

Abstract
Oxidative stress is a commonly cited mechanism of toxicity of environmental agents. Ubiquitous environmental chemicals such as the diesel exhaust component 1,2-naphthoquinone (1,2-NQ) induce oxidative stress by redox cycling, which generates hydrogen peroxide (H2O2). Cysteinyl thiolate residues on regulatory proteins are subjected to oxidative modification by H2O2 in physiological contexts and are also toxicological targets of oxidant stress induced by environmental contaminants. We investigated whether exposure to environmentally relevant concentrations of 1,2-NQ can induce H2O2-dependent oxidation of cysteinyl thiols in regulatory proteins as a readout of oxidant stress in human airway epithelial cells. BEAS-2B cells were exposed to 0–1000 μM 1,2-NQ for 0–30 min, and levels of H2O2 were measured by ratiometric spectrofluorometry of HyPer. H2O2-dependent protein sulfenylation was measured using immunohistochemistry, immunoblotting, and isotopic mass spectrometry. Catalase overexpression was used to investigate the relationship between H2O2 generation and protein sulfenylation in cells exposed to 1,2-NQ. Multiple experimental approaches showed that exposure to 1,2-NQ at concentrations as low as 3 μM induces H2O2-dependent protein sulfenylation in BEAS-2B cells. Moreover, the time of onset and duration of 1,2-NQ-induced sulfenylation of the regulatory proteins GAPDH and PTP1B showed significant differences. Oxidative modification of regulatory cysteinyl thiols in human lung cells exposed to relevant concentrations of an ambient air contaminant represents a novel marker of oxidative environmental stress.

INTRODUCTION
A number of epidemiological and toxicological studies conducted in recent years have shown an association between exposure to diesel exhaust and the incidence of cardiovascular and respiratory morbidity and mortality.1–6 Diesel exhaust is a complex mixture of gaseous and particulate components, including prooxidant organic compounds such as quinones.7 Quinones, as exemplified by 1,2-naphthoquinone (1,2-NQ), are toxic via two mechanisms of action. 1,2-NQ can form covalent adducts with macromolecules through Michael addition.8–10 It can also undergo redox cycling reactions to form reactive oxygen species (ROS) such as hydrogen peroxide (H2O2) that contribute to oxidative stress.11,12 Investigations to date have predominantly focused on the effect of 1,2-NQ adduction, while the biological ramifications of 1,2-NQ-induced oxidative stress have yet to be fully investigated.
Oxidative stress has been identified as a mechanistic feature of a broad range of environmental exposures.13 Evidence of environmental oxidative stress typically involves the analysis of the oxidant-damaged biomolecules, such as the oxidation of protein thiols to sulfonates (Figure 1) as well as the formation of ROS such as H2O2. However, there is a growing appreciation that intracellular H2O2 is a closely regulated second messenger with pivotal roles in cellular processes ranging from the regulation of cytoskeletal function to bioenergetics and signaling.14–18 Specifically, H2O2 has been shown to hydroxylate cysteinyl thiols to form protein sulfenic acids. Sulfenylation of cysteines is now regarded as a critical step in the formation of inter- and intramolecular disulfide bonds as well as the formation of mixed disulfides with glutathione (Figure 1). Importantly, these formed disulfides can be reduced to the basal thiol level through the activity of proteins like glutaredoxin or thioredoxin, supporting the role of thiol oxidation as a hub of redox signaling.15,18 The oxidation of protein cysteinyls to sulfenates has been directly linked to the regulation of signaling and metabolic processes.19–25
Figure 1.
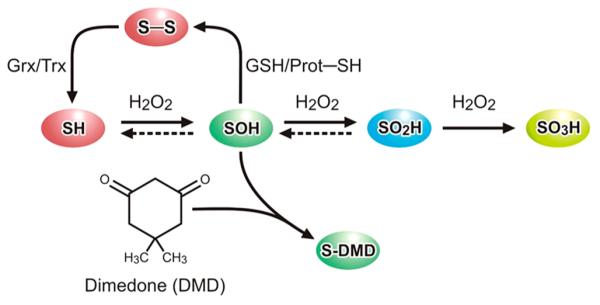
Schematic of cysteinyl-thiol post-translational modifications. A dashed line indicates the possible reversibility of thiol oxidation. Dimedone (shown) and dimedone-like molecules, including DAz-2 and DYn-2, specifically and irreversibly label sulfenic acids (SOH). Glutaredoxin (Grx) and thioredoxin (Trx) reduce disulfide bonds or mixed disulfide bonds with glutathione to the thiol. The continual presence of H2O2 hyperoxidizes the sulfenic acid to the sulfinic (SO2H) and sulfonic (SO3H) acids, which are largely irreversible under physiological conditions.
The inventory of cellular proteins known to be sulfenylated, recently termed the “sulfenome”, continues to expand with the development of new techniques and analytical strategies,26–29 yet to the best of our knowledge, the effect of an environmental exposure on protein sulfenylation has not been investigated. We report here that exposure to 1,2-NQ at concentrations that are theoretically achievable in real world scenarios12 induces sulfenylation of regulatory proteins in human airway epithelial cells (HAEC).
MATERIALS AND METHODS
Reagents
Tissue culture media and supplements were purchased from Lonza (Walkersville, MD). Phenol red-free keratinocyte basal medium (KBM) was acquired from Cell Applications, Inc. (San Diego, CA). Adenoviral vectors were obtained from the Gene Therapy Center Virus Vector Core Facility (University of North Carolina at Chapel Hill). The following chemicals were obtained from Sigma-Aldrich (St. Louis, MO): hydrogen peroxide (H2O2), 1,2-naphthoquinone (1,2-NQ), Me2SO, dimedone, DTT, tert-butyl 2,2,2-trichloroacetimidate (TBTA), copper sulfate (Cu2SO4), and sodium ascorbate (SA). Biotin azide (PEG4 carboxamide-6-azidohexanyl biotin) was obtained through Life Technologies (Grand Island, NY). Basic laboratory supplies were obtained from Fisher Scientific (Raleigh, NC).
Cell Culture
SV40 large T antigen-transformed HAEC (BEAS-2B, subclone S6, passage 54–60) were cultured as previously described30 in keratinocyte growth medium (KGM). Cells at 80% confluence were deprived of growth factors overnight by changing cell medium to KBM. Cells were exposed to KBM containing H2O2 or 1,2-NQ (dissolved in Me2SO, final Me2SO concentration ratio of <1:1000) at the indicated concentrations for 10 min or as otherwise noted. Cells were then quickly rinsed with PBS and then labeled in KBM with 5 mM dimedone or a dimedone analogue (DYn-2 or DAz-2) with Me2SO at a 1:200 dilution for 1 h.
Viral Transduction
The plasmid for the genetically encoded H2O2 sensor, HyPer, was purchased from Evrogen (Axxora, Farmingdale, NY). The genetically encoded pH sensor, SypHer, was created as described by Poburko et al.31 through a single-point Cys199Ser mutation of HyPer. Both plasmids were introduced into lentiviral vectors as described previously.12 Stable expression of Hyper and SyPher in the cytosolic compartment of BEAS-2B cells was accomplished using lentiviral transduction. For cytosolic catalase overexpression, cells were transduced with an adenoviral vector encoding human catalase driven by a cytomegalovirus promoter at a MOI of 500 for 4 h, followed by a 1 day incubation in KGM.32 The pH-specific fluorogenic sensor pHred, created by the laboratory of Yellen,33 was obtained as a construct through Addgene (Cambridge, MA) for expression in BEAS-2B cells via transient transfection of 1–2 μg of plasmid DNA using the suggested X-tremeGENE 9 protocol (Roche Applied Science, Indianapolis, IN).
Detection of Intracellular Sulfenylation
Staudinger Ligation
Cells labeled with DAz-2 underwent Staudinger ligation for fluorescent detection utilizing the recommended protocol of the Sulfenylated Protein Cell-Based Dectection Kit (Cayman Chemical, Ann Arbor, MI). Labeled cells were then detected by fluorescence microscopy using a Nikon Eclipse C1si instrument with laser excitation at 488 nm and 525/30 nm emission with identical laser settings. Images were then imported into ImageJ (National Institutes of Health, Bethesda, MD) for Lookup Table editing to enhance the visual determination of fluorescence by converting images to 16-bit monochromatic images, implementing Green Lookup Table edited to an interpolated 4 color Table (0 Green, 150 Green, 175 Green, 255 Green) followed by a median filter with a 2.0 pixel radius.
Immunoblotting
Cells labeled with dimedone were washed three times with ice-cold PBS and lysed in a mild detergent buffer [1% NP40, 150 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 5 mM DTT supplemented with Calbiochem protease inhibitor cocktail set III (EMD Millipore, Temecula, CA)] for 20 min and then centrifuged at 4 °C and 12000g for 10 min. The supernatant was collected and normalized for protein concentration via the Bradford assay. Samples were prepared for Western blotting with 4× Laemmli Sample Buffer and boiled for 10 min before being loaded into Mini-PROTEAN TGX Precast Gels (Bio-Rad, Hercules, CA) alongside Precision Plus Protein Kaleidoscope Standards (Bio-Rad) and then gel electrophoresed for size separation. Gels were transferred using the Trans-Blot Turbo Transfer System onto nitrocellulose membranes (Bio-Rad). Membranes were then blocked with 5% milk in TBST for 1 h at room temperature followed by incubation with the primary antibody overnight at 4 °C and then secondary antibody for 1 h at room temperature. The following antibodies were used: anti-sulfenic acid-modified cysteine (2-Thiodimedone-Specific Ig) antibody (Millipore) and anti-GAPDH (6C5), anti-catalase (A-7), goat anti-mouse IgG-HRP, and goat anti-rabbit IgG-HRP (all from Santa Cruz, Dallas, TX). After antibody incubation, membranes were set in Clarity Western ECL Blotting Substrate for 5 min followed by detection with a LAS-3000 FujiFilm Imager.
Copper-Catalyzed Azide Alkyne Cycloaddition
Cells labeled with DYn-2, which was prepared as described by Paulsen et al.,22 were washed three times with ice-cold PBS, then lysed with mild detergent buffer described in Immunoblotting for 20 min, and centrifuged at 4 °C and 12000g for 10 min. The protein supernatant was normalized to 1.5 mg/mL and precleared of endogenous biotin by agitation in a 150 μL slurry of Pierce NeutrAvidin agarose (N-agarose, Life Technologies). Cleared samples were labeled with biotin using copper-catalyzed azide alkyne cycloaddition (CuAAC) by agitation for 1 h in a buffer containing the following (final concentrations): biotin azide (0.2 mM), TBTA (0.1 mM), Cu2SO4 (1.0 mM), and SA (1.0 mM). An aliquot of each sample was immunoblotted for total sulfenylation probed against Pierce High Sensitivity NeutrAvidin-HRP (N-HRP, Life Technologies). The remaining post-CuAAC sample was separated into equal aliquots for immunoprecipitation overnight at 4 °C with anti-GAPDH (6C5) (Santa Cruz) or anti-PTP1B (H-135) (Santa Cruz) followed by rotation for 2 h with Protein A-agarose. Samples were then prepared for immunoblotting and probed against N-HRP (Life Technologies), and anti-GAPDH (6C5) or anti-PTP1B (H-135) (both from Santa Cruz).
Detection of Recombinant Protein Sulfenylation
Deuterated dimedone (d6-dimedone) and iodo-dimedone (I-dimedone) were synthesized as described by Seo and Carroll.34 Recombinant GAPDH from rabbit muscle (Sigma-Aldrich) was reduced in a solution of Tris-HCl (50 mM, pH 7.4) with 10 mM DTT on ice for 30 min and then purified with Micro Bio-Spin Columns Bio-Gel P-6 (Bio-Rad) before use in sulfenylation assays. GAPDH (25 μM) was stimulated with a range of molar equivalents of H2O2 or 1,2-NQ dissolved in Tris-HCl (50 mM, pH 7.4) in the presence of 10 mM d6-dimedone. Samples were then reduced with DTT, isolated with P-6 column, and labeled with 20 mM I-dimedone. Samples of modified GAPDH were digested at 37 °C overnight using Trypsin Gold (Promega, Madison, WI) and RapiGest surfactant (Waters Corp., Milford, MA) according to the manufacturer’s specifications. The resulting mixture was analyzed for the presence of modified peptides by injection onto an Agilent (Milford, MA) 1200 HPLC system coupled to an Agilent 6520 Accurate Mass Q-TOF mass spectrometer. Peptides were separated on a 150 mm × 2.1 mm Agilent PLRP-S 5 μm analytical column using a gradient of 0.1% formic acid in water and 0.1% formic acid in acetonitrile at a flow rate of 200 μL/min, and data were collected in positive mode while scanning from m/z 100 to 3200.
Live Cell Imaging
Immediately before exposure, HyPer- or SypHer-expressing cells were placed in KBM without phenol red. Fluorescence in cell cultures was imaged using a Nikon Eclipse C1si spectral confocal imaging system under illumination with 404, 488, or 561 nm primary laser lines (Nikon Instruments Corp., Melville, NY). Sequential scans of each laser line were performed at a frequency of 60 s with 10 cells expressing the biosensor in the field of view and results calculated as a ratio of the respective 525/30 nm emission for the 404 and 488 nm excitation of each sensor. Baseline fluorescence was established for 5 min prior to the addition of 0–10 μM 1,2-NQ. To normalize for variability in the dynamic range of the sensors expressed in individual cells, 100 μM H2O2 was added at min 30, 1 mM H2O2 was added at min 33, and 5 mM DTT was added at min 35. Data were expressed normalized to the maximal sensor response (percent oxidized HyPer) or as raw ratiometric values normalized to baseline. HyPer cells expressing pHred were analyzed similarly but with an additional excitation at 561 nm allowing for a ratiometric analysis of pH between the 404 and 561 nm exciations and 605/75 nm emission.
Statistical Analyses
All imaging data were quantified using NIS-Elements AR software (Nikon). Data are expressed as means ± SEM of at least three separate experiments. Statistical significance (p < 0.05) of immunoblot results was determined through one-way ANOVA with Dunnett’s post-test. PRISM (Graphpad Software, La Jolla, CA) was used for statistical analyses.
RESULTS
Exposure to 1,2-Naphthoquinone Induces Protein Sulfenylation in BEAS-2B Cells
Dimedone is a cell permeable molecule that can be used to label sulfenic acids specifically and irreversibly (Figure 1). A number of dimedone analogues have been generated to meet a range of analytical goals.35 We used an azide-based dimedone derivative, DAz-2, to biotinylate protein sulfenic acids using a commercially available assay that allows for their detection as a fluorescent readout in fixed BEAS-2B cells exposed to 3–100 μM 1,2-NQ. As shown in Figure 2A, exposure of BEAS-2B cells to concentrations of 1,2-NQ as low as 3 μM for 10 min resulted in a marked increase in the concentration of protein sulfenylation, the magnitude of which approximated that induced by exposure to 1000 μM H2O2. In accord with our previous report,12 exposure of BEAS-2B cells to 0–100 μM 1,2-NQ for 10 min did not result in cytotoxicity.
Figure 2.

1,2-Naphthoquinone induces intracellular protein sulfenylation. (A) BEAS-2B cells were exposed to H2O2 (0–1000 μM) or 1,2-NQ (0–100 μM) for 10 min and labeled with DAz-2 followed by conjugation to FITC by Staudinger ligation. (B) BEAS-2B cells were exposed to the indicated concentration of 1,2-NQ for 10 min and then immunoblotted for sulfenylation using the α-thiodimedone antibody. GAPDH was utilized as a loading control.
To assess the range of proteins sulfenylated in response to 1,2-NQ exposure, we next subjected protein extracts of BEAS-2B cells treated with 3–1000 μM 1,2-NQ for 10 min to immunoblotting using an antibody that detects the dimedone– protein thioether complex.36 The results (Figure 2B) show that 1,2-NQ induces a dose-dependent increase in protein sulfenylation of multiple proteins varying in molecular weight between 37 and 250 kDa.
1,2-Naphthoquinone-Induced Protein Sulfenylation Is Dependent on H2O2
Sulfenylation of proteins can occur by the reaction of peroxynitrite, hypohalous acids, haloamines, and hydroperoxides with cysteine residues.37 We have previously reported that exposure to environmentally relevant concentrations of 1,2-NQ results in elevation of H2O2 levels in BEAS-2B cells.12 We therefore examined the role of H2O2 generation in 1,2-NQ-induced protein sulfenylation in BEAS-2B cells. In agreement with our previous studies using the H2O2 sensor, HyPer, 10 μM 1,2-NQ induced a rapid increase in the level of cytosolic H2O2 (Figure S1). The responsiveness of HyPer to H2O2 is known to be influenced by pH. We, therefore, conducted control experiments using BEAS-2B cells expressing the pH sensor SypHer or pHred, which showed that the HyPer fluorescence intensity changes observed in response to 10 μM 1,2-NQ over a 30 min time period were not attributable to changes in cytosolic pH (Figure S1).
We next examined the role of H2O2 in 1,2-NQ-induced protein sulfenylation by increasing the rate of catabolism of H2O2 by overexpressing catalase in BEAS-2B cells. Catalase overexpression ablated the increase in H2O2 concentration induced by treatment of BEAS-2B cells with either 3 or 10 μM 1,2-NQ throughout the experiments, confirming the efficacy of catalase overexpression as a means to blunt H2O2 levels induced by 1,2-NQ exposure in BEAS-2B cells. Immunoblotting assays showed that catalase overexpression blunted protein sulfenylation induced by treatment of BEAS-2B cells with 3 or 10 μM 1,2-NQ (Figure S2). We then examined the temporal relationship between 1,2-NQ-induced production of H2O2 measured using HyPer and protein sulfenylation levels quantified using immunoblotting. Exposure to 10 μM 1,2-NQ induced a rapid peak in protein sulfenylation that coincided with the sharp rise in cytosolic H2O2 levels reported by HyPer (Figure 3). Protein sulfenylation then appeared to decrease at a rate faster than that observed for H2O2 concentrations, although protein sulfenylation rebounded with 10 μM 1,2-NQ (but not with 3 μM) reaching its highest level at 30 min of exposure. In support of the earlier experiments (Figure S2), BEAS-2B cells overexpressing catalase showed diminished levels of protein sulfenylation induced by exposure to 3 or 10 μM 1,2-NQ (Figure 3). These results established that 1,2-NQ-induced protein sulfenylation depends on the generation of H2O2 in BEAS-2B cells.
Figure 3.

1,2-Naphthoquinone-induced protein sulfenylation is H2O2-dependent. Control BEAS-2B cells (■) and BEAS-2B cells overexpressing catalase (empty blue squares) were exposed to 3 μM (A) or 10 μM (B) 1,2-NQ for the indicated time, labeled with dimedone, and harvested for immunoblotting to detect protein sulfenylation (SOH). Values are presented as means ± SEM (n = 3). An asterisk indicates p < 0.05 compared to the control. Cytosolic H2O2 was monitored in BEAS-2B cells expressing HyPer (●) exposed to 3 μM (A) or 10 μM (B) 1,2-NQ over a time course of 30 min. HyPer-expressing cells overexpressing catalase (empty blue circles) were exposed in the same manner. Intracellular H2O2 levels are reported every minute as an average of the percent maximal oxidation of HyPer induced by 1,2-NQ exposure (n = 3; error bars omitted for the sake of clarity).
1,2-Naphthoquinone-Induced Sulfenylation of Regulatory Proteins
To examine the effect of 1,2-NQ on the sulfenylation of specific proteins of interest in BEAS-2B cells, we used a copper-catalyzed azide–alkyne cycloaddition (CuAAC) labeling strategy to biotinylate protein sulfenic acids, to gain the analytical sensitivity and specificity afforded by the strong avidin–biotin interaction (Figure S3A). CuAAC-based detection of protein sulfenylation in BEAS-2B showed elevations at 1,2-NQ concentrations as low as 1 μM for 10 min (Figure S3B,C). We next examined specific proteins sulfenylated as a result of exposure of BEAS-2B cells to 1,2-NQ.
We tested the effect of 1,2-NQ exposures on the sulfenylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) because of its integral role in cellular bioenergetics,38,39 and of protein tyrosine phosphatase 1B (PTP1B) for its pivotal role in signaling processes.20,40 Levels of sulfenylation of GAPDH and PTP1B were measured in immunoprecipitates of the same protein extract samples prepared from BEAS-2B cells exposed to 10 μM 1,2-NQ using the CuAAC biotinylation protocol. Exposure of BEAS-2B cells to 1,2-NQ induced GAPDH sulfenylation that increased steadily over a 30 min exposure time course (Figure 4A,C). In contrast, the level of sulfenylated PTP1B in the same samples attained a maximum after exposure for 1 min and decreased rapidly thereafter (Figure 4B,D). The same marked difference in the time courses of 1,2-NQ-induced sulfenylation was observed when GAPDH and PTP1B were immunoblotted from the CuAAC-biotinylated protein fraction (Figure S4).
Figure 4.

1,2-Naphthoquinone-induced sulfenylation of regulatory proteins with differential kinetics. BEAS-2B cells were exposed to 10 μM 1,2-NQ from 0 to 30 min followed by CuAAC. Lysates were subjected to immunoprecipitation using specific antibodies GAPDH (A and C) and PTP1B (B and D). Immunoblots were probed against neutravidin-HRP (N-HRP) and then GAPDH (A) or PTP1B (B). Data are shown as a representative immunoblot of three experiments, quantified respectively as a ratio of the sulfenylated protein to total GAPDH and PTP1B, (C) N-HRP/GAPDH and (D) N-HRP/PTP1B. Values shown as means ± SEM normalized to the unexposed control. An asterisk indicates p < 0.05 compared to the control.
1,2-Naphthoquinone Induces Sulfenylation of the Catalytic Cysteine (150C) in GAPDH
To examine the site specificity of 1,2-NQ-induced sulfenylation, we utilized an isotope labeling strategy devised by Seo and Carroll34 to detect sulfenic groups in trypsin digests of recombinant GAPDH treated with 1,2-NQ in vitro (Figure 5A). The catalytic cysteine (150C) of GAPDH serves as the peroxide-susceptible thiol that becomes sulfenylated upon oxidation, inactivating GAPDH.41 Mass spectrometric analysis of the isotopically coded, dimedone-labeled GAPDH peptides showed maximal sulfenylation of 150C in GAPDH treated with 1.0 molar equiv of 1,2-NQ. In contrast, H2O2 exposure induced increasing sulfenylation of 150C with exposure to up to 2.0 molar equiv (Figure 5B).
Figure 5.

1,2-Naphthoquinone induces sulfenylation of the GAPDH catalytic cysteine. (A) General scheme of isotope-encoded dimedone iododimedone (ICDID) strategy for quantifying sulfenic acids relative to total thiols. Deuterated dimedone (d6-DMD) labels all sulfenic acids, while subsequent treatment with iodo-dimedone (I-DMD) labels all remaining thiols (d0-DMD). Samples are then trypsinized and subjected to QTOF-MS analysis for the d6/d0 ratio of the indicated GAPDH peptide sequence (center) containing 150C. (B) Recombinant GAPDH (25 μM) was incubated with either H2O2 or 1,2-NQ at indicated molar equivalencies and then subjected to ICDID, and values are reported as a ratio of sulfenylated 150C to reduced 150C. Values are presented as means ± SEM (n = 3). An asterisk indicates p < 0.05 compared to the control.
DISCUSSION
Toxicological studies have long equated oxidative stress with the production of ROS and damage to DNA, lipids, and proteins, leading to a loss of function and cell death. However, there are now numerous examples of physiological redox reactions such as reversible cysteine sulfenylation that are involved in pivotal regulatory functions in the cell, from signaling to energy metabolism.17,21,24,42 These processes themselves represent potential targets of oxidant stress induced by xenobiotics. This study demonstrates that exposure to environmentally relevant concentrations of a ubiquitous redox-active environmental pollutant can induce H2O2-dependent protein sulfenylation in a dose- and time-dependent manner.
Although 1,2-NQ toxicity has been thought to predominantly occur through covalent adduction,7 there is evidence to suggest that induction of oxidative stress is a major pathway of 1,2-NQ toxicity. Our previous study implicated 1,2-NQ-induced H2O2 generation in the induction of inflammatory gene expression, suggesting a mechanism of toxicity that does not require the adduction of proteins.12 This study further supports the importance of 1,2-NQ-induced H2O2, as it was shown to induce sulfenylation of thiols on regulatory proteins. Given the critical role of protein sulfenylation in many redox-dependent physiological processes,19,22,43 the inappropriate induction of sulfenylation by 1,2-NQ exposure represents a significant, previously unrecognized mechanism of cellular toxicity. Work by Rhee,20 Tonks,44 and others40 has shown that the activity of protein tyrosine phosphatases (PTP) is redox-regulated by reversible sulfenylation in response to physiological stimuli, and further, PTP activity has been shown to be inhibited in response to 1,2-NQ exposure.8 Of direct relevance to diesel exhaust, our laboratory has shown that DEP exposure inhibits PTP activity, induces a pan-activation of intracellular signaling, and specifically impairs the dephosphorylation of the epidermal growth factor receptor.45 Ongoing work in our laboratory is aimed at examining the relationship among protein sulfenylation, loss of PTP activity, and inflammatory and adaptive gene expression in human airway epithelial cells.
In general, redox-dependent signaling is associated with a classic negative-feedback process as the rapid activation of redox-dependent gene expression also leads to induction of expression of antioxidant mediators.16,46 Our finding of a biphasic response in total protein sulfenylation is in accord with the induction of redox-dependent gene expression observed in anticancer therapies such as ionizing radiation47 and nitric oxide.48,49 The early induction and subsequent decline of protein sulfenylation observed in this study may reflect the regulation of redox-dependent transcriptional activation via a biphasic response of signaling through protein sulfenylation. The second, later increase in protein sulfenylation resulting from 10 μM 1,2-NQ exposure may reflect loss of cell viability caused by prolonged exposure to 1,2-NQ.
GAPDH and PTP1B were selected in this study as examples of regulatory proteins whose activity is known to be controlled through cysteine thiol oxidation50 and are also toxicologically important as targets in cellular bioenergetics and inflammatory signaling, respectively. The differences in the time course of sulfenylation of GAPDH and PTP1B observed in this study were unexpected, as increasing H2O2 levels could be expected to oxidize GAPDH and PTP1B with similar efficacy. One possible explanation for the differential kinetics may be that some proteins become sulfenylated at different rates as a consequence of localized induction of intracellular H2O2 produced under different exposure conditions.11,51 Oxidants, including 1,2-NQ, can also target cysteinyl thiols, most notably glutathione, that usually serve as a “first line” of defense against ROS, allowing GAPDH and PTP1B to be more readily targeted.12,46 Susceptible cysteine such as those in PTP1B and GAPDH that are subject to redox regulation in physiological contexts may be oxidized differentially by reactive electrophiles. Alternatively, these proteins may inherently have different susceptibilities to oxidation due to steric constraints or differences in the pKa of their cysteinyl thiols. In the case of tyrosine phosphatases such as PTP1B, the formation of the sulfenic acid is likely to be an intermediate step in the formation of the more stable sulfenamide20 or to conjugation with glutathione.52 This would be supported by the relatively short duration of detectable PTP1B sulfenylation, as the high specificity of dimedone for sulfenic acids would not detect the sulfenamides, gluthionyl-ethers, or higher oxidation states such as the sulfinic (SO2H) or sulfonic (SO3H) acids.35,53,54 In contrast, sulfenylation of the GAPDH active site cysteine generates a more stable and long-lived species and as such would allow for labeling with dimedone.38,39,55 Additional studies will be needed to elucidate the mechanistic basis for the differential kinetics of GAPDH and PTP1B sulfenylation.
Our observation that, on a molar basis, 1,2-NQ appears to be 100 times more potent than H2O2 in inducing protein sulfenylation may be explained by differences in the targets with which each stimulus preferentially interacts. This would be consistent with reports that the pattern of EGF-stimulated protein sulfenylation is significantly different relative to that induced by H2O2.17,37 H2O2 is subject to catabolism by multiple enzymes, including catalase, glutathione peroxidase, and peroxiredoxin. In addition to 1,2-NQ generation of ROS through redox cycling,11 1,2-NQ also generates H2O2 in mitochondria through uncoupling of the respiratory chain.12 Additionally, our cell-free studies demonstrate the interaction between GAPDH and 1,2-NQ, as proposed by Kumagai and his colleagues,7 is yet another nonenzymatic mechanism through which 1,2-NQ can lead to elevated ROS levels. Thus, multiple sources of ROS may act synergistically to induce a sustained and localized elevation of the level of H2O2 in cells exposed to 1,2-NQ at a rate that overcomes the capacity of antioxidant mechanisms. These considerations would suggest that 1,2-NQ exposure could be more effective in driving sulfenylation than addition of an exogenous bolus of H2O2.
In summary, through the use of multiple analytical approaches, this study provides the first evidence that exposure of a lung epithelial cell line to environmental concentrations of a ubiquitous redox-active pollutant can induce cysteinyl sulfenylation of critical regulatory proteins, a novel biomarker of xenobiotic oxidant stress. Exposure to the ubiquitous environmental oxidant 1,2-NQ induces H2O2-dependent sulfenylation of cysteine residues in proteins, including GAPDH and PTP1B, proteins involved in bioenergetic and signaling regulation, respectively. The work presented in this study shows that protein sulfenylation is a novel readout of oxidant stress induced by exposure to environmental agents.
ACKNOWLEDGMENTS
We express sincere gratitude for insights regarding CuAAC provided by Drs. Daniel Liebler, Simona Codreanu, and Jing Yang. We also thank Dr. Wanda Bodnar and Mr. Leonard Collins of the UNC Biomarker Mass Spectrometry Core for assistance analyzing mass spectrometry samples. We are also grateful for the graphical expertise provided by Mr. John Havel.
Funding
P.A.W. was supported as a predoctoral candidate in part by NIEHS Toxicology Training Grant T32 ES007126 and UNCEPA Training Agreement CR-83515201-0. This research was supported in part by a grant from the National Institute of Environmental Health Sciences (P30ES010126).
ABBREVIATIONS
- 1,2-NQ
1,2-naphthoquinone
- 150C
catalytic cysteine of rabbit glyceraldehyde-3-phophate dehydrogenase
- CuAAC
copper-catalyzed azide alkyne cycloaddition
- Cu2SO4
copper sulfate
- d6-dimedone
deuterated dimedone
- GAPDH
glyceraldehyde-3-phophate dehydrogenase
- HAEC
human airway epithelial cells
- H2O2
hydrogen peroxide
- ICDID
isotope-coded dimedone iodo-dimedone
- I-dimedone
iodo-dimedone
- KBM
keratinocyte basal medium
- KGM
keratinocyte growth medium
- PTP1B
protein tyrosine phosphatase 1b
- ROS
reactive oxygen species
- SA
sodium ascorbate
- TBTA
tert-butyl 2,2,2-trichloroacetimidate
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.chemrestox.5b00424.
Technical controls as well as representative immunoblots and supporting data (PDF)
Notes
The research described in this article has been reviewed by the National Health and Environmental Effects Research Laboratory, U.S. EPA, and approved for publication. The contents of this article should not be construed to represent Agency policy, nor does the mention of trade names or commercial products constitute endorsement or recommendation for use.
The authors declare no competing financial interest.
REFERENCES
- (1).Cosselman KE, M. Krishnan R, Oron AP, Jansen K, Peretz A, Sullivan JH, Larson TV, Kaufman JD. Blood pressure response to controlled diesel exhaust exposure in human subjects. Hypertension. 2012;59:943–948. doi: 10.1161/HYPERTENSIONAHA.111.186593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Krishnan RM, Sullivan JH, Carlsten C, Wilkerson HW, Beyer RP, Bammler T, Farin F, Peretz A, Kaufman JD. A randomized cross-over study of inhalation of diesel exhaust, hematological indices, and endothelial markers in humans. Part. Fibre Toxicol. 2013;10:7. doi: 10.1186/1743-8977-10-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Peretz A, Sullivan JH, Leotta DF, Trenga CA, Sands FN, Allen J, Carlsten C, Wilkinson CW, Gill EA, Kaufman JD. Diesel exhaust inhalation elicits acute vasoconstriction in vivo. Environ. Health Perspect. 2008;116:937–942. doi: 10.1289/ehp.11027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Tong H, Rappold AG, Caughey M, Hinderliter AL, Graff DW, Berntsen JH, Cascio WE, Devlin RB, Samet JM. Cardiovascular effects caused by increasing concentrations of diesel exhaust in middle-aged healthy GSTM1 null human volunteers. Inhalation Toxicol. 2014;26:319–326. doi: 10.3109/08958378.2014.889257. [DOI] [PubMed] [Google Scholar]
- (5).Alexis NE, Carlsten C. Interplay of air pollution and asthma immunopathogenesis: a focused review of diesel exhaust and ozone. Int. Immunopharmacol. 2014;23:347–355. doi: 10.1016/j.intimp.2014.08.009. [DOI] [PubMed] [Google Scholar]
- (6).Ghio AJ, Smith CB, Madden MC. Diesel exhaust particles and airway inflammation. Curr. Opin. Pulm. Med. 2012;18:144–150. doi: 10.1097/MCP.0b013e32834f0e2a. [DOI] [PubMed] [Google Scholar]
- (7).Kumagai Y, Shinkai Y, Miura T, Cho AK. The chemical biology of naphthoquinones and its environmental implications. Annu. Rev. Pharmacol. Toxicol. 2012;52:221–247. doi: 10.1146/annurev-pharmtox-010611-134517. [DOI] [PubMed] [Google Scholar]
- (8).Iwamoto N, Sumi D, Ishii T, Uchida K, Cho AK, Froines JR, Kumagai Y. Chemical knockdown of protein-tyrosine phosphatase 1B by 1,2-naphthoquinone through covalent modification causes persistent transactivation of epidermal growth factor receptor. J. Biol. Chem. 2007;282:33396–33404. doi: 10.1074/jbc.M705224200. [DOI] [PubMed] [Google Scholar]
- (9).Miura T, Kumagai Y. Immunochemical method to detect proteins that undergo selective modification by 1,2-naphthoquinone derived from naphthalene through metabolic activation. J. Toxicol. Sci. 2010;35:843–852. doi: 10.2131/jts.35.843. [DOI] [PubMed] [Google Scholar]
- (10).Takayama N, Iwamoto N, Sumi D, Shinkai Y, Tanaka-Kagawa T, Jinno H, Kumagai Y. Peroxiredoxin 6 is a molecular target for 1,2-naphthoquinone, an atmospheric electrophile, in human pulmonary epithelial A549 cells. J. Toxicol. Sci. 2011;36:817–821. doi: 10.2131/jts.36.817. [DOI] [PubMed] [Google Scholar]
- (11).Shinkai Y, Iwamoto N, Miura T, Ishii T, Cho AK, Kumagai Y. Redox cycling of 1,2-naphthoquinone by thioredoxin1 through Cys32 and Cys35 causes inhibition of its catalytic activity and activation of ASK1/p38 signaling. Chem. Res. Toxicol. 2012;25:1222–1230. doi: 10.1021/tx300069r. [DOI] [PubMed] [Google Scholar]
- (12).Cheng WY, Currier J, Bromberg PA, Silbajoris R, Simmons SO, Samet JM. Linking oxidative events to inflammatory and adaptive gene expression induced by exposure to an organic particulate matter component. Environ. Health Perspect. 2011;120:267–274. doi: 10.1289/ehp.1104055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Braconi D, Bernardini G, Santucci A. Linking protein oxidation to environmental pollutants: redox proteomic approaches. J. Proteomics. 2011;74:2324–2337. doi: 10.1016/j.jprot.2011.06.029. [DOI] [PubMed] [Google Scholar]
- (14).Schwarzlander M, Dick TP, Meye AJ, Morgan B. Dissecting Redox Biology using Fluorescent Protein Sensors. Antioxid. Redox Signaling. 2015 doi: 10.1089/ars.2015.6266. DOI: 10.1089/ars.2015.6266. [DOI] [PubMed] [Google Scholar]
- (15).Poole LB. The basics of thiols and cysteines in redox biology and chemistry. Free Radical Biol. Med. 2015;80:148–157. doi: 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol. 2015;4:180–183. doi: 10.1016/j.redox.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Go YM, Chandler JD, Jones DP. The cysteine proteome. Free Radical Biol. Med. 2015;84:227–245. doi: 10.1016/j.freeradbiomed.2015.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Groitl B, Jakob U. Thiol-based redox switches. Biochim. Biophys. Acta, Proteins Proteomics. 2014;1844:1335–1343. doi: 10.1016/j.bbapap.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Dotsey EY, Jung KM, Basit A, Wei D, Daglian J, Vacondio F, Armirotti A, Mor M, Piomelli D. Peroxide-Dependent MGL Sulfenylation Regulates 2-AG-Mediated Endocannabinoid Signaling in Brain Neurons. Chem. Biol. 2015;22:619–628. doi: 10.1016/j.chembiol.2015.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Lee SR, Kwon KS, Kim SR, Rhee SG. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- (21).Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2014;2:123–139. doi: 10.1016/j.redox.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Paulsen CE, Truong TH, Garcia FJ, Homann A, Gupta V, Leonard SE, Carroll KS. Peroxide-dependent sulfenylation of the EGFR catalytic site enhances kinase activity. Nat. Chem. Biol. 2011;8:57–64. doi: 10.1038/nchembio.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Randall LM, Ferrer-Sueta G, Denicola A. Peroxiredoxins as preferential targets in H2O2-induced signaling. Methods Enzymol. 2013;527:41–63. doi: 10.1016/B978-0-12-405882-8.00003-9. [DOI] [PubMed] [Google Scholar]
- (24).Lo Conte M, Carroll KS. The redox biochemistry of protein sulfenylation and sulfinylation. J. Biol. Chem. 2013;288:26480–26488. doi: 10.1074/jbc.R113.467738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Saurin AT, Neubert H, Brennan JP, Eaton P. Widespread sulfenic acid formation in tissues in response to hydrogen peroxide. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17982–17987. doi: 10.1073/pnas.0404762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Leonard SE, Reddie KG, Carroll KS. Mining the thiol proteome for sulfenic acid modifications reveals new targets for oxidation in cells. ACS Chem. Biol. 2009;4:783–799. doi: 10.1021/cb900105q. [DOI] [PubMed] [Google Scholar]
- (27).Spickett CM, Pitt AR. Protein oxidation: role in signalling and detection by mass spectrometry. Amino Acids. 2012;42:5–21. doi: 10.1007/s00726-010-0585-4. [DOI] [PubMed] [Google Scholar]
- (28).Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang J, Gupta V, Tallman KA, Porter NA, Carroll KS, Liebler DC. Global, in situ, site-specific analysis of protein S-sulfenylation. Nat. Protoc. 2015;10:1022–1037. doi: 10.1038/nprot.2015.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tal TL, Simmons SO, Silbajoris R, Dailey L, Cho SH, Ramabhadran R, Linak W, Reed W, Bromberg PA, Samet JM. Differential transcriptional regulation of IL-8 expression by human airway epithelial cells exposed to diesel exhaust particles. Toxicol. Appl. Pharmacol. 2010;243:46–54. doi: 10.1016/j.taap.2009.11.011. [DOI] [PubMed] [Google Scholar]
- (31).Poburko D, Santo-Domingo J, Demaurex N. Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem. 2011;286:11672–11684. doi: 10.1074/jbc.M110.159962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Erzurum SC, Lemarchand P, Rosenfeld MA, Yoo JH, Crystal RG. Protection of human endothelial cells from oxidant injury by adenovirus-mediated transfer of the human catalase cDNA. Nucleic Acids Res. 1993;21:1607–1612. doi: 10.1093/nar/21.7.1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Tantama M, Hung YP, Yellen G. Imaging intracellular pH in live cells with a genetically encoded red fluorescent protein sensor. J. Am. Chem. Soc. 2011;133:10034–10037. doi: 10.1021/ja202902d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Seo YH, Carroll KS. Quantification of protein sulfenic acid modifications using isotope-coded dimedone and iododimedone. Angew. Chem., Int. Ed. 2011;50:1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- (35).Bachi A, Dalle-Donne I, Scaloni A. Redox proteomics: chemical principles, methodological approaches and biological/biomedical promises. Chem. Rev. 2013;113:596–698. doi: 10.1021/cr300073p. [DOI] [PubMed] [Google Scholar]
- (36).Seo YH, Carroll KS. Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16163–16168. doi: 10.1073/pnas.0903015106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radical Biol. Med. 2008;45:549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- (38).Ishii T, Sunami O, Nakajima H, Nishio H, Takeuchi T, Hata F. Critical role of sulfenic acid formation of thiols in the inactivation of glyceraldehyde-3-phosphate dehydrogenase by nitric oxide. Biochem. Pharmacol. 1999;58:133–143. doi: 10.1016/s0006-2952(99)00060-x. [DOI] [PubMed] [Google Scholar]
- (39).Schmalhausen EV, Muronetz VI, Nagradova NK. Rabbit muscle GAPDH: non-phosphorylating dehydrogenase activity induced by hydrogen peroxide. FEBS Lett. 1997;414:247–252. doi: 10.1016/s0014-5793(97)01044-2. [DOI] [PubMed] [Google Scholar]
- (40).Salmeen A, Barford D. Functions and mechanisms of redox regulation of cysteine-based phosphatases. Antioxid. Redox Signaling. 2005;7:560–577. doi: 10.1089/ars.2005.7.560. [DOI] [PubMed] [Google Scholar]
- (41).Peralta D, Bronowska AK, Morgan B, Doka E, Van Laer K, Nagy P, Grater F, Dick TP. A proton relay enhances H2O2 sensitivity of GAPDH to facilitate metabolic adaptation. Nat. Chem. Biol. 2015;11:156–163. doi: 10.1038/nchembio.1720. [DOI] [PubMed] [Google Scholar]
- (42).Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid. Redox Signaling. 2009;11:997–1014. doi: 10.1089/ars.2008.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Cai Z, Yan LJ. Protein Oxidative Modifications: Beneficial Roles in Disease and Health. J. Biochem. Pharmacol. Res. 2013;1:15–26. [PMC free article] [PubMed] [Google Scholar]
- (44).Schwertassek U, Haque A, Krishnan N, Greiner R, Weingarten L, Dick TP, Tonks NK. Reactivation of oxidized PTP1B and PTEN by thioredoxin 1. FEBS J. 2014;281:3545–3558. doi: 10.1111/febs.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Tal TL, Bromberg PA, Kim Y, Samet JM. Epidermal growth factor receptor activation by diesel particles is mediated by tyrosine phosphatase inhibition. Toxicol. Appl. Pharmacol. 2008;233:382–388. doi: 10.1016/j.taap.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Antelmann H, Helmann JD. Thiol-based redox switches and gene regulation. Antioxid. Redox Signaling. 2011;14:1049–1063. doi: 10.1089/ars.2010.3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Patwardhan RS, Sharma D, Checker R, Thoh M, Sandur SK. Spatio-temporal changes in glutathione and thioredoxin redox couples during ionizing radiation-induced oxidative stress regulate tumor radio-resistance. Free Radical Res. 2015;49:1218–1232. doi: 10.3109/10715762.2015.1056180. [DOI] [PubMed] [Google Scholar]
- (48).Levonen AL, Hill BG, Kansanen E, Zhang J, Darley-Usmar VM. Redox regulation of antioxidants, autophagy, and the response to stress: implications for electrophile therapeutics. Free Radical Biol. Med. 2014;71:196–207. doi: 10.1016/j.freeradbiomed.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Singh S, Gupta AK. Nitric oxide: role in tumour biology and iNOS/NO-based anticancer therapies. Cancer Chemother. Pharmacol. 2011;67:1211–1224. doi: 10.1007/s00280-011-1654-4. [DOI] [PubMed] [Google Scholar]
- (50).Kettenhofen NJ, Wood MJ. Formation, reactivity, and detection of protein sulfenic acids. Chem. Res. Toxicol. 2010;23:1633–1646. doi: 10.1021/tx100237w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Miura T, Shinkai Y, Hirose R, Iwamoto N, Cho AK, Kumagai Y. Glyceraldehyde-3-phosphate dehydrogenase as a quinone reductase in the suppression of 1,2-naphthoquinone protein adduct formation. Free Radical Biol. Med. 2011;51:2082–2089. doi: 10.1016/j.freeradbiomed.2011.09.008. [DOI] [PubMed] [Google Scholar]
- (52).Barrett WC, DeGnore JP, Konig S, Fales HM, Keng YF, Zhang ZY, Yim MB, Chock PB. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- (53).Freeman F, Adesina IT, La JL, Lee JY, Poplawski AA. Conformers of cysteine and cysteine sulfenic acid and mechanisms of the reaction of cysteine sulfenic acid with 5,5-dimethyl-1,3-cyclohexanedione (dimedone) J. Phys. Chem. B. 2013;117:16000–16012. doi: 10.1021/jp409022m. [DOI] [PubMed] [Google Scholar]
- (54).Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, Daniel LW, Poole LB. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Maller C, Schroder E, Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid. Redox Signaling. 2011;14:49–60. doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]