figure 5). This may be a cellular compensatory attempt to increase fusion of autophagosome and lysosome, since autophagy flux has been decreased due to the silencing of SQSTM1/p62 (Viiri et al., 2010; Viiri et al., 2013). In addition to regulation of the fusion process, LAMP2 is a central molecule in CMA. Alternative splicing of the LAMP2 gene produces three variants - LAMP-2A, LAMP-2B and LAMP-2C. The only isoform that has a role in CMA is LAMP2A (Cuervo and Dice 1996). It is the receptor for translocation of proteins into the lysosomes during CMA. Although CMA was not studied, we have observed upregulated LAMP2 expression accompanies the formation of ubiquitin and heat shock protein 70 positive protein aggregates following proteasome inhibition (see later; Ryhänen et al., 2009, Viiri et al., 2013; Figure 6).
Figure 5. Rab7 expression with p62 silencing and proteasome inhibition.
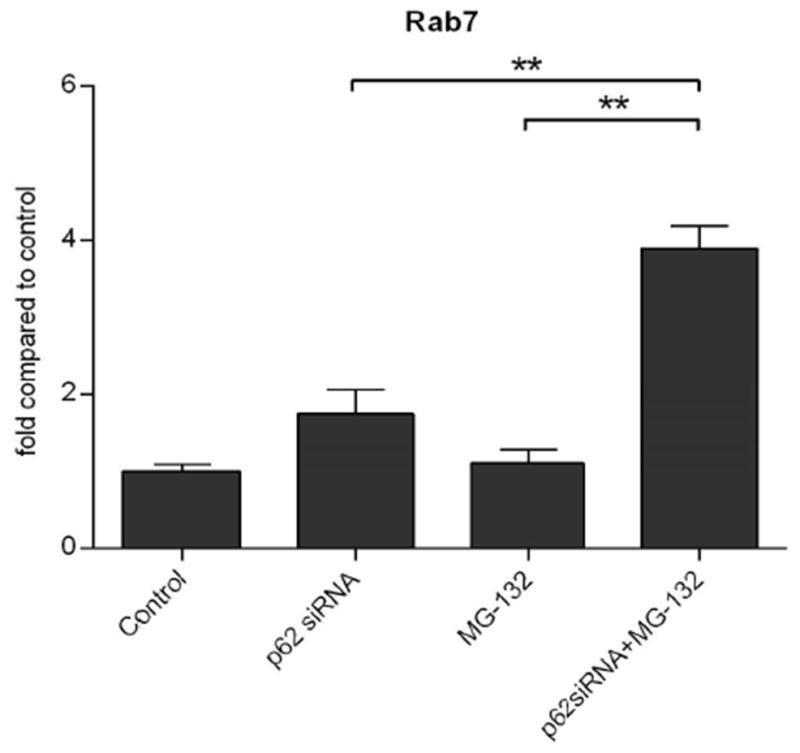
Summary of densitometry from Western blots of Rab7 in homogenates of WT or SQSTM1/p62 silenced ARPE-19 cells after exposure to 1 μM MG-132 for 24 h. Results are expressed as means ± S.E.M. **p < 0.05 by Mann-Whitney test for the indicated comparisons. Unpublished data.
Figure 6. LAMP-2 expression under proteasome inhibition.
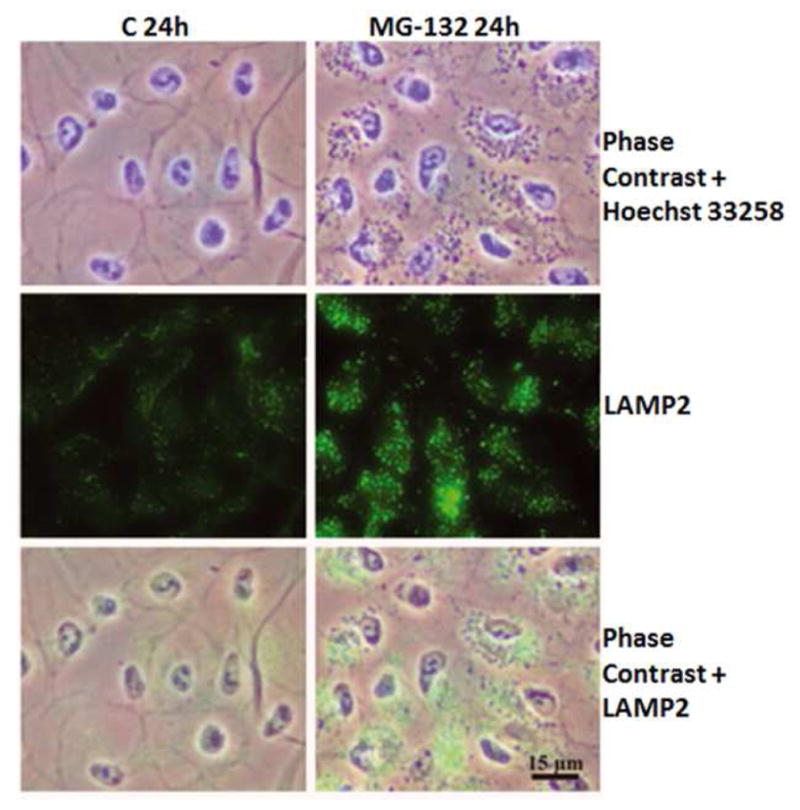
Phase contrast microscopy and immunofluorescence staining of LAMP-2 in control cells, and cells exposed to 10 μM MG-132 for 24 hrs. Nuclei were stained with Hoechst 33258 dye (blue). Reprinted with permission of Journal of Cellular and Molecular Medicine.
SNAREs regulate fusion of autophagosomes and lysosomes as well. SNAREs increase the permeability of autophagosome and lysosome membranes and drive the actual membrane opening leading to fusion of the two organelles. Defects in any of the steps in fusion could potentially impair autophagy flux. However, whether these defects play a role in the pathogenesis of AMD has not been studied.
3.2 Mitophagy: Selective autophagy of mitochondria
Macroautophagy that targets specific cellular components (i.e., mitochondria, protein aggregates, lipids, ribosomes) have been assigned names that reflect their cargo, such as mitophagy, aggrephagy, lipophagy, ribophagy, reticulophagy, granulophagy and others (Rogov et al., 2014). Mitophagy is the process by which mitochondria are selectively degraded in response to hypoxic conditions as a pro-survival strategy, or in response to the presence of damaged or defective mitochondria. As discussed in Section 3.3, there is good evidence that mitophagy is diminished in the RPE at an early stage of AMD.
The purpose of mitophagy is to maintain mitochondrial function under changing cellular conditions. Maintenance of mitochondrial function also depends on fusion and fission, which are dynamic processes that facilitate the constant remodeling of mitochondrial architecture and the mixing of contents from different mitochondrial networks (Pollanck, 2010). Fission is one mechanism for ridding the cell of defective mitochondria, whereby damaged segments are sequestered away from the mitochondrial network (Figure 7). Key proteins of the fission machinery include dynamin-1-like protein (DRP) and mitochondrial fission 1 protein (FIS1). Knockdown of these proteins result in accumulation of oxidized mitochondrial proteins and reduced respiratory capacity (Twig et al., 2008), thus highlighting the importance of this initial step in mitophagy.
Figure 7. Mitophagic elimination of damaged mitochondria.

Steps for segregation and elimination of damaged mitochondria includes: (1) Mitochondrial damage (star) followed by fission isolates damaged mitochondria. (2) Pink1 accumulates on damaged mitochondria and recruits Parkin, which ubiquitinates mitofusion. (3) Ub-mitofusion is degraded by the proteasome. (4) Reduced mitofusion prevent fusion of damaged with healthy mitochondria. (5) Damaged mitochondria are targeted for mitophagy.
Fission followed by selective fusion of adjacent mitochondria occurs under the regulation of the membrane GTPase proteins mitofusin and optic atrophy protein 1. A defect in optic atrophy protein 1 causes dominant optic atrophy, which is an inherited degeneration of the retinal ganglion cells (Ban et al., 2010). Fusion is impaired in mitochondria with reduced membrane potential or those containing damaged mtDNA, thereby isolating the unhealthy mitochondrial fragment and promoting its mitophagic degradation. Thus, the coordinate action of fission and fusion segregates dysfunctional mitochondria prior to their selective removal from the cell (Twig et al., 2008).
Initiation of mitophagy requires recognition of targeted mitochondria and this step is mediated either by mitophagy receptors (Nix, Bcl-2/E1B-19 kDa interacting protein-3 (Bnip3), FUN14 domain-containing protein 1 (FUNDC1)), or PTEN-induced kinase 1 (Pink1) and Parkin signaling (Ding and Yin, 2012). Nix, Bnip3, and FUNDC1 are proteins that reside in the outer mitochondrial membrane and contain an LIR sequence that is recognized by the autophagy machinery. These mitophagy receptors are only negligibly expressed under normoxic conditions, but under conditions of hypoxia, their expression is significantly upregulated, leading to mitophagic elimination of mitochondria. Under hypoxic conditions, the selective removal of mitochondria is thought to be a pro-survival response that prevents excessive production of damaging reactive oxygen species by the mitochondria.
Mitochondrial dysfunction, specifically depolarization of the inner membrane, signals the selective and efficient turnover of damaged mitochondria via Pink1-Parkin signaling (Figure 7). Pink1 contains a mitochondrial targeting sequence and is imported into the inner mitochondrial membrane through TOM and TIM, import complexes located on the outer and inner membrane, respectively (Ding and Yin, 2012). In healthy mitochondria, Pink1 is continually degraded, but in unhealthy mitochondria where the mitochondrial membrane potential dissipates, Pink1 import stops and it accumulates on the outer mitochondrial membrane (Durcan and Fon, 2015). Parkin, an E3 Ubiquitin Ligase, is recruited to the mitochondria where it is phosphorylated and activated by Pink1. Parkin then ubiquitinates proteins that reside on the mitochondrial outer membrane, including mitofusin. Ubiquitin-modified mitofusin is targeted for degradation by the proteasome, thereby reducing mitochondrial mitofusin content and preventing fusion of this damaged mitochondrial fragment with healthy mitochondria. The fragment of damaged mitochondria is then escorted by autophagy receptors SQSTM1/p62 and NBR1 to the autophagic machinery where mitophagy ensues (Ding and Yin, 2012). As highlighted above, mitophagic clearance of dysfunctional mitochondria requires the coordinate action of both the proteasome and autophagy pathways.
3.3 Autophagy in the pathogenesis of AMD
Data from cultured cells and transgenic mice have provided valuable insight into the molecular details of the autophagic pathway and the consequences of interrupting key steps or upsetting cellular homeostasis beyond the capacity of the system to sustain normal function. For example, downregulation of Atg5 specifically in the RPE resulted in decreased photoreceptor response (Kim et al., 2013). Thus, data from a variety of model systems have helped generate well-reasoned hypotheses about the role of autophagy in AMD pathology.
Hypoxia, oxidative stress, UPR, and inflammation are typical inducers of autophagy (Ryhänen et al., 2009; Viiri et al., 2010; Kaarniranta et al., 2013; Klettner et al., 2013; Blasiak et al., 2014; Mitter et al., 2014; Valapala et al., 2014a). These conditions are also associated with RPE aging and AMD, so one could speculate that autophagy should be increased. Additionally, autophagy controls the turnover of aggregate-prone proteins, a process that is extremely important in post-mitotic cells, including the RPE (Kaarniranta et al., 2010; Viiri et al., 2013; Kim et al., 2013). Considering the harsh environment where RPE reside, it seems reasonable to expect that these cells have an enormous capacity to maintain the ability to process and remove cellular debris.
Whether autophagy increases or declines with aging and disease remains controversial; the direction seems to depend on both the cell as well as the disease (Salminen and Kaarniranta, 2009; McCray and Taylor, 2008; Rodriguez-Muela et al., 2013). It can be reasoned that proteolytic capacity is elevated in aged cells due to the increased burden of damaged proteins and organelles that must be cleared to maintain function. Results from Rodriguez-Muela and colleagues provide a relevant example of the complexity of changes in proteolytic capacity in the aged retina. They report that a marked age-related reduction in macroautophagy was accompanied by a corresponding increase in CMA (Rodriguez-Muela et al., 2013) suggesting there is a compensatory response to accommodate defects in specific proteolytic pathways.
In aged RPE of mice an in human donors without AMD, evidence for elevated autophagy includes the increased content of autophagy proteins and autophagic vesicles (Wang et al., 2009; Mitter et al., 2014). With the onset of AMD, the autophagic capacity of the RPE, which is already taxed by age-related challenges, becomes overwhelmed by the proteolytic load and autophagy declines (Mitter et al., 2014). It is also possible that lysosomal enzyme activity is suppressed due to the excessive accumulation of lipofuscin in aged RPE (Kaemmerer et al., 2007; Ryhänen et al., 2009; Viiri et al., 2013, Mitter et al., 2014; Valapala et al., 2014). Evidence of declines in autophagy with AMD include the reported decreased number of autophagic vesicles in AMD donor RPE compared with age-matched controls (Mitter et al., 2014) and the accumulation of SQSTM1/p62 in the macula of AMD donors (Viiri et al., 2013; Figure 8). Degradation of SQSTM1/p62 occurs via autophagy, so its accumulation could be due to decreased autophagic flux (Viiri et al., 2013, Kivinen et al, 2013).
Figure 8. Immunostaining for SQSTM1/p62 in human AMD samples.

The extent of cytoplasmic immunopositivity in the retinal pigment epithelial cells (RPE, shown by arrows) and in the drusen was evaluated microscopically (no staining or positive staining) by selecting 5 mm long areas of foveomacular (A), perimacular (B) and peripheral (C) regions. The drusen (shown by asterisks) were mostly SQSTM1/p62 negative. The nuclei of RPE cells were SQSTM1/p62 negative. (Original magnifications of x 200 and in insets x 400; Bruch’s membrane shown by arrow heads). Reprinted with permission of PlosOne.
There is also substantial evidence suggesting that RPE mitophagic processes, including fission and fusion, begins to fail with AMD. Indicators of potential defects in mitophagy include the accumulation of mtDNA damage in the RPE of human donors with AMD (Karunadharma et al., 2010; Terluk et al, 2015), suggesting that damaged mitochondria are not being effectively eliminated from the diseased cell. Other evidence of damaged mitochondria accumulating in the diseased RPE includes the AMD-related disruption and disorganization in mitochondrial cristae in electron micrographs from AMD donors (Feher et al., 2006), and the increased content of mitofilin at an early stage of AMD (Nordgaard et al., 2008). Mitofilin is involved in stabilizing cristae, so its upregulation could be a compensatory response to altered mitochondrial remodeling due to defects in fission and fusion. These observations in human donor tissue are consistent with results in model systems where the genetic disruption of autophagy by knocking out the mitophagy receptor Nix or Atg5 results in the accumulation of defective mitochondria (Sandoval et al., 2008; Stephenson et al., 2009).
The preservation of autophagic activity, together with functional lysosomal enzymes, is a prerequisite if one wishes to prevent detrimental intracellular accumulation of damaged molecules (Ryhänen et al., 2009; Viiri et al., 2010). A well-functioning proteolytic machine guarantees that there is sufficient capacity to handle damaged protein and organelles. Improving lysosomal and autophagic function may be a viable target for therapeutic interventions aimed at improving RPE cell function, retarding the aging process, and combating AMD (Ryhänen et al., 2009; Chen et al., 2013; Viiri et al., 2013).
4.0 Proteasomal pathway and proteostasis
The proteasome degrades 80–90% of cellular proteins in eukaryotes, including short-lived, abnormal, denatured or damaged soluble proteins (Ciechanover, 2012). This proteolytic system also controls processes essential for cell viability, such as cell-cycle regulation, signal transduction, and synaptic plasticity (Coux, 1996; Ferrington and Gregerson, 2012). In the retina, the proteasome regulates proteins involved in circadian cycles; for example, it orchestrates the light-dependent degradation of proteins involved in melatonin synthesis (arylalkylamine-N-acetyltransferase, 14-3-3) and the molecular clock proteins (i.e., timeless) (Pozdeyev et al., 2006; Knowles et al., 2009). Up to 1 % of cellular proteins are members of the ubiquitin-proteasome system (UPS), highlighting the importance of this pathway in maintaining cell function and viability (Coux et al., 1996).
The proteasome exists as different oligomeric assemblies, including the 20S, 26S, hybrid, and immunoproteasome (Figure 9). These different subtypes exhibit some specificity in function and are defined by the composition of their catalytic subunits and the regulatory complex associated with the catalytic core (Ciechanover, 2012; Ferrington and Gregerson, 2012). The 20S catalytic core is composed of four stacked rings of seven subunits each. The two outer rings contain the constitutively-expressed α subunits that interact with regulatory complexes. The inner two rings contain the β subunits. Three of the β subunits (β1, β2, β5) contain the catalytic sites that perform distinct proteolytic activities referred to as caspase-like (β1), trypsin-like (β2), and chymotrypsin-like (β5). The catalytic subunits β1, β2, and β5 that form the standard proteasome can be replaced in nascent proteasomes by the subunits LMP2 (β1i), MECL-1 (β2i), and LMP7 (β5i), respectively. These subunits form the core of the immunoproteasome. A third type of catalytic core, referred to as the intermediate-type proteasome, contains a mixture of the standard and immunoproteasome catalytic subunits (Dahlmann et al., 2000; Klare et al., 2007). All three 20S cores have been shown to co-associate with both PA28 and PA700 to form the hybrid proteasome. The ATP-independent degradation of proteins by the 20S core proteasome has been suggested as the primary mechanism for degrading oxidized proteins following an oxidative insult (Reinheckel et al., 1998; Davies, 2001; Jung et al., 2013). The 26S proteasome requires ATP for activation and is responsible for the degradation of many ubiquitinated (Hochstrasser, 1996), and some non-ubiquitinated proteins (Kisselev et al., 1999).
Figure 9. Proteasome structure and regulatory proteins.

The 20S core particle contains the standard (β1, β2, β5), immunoproteasome (LMP2, MECL-1, LMP7) catalytic subunits, or a mixture of both types of catalytic subunits. The 20S can bind to the regulatory complexes PA700 and/or PA28 to form the 26S, hybrid proteasome, or the immunoproteasome.
4.1 Immunoproteasome and the cellular stress response
Analysis of the three subtypes of catalytic cores has shown that they differ substantially in their enzymatic characteristics and cleavage of model protein substrates (Dahlmann et al, 2000; Klare et al., 2007). While most cells contain a heterogenous population of 20S cores, the relative ratio of different subtypes is cell-specific (Dahlmann et al., 2000) and can be altered under different cellular conditions (Ferrington and Gregerson, 2012). The standard proteasome is constitutively expressed in nearly all mammalian cells. In contrast, immunoproteasome is abundantly expressed in immune cells, where it performs functions associated with immune surveillance. Immunoproteasome is also present, albeit in low concentrations, in the uninjured retina and RPE. Notably, we have shown that this proteasome subtype can be significantly upregulated under conditions of stress, including retinal injury induced by cytotoxic T- lymphocytes or optic nerve crush (Ferrington et al., 2008; Schuld et al., 2015), and with aging (Hussong et al., 2011). Immunoproteasome expression is also induced in RPE under chronic oxidative stress (Hussong et al., 2010), following exposure to pro-inflammatory cytokines (Gregerson et al., 2006), and with induction of ER stress (Figure 10). These conditions occur in the diseased retina. We have also found RPE cells deficient in the LMP7 and MECL-1 immunoproteasome subunits have reduced cell survival under conditions of oxidative stress (Hussong et al., 2010) and ER stress (Figure 10). In contrast, KO of the LMP2 immunoproteasome subunit confers protection from ganglion cell death in the murine retina after partial optic nerve crush (Schuld et al., 2015). Results from experiments in cultured RPE isolated from immunoproteasome KO mice suggest a previously unrecognized role for immunoproteasome in modulating NFkB and Akt signaling. These are two key pathways involved in the cellular stress response. Collectively, these findings support the ideas that (1) the proteasome population is highly dynamic, (2) each proteasome subtype performs specialized functions that allow the cell to respond to changing conditions, and (3) the immunoproteasome is involved in the cellular stress response (Ferrington and Gregerson, 2012).
Figure 10. RPE Immunoproteasome induction by conditions of stress.

Immunoproteasome content was upregulated in cultures of WT murine RPE following exposure to (A) chronic, low levels of hydrogen peroxide (0.5 mM), (B) TNFα (1 ng/ml) to mimic the inflammatory response, and (C) tunicamycin (5 μg/ml) to induce ER stress. Blots show antibody reaction to immunoproteasome subunits LMP2 and LMP7. Total proteasome content was estimated from reactions of alpha subunits (α6, α7), which are present in all proteasome subtypes. (A, B) Graph and numbers below blots indicate the densitometry of immune reactions relative to untreated controls. (C) Graph shows the dose-dependent response of RPE cell viability for WT and cells lacking the LMP7 and MECL-1 (L7M1) immunoproteasome subunits. Unpublished data.
The idea that the immunoproteasome performs functions that are unrelated to its role in immune surveillance is also supported by the growing list of human genetic diseases linked to immunoproteasome deregulation. For example, missense and nonsense mutations in the LMP7 gene, resulting in a truncated or non-functional protein, have been associated with diseases characterized by joint contractures, microcytic anemia, muscle dystrophy and/or lipodystrophy phenotypes (Agarwal et al., 2010, Liu et al, 2012, Arima et al., 2011, Kitamura et al., 2011). A single nucleotide polymorphism in immunoproteasome subunits of either LMP7 or LMP2 have been associated with a higher risks for diabetes and ankylosing spondylitis (Zaiss et al., 2011, Haroon et al., 2012). Manifestation of these diseases in non-immune tissues provides strong rationale for an expanded role for specific proteasome subtypes.
Of particular importance to this review, higher immunoproteasome content was observed in the retina of human donors with age-related macular degeneration (Ethen et al., 2007). Although we did not track immunoproteasome content in human donor RPE, we did find that total proteasome content was elevated 3-fold in AMD donors compared with age-matched controls (Decanini et al., 2007). We interpret the upregulation of proteasome as a compensatory response to cellular conditions that place an increasing burden on all the systems involved in protein quality control.
4.2 Mechanisms targeting proteins for proteasomal degradation
There are a number of mechanisms whereby proteins destined for proteolytic destruction are recognized by the proteasome. One of the best described mechanisms involves marking proteins with ubiquitin, a 76-amino acid polypeptide that is covalently attached to the protein substrate via a three-step process involving different enzymes (Figure 11). The ubiquitin-activating (E1) and ubiquitin-conjugating (E2) enzymes activate ubiquitin and transfer it to the protein substrate that is chaperoned to the E1/E2 complex by the ubiquitin ligase enzyme (E3). Of note, there are hundreds of different E3 ligases in the human genome and each E3 recognizes a subset of specific protein substrates. The E3 enzyme not only recognizes the protein substrates, but also catalyzes the reaction for the covalent attachment of ubiquitin to a lysine residue (denoted as a K in Figure 11) in the substrate. Of potential relevance to AMD, these three enzymes utilize an active site cysteine and therefore, are particularly vulnerable to oxidative inactivation. Additional ubiquitin molecules are conjugated to the first ubiquitin moiety to form an ubiquitin chain; four or more are the signal recognized by the proteasome. Once the ubiquitin-modified protein is docked at the proteasome, the ubiquitin conjugates are released by deubiquitinating enzymes that are part of the 26S regulatory complex. The protein substrate is then unfolded and translocated into the interior chamber of the 20S core where it gains access to the catalytic sites.
Figure 11. Ubiquitin conjugation marks proteins for proteasome degradation.

The coordinate action of the enzymes E1, E2, and E3 actively conjugate ubiquitin proteins to a lysine (K) residue on the protein substrate to mark it for proteasome degradation. An ubiquitin chain of at least 4 proteins is required for recognition by the regulatory complex of the 26S proteasome. Following binding of the Ub-protein, the regulatory complex deubiquitinates the substrate prior to protein unfolding and translocation into the core, where it is degraded to small peptides. The ubiquitin is recycled for another round of protein marking.
Another signal for proteasome degradation is the exposure of an intrinsic degradation signal, a “degron”, that is a cryptic amino acid sequence buried within the proteins three-dimensional structure. Once the protein is modified, often via phosphorylation, the protein undergoes a structural change that exposes the degron to the solvent where it is recognized by an E3 ligase and targeted for proteasome degradation (Figure 12). Many signaling pathways utilize this mechanism to elicit either complete degradation of the protein or partial proteolysis, which releases the active portion of the protein (Figure 13). The partial proteolysis of the NFkB subunits p100 and p105 to their active transcription factors, p52 and p50, are examples of proteins that use this mechanism to regulate the response.
Figure 12. Signals for degradation by the proteasome.
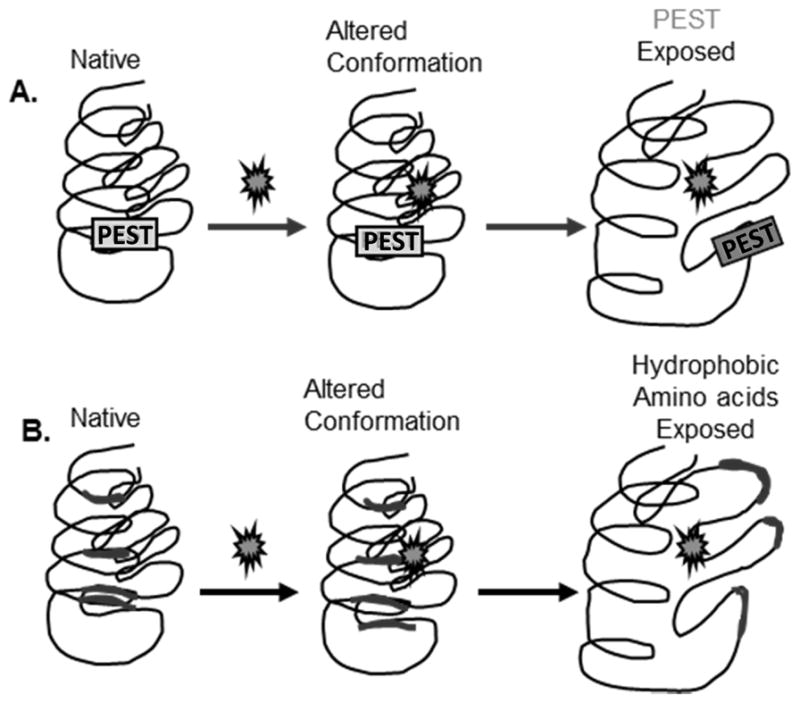
(A) PEST sequences are degrons that are buried within the proteins interior. With modification of the protein, such as phosphorylation, there is a change in protein structure that causes the cryptic sequence to become solvent exposed. The degron is recognized by a specific E3 ligase, which targets the protein for proteasome degradation. (B) Protein modification, such as oxidation, causes a change in protein structure that exposes hydrophobic residues that are normally buried within the protein. Exposure of hydrophobic patches targets this molecule for proteasome degradation through a mechanism that is still unclear.
Figure 13. Proteolytic degradation of protein substrates by the proteasome.
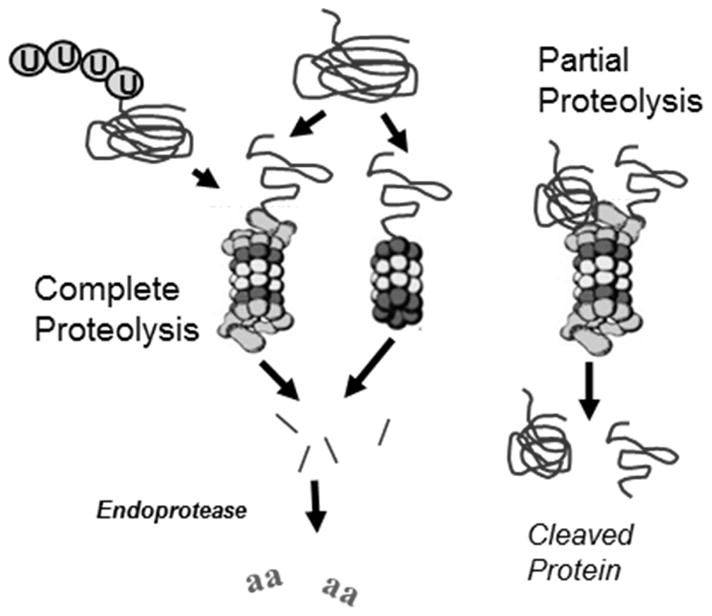
Proteins containing a degradation signal (eg., ubiquitin modification or exposed hydrophobic amino acids) are targeted to the proteasome. Complete proteolysis results in the release of multiple peptides of varying size. These peptides are further degraded to amino acids by cellular endopeptidases. Partial proteolysis occurs when an unstructured region of a protein gains access to catalytic sites within the 20S core. Enzymatic cleavage cuts the protein so that two partial pieces of the protein are released.
Degrons assume varied positions within the protein, but most often occur at either the N- or C-terminus. A well-studied degron is the PEST sequence, which contains proline, glutamic acid, serine and threonine, flanked by charged amino acids. PEST sequences exhibit considerable heterogeneity between proteins. For example, PEST sequences (critical components are underlined) for Fos and ornithine decarboxylase are KVEQLSPEEEEK and HGFPPEVEEQDDGTLPMSCAQESGMDR, respectively (Rechsteiner and Rogers, 1996). Modifications to the amino acids within the PEST sequence, such as addition of an oxygen to methionine or glutathione to cysteine, could inappropriately either stimulate or prevent degradation of the protein. Conformational changes to protein structure caused by modifications, including oxidative damage, can also signal for proteasome degradation via the exposure of hydrophobic residues that are buried in the native protein (Figure 12). These hydrophobic patches are also recognized by heat shock proteins and other chaperones that attempt to refold the protein back to its native conformation. If that effort fails, the damaged protein is escorted to the proteasome for degradation.
Balanced degradation of proteins is essential for maintaining cell health. When this balance goes awry, due to either genetic mutation, oxidative inactivation of the Ub-proteasome pathway, or oxidative damage to protein substrates, pathology can occur. For example, mutations that either inactivate or promote the accumulation of specific E3 ligases can cause decreased (eg., E6-AP causing Angelman’s syndrome) or accelerated (eg., Skp2 causing cancer via degradation of p27) degradation of their specific protein targets. Correcting the pathologic over- or under-activation of E3 ligases is a recent strategy for pharmaceutical intervention in multiple diseases where the specific substrate of an E3 ligase has been discovered (Skaar et al., 2014).
4.3 Proteasomes, heat shock proteins, and AMD
The accumulation of ubiquitin-marked proteins is often used as evidence for proteasome inhibition. This assay has been validated in RPE cultured cells, as demonstrated by the ubiquitinated protein conjugates that accumulate after proteasome inhibition (Figure 14). The observation of ubiquitin in drusen localized under the macula from donors with AMD (Mullins et al., 2000; Ryhänen et al., 2009; Viiri et al., 2010; Viiri et al., 2013; Figure 15) implicates proteasome dysfunction in AMD pathology. While this idea has not been tested experimentally in the RPE, measurement of proteasome activity in the neural retina shows activity was elevated in the neural retina of the human macula in donors with progressively more severe stages of AMD (Ethen et al, 2007). These results suggest proteasome activity in the neural retinal is upregulated in response to changing cellular conditions associated with advancing disease. An important consideration is that there may be tissue-specific differences in how AMD affects the major proteolytic systems in each cell type, which ultimately could affect the extent of damaged molecules that accumulate. Evidence supporting the idea of tissue-specific differences is provided by a recent study where we evaluated mtDNA damage in the RPE and retina from individual human donors with AMD. We found that mtDNA damage increased with disease progression only in the RPE (Terluk et al., 2015). The above discussion raises a cautionary note about the use of a single assay to gauge the functional health of a complex biological system, especially in light of the observed tissue-specific differences in accumulated damage.
Figure 14. Ubiquitinated protein conjugates accumulate under proteasome inhibition.
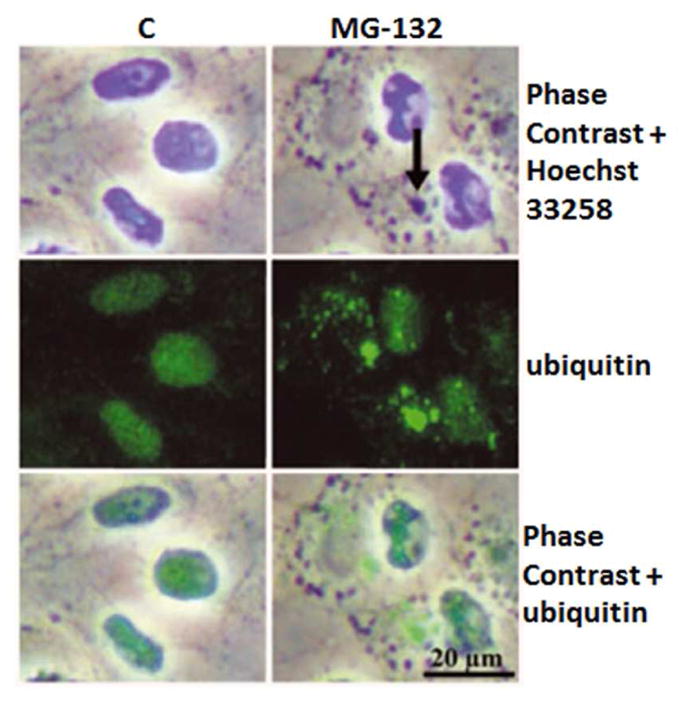
Phase contrast microscopy and immunofluorescence analysis for ubiquitin (green) in control cells, and cells exposed to 10 μM MG-132 proteasome inhibition for 24 hrs. Nuclei were stained with Hoechst 33258 dye (blue). Arrows point to perinuclear protein aggregates. Reprinted with permission of Journal of Cellular and Molecular Medicine.
Figure 15. Immunostaining for ubiquitin in human AMD samples.

The ubiquitin immune reaction in the retinal pigment epithelial cells (RPE, shown by arrows) and in the drusen (asterisks) was evaluated microscopically by selecting foveomacular (A), perimacular (B) and peripheral (C) regions. Most of the drusen were strongly ubiquitin-positive (asterisks). Bruch’s membrane is shown by arrow heads. Original magnification was 200x and in insets 400x. Reprinted with permission of PlosOne.
As discussed in detail in Section 4.4, there is coordinate interaction between heat shock proteins (Hsp) and the proteasome in maintaining protein quality control. We have shown that human AMD donor RPE exhibited significantly higher content of the proteasome as well as the heat shock proteins 27 and 90 (Decanini et al., 2007), supporting the idea that there is increased oxidative stress and more damaged proteins in the RPE with AMD. However, the increase in Hsps was not universal; lower content of αA crystallin as well as resident Hsp in the mitochondria were also observed (Nordgaard et al., 2006 & 2008). The mitochondrial Hsp are involved in the import and refolding of nuclear encoded protein into the mitochondria, so the decrease in these chaperones could have detrimental consequences on mitochondrial function.
4.4 SQSTM1/p62 and Hsp70 link Proteasomes and Autophagy in RPE cells
All functional proteins have their distinctive tertiary and quaternary structures. These three-dimensional structures are often altered in response to different stresses resulting in an inappropriately folded protein (Pierpaoli et al., 2005). One of the most serious consequences associated with protein unfolding is the exposure of normally buried hydrophobic amino acids that promote the formation of protein aggregates. Molecular chaperones and Hsps help maintain the correct protein conformation and therefore reduce protein aggregation in response to increased stress and damage (Kaarniranta et al., 2009). They are also important regulators for protein trafficking through the cell organelle membranes and are part of the process of chaperoning damaged proteins to either the proteasome or lysosome for degradation.
SQSTM1/p62 and Hsp70 work in concert between the Ub-proteasome pathway and autophagy to help regulate cellular proteolysis. Both proteins are upregulated under conditions that cause protein damage and unfolding and thus, they are considered sensors of proteotoxic stress. The main roles of Hsp70 are to refold partially denatured proteins and to chaperone proteins to either the lysosomes or proteasomes for degradation. SQSTM1/p62 is a multifunctional protein that plays an essential role in cell signaling. It also promotes the formation and selective degradation of protein aggregates by utilizing its multiple protein-protein interaction domains by (1) binding to nonfunctional ubiquitinated proteins via its ubiquitin association domain, (2) reducing the toxicity of soluble unfolded proteins by promoting their aggregation through its protein binding domain, and (3) delivering aggregates to the autophagy machinery using the protein binding domain and LIR domains (Su and Wang, 2011). Recent evidence also suggests active crosstalk between proteasome-mediated degradation and selective autophagy, with SQSTM1/p62 bridging these two proteolytic pathways. For example, when proteasome is inhibited, SQSTM1/p62 binds to Atg8/LC3 and facilitates degradation of ubiquitin (Ub) positive protein aggregates after autophagy induced by AICAR (Pankiv et al., 2007; Ryhänen et al., 2009; Viiri et al., 2010, Viiri et al., 2013; Figure 16).
Figure 16. Co-localization of LC3 and SQSTM1/p62.
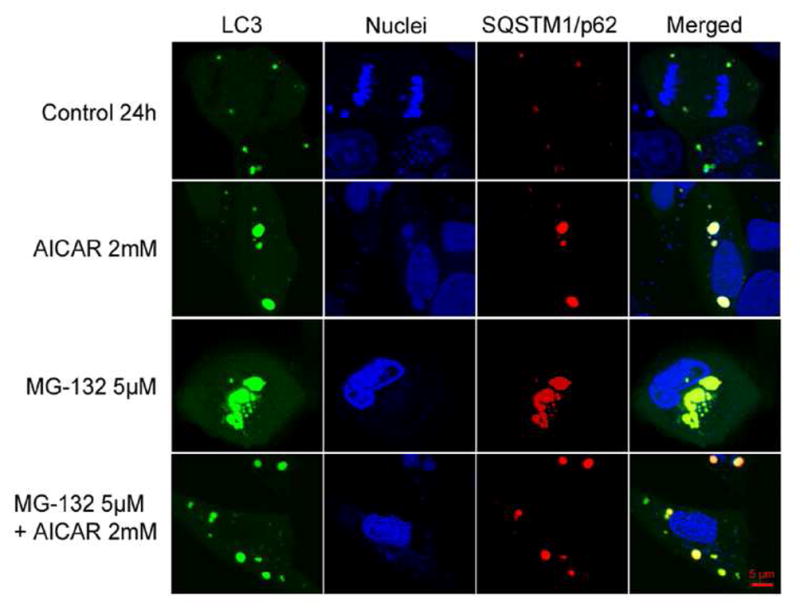
Confocal images show staining for LC3 (pDendra2-hLC3, green) and SQSTM1/p62 (pDsRed2-hp62, red), and nuclei (Hoechst 33258 dye, blue) in ARPE-19 cells. An orange/yellow signal in the merged image indicates co-localization of LC3 and SQSTM1/p62 in untreated control ARPE-19 cells and cells exposed to 2 mM AICAR or/and 5 μM MG-132 for 24 h. Reprinted with permission of PlosOne.
Extensive study of SQSTM1/p62 and Hsp70 in cultured RPE cells has elucidated characteristics that are both shared and distinct for each protein. For example, both Hsp70 (Li et al., 2008; Fernandes et al., 2008) and SQSTM1/p62 (Bjørkøy et al., 2005 & 2006; Pankiv et al., 2007; Clausen et al., 2010; Viiri et al., 2010) are upregulated under conditions of oxidative stress and with proteasome inhibition. In addition to its role in refolding and chaperoning damaged proteins, Hsp70 can modify the permeability of lysosomes (Daugaard et al., 2007). Thus, the inducible Hsp70 may be an important regulator not only for repairing misfolded proteins, but also for regulating lysosomal proteolysis. In cultured RPE, abrogation of proteasomes evokes an intense accumulation of ubiquitinated protein conjugates, Hsp70, LAMP-1 and- 2, and SQSTM1/p62, which co-localize with perinuclear deposits in RPE cells (Ryhänen et al., 2009; Viiri et al., 2013; Figure 17A). Under these conditions, Hsp70 is detected in lysosomal extracts, which supports the idea that the complex of Hsp70- and lysosomal specific LAMP-1 and- 2 associated perinuclear protein aggregates are targeted to autophagy in human RPE cells (Ryhänen et al., 2009; Viiri et al., 2013, Figure 17B).
Figure 17. Hsp70 expression under proteasome inhibition.
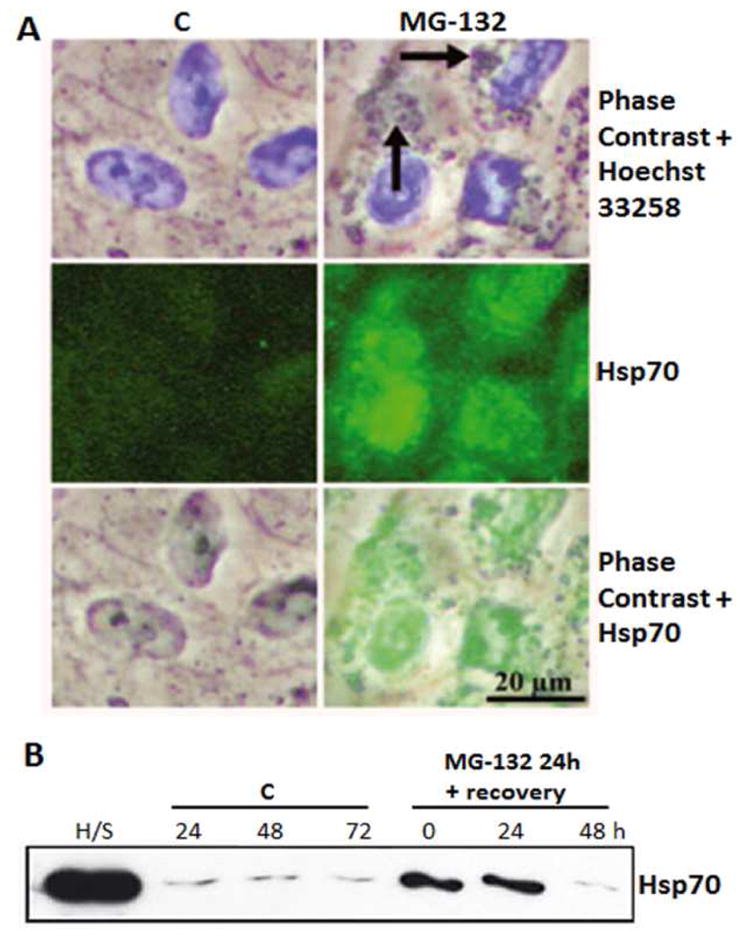
(A) Phase contrast microscopy and immunofluorescence analysis for Hsp70 (green) in control cells, and cells exposed to 10 μM MG-132 proteasome inhibition for 24 hrs. Nuclei were stained with Hoechst 33258 dye (blue). Arrows point to perinuclear protein aggregates. (B) Western blotting analysis (10 μg protein/lane) of the Hsp70 content in isolated lysosome fractions from the control cells (lanes for C) or cells exposed to 10 μM MG-132 for 24hrs (lane 0) or cells exposed to 10 μM MG-132 for 24 and then allowed to recover for up to 48 hrs (lanes 24 and 48). Lysate from heat shocked cells (H/S) are the positive control. Reprinted with permission of Journal of Cellular and Molecular Medicine.
One characteristic that is distinct for Hsp70 and SQSTM1/p62 is their fate after dealing with perinuclear protein aggregates. The autophagy receptor SQSTM1/p62 binds irreversibly to proteasome inhibitor–induced perinuclear protein aggregates and undergoes autophagic clearance in RPE cell cultures (Viiri et al., 2013). In contrast to SQSTM1/p62, Hsp70 binds reversibly to proteasome inhibitor–induced perinuclear protein aggregates and is not degraded by autophagy in ARPE-19 cells (Kivinen et al., 2014). Rather, the proteasome ultimately regulates Hsp70 degradation.
The therapeutic value of upregulating Hsp70 has been explored in RPE cultures. We have recently shown that either the induction of Hsp70 via pharmacological inhibition of NFkB by celastrol, or addition of exogenous recombinant human Hsp70 offers protection against oxidative stress and suppresses inflammation in RPE cells (Paimela et al., 2011; Subrizi et al., 2015). Importantly, exogenously delivered rhHsp70 was taken up by RPE cells and localized in late endosomes and lysosomes. This work provides the basis for a Hsp70 chaperone therapeutic strategy to target protein aggregates in AMD.
In summary, the above findings show that both Hsp70 and SQSTM1/p62 are involved in the regulation of proteostasis, but have different functions in the clearance system. However, there is considerable overlap in their processing of protein aggregates.
5.0 Dysregulation of clearance systems: Progression from para- to chronic- inflammation triggers AMD pathology
The repercussions of a loss in proteolytic capacity that have been discussed in this review include the accumulation of oxidized proteins and toxic protein aggregates, formation of lipofuscin, and the increase in dysfunctional mitochondria due to impaired mitophagy. This cumulative damage over a person’s lifetime could eventually tip the balance toward pathology, thus providing an explanation for the aging component of AMD. Another concept that incorporates the long term consequence of cumulative damage is the notion that AMD is a chronic inflammatory disease not caused by the classical instigators, such as infections or injury. Rather, low grade inflammation, also called para-inflammation, that is present in the aged retina is an adaptive response to tissue malfunction (Medzhitove, 2008; Xu et al., 2009; Parmeggiani et al., 2012). The goal of para-inflammation is to obtain a new set-point of homeostasis and restore tissue function. However, if tissue malfunction is maintained for an extended time, the para-inflammatory state becomes chronic and can promote disease progression.
We propose that the basis for the transition from para- to chronic inflammation could include the age-related increase in toxic compounds, such as protein aggregates and lipofuscin, that accumulate due to defects in RPE proteolysis. Lipofuscin is localized in lysosomal storage bodies or in melanosomes of the RPE cell. Autofluorescent lipofuscin contains a complex heterogeneous mixture of lipid–protein-carbohydrate aggregates and undigested mitochondria that form clusters of granules mainly in aged postmitotic cells, such as the RPE (Kaarniranta et al., 2010). The accumulation of lipofuscin is a hallmark of RPE senescence and it is a major factor in lysosomal dysfunction (Holz et al., 1999; Bergmann et al., 2004). With lipofuscin accumulation, both phagocytotic capacity (heterophagy) and processes associated ith degradation of damaged mtDNA and cytoplasmic proteins via autophagy may become disturbed, which subsequently leads to increased transcytosis and exocytosis of proteins and drusen formation outside RPE cells (Krohne et al., 2010; Viiri et al., 2013; Kaarniranta et al., 2013; Kay et al., 2014; Kim et al., 2013; Valapala et al., 2014b; Sundelin et al., 1998).
Human retinal lipofuscin contains bis-retinoid pyridinium compounds, A2E and the 13-cis isomer iso-A2E (Eldred 1993; Eldred and Lasky 1993; Sakai et al., 1996). A2E is formed by the hydrolytic cleavage from A2-PE, a phosphatidyl ester derivative. In the presence of oxygen, A2E has been shown to photochemically initiate auto-oxidation to produce superoxide and peroxyl radicals and evoke cellular membrane damage (Boulton et al., 1993; Reszka et al., 1995; Sparrow et al., 1999; Ragauskaite et al., 2001). A2E exerts a direct effect on the autophagy/lysosomal system by inhibiting the ATP-driven protein pump in RPE lysosomes (Bergmann et al., 2004), and elevating lysosomal pH (Lamb and Simon, 2004). Both processes result in alkalinisation of the lysosomal lumen, which has an inhibitory effect on lysosomal proteolytic enzymes. A2E also impairs the phagocytosis of photoreceptor outer segments by slowing the degradation of phospholipids, which then remain in lysosomes and result in a build-up of undigested lipids in the RPE (Finnemann et al., 2002; Vives-Bauza et al., 2008).
In addition to accumulation of lysosomal lipofuscin, melanosomes also collect lipofuscin in RPE cells. Constant retinal oxidative stress and decreased melanin content reduces melanin’s antioxidant capacity during aging (Sarna et al., 2003; Burke et al., 2007). Moreover, increased melanosomal oxygen consumption and reactive oxygen species production evoke melanosomal lipofuscin accumulation and lead to detrimental effects in RPE cells (Rózanowska et al., 2002; Rózanowski et al., 2008). Melanolipofuscin accumulation is associated with retinal degeneration and increased amount of complement proteins (Wolkow et al., 2011).
5.1 Dysregulated complement and the transition to chronic inflammation
A2E in RPE lipofuscin activates the complement system and thus connects oxidative stress and impaired proteolysis with inflammation (Zhou et al., 2006; Zhou et al., 2009; Sparrow, 2010; Radu et al., 2011; Ma et al., 2013, Nowak et al., 2013). The complement system can be activated through three pathways; the classical pathway, the alternative pathway, and the lectin pathway (Donoso et al., 2006; Kaarniranta and Salminen, 2009). All the complement pathways involve the following steps: initiation, formation of a C3 convertase, cleavage of C3, formation of a C5 convertase, cleavage of C5, and formation of the membrane attack complex (C5b, 6, 7, 8, 9), that terminates the complement cascade in cellular lysis (Bradley et al., 2011). All three pathways result in the production of C3b, and therefore complement factors (CF) that act at this step of the cascade, such as CFB, D, H, and I, are crucial regulators of the whole complement system (Ohno-Matsui, 2011). Thus, events resulting in complement dysregulation could contribute to the chronic inflammation, drusen formation, and loss of RPE that underlie the AMD pathogenesis (Kaarniranta et al., 2011).
Proteins that have been identified in drusen are present in the RPE, photoreceptors, and blood, and accordingly, they serve as an origin of drusen components for both the local RPE and the choroidal vasculature (Johnson et al, 2000; Mullins et al., 2000; Hageman et al., 2001; Johnson et al., 2001, Wang et al., 2010). The presence of plasma proteins in drusen that are up-regulated during an inflammatory response has led to the proposal that immune-mediated, complement activation events, triggered by signals from the RPE, are causally involved in the formation of drusen. AMD can also be considered an aggregation disease, since drusen components resemble other inflammation-related aggregates found in Alzheimer’s disease, amyloidosis, atherosclerosis and elastosis (Ohno-Matsui, 2011; Kaarniranta et al., 2011; Mullins et al., 2000; Machalińska et al., 2011; Machalińska et al., 2012).
AMD-associated drusen contain nearly all alternative complement pathway proteins, including CFH, C3, and the products of its activation and degradation, and the terminal pathway MAC proteins (van der Schaft et al., 1993; Johnson et al, 2000; Mullins et al., 2000; Hageman et al., 2001; Johnson et al., 2001, Wang et al., 2010). This supports the idea that there is local, complement-mediated inflammation between the RPE cell layer and Bruch’s membrane in AMD. In addition to biomarkers localized in and around the RPE, proteins involved in complement activation can also be detected in AMD patient blood, which suggests a systemic immunological response (Scholl et al., 2008). To what extent systemic complement activation reflects pathological events in the eye, and to what extent systemic inflammation may be manifest in the eye, is not yet clear.
Various polymorphisms in different complement factors increase the risk for AMD (Fritsche et al., 2014). The polymorphic variant of CFH with the substitution of histidine for tyrosine at codon Y420H has been well-characterized in AMD. CFH is a complement regulator protein whose main function is to inhibit the activation of the alternative complement pathway (Donoso et al., 2006; Kaarniranta and Salminen, 2009; Bradley et al., 2011). CFH protein is composed of 20 short consensus folding domain repeats. The Y420H substitution is located in short consensus folding domain-7 and affects the binding properties of CFH, e.g. to the C-reactive pentraxin protein. It can potentiate drusen formation by inhibiting phagocytosis by macrophages and microglia to clear cellular debris present between RPE cells and Bruchs’membrane (Donoso et al., 2006, Buschini et al., 2011; Weismann et al., 2011). Impaired phagocytosis of oxidised photoreceptor OS, lipofuscin accumulation and decreased proteolysis in RPE cells connect oxidative stress to inflammation that is regulated by the complement system in AMD (Kaarniranta et al., 2013; Piippo et al., 2014; Valapala et al., 2014a).
Recent findings reveal that macrophages and microglia are essential regulators of the local complement system in the retina. They have an important role in clearance of debris and in the regulation of inflammatory signalling and tissue homeostasis (Xu et al., 2008; Wang et al., 2009; Xu et al., 2009; Zhao et al., 2013). In young and healthy animals, the number of macrophages and microglia are low in the retinal space and they have beneficial effects on the clearance process and in the regulation of para-inflammation (Xu et al., 2009). During aging and in pathological processes, they accumulate in the retinal space and progressively contribute to detrimental inflammation. Concurrently, their capacity to clear foreign material decreases. Similar sub-retinal macrophage and microglia accumulation has also been characterized in human AMD (Penfold et al., 2001; Anderson et al., 2010). These findings imply that these accumulations of senescent macrophage and microglia may dysregulate immune interactions and drive AMD progression (Ma et al., 2009), linking the cellular mechanisms of aging with chronic inflammation, including inflammasome activation in the outer retina (Buschini et al., 2011). However, it is not known whether inflammasome activation can be interpreted as a beneficial para-inflammation process or detrimental chronic inflammation in AMD pathogenesis (Doyle et al., 2012; Kauppinen et al., 2012; Tarallo et al., 2012; Klettner et al., 2013; Tseng et al., 2013; Doyle et al., 2014).
6.0 Impaired Proteostasis Evokes AMD Pathology
We propose a model that includes defects in RPE proteolysis and the consequent accumulation of toxic compounds, such as protein aggregates and lipofuscin, as a potential mechanism underlying AMD pathology (Figure 18). The unique and harsh retinal environment sets the stage for many of the age-related changes that eventually tip the balance from the para-inflammatory state to the more destructive chronic inflammatory state. Figure 18 summarizes the oxidative challenge that RPE cells are constantly exposed to due to heterophagy, light exposure, and high cellular metabolism. One of the consequences of elevated oxidative stress is damage to proteins, leading to their structural unfolding. As a first line of defense, Hsps refold ROS-damaged proteins that are misfolded. However, if the Hsps are unsuccessful in salvaging the damaged protein, then the soluble proteins are targeted to the proteasome for degradation. Impaired proteasome-dependent degradation can lead to the formation of protein aggregates, which undergo autophagic clearance.
Figure 18. Dysregulated proteostasis in aged RPE cells evokes AMD pathology.

RPE cells are constantly exposed to oxidative stress. One consequence is that damage to proteins causes their unfolding. Heat-shock proteins (Hsps) attempt to refold damaged proteins, but if not successful, the misfolded proteins are ubiquitinated (U) and targeted for proteasomal clearance. If proteasome activity is decreased, proteins aggregate and are degraded via autophagy with the assistance of p62. Autophagy is also used to digest damaged mitochondria (mitophagy). Photoreceptor outer segments that are phagocytosed by the RPE (heterophagy) are degraded in the lysosomes in a recently described process of LC3-Associated Phagocytosis. In aged RPE cells, lipofuscin accumulates in lysosomes as a result of the coincident decline of lysosomal enzyme activity. The impaired lysosomal enzyme activity inhibits autophagic flux and non-digested mitochondria, protein aggregates, and lipofuscin accumulate. Thus, in aged RPE cells, disturbed proteostasis and accumulated toxic compounds trigger the progression from para-inflammation to chronic inflammation and evoke the AMD-associated formation of extracellular drusen formation and complement activation.
Damaged mitochondria are degraded in autolysosomes; this specialized version of autophagy is called mitophagy. The autophagy machinery and lysosomes are also utilized for the digestion of photoreceptor OS in a recently described process of LC3-Associated Phagocytosis (LAP). In aged RPE cells, auto-oxidative lipofuscin accumulates in lysosomes as a result of the coincident decline of lysosomal enzyme activity. Decreased activity of lysosomal enzymes inhibits the autophagic clearance of protein aggregates, damaged mitochondria, and photoreceptor OS in the lysosomes thereby accelerating the accumulation of lipofuscin. When damaged cell components exceed the capacity of chaperones and proteolytic machines to repair and remove the damage, toxic compounds accumulate and trigger the inflammatory response in RPE cells. Additionally, these intracellular aggregates can be exocytosed from the RPE, where they contribute to drusen formation and complement activation.
7.0 Future directions
Failure in proteostasis, which can be initiated when the degradative systems are overwhelmed, may be one of the underlying mechanisms responsible for the cascade of events leading to AMD. Specific areas that need to be developed in order to first further understand the molecular bases of AMD include a comprehensive analysis of the two major proteolytic systems- the proteasome and autophagy/lysosomes- in both the retina and RPE. Importantly, we need to first understand how these systems change with aging so that we can distinguish normal aging from the pathologic aging that is associated with disease. Continued work using in vitro and in vivo model systems, coupled with validating specific hypotheses in tissue from human donors across a broad age range and with AMD, will provide a more accurate and detailed picture of how failure in RPE proteolysis contribute to AMD pathology. Importantly, a more thorough investigation of the mechanistic details of AMD is required before better prevention or targeted interventions can be developed.
Highlights.
RPE proteolysis involves the Ub-proteasome and lysosome/autophagy pathways
The proteasome and heat shock proteins are upregulated in AMD donor retinas
Autophagy rids the cell of toxic protein aggregates and defective mitochondria
Lipofuscin accumulates due to an age-related decrease in lysosomal enzymes
Defects in proteolysis facilitates the transition from para- to chronic inflammation
Acknowledgments
The authors would like to acknowledge funding support from National Institutes of Health EY019037 (DS), EY019037-S (DS), The Finnish Eye Foundation (KK), The VTR grant (5503743) for Kuopio University Hospital (KK), Foundation Fighting Blindness (0613-0620-UMN, DF), Arnold and Mabel Beckman Initiative for Macular Research (1303, DF), an anonymous benefactor for AMD Research (DF), Minnesota Lions Vision Foundation (DF), and an unrestricted grant to the Department of Ophthalmology and Visual Neuroscience (U of MN) from the Research to Prevent Blindness.
DF is the recipient of the Elaine and Robert Larson Endowed Vision Research Chair. DS is a recipient of the Carolyn K. McGillvray Memorial Award for Macular Degeneration Research from BrightFocus Foundation and the Sybil B. Harrington Special Scholar Award for Macular Degeneration from Research to Prevent Blindness.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Deborah A. Ferrington, Email: ferri013@umn.edu.
Debasish Sinha, Email: debasish@jhmi.edu.
Kai Kaarniranta, Email: kai.kaarniranta@uef.fi.
References
- Ambati J, Atkinson JP, Gelfand BD. Immunology of age-related macular degeneration. Nat Rev Immunol. 2013;13:438–451. doi: 10.1038/nri3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112. doi: 10.1016/j.preteyeres.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, Martinez de Villarreal L, dos Santos HH, Garg A. PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet. 2010;87:866–872. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T, Mizushima T, et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc Natl Acad Sci U S A. 2011;108:14914–14919. doi: 10.1073/pnas.1106015108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban T, Heymann JAW, Song Z, Hinshaw JE, Chan DC. OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tabulation. Hum Mol Genet. 2010;19:2113–2122. doi: 10.1093/hmg/ddq088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann M, Schutt F, Holz FG, Kopitz J. Inhibition of the ATP-driven proton pump in RPE lysosomes by the major lipofuscin fluorophore A2-E may contribute to the pathogenesis of age-related macular degeneration. FASEB J. 2004;18:562–564. doi: 10.1096/fj.03-0289fje. [DOI] [PubMed] [Google Scholar]
- Besharse JC, Hollyfield JG, Rayborn ME. Photoreceptor outer segments: accelerated membrane renewal in rods after exposure to light. Science. 1977;196:536–538. doi: 10.1126/science.300504. [DOI] [PubMed] [Google Scholar]
- Birgisdottir ÅB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci. 2013;126:323732–47. doi: 10.1242/jcs.126128. [DOI] [PubMed] [Google Scholar]
- Blasiak J, Petrovski G, Vereb Z, Facsko A, Kaarniranta K. Oxidative stress, hypoxia, and autophagy in the neovascular processes of age-related macular degeneration. Biomed Res Int. 2014;2014:768026. doi: 10.1155/2014/768026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Johansen T. p62/SQSTM1: a missing link between protein aggregates and the autophagy machinery. Autophagy. 2006;2:138–139. doi: 10.4161/auto.2.2.2405. [DOI] [PubMed] [Google Scholar]
- Bobu C, Hicks D. Regulation of retinal photoreceptor phagocytosis in a diurnal mammal by circadian clocks and ambient lighting. Invest Ophthalmol Vis Sci. 2009;50:3495–3502. doi: 10.1167/iovs.08-3145. [DOI] [PubMed] [Google Scholar]
- Bosch E, Horwitz J, Bok D. Phagocytosis of outer segments by retinal pigment epithelium: phagosome-lysosome interaction. J Histochem Cytochem. 1993;41:253–263. doi: 10.1177/41.2.8419462. [DOI] [PubMed] [Google Scholar]
- Boulton M, Dontsov A, Jarvis-Evans J, Ostrovsky M, Svistunenko D. Lipofuscin is a photoinducible free radical generator. J Photochem Photobiol B. 1993;19:201–204. doi: 10.1016/1011-1344(93)87085-2. [DOI] [PubMed] [Google Scholar]
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol. 2013;15:713–720. doi: 10.1038/ncb2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley DT, Zipfel PF, Hughes AE. Complement in age-related macular degeneration: a focus on function. Eye (Lond) 2011;25:683–693. doi: 10.1038/eye.2011.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke JM, Henry MM, Zareba M, Sarna T. Photobleaching of melanosomes from retinal pigment epithelium: I. Effects on protein oxidation. Photochem Photobiol. 2007;83:920–924. doi: 10.1111/j.1751-1097.2007.00081.x. [DOI] [PubMed] [Google Scholar]
- Burnside B, Bost-Usinger L. The retinal pigment epithelium cytoskeleton. In: Marmor MF, Wolfensberger TJ, editors. The retinal pigment epithelium: function and disease. Oxford University Press; New York: 1998. pp. 41–67. [Google Scholar]
- Buschini E, Piras A, Nuzzi R, Vercelli A. Age related macular degeneration and drusen: neuroinflammation in the retina. Prog Neurobiol. 2011;95:14–25. doi: 10.1016/j.pneurobio.2011.05.011. [DOI] [PubMed] [Google Scholar]
- Caberoy NB, Zhou Y, Li W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010;29:3898–3910. doi: 10.1038/emboj.2010.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Biochim Biophys Acta. 2012;1824:3–13. doi: 10.1016/j.bbapap.2011.03.007. [DOI] [PubMed] [Google Scholar]
- Ciechanover A, Stanhill A. The complexity of recognition of ubiquitinated substrates by the 26S proteasome. Biochim Biophys Acta. 2014;1843:86–96. doi: 10.1016/j.bbamcr.2013.07.007. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sawada O, Kohno H, Le YZ, Subauste C, Maeda T, Maeda A. Autophagy protects the retina from light-induced degeneration. J Biol Chem. 2013;288:7506–7518. doi: 10.1074/jbc.M112.439935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Øvervatn A, Stenmark H, Bjørkøy G, Simonsen A, Johansen T. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010;6:330–344. doi: 10.4161/auto.6.3.11226. [DOI] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Ann Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Dice JF. A receptor for the selective uptake and degradation pf proteins by lysosomes. Science. 1996;273:501–503. doi: 10.1126/science.273.5274.501. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24:92–104. doi: 10.1038/cr.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlmann BT, Ruppert T, Kuehn L, Merforth S, Kloetzel PM. Different proteasome subtypes in a single tissue exhibit different enzymatic properties. J Mol Biol. 2000;303:643–653. doi: 10.1006/jmbi.2000.4185. [DOI] [PubMed] [Google Scholar]
- Damek-Poprawa M, Diemer T, Lopes VS, Lillo C, Harper DC, Marks MS, Wu Y, Sparrow JR, Rachel RA, Williams DS, Boesze-Battaglia K. Melanoregulin (MREG) modulates lysosome function in pigment epithelial cells. J Biol Chem. 2009;284:10877–10889. doi: 10.1074/jbc.M808857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Kirkegaard-Sørensen T, Ostenfeld MS, Aaboe M, Høyer-Hansen M, Orntoft TF, Rohde M, Jäättelä M. Lens epithelium derived growth factor is an Hsp70–2 regulated guardian of lysosomal stability in human cancer. Cancer Res. 2007;67:2559–2567. doi: 10.1158/0008-5472.CAN-06-4121. [DOI] [PubMed] [Google Scholar]
- Davies KJA. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- D’Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D. Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic RCS rat. Hum Mol Gen. 2000;9:645–651. doi: 10.1093/hmg/9.4.645. [DOI] [PubMed] [Google Scholar]
- Decanini A, Nordgaard CL, Feng X, Ferrington DA, Olsen TW. Changes in select redox proteins of the retinal pigment epithelium in age-related macular degeneration. Am J Ophthalmol. 2007;143:607–615. doi: 10.1016/j.ajo.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deguchi J, Yamamoto A, Yoshimori T, Sugasawa K, Moriyama Y, Futai M, Suzuki T, Kato K, Uyama M, Tashiro Y. Acidification of phagosomes and degradation of rod outer segments in rat retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1994;35:568–579. [PubMed] [Google Scholar]
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem. 2012;393:547–564. doi: 10.1515/hsz-2012-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donoso LA, Kim D, Frost A, Callahan A, Hageman G. The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2006;51:137–152. doi: 10.1016/j.survophthal.2005.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, Farrar GJ, Kiang AS, Humphries MM, Lavelle EC, O’Neill LA, Hollyfield JG, Humphries P. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012;18:791–798. doi: 10.1038/nm.2717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle SL, Ozaki E, Brennan K, Humphries MM, Mulfaul K, Keaney J, Kenna PF, Maminishkis A, Kiang AS, Saunders SP, Hams E, Lavelle EC, Gardiner C, Fallon PG, Adamson P, Humphries P, Campbell M. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci Transl Med. 2014;6:230ra44. doi: 10.1126/scitranslmed.3007616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Fon EA. The three P’s of mitophagy: PARKIN, PINK1, and post-translatioal modifications. Genes Dev. 2015;29:989–99. doi: 10.1101/gad.262758.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebermann I, Walger M, Scholl HP, Charbel Issa P, Luke C, Nurnberg G, Lang-Roth R, Becker C, Nurnberg P, Boltz HJ. Truncating mutation of the DFNB59 gene causes cochlear hearing impairment and central vestibular dysfunction. Hum Mutat. 2007;28:571–577. doi: 10.1002/humu.20478. [DOI] [PubMed] [Google Scholar]
- Eldred GE. Age pigment structure. Nature. 1993;364:396. doi: 10.1038/364396a0. [DOI] [PubMed] [Google Scholar]
- Eldred GE, Lasky MR. Retinal age pigments generated by self-assembling lysosomotropic detergents. Nature. 1993;361:724–726. doi: 10.1038/361724a0. [DOI] [PubMed] [Google Scholar]
- Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen EL. New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol. 2008;266:207–247. doi: 10.1016/S1937-6448(07)66005-5. [DOI] [PubMed] [Google Scholar]
- Ethen CM, Hussong SA, Reilly C, Feng X, Olsen TW, Ferrington DA. Transformation of the proteasome with age-related macular degeneration. FEBS Lett. 2007;581:885–890. doi: 10.1016/j.febslet.2007.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feher J, Kovacs I, Artico M, et al. Mitochondrial alterations of RPE in AMD. Neurobiol Aging. 2006;27:983–993. doi: 10.1016/j.neurobiolaging.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Ferguson TA, Green DR. Autophagy and phagocytosis converge for better vision. Autophagy. 2014;10:165–167. doi: 10.4161/auto.26735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes AF, Zhou J, Zhang X, Bian Q, Sparrow J, Taylor A, Pereira P, Shang F. Oxidative inactivation of the proteasome in retinal pigment epithelial cells. A potential link between oxidative stress and up-regulation of interleukin-8. J Biol Chem. 2008;283:20745–20753. doi: 10.1074/jbc.M800268200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Hussong SA, Roehrich H, Kapphahn RJ, Kavanaugh SM, Heuss ND, Gregerson DS. Immunoproteasome responds to injury in the retina and brain. J Neurochem. 2008;106:158–169. doi: 10.1111/j.1471-4159.2008.05345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function and antigen presentation. Prog Mol Biol Transl Sci. 2012;109:75–112. doi: 10.1016/B978-0-12-397863-9.00003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnemann SC, Leung LW, Rodriguez-Boulan E. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipid by the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2002;99:3842–3847. doi: 10.1073/pnas.052025899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SK, Pfeffer BA, Anderson DH. Both rod and cone disc shedding are related to light onset in the cat. Invest Ophthalmol Vis Sci. 1983;24:844–856. [PubMed] [Google Scholar]
- Freeman GJ, Casasnovas JM, Umetsu DT, Dekruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235:172–189. doi: 10.1111/j.0105-2896.2010.00903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker M, Neher JJ, Zhao JW, Thery C, Tolkovsky AM, Brown GC. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci. 2012;32:2657–2666. doi: 10.1523/JNEUROSCI.4837-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche LG, Fariss RN, Stambolian D, Abecasis GR, Curcio CA, Swaroop A. Age-related macular degeneration: genetics and biology coming together. Annu Rev Genomics Hum Genet. 2014;15:151–171. doi: 10.1146/annurev-genom-090413-025610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D. Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy gene, cause retinitis pigmentosa. Nat Gene. 2000;26:270–271. doi: 10.1038/81555. [DOI] [PubMed] [Google Scholar]
- Gibbs D, Kitamoto J, Williams DS. Abnormal phagocytosis by retinal pigmented epithelium that lacks myosin VIIa, the usher syndrome 1B protein. Proc Natl Acad Sci U S A. 2003;100:6481–64. doi: 10.1073/pnas.1130432100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregerson DS, Lew KL, McPherson SW, Heuss ND, Ferrington DA. RPE cells resist bystander killing by CTLs, but are highly susceptible to antigen-dependent CTL killing. Invest Ophthalmol Vis Sci. 2006;47:5385–5396. doi: 10.1167/iovs.06-0636. [DOI] [PubMed] [Google Scholar]
- Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–732. doi: 10.1016/s1350-9462(01)00010-6. [DOI] [PubMed] [Google Scholar]
- Haroon N, Maksymowych WP, Rahman P, Tsui FW, O’Shea FD, Inman RD. Radiographic severity of ankylosing spondylitis is associated with polymorphism of the large multifunctional peptidase 2 gene in the Spondyloarthritis Research Consortium of Canada cohort. Arthritis Rheum. 2012;64:1119–1126. doi: 10.1002/art.33430. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Ubiquitin-dependent protein degradation. Genetics. 1996;30:405–439. doi: 10.1146/annurev.genet.30.1.405. [DOI] [PubMed] [Google Scholar]
- Holz FG, Schütt F, Kopitz J, Eldred GE, Kruse FE, Völcker HE, Cantz M. Inhibition of lysosomal degradative functions in RPE cells by a retinoid component of lipofuscin. Invest Ophthalmol Vis Sci. 1999;40:737–743. [PubMed] [Google Scholar]
- Hoppe G, O’Neil J, Hoff HF, Sears J. Accumulation of oxidized lipidprotein complexes alters phagosome maturation in retinal pigment epithelium. Cell Mol Life Sci. 2004;61:1664–1674. doi: 10.1007/s00018-004-4080-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussong SA, Kapphahn RJ, Phillips SL, Maldonado M, Ferrington DA. Immunoproteasome deficiency alters retinal proteasome’s response to stress. J Neurochem. 2010;113:1481–90. doi: 10.1111/j.1471-4159.2010.06688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussong SA, Roehrich H, Kapphahn RJ, Maldonato M, Pardue MT, Ferrongton DA. A novel role for the immunoproteasome in retinal function. Invest Ophthalmol Vis Sci. 2011;52:714–23. doi: 10.1167/iovs.10-6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyttinen JM, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev. 2014;18:16–28. doi: 10.1016/j.arr.2014.07.002. [DOI] [PubMed] [Google Scholar]
- Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta. 2013;1833:503–510. doi: 10.1016/j.bbamcr.2012.11.018. [DOI] [PubMed] [Google Scholar]
- Immel JH, Fisher SK. Cone photoreceptor shedding in the tree shrew (Tupaia belangerii) Cell Tissue Res. 1985;239:667–675. doi: 10.1007/BF00219247. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Leitner WP, Staples MK, Anderson DH. Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res. 2001;73:887–896. doi: 10.1006/exer.2001.1094. [DOI] [PubMed] [Google Scholar]
- Johnson LV, Ozaki S, Staples MK, Erickson PA, Anderson DH. A potential role for immune complex pathogenesis in drusen formation. Exp Eye Res. 2000;70:441–449. doi: 10.1006/exer.1999.0798. [DOI] [PubMed] [Google Scholar]
- Jung T, Höhn A, Grune T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol. 2013;2:99–104. doi: 10.1016/j.redox.2013.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A. Age-related macular degeneration: activation of innate immunity system via pattern recognition receptors. J Mol Med (Berl) 2009;87:117–123. doi: 10.1007/s00109-008-0418-z. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A, Eskelinen EL, Kopitz J. Heat shock proteins as gatekeepers of proteolytic pathways-Implications for age-related macular degeneration (AMD) Ageing Res Rev. 2009;8:128–139. doi: 10.1016/j.arr.2009.01.001. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Hyttinen J, Ryhänen T, Viiri J, Paimela T, Toropainen E, Sorri I, Salminen A. Mechanisms of protein aggregation in the retinal pigment epithelial cells. Front Biosci (Elite Ed) 2010;2:1374–84. doi: 10.2741/e198. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M. Age-related macular degeneration (AMD): Alzheimer’s disease in the eye? J Alzheimers Dis. 2011;24:615–631. doi: 10.3233/JAD-2011-101908. [DOI] [PubMed] [Google Scholar]
- Kaarniranta K, Sinha D, Blasiak J, Kauppinen A, Vereb Z, Salminen A, Boulton ME, Petrovski G. Autophagy and heterophagy dysregulation leads to retinal pigment epithelium dysfunction and development of age-related macular degeneration. Autophagy. 2013;9:973–984. doi: 10.4161/auto.24546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaemmerer E, Schütt F, Krohne TU, Holz FG, Kopitz J. Effects of lipid peroxidation-related protein modifications on RPE lysosomal functions and POS phagocytosis. Invest Ophthalmol Vis Sci. 2007;48:1342–1347. doi: 10.1167/iovs.06-0549. [DOI] [PubMed] [Google Scholar]
- Karunadharma PP, Nordgaard CL, Olsen TW, Ferrington DA. Mitochondrial DNA damage as a potential mechanism for AMD. Invest Ophthalmol Vis Sci. 2010;51:5470–5479. doi: 10.1167/iovs.10-5429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen A, Niskanen H, Suuronen T, Kinnunen K, Salminen A, Kaarniranta K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells--implications for age-related macular degeneration (AMD) Immunol Lett. 2012;147:29–33. doi: 10.1016/j.imlet.2012.05.005. [DOI] [PubMed] [Google Scholar]
- Kay P, Yang YC, Hiscott P, Gray D, Maminishkis A, Paraoan L. Age-related changes of cystatin C expression and polarized secretion by retinal pigment epithelium: potential age-related macular degeneration links. Invest Ophthalmol Vis Sci. 2014;55:926–934. doi: 10.1167/iovs.13-13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kevany BM, Palczewski K. Phagocytosis of retinal rod and cone photoreceptors. Physiology. 2010;25:8–15. doi: 10.1152/physiol.00038.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, Ferguson TA. Noncanonical autophagy promotes the visual cycle. Cell. 2013;154:365–376. doi: 10.1016/j.cell.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, Akopian TN, Woo KM, Goldberg AL. The size of peptides generated from protein by mammalian 26S and 20S proteasome. Implications for understanding the degenerative mechanism and antigen presentation. J Biol Chem. 1999;274:3363–3371. doi: 10.1074/jbc.274.6.3363. [DOI] [PubMed] [Google Scholar]
- Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J Clin Invest. 2011;121:4150–4160. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivinen N, Hyttinen JMT, Viiri J, Paterno JJ, Felszheghy S, Kauppinen A, Salminen A, Kaarniranta K. Hsp70 binds reversibly to proteasome inhibitor-induced protein aggregates and evades autophagic clearance in ARPE-19 cells. J Biochem Pharm Res. 2014;2:1–7. [Google Scholar]
- Klare N, Seeger M, Janek K, Jungblut PR, Dahlmann B. Intermediate-type 20S proteasome in HeLa cells: “asymmetric” subunit composition, diversity and adaptation. J Mol Biol. 2007;373:1–10. doi: 10.1016/j.jmb.2007.07.038. [DOI] [PubMed] [Google Scholar]
- Klettner A, Kauppinen A, Blasiak J, Roider J, Salminen A, Kaarniranta K. Cellular and molecular mechanisms of age-related macular degeneration: from impaired autophagy to neovascularization. Int J Biochem Cell Biol. 2013;45:1457–1467. doi: 10.1016/j.biocel.2013.04.013. [DOI] [PubMed] [Google Scholar]
- Knowles A, Koh K, Wu JT, Chien CT, Chamovitz DA, Blau J. The COP9 signalsome is required for light-dependent timeless degradation and Drosophila clock resetting. J Neurosci. 2009;29:1152–1162. doi: 10.1523/JNEUROSCI.0429-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krohne TU, Kaemmerer E, Holz FG, Kopitz J. Lipid peroxidation products reduce lysosomal protease activities in human retinal pigment epithelial cells via two different mechanisms of action. Exp Eye Res. 2010;90:261–266. doi: 10.1016/j.exer.2009.10.014. [DOI] [PubMed] [Google Scholar]
- Lai CM, Robertson T, Papadimitriou J, Shen WY, Daw N, Constable IJ, Rakoczy PE. Controlled production of active Cathepsin D in Retinal Pigment Epithelial cells following adenovirus-mediated gene delivery. Mol Ther. 2000;2:476–484. doi: 10.1006/mthe.2000.0195. [DOI] [PubMed] [Google Scholar]
- Lamb LE, Simon JD. A2E: a component of ocular lipofuscin. Photochem Photobiol. 2004;79:127–136. doi: 10.1562/0031-8655(2004)079<0127:aacool>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- LaVail MM. Rod outer segment disc shedding in rat retina: relationship to cyclic lighting. Science. 1976;194:1071–1074. doi: 10.1126/science.982063. [DOI] [PubMed] [Google Scholar]
- Li W. Phagocyte dysfunction, tissue aging and degeneration. Ageing Res Rev. 2013;12:1005–1012. doi: 10.1016/j.arr.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang YS, Shen XF, Hui YN, Han J, Zhao W, Zhu J. Alterations of activity and intracellular distribution of the 20S proteasome in ageing retinal pigment epithelial cells. Exp Gerontol. 2008;43:1114–1122. doi: 10.1016/j.exger.2008.08.052. [DOI] [PubMed] [Google Scholar]
- Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit β-type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64:895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Coon S, Zhao L, Fariss RN, Wong WT. A2E accumulation influences retinal microglial activation and complement regulation. Neurobiol Aging. 2013;34:943–960. doi: 10.1016/j.neurobiolaging.2012.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Zhao L, Fontainhas AM, Fariss RN, Wong WT. Microglia in the mouse retina alter the structure and function of retinal pigmented epithelial cells: a potential cellular interaction relevant to AMD. PLoS One. 2009;4:e7945. doi: 10.1371/journal.pone.0007945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machalinska A, Kawa MP, Marlicz W, Machalinski B. Complement system activation and endothelial dysfunction in patients with age-related macular degeneration (AMD): possible relationship between AMD and atherosclerosis. Acta Ophthalmol. 2012;90:695–703. doi: 10.1111/j.1755-3768.2011.02295.x. [DOI] [PubMed] [Google Scholar]
- Machalinska A, Safranow K, Dziedziejko V, Mozolewska-Piotrowska K, Paczkowska E, Klos P, Pius E, Grymula K, Wiszniewska B, Karczewicz D, Machalinski B. Different populations of circulating endothelial cells in patients with age-related macular degeneration: a novel insight into pathogenesis. Invest Ophthalmol Vis Sci. 2011;52:93–100. doi: 10.1167/iovs.10-5756. [DOI] [PubMed] [Google Scholar]
- Maldonado M, Kapphahn RJ, Terluk MR, Heuss ND, Yuan C, Gregerson DS, Ferrington DA. Immunoproteasome deficiency modifies the alternative pathway of NFkB signaling. PLoS ONE. 2013;8:e56187. doi: 10.1371/journal.pone.0056187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques FC, Volovik Y, Cohen E. The roles of cellular and organismal aging in the development of late-onset maladies. Annu Rev Pathol Mech Dis. 2015;10:1–23. doi: 10.1146/annurev-pathol-012414-040508. [DOI] [PubMed] [Google Scholar]
- McCray BA, Taylor JP. The role of autophagy in age-related neurodegeneration. Neurosignals. 2008;16:75–84. doi: 10.1159/000109761. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- Mitter SK, Song C, Qi X, Mao H, Rao H, Akin D, Lewin A, Grant M, Dunn W, Jr, Ding J, Rickman CB, Boulton M. Dysregulated autophagy in the RPE is associated with increased susceptibility to oxidative stress and AMD. Autophagy. 2014;10:1989–2005. doi: 10.4161/auto.36184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- Mullins RF, Russell SR, Anderson DH, Hageman GS. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. FASEB J. 2000;14:835–846. [PubMed] [Google Scholar]
- Mustafi D, Kevany BM, Genoud C, Okano K, Cideciyan AV, Sumaroka A, Roman AJ, Jacobson SG, Engel A, Adams MD, Palczewski K. Defective photoreceptor phagocytosis in a mouse model of enhanced s-cone syndrome causes progressive retinal degeneration. FASEB J. 2011;25:3157–3176. doi: 10.1096/fj.11-186767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandrot EF. Animal models, in “the Quest to decipher RPE phagocytosis”. In: Ash JD, editor. Retinal Degenerative Diseases, Adv Exper Med Biol. Vol. 10. 2014. pp. 77–83. [DOI] [PubMed] [Google Scholar]
- Nordgaard CL, Karunadharma PP, Feng X, Olsen TW, Ferrington DA. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of AMD. Invest Ophthalmol Vis Sci. 2008;49:2848–2855. doi: 10.1167/iovs.07-1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordgaard CL, Berg KM, Kapphahn RJ, Reilly C, Feng X, Olsen TW, Ferrington DA. Proteomics of the retinal pigment epithelium reveals altered protein expression at progressive stages of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2006;47:815–22. doi: 10.1167/iovs.05-0976. [DOI] [PubMed] [Google Scholar]
- Nowak JZ. Oxidative stress, polyunsaturated fatty acids-derived oxidation products and bisretinoids as potential inducers of CNS diseases: focus on age-related macular degeneration. Pharmacol Rep. 2013;65:288–304. doi: 10.1016/s1734-1140(13)71005-3. [DOI] [PubMed] [Google Scholar]
- O’Day WT, Young RW. Rhythmic daily shedding of outer segment membranes by visual cells in the goldfish. J Cell Biol. 1978;76:593–604. doi: 10.1083/jcb.76.3.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Matsui K. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog Retin Eye Res. 2011;30:217–238. doi: 10.1016/j.preteyeres.2011.02.004. [DOI] [PubMed] [Google Scholar]
- Paimela T, Hyttinen JM, Viiri J, Ryhänen T, Karjalainen RO, Salminen A, Kaarniranta K. Celastrol regulates innate immunity response via NF-κB and Hsp70 in human retinal pigment epithelial cells. Pharmacol Res. 2011;64:501–508. doi: 10.1016/j.phrs.2011.05.027. [DOI] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock 180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. [DOI] [PubMed] [Google Scholar]
- Park SY, Kang KB, Thapa N, Kim SY, Lee SJ, Kim IS. Requirement of adaptor protein GULP during stabilin-2 mediated cell corpse engulfment. J Biol Chem. 2008;283:10593–10600. doi: 10.1074/jbc.M709105200. [DOI] [PubMed] [Google Scholar]
- Parmeggiani F, Romano MR, Costagliola C, Semeraro F, Incorvaia C, D’Angelo S, Perri P, De Palma P, De Nadai K, Sebastiani A. Mechanism of inflammation in age-related macular degeneration. Mediators Inflamm. 2012;2012:546786. doi: 10.1155/2012/546786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penfold PL, Madigan MC, Gillies MC, Provis JM. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res. 2001;20:385–414. doi: 10.1016/s1350-9462(00)00025-2. [DOI] [PubMed] [Google Scholar]
- Pierpaoli EV. The role of Hsp70 in age-related diseases and the prevention of cancer. Ann NYA Sci. 2005;1057:206–219. doi: 10.1196/annals.1356.014. [DOI] [PubMed] [Google Scholar]
- Piippo N, Korkmaz A, Hytti M, Kinnunen K, Salminen A, Atalay M, Kaarniranta K, Kauppinen A. Decline in cellular clearance systems induces inflammasome signaling in human ARPE-19 cells. Biochim Biophys Acta. 2014;1843:3038–3046. doi: 10.1016/j.bbamcr.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Pollanck LJ. Culling sick mitochondria from the herd. J Cell Biol. 2010;191:1225–1227. doi: 10.1083/jcb.201011068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozdeyev N, Taylor C, Haque R, et al. Photic regulation of arylakkylamine N-acetyltransferase binding to 14-3-3 prtoeins in retinal photoreceptor cells. J Neurosci. 2006;26:9153–9161. doi: 10.1523/JNEUROSCI.1384-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radu RA, Hu J, Yuan Q, Welch DL, Makshanoff J, Lloyd M, McMullen S, Travis GH, Bok D. Complement system dysregulation and inflammation in the retinal pigment epithelium of a mouse model for Stargardt macular degeneration. J Biol Chem. 2011;286:18593–601. doi: 10.1074/jbc.M110.191866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragauskaite L, Heckathorn RC, Gaillard ER. Environmental effects on the photochemistry of A2-E, a component of human retinal lipofuscin. Photochem Photobiol. 2001;74:483–488. doi: 10.1562/0031-8655(2001)074<0483:eeotpo>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Rakoczy PE, Lai CM, Baines M, Di Grandi S, Fitton JH, Constable IJ. Modulation of cathepsin D activity in retinal pigment epithelial cells. Bioche J. 1997;324:935–940. doi: 10.1042/bj3240935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends Biochem Sci. 1999;21:267–71. [PubMed] [Google Scholar]
- Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. Int J Cell Biol. 2012:219625. doi: 10.1155/2012/219625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinheckel T, Sitte M, Ullrich KO, Kuckelkorn U, Davies KJA, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335:637–643. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reszka K, Eldred GE, Wang RH, Chignell C, Dillon J. The photochemistry of human retinal lipofuscin as studied by EPR. Photochem Photobiol. 1995;62:1005–1008. doi: 10.1111/j.1751-1097.1995.tb02400.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Muela N, Koga H, Garcia-Ledo L, de la Villa P, de la Rosa EJ, Cuervo AM, Boya P. Balance between autophagic pathways preserves retinal homeostasis. Aging Cell. 2013;12:478–88. doi: 10.1111/acel.12072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov V, Dotsch V, Johansen T, Kirkin V. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014;53:167–178. doi: 10.1016/j.molcel.2013.12.014. [DOI] [PubMed] [Google Scholar]
- Rozanowska M, Korytowski W, Rozanowski B, Skumatz C, Boulton ME, Burke JM, Sarna T. Photoreactivity of aged human RPE melanosomes: a comparison with lipofuscin. Invest Ophthalmol Vis Sci. 2002;43:2088–2096. [PubMed] [Google Scholar]
- Rozanowski B, Burke JM, Boulton ME, Sarna T, Rozanowska M. Human RPE melanosomes protect from photosensitized and iron-mediated oxidation but become pro-oxidant in the presence of iron upon photodegradation. Invest Ophthalmol Vis Sci. 2008;49:2838–2847. doi: 10.1167/iovs.08-1700. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- Ruggiero L, Finnemann SC. Rhythmicity of the retinal pigment epithelium. In: Tosini G, Iuvonne PM, McMahon DG, editors. The Retina and Circadian Rhythms, Springer Series in Vision Research. Springer press; 2014. [Google Scholar]
- Ryhänen T, Hyttinen JM, Kopitz J, Rilla K, Kuusisto E, Mannermaa E, Viiri J, Holmberg CI, Immonen I, Meri S, Parkkinen J, Eskelinen EL, Uusitalo H, Salminen A, Kaarniranta K. Crosstalk between Hsp70 molecular chaperone, lysosomes and proteasomes in autophagy-mediated proteolysis in human retinal pigment epithelial cells. J Cell Mol Med. 2009;13:3616–3631. doi: 10.1111/j.1582-4934.2008.00577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryhänen T, Viiri J, Hyttinen JM, Uusitalo H, Salminen A, Kaarniranta K. Influence of Hsp90 and HDAC inhibition and tubulin acetylation on perinuclear protein aggregation in human retinal pigment epithelial cells. J Biomed Biotechnol. 2011:798052. doi: 10.1155/2011/798052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Decatur J, Nakanishi K, Eldred KE. Ocular age pigment “A2-E”: an unprecedented pyridinium bisretinoid. Journal of the American Chemistry Society. 1996;118:1559–1560. [Google Scholar]
- Salminen A, Kaarniranta K. Regulation of the aging process by autophagy. Trends Mol Med. 2009;15:217–224. doi: 10.1016/j.molmed.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Sandoval H, Thiagarajan P, Dasgupta SK, Shumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in augophagic maturation of erythroid cells. Nature. 2008;454:232–5. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarna T, Burke JM, Korytowski W, Rozanowska M, Skumatz CM, Zareba A, Zareba M. Loss of melanin from human RPE with aging: possible role of melanin photooxidation. Exp Eye Res. 2003;76:89–98. doi: 10.1016/s0014-4835(02)00247-6. [DOI] [PubMed] [Google Scholar]
- Scholl HP, Charbel Issa P, Walier M, Janzer S, Pollok-Kopp B, Borncke F, Fritsche LG, Chong NV, Fimmers R, Wienker T, Holz FG, Weber BH, Oppermann M. Systemic complement activation in age-related macular degeneration. PLoS One. 2008;3:e2593. doi: 10.1371/journal.pone.0002593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuld NJ, Hussong SA, Kapphahn RJ, Lehmann U, Roehrich H, Rageh A, Heuss N, Gregerson DS, Ferrington DA. Immunoproteasome deficiency protects the retina after optic nerve crush. PLOS One. 2015 doi: 10.1371/journal.pone.0126768. (in revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nature Rev. 2014;13:889–903. doi: 10.1038/nrd4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparrow J. Bisretinoids of RPE lipofuscin: Trigger for complement activation in age-related macular degeneration. Adv Exp Med Biol. 2010;703:63–74. doi: 10.1007/978-1-4419-5635-4_5. [DOI] [PubMed] [Google Scholar]
- Sparrow JR, Parish CA, Hashimoto M, Nakanishi K. A2E, a lipofuscin fluorophore, in human retinal pigmented epithelial cells in culture. Invest Ophthalmol Vis Sci. 1999;40:2988–2995. [PubMed] [Google Scholar]
- Sparrow JR, Hicks D, Hamel CP. The retinal pigment epithelium in health and disease. Curr Mol Med. 2010;10:802–823. doi: 10.2174/156652410793937813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson LM, Miller BC, Ng A, Eisenberg J, Zhao Z, Cadwell K, Graham DB, Mizushima NN, Xavier R, Virgin HW, Swat W. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–35. doi: 10.4161/auto.5.5.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su H, Wang X. P62 stages an interplay between the ubiquitin-proteasome system and autophagy in the heart of defense against proteotoxic stress. Trends Cardiovasc Med. 2011;21:224–228. doi: 10.1016/j.tcm.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subrizi A, Toropainen E, Ramsay E, Airaksinen AJ, Kaarniranta K, Urtti A. Oxidative Stress Protection by Exogenous Delivery of rhHsp70 Chaperone to the Retinal Pigment Epithelium (RPE), a Possible Therapeutic Strategy Against RPE Degeneration. Pharm Res. 2015;32:211–221. doi: 10.1007/s11095-014-1456-6. [DOI] [PubMed] [Google Scholar]
- Sundelin S, Wihlmark U, Nilsson SE, Brunk UT. Lipofuscin accumulation in cultured retinal pigment epithelial cells reduces their phagocytic capacity. Curr Eye Res. 1998;17:851–857. [PubMed] [Google Scholar]
- Suzuki K, Kirisako T, Kamada Y, Mizushima N, Noda T, Ohsumi Y. The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 2001;20:5971–5981. doi: 10.1093/emboj/20.21.5971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenning S, Johansen T. Selective autophagy. Essays Biochem. 2013;55:79–92. doi: 10.1042/bse0550079. [DOI] [PubMed] [Google Scholar]
- Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, Cho WG, Kaneko H, Fowler BJ, Bogdanovich S, Albuquerque RJ, Hauswirth WW, Chiodo VA, Kugel JF, Goodrich JA, Ponicsan SL, Chaudhuri G, Murphy MP, Dunaief JL, Ambati BK, Ogura Y, Yoo JW, Lee DK, Provost P, Hinton DR, Núñez G, Baffi JZ, Kleinman ME, Ambati J. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149:847–859. doi: 10.1016/j.cell.2012.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terluk MR, Kapphahn RJ, Soukup LM, Gong H, Gallardo C, Montezuma SR, Ferrington DA. Investigating mitochondria as a target for treating age-related macular degeneration. J Neuroscience. 2015;35:7304–7311. doi: 10.1523/JNEUROSCI.0190-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschernutter M, Jenkins SA, Waseem NH, Saihan Z, Holder GE, Bird AC, Bhattacharya SS, Ali RR, Webster AR. Clinical characterization of a family with retinal dystrophy caused by mutation in the Mertk gene. Br J Ophthalmol. 2006;90:718–723. doi: 10.1136/bjo.2005.084897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng WA, Thein T, Kinnunen K, Lashkari K, Gregory MS, D’Amore PA, Ksander BR. NLRP3 Inflammasome Activation in Retinal Pigment Epithelial Cells by Lysosomal Destabilization: Implications for Age-Related Macular Degeneration. Invest Ophthalmol Vis Sci. 2013;54:110–120. doi: 10.1167/iovs.12-10655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–46. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valapala M, Edwards M, Hose S, Grebe R, Bhutto IA, Cano M, Berger T, Mak TW, Wawrousek E, Handa JT, Lutty GA, Samuel Zigler J, Jr, Sinha D. Increased Lipocalin-2 in the retinal pigment epithelium of Cryba1 cKO mice is associated with a chronic inflammatory response. Aging Cell. 2014a;13:1091–1094. doi: 10.1111/acel.12274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valapala M, Wilson C, Hose S, Bhutto IA, Grebe R, Dong A, Greenbaum S, Gu L, Sengupta S, Cano M, Hackett S, Xu G, Lutty GA, Dong L, Sergeev Y, Handa JT, Campochiaro P, Wawrousek E, Zigler JS, Jr, Sinha D. Lysosomal-mediated waste clearance in retinal pigment epithelial cells is regulated by CRYBA1/betaA3/A1-crystallin via V-ATPase-MTORC1 signaling. Autophagy. 2014b;10:480–496. doi: 10.4161/auto.27292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaft TL, Mooy CM, de Bruijn WC, de Jong PT. Early stages of age-related macular degeneration: an immunofluorescence and electron microscopy study. Br J Ophthalmol. 1993;77:657–661. doi: 10.1136/bjo.77.10.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D’Agostino VG, Govoni S, Pascale A, Agostini H, Petrovski G, Salminen A, Kaarniranta K. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One. 2013;8:e69563. doi: 10.1371/journal.pone.0069563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viiri J, Hyttinen JM, Ryhanen T, Rilla K, Paimela T, Kuusisto E, Siitonen A, Urtti A, Salminen A, Kaarniranta K. P62/sequestosome 1 as a Regulator of Proteasome Inhibitor-Induced Autophagy in Human Retinal Pigment Epithelial Cells. Mol Vis. 2010;16:1399–1414. [PMC free article] [PubMed] [Google Scholar]
- Vives-Bauza C, Anand M, Shiraz AK, Magrane J, Gao J, Vollmer-Snarr HR, Manfredi G, Finnemann SC. The age lipid A2E and mitochondrial dysfunction synergistically impair phagocytosis by retinal pigment epithelial cells. J Biol Chem. 2008;283:24770–24780. doi: 10.1074/jbc.M800706200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AL, Lukas TJ, Yuan M, Du N, Tso MO, Neufeld AH. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009;4:e4160. doi: 10.1371/journal.pone.0004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ohno-Matsui K, Yoshida T, Shimada N, Ichinose S, Sato T, Mochizuki M, Morita I. Amyloid-beta up-regulates complement factor B in retinal pigment epithelial cells through cytokines released from recruited macrophages/microglia: Another mechanism of complement activation in age-related macular degeneration. J Cell Physiol. 2009;220:119–128. doi: 10.1002/jcp.21742. [DOI] [PubMed] [Google Scholar]
- Wang L, Clark ME, Crossman DK, Kojima K, Messinger JD, Mobley JA, Curcio CA. Abundant lipid and protein components of drusen. PLoS One. 2010;5:e10329. doi: 10.1371/journal.pone.0010329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, Cano M, Brandstatter H, Tsimikas S, Skerka C, Superti-Furga G, Handa JT, Zipfel PF, Witztum JL, Binder CJ. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76–81. doi: 10.1038/nature10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcup SM, Sodhi A, Atkinson JP, Holers M, Sinha D, Rohrer B, Dick AD. The role of the immune response in age-related macular degeneration. Int J Inflam. 2013:348092. doi: 10.1155/2013/348092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DS, Fisher SK. Prevention of rod disc shedding by detachment from the retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1987;28:184–187. [PubMed] [Google Scholar]
- Wolkow N, Song Y, Wu TD, Qian J, Guerquin-Kern JL, Dunaief JL. Aceruloplasminemia: retinal histopathologic manifestations and iron-mediated melanosome degradation. Arch Ophthalmol. 2011;129:1466–1474. doi: 10.1001/archophthalmol.2011.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong E, Cuervo AM. Integration of clearance mechanisms: the proteasome and autophagy. Cold Spring Harb Perspect Biol. 2010;12:a006734. doi: 10.1101/cshperspect.a006734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Tibrewal N, Birge RB. Phosphatidylserine recognitionby phagocytes: a view to a kill. Trends Cell Biol. 2006;16:189–197. doi: 10.1016/j.tcb.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Forrester JV. Para-inflammation in the aging retina. Prog Retin Eye Res. 2009;28:348–368. doi: 10.1016/j.preteyeres.2009.06.001. [DOI] [PubMed] [Google Scholar]
- Xu H, Chen M, Manivannan A, Lois N, Forrester JV. Age-dependent accumulation of lipofuscin in perivascular and subretinal microglia in experimental mice. Aging Cell. 2008;7:58–68. doi: 10.1111/j.1474-9726.2007.00351.x. [DOI] [PubMed] [Google Scholar]
- Young RW. The renewal of photoreceptor cell outer segments. J Cell Biol. 1967;33:61–72. doi: 10.1083/jcb.33.1.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW. The daily rhythm of shedding and degradation of cone outer segment membranes in the lizard retina. J Ultrastruct Res. 1977;61:172–185. doi: 10.1016/s0022-5320(77)80084-1. [DOI] [PubMed] [Google Scholar]
- Young RW. The daily rhythm of shedding and degradation of rod and cone outer segment membranes in the chick retina. Invest Ophthalmol Vis Sci. 1978;17:105–116. [PubMed] [Google Scholar]
- Young RW, Bok D. Participation of the retinal pigment epithelium in the rod outer segment renewal process. J Cell Biol. 1969;42:392–403. doi: 10.1083/jcb.42.2.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RW, Droz B. The renewal of protein in retinal rods and cones. J Cell Biol. 1968;39:169–184. doi: 10.1083/jcb.39.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaiss DM, Bekker CP, Gröne A, Lie BA, Sijts AJ. Proteasome immunosubunits protect against the development of CD8 T cell-mediated autoimmune diseases. J Immunol. 2011;187:2302–2309. doi: 10.4049/jimmunol.1101003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Roychoudhury J, Doggett TA, Apte RS, Ferguson TA. Age-dependent changes in FasL (CD95L) modulate macrophage function in a model of age-related macular degeneration. Invest Ophthalmol Vis Sci. 2013;54:5321–5331. doi: 10.1167/iovs.13-12122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Jang YP, Kim SR, Sparrow JR. Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A. 2006;103:16182–16187. doi: 10.1073/pnas.0604255103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Kim SR, Westlund BS, Sparrow JR. Complement activation by bisretinoid constituents of RPE lipofuscin. Invest Ophthalmol Vis Sci. 2009;50:1392–1399. doi: 10.1167/iovs.08-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Yu X. Phagosome maturation during the removal of apoptotic cells: receptors lead the way. Trends Cell Biol. 2008;18:474–485. doi: 10.1016/j.tcb.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zigler JS, Jr, Zhang C, Grebe R, Sehrawat G, Hackler L, Jr, Adhya S, Hose S, McLeod DS, Bhutto I, Barbour W, Parthasarathy G, Zack DJ, Sergeev Y, Lutty GA, Handa JT, Sinha D. Mutation in the βA3/A1-crystallin gene impairs phagosome degradation in the retinal pigmented epithelium of the rat. J Cell Sci. 2011;124:523–531. doi: 10.1242/jcs.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]