

Abstract
Nitric oxide (NO) is a fascinating and important endogenous free-radical gas with potent antimicrobial, vasodilating, smooth muscle relaxant, and growth factor stimulating effects. However, its wider biomedical applicability is hindered by its cumbersome administration, since NO is unstable especially in biological environments. In this work, to ultimately develop site-specific controlled release vehicles for NO, the NO donor S-nitroso-N-acetyl-D-penicillamine (SNAP) was encapsulated within poly(lactic-co-glycolic acid) 50:50 (PLGA) microspheres by using a solid-in-oil-in-water emulsion solvent evaporation method. The highest payload was 0.56(±0.01) μmol SNAP/mg microspheres. The release kinetics of the donor were controlled by the bioerosion of the PLGA microspheres. By using an uncapped PLGA (Mw = 24,000 – 38,000) SNAP was slowly released for over 10 days, whereas by using the ester capped PLGA (Mw = 38,000 – 54,000) the release lasted for over 4 weeks. The presence of copper ions and/or ascorbate in solution was necessary to efficiently decompose the released NO donor and obtain sustained NO release. It was also demonstrated that light can be used to induce rapid NO release from the microspheres over several hours. SNAP exhibited excellent storage stability when encapsulated in the PLGA microspheres. These new microsphere formulations may be useful for site-specific administration and treatment of pathologies associated with dysfunction in endogenous NO production, e.g. treatment of diabetic wounds, or in diseases involving other biological functions of NO including vasodilation, antimicrobial, anticancer, and neurotransmission.
Graphical Abstract
Introduction
Since its initial discovery as the endothelium-derived relaxing factor in 1987 by Ignarro and co-workers [1], several crucial roles of nitric oxide (NO) have been identified in the body. These include: regulation of blood pressure via its smooth muscle relaxant activity [2], mediating immune response [3–5], controlling cell proliferation [6], modulating apoptosis [7], promoting growth factor-induced angiogenesis [8], accelerating wound healing [9–11], and functioning as a neurotransmitter [12].
Although NO has been shown to have pharmacological activity for many years, such effects are highly dependent on the location of NO production and its concentration in physiological systems [6]. Endothelial cells lining the interior surface of all blood vessels and lymphatic vessels produce an estimated NO surface flux of 0.5–4.1×10−10 mol cm−2 min−1 [7].
The pharmaceutical application of NO is further complicated because of NO’s extreme reactivity, short half-life and associated relatively short diffusional distance [8,9]. The therapeutical application of NO in its native gaseous form is complicated, expensive and very limited. Nitric oxide is, however, used as an inhaled gas at 5–20 ppmV concentrations to reduce pulmonary hypertension in neonates and to improve their gas exchange rates [10].
Beyond inhaled NO, a wide range of NO donor molecules have been studied for NO delivery. The oldest NO donors employed to date are organic nitrates, such as glyceryl trinitrate (GTN) which has been used for more than 100 years for treating angina pectoris [11,12]. However, short term oral application of GTN causes measurable NO release into the body [13], and it has also been reported that the actual action of GTN is unrelated to its bioconversion to NO [14]. Other NO donors such as nitrites, metal ion-NO complexes, diazeniumdiolates (NONOates), N-nitroso compounds, C-nitroso compounds, and S-nitroso (RSNO) compounds [15,16] all require different interactions to produce NO. Their NO release behavior is further complicated by their different sensitivity to changes in light intensity (and wavelength), temperature, trace metal ions and pH. Recently, the two most promising groups of small molecule NO donors are the NONOates and nitrosothiols (RSNOs). Endogenous RSNOs (e.g., S-nitrosocysteine, S-nitrosoglutathione and S-nitrosoalbumin) are also considered as NO storage reservoirs in vivo [17,18].
Several research groups have attempted to develop novel NO delivery and storage systems whereby the NO donors are blended into or covalently attached to macromolecular frameworks. Pulfer et al. prepared 10–50 μm diameter polyethyleneimine microspheres and derivatized them with a NONOate moiety [19]. These microspheres released 194 nmol NO/mg with a half-life of over 66 h under physiological conditions. Jeh et al. encapsulated diazeniumdiloated proline (PROLI/NO) as a small molecule NO donor within biodegradable PLGA and polyethylene oxide-co-lactic acid (PELA) microspheres for inhalation delivery of NO [20]. The PELA particles were found to be better, but the half-life of NO release was only several minutes. Our group prepared fumed silica particles that release NO for several hours by first tethering alkylamines onto the surface of the silica, then converting these amine groups to corresponding N-diazeniumdiolate groups via reaction with NO gas at high pressure [21]. Similarly, Schoenfisch and co-workers formed NONOate moieties on amine functionalized gold [22] and silica [23] nanoparticles, as well polypropyleneimine dendrimers [24] that typically released NO for several hours. The longest release time was achieved by employing O2-protected diazeniumdiolate-modified mesoporous silica nanoparticles, which had a half-life of 3 weeks at pH 7.4 [25]. Yoo et al. encapsulated diethylenetriamine (DETA) NONOate into PLGA successfully and the loaded PLGA released ~0.3 μmol NO/mg polymer in a controlled manner for just over 6 h. [26] The drawback of the these particles in general is that formation of N-diazeniumdiolates using polyamine compounds may also yield carcinogenic N-nitrosamines as side-products [27].
S-Nitrosothiols have also been utilized as NO donors within polymeric matrices. Frost et al. anchored RSNOs to fumed silica particles for embedding them into polymeric films [28]. Such particles were capable of NO release via addition of copper(II) ions or ascorbic acid to the media, or by shining light on the particles. Stasko et al. developed an RSNO modified polyamidoamine (PAMAM) dendrimer type NO delivery vehicles, that were able to store up to 2 μmol NO/mg dendrimer and release NO for 10 min - several hours via triggering with copper(II) or by light [29]. One issue with these prior efforts to make particle type NO donors is that since the NO donors are immobilized on the surface of the particles, they are exposed to the matrix environment. Hence, the presence of small amounts of ascorbate or copper ions can induce NO burst type release, which can potentially cause adverse effects.
The aim of this work was to develop a particle-based NO release material that could yield physiologically relevant levels of NO for extended periods (e.g., 1–2 weeks), without any risk of toxic by-products. This is possible since the product of NO loss from S-nitroso-N-acetyl-D-penicillamine (SNAP), the NO donor used in this work, is N-acetyl-L-penicillamine (NAC), which is non-toxic at low levels [30]. Further, penicillamine itself (after loss of acetate group from NAC) is FDA approved for reversing heavy metal ion poisoning [31]. Herein, we demonstrate that the duration and rate of NO release from the proposed PLGA microparticles can be tuned by the nature of the PLGA and degree of SNAP loading within the particles. Examples of light activated release are also explored.
Materials and methods
PLGA (RG 503H and RG 504) was obtained from Evonik Industries (Essen, Germany). Reagents, buffer salts, poly(vinyl alcohol) (PVA, Mw: 9,000–10,000, 80% hydrolyzed) and solvents were ordered from Sigma-Aldrich. All chemicals were used as received. All aqueous solutions were prepared with ultrapure deionized water (18.2 MΩ cm resistivity, Millipore). Composition of PBS buffer was 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride, pH 7.4. The PBSACu buffer contained 50 μM CuCl2 and 1 mM ascorbate in PBS. PBSE contained 0.1 mM EDTA in PBS. 50 mL amber polypropylene centrifuge tubes were used for in-vitro NO release experiments (Greiner Bio-One International, Kremsmünster, Austria).
S-Nitroso-N-acetyl-D-penicillamine (SNAP) synthesis
Two g of N-acetyl-D-penicillamine (NAP) (Fluka) was dissolved in a mixture of 50 mL methanol, 16.7 mL H2O, 8.3 mL cc. HCl and 2.5 mL cc. H2SO4. Twenty mL of 1 M NaNO2 was then slowly added to the mixture. The color of the solution turned dark red. The solution was placed onto an ice bath and with blowing N2 over the solution so that the methanol can be evaporated until crystals formed (~3 h). The green crystals were collected by solution filtration, washed with ice-cold water and vacuum dried. The entire process was performed while protecting the solutions from light to avoid photo-decomposition of SNAP. Before encapsulation into PLGA particles, the SNAP was ground by a CryoMill (Retsch, Düsseldorf, Germany). Five hundred mg of SNAP was ground in two 5 mL stainless steel jars, with 16 stainless steel (3 mm diameter) balls in each. After 8 min long precooling with liquid nitrogen at 5 Hz, the SNAP particles were ground for 40 min at 20 Hz, then freeze-dried (−41 °C, 0.160 mbar) for 1 d after flash freezing in liquid nitrogen.
SNAP loaded PLGA microsphere preparation
SNAP loaded microspheres were prepared by a solid-in-oil-in-water emulsion solvent evaporation technique [32]. PLGA polymer was dissolved in 1 mL of methylene-chloride in a 100 mm × 16 mm glass culture tube. The cryomilled SNAP was added to this solution and homogenized at 10 000 rpm for 1 min with a Tempest IQ2 homogenizer (The VirTis Co., Gardiner, NY, USA) equipped with a 10 mm shaft. Four mL of a 5% (w/v) PVA in DI water was immediately pipetted in it and vortexed (Genie 2, Fisher-Scientific Industries, Inc., Bohemia, NY, USA) at the highest speed for 1 min, then poured into 100 mL of 0.5% (w/v) PVA in DI water under rapid stirring with a magnetic stir-bar, and hardened for 3 h. The resulting microspheres were sieved and washed with 500 mL DI water. The fraction of particles sieved in the range of 20 – 125 μm was collected and freeze-dried (−41 °C, 0.160 mbar) for 2 d after flash freezing in liquid nitrogen.
Measurement of loading
To determine the SNAP loading in given lot of microspheres, 5 mg of the microspheres were dissolved in 1 mL acetonitrile and the concentration of SNAP was measured by absorbance at 340 nm using a Synergy Neo Microplate Reader (BioTek U.S., Winooski, VT, USA). The encapsulation efficiency was calculated as the ratio of the actual to the theoretical loading (based on total amount of SNAP used in the preparation).
To measure the SNAP remaining within the microspheres after soaking in buffer for given time periods, the microspheres were washed three times with 100 μM EDTA containing buffer and DI water to remove residual copper and buffer salts prior to dissolving them in acetonitrile, and measuring the absorbance at 340 nm.
Morphology and size distribution of microspheres
For morphology and size distribution analysis, microspheres were coated with 40 nm of gold using a sputter coater (Desk II, Denton Vacuum Inc., Hill, NJ, USA) for 120 s. Secondary electron micrographs were taken by a Hitachi S-3200N Variable Pressure SEM (Hitachi High-Technologies Corp., Tokyo, Japan). The applied accelerating voltage was 15 kV. For size distribution measurements, the diameter of at least 500 particles was measured and analyzed with ImageJ software [33]. For cross-sectional images, the microspheres were cut using a razor-blade on glass slide prior to coating with gold for the SEM analysis.
To observe the morphology changes of the microspheres after soaking in buffer, the microspheres were washed three times with DI water after a given soaking time in order to remove salts prior to SEM analysis.
Nitric oxide release measurement by Nitric Oxide Analyzer
For NO release experiments, 10 mg of microspheres were incubated in 5 mL of release media at 37°C in an amber 50 mL centrifuge vial placed onto a rocker for gentle shaking during incubation at 37°C. Nitric oxide release was quantitated by using an ozone-based chemiluminescence technology with a Sievers Nitric Oxide Analyzer (NOA, Model 280i, GE Analytical Instruments, Boulder, CO, USA). The amber incubation vials were thermostated at 37 °C using a water bath. Nitric oxide released from the suspension of particles was purged from the media via a N2 purge gas (at ca. 50 mL/min) into the NOA system that had been pre-calibrated. The release media was changed every second day whenever NO release experiments were performed. The NO levels in ppb unit measured every other day were converted to NO release rate in mol min−1 mg−1 unit using the NOA instrument constant determined by quantitative reduction of a known amount of nitrite. The amount of NO released was calculated by integration of NO release rate curves for given time periods, and the fraction of NO release was determined based on the initial predetermined SNAP loading into the microspheres.
Light modulated NO release measurements were performed by placing a given amount of the SNAP loaded PLGA particles into a transparent vial irradiated with a tungsten halogen lamp using a Fostec DCR II EKE cold light source (Schott-Fostec, LLC, New York, USA) set at the highest intensity.
Results and Discussion
Encapsulation of SNAP into PLGA microspheres
Before encapsulation, the dark green crystalline SNAP was micronized at −196°C in a ball mill, resulting in a pale green powder of SNAP with particle dimensions typically < 25 μm (see Figure 1). For encapsulation, we used a faster degrading acid terminated PLGA (RG 503H) as well as a slower degrading ester capped PLGA (RG 504), and both were 50:50 of glycolic acid and lactic acid polymers. The polymers were dissolved in methylene chloride, which does not dissolve the NO donor SNAP. The solid in oil in water emulsion and solvent evaporation method (S/O/W) described in the Materials and Methods section provided encapsulation efficiencies of ~48–62% for SNAP (see Table 1), yielding pale green PLGA microspheres with diameters in the 20–125 μm size range (see Figure S1) after sieving. The SNAP loading of the PLGA microspheres was determined by UV/VIS absorption after dissolving the microspheres in acetonitrile. Increasing the amount of micronized SNAP in the formulation yielded increased SNAP loading with both PLGA polymers with values up to ~12% (w/w).
Figure 1.

SEM images of SNAP crystals before (A) and after (B) cryomilling
Table 1.
Formulations (S/O/W), loadings, encapsulation efficiency and particle size distribution
| Formulation | Molecular weight | End group | PLGA | SNAP | Loadinga | Encapsulation efficiency | D[4,3]b |
|---|---|---|---|---|---|---|---|
|
| |||||||
| kDa | mg/mL | mg | (w/w)% | % | μm | ||
| RG 503H blank | 24–38 | Free carboxylic acid | 400 | 0 | n.a. | n.a. | 68 |
| RG 503H 60 | 24–38 | Free carboxylic acid | 400 | 60 | 7.4(±0.3) | 56.3 | 88 |
| RG 503H 80 | 24–38 | Free carboxylic acid | 400 | 80 | 9.1(±0.3) | 53.9 | 75 |
| RG 503H 100 | 24–38 | Free carboxylic acid | 400 | 100 | 12.4(±0.2) | 61.9 | 76 |
| RG 504 blank | 38–54 | Ester | 400 | 0 | n.a. | n.a. | 84 |
| RG 504 60 | 38–54 | Ester | 400 | 60 | 6.3(±0.2) | 48.2 | 80 |
| RG 504 80 | 38–54 | Ester | 400 | 80 | 8.5(±0.7) | 50.7 | 88 |
| RG 504 100 | 38–54 | Ester | 400 | 100 | 11.7(±0.8) | 58.5 | 81 |
The amount of encapsulated SNAP was approximately 0.3–0.6 μmol per mg microsphere. Figure 2 shows the cross sectional SEM images of the microspheres with increasing SNAP loadings. The SNAP crystals are embedded typically in the center portion of the microspheres.
Figure 2.

Cross sectional secondary electron micrographs of RG 503H (A–D) and RG 504 (E–H) PLGA-SNAP microspheres with increasing SNAP loadings: (A, E) blank, (B) 7.4(±0.3)%, (C) 9.1(±0.3%), (D) 12.4(±0.2)%, (F) 6.3(±0.2%), 8.5(±0.7%), 11.7(±0.8%) (all as wt%). Scale bar is 10 μm.
For in vitro monitoring of the NO release from the microspheres, the microspheres were dispersed in the release media within a 50 mL polypropylene centrifuge tube that was placed into a water bath thermostated at 37°C. An amber centrifuge tube was employed to avoid light induced NO photo-release. The emitted NO was purged out from the release media with a continuous nitrogen stream and detected by a chemiluminescence method, which is the gold standard for NO release measurements [34,35]. The NO concentration of the purged gas was measured until a plateau occurred in the detected NO concentration, which usually took about 0.5 h. As shown on Figure 3, the microspheres did not show an initial burst release even in the presence of copper(II) ions and ascorbic acid in the release media (PBSACu), which are known to be initiators for SNAP decomposition [36–38]. However, in the presence of EDTA (PBSE), that chelates the copper(II) ions, the NO release was negligible.
Figure 3.

Instantaneous NO release rates of highest loaded (A) RG 503H 100 and (B) RG 504 100 PLGA-SNAP microspheres in PBSACu and PBSE.
The instantaneous NO release of the PLGA microspheres with different loadings at different time points is shown on Figure 4. In all cases, a significantly higher release rate can be observed just before the measured NO release rates begin to decrease with time. Indeed, the overall NO release is controlled, shown by the estimated cumulative NO release curves (Fig. S2), which were estimated by integrating the instantaneous NO release rates with the trapezoidal rule. The duration of NO release was typically 10–14 days in case of microspheres prepared with the RG 503H polymer and ca. 30 days in case of the RG 504 microspheres. This correlates well with the expected degradation rates of these two different PLGA polymers. Since the RG 503H has a smaller molecular weight and it is free acid terminated (which increases water content and speeds up the acid catalyzed hydrolysis of the PLGA copolymer) its degradation rate is faster compared to the RG 504, which has a higher molecular weight and an ester capped terminus. In addition, the absolute value for the NO release rate was tunable as a function of the amount of encapsulated SNAP. It should be pointed out that the estimated cumulative NO release was significantly overestimated in the case of the RG 503H microparticles (see also Figure S2 A). This is probably due to the different conditions used during the NOA measurements (purging with nitrogen) vs. during the incubation of the microspheres in just buffer solution, without purging the NO. To measure the NO release rate, the particles are more intensively mixed and there is essentially no dissolved NO in the solution at any given time (nitrogen purge) that might further decrease the rate of SNAP decomposition. Measuring the remaining SNAP loading in the particles over time demonstrates very similar NO release kinetics, but without the aforementioned measurement error (Figure 5). The loading change over time is not significantly different in the PBSACu vs. the PBSE buffer.
Figure 4.

Instantaneous NO release rates of (A) RG 503H and (C) RG 504 PLGA-SNAP microspheres in PBSACu release media and estimated amount of released NO from (B) RG 503H and (D) RG 504 PLGA-SNAP microspheres in PBSACu release media. Note: the NO release is overestimated for RG 503H (see Figure S2)
Figure 5.
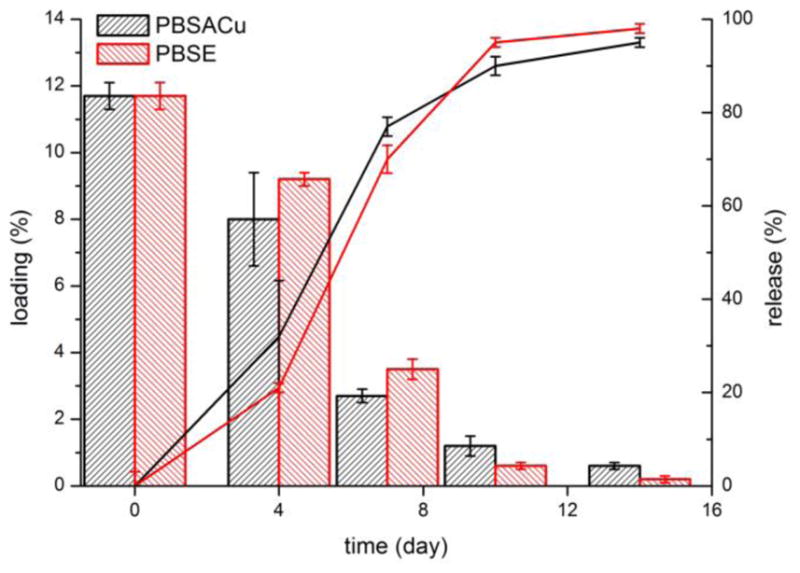
SNAP loading change (bars) and the SNAP release (lines) of RG 503H 100 PLGA-SNAP incubated in PBSACu and PBSE buffers
The concentration of decomposition product N-acetyl-D-penicillamine disulfide ((NAP)2) in the release media was also monitored by RP-HPLC-MS (see Supporting Information). (NAP)2 leached out continuously during the NO release and after the NO release stopped we did not see a significant amount of (NAP)2 in the release media after 16 and 30 days for RG 503H 100 and RG 504 100 formulations, respectively (data not shown). The overall kinetic pattern of release was similar to that described in Figs 4, 5, and S2, and suggests that the release of (NAP)2 happens approximately at the same time with the NO release. Thus, it seems likely that the location of the SNAP decomposition is within the pores of microspheres formed during the degradation process. Indeed, the degradation of the microspheres was monitored by SEM and correlates well with the release data (see Figure 6). Therefore, in practice, the NO release rate can be controlled by the degradation rate of the PLGA microsphere, since exposure of the encapsulated SNAP species to the aqueous media containing trace copper ions and/or ascorbic acid, is required for NO release. Indeed, physiological levels of copper (1 μM) and ascorbate (100 μM) are sufficient to measure physiologically relevant NO release from RG 503H 100 microspheres, that is 10 mg RG 503H 100 microsphere spread over a 1 cm2 area can deliver NO well above the endothelial NO flux rate for about two weeks under physiological conditions (data not shown).
Figure 6.

Secondary electron micrographs of the surface (left side) and the cross section (right side) of RG 503H PLGA-SNAP microspheres before (A) and after incubation in release media at 37°C for 4 days (B), 7 days (C), 10 days (D) and 14 days (E), as well RG 504 PLGA-SNAP microspheres before (F) and after incubation in release media at 37°C for 1 week (G), 2 weeks (H), 3 weeks (I) and 4 weeks (J). White colored scale bar is 20 μm.
The data provided above suggests that the main mechanism of NO release is the controlled release of the SNAP species and its immediate decomposition at the interfaces of the microspheres where the necessary copper(II) ions and/or ascorbate are present. However, by shining light directly on the microspheres another release mechanism is also possible. Figure 7 shows the instantaneous NO release by shining light on the dispersed microspheres (A) in two different solutions as well as on the intact dry microspheres. Clearly, in this case, no exposure of the SNAP to the soaking solution is required, and the crystals of SNAP within the microspheres are sensitive to the photolysis reaction.
Figure 7.

Light modulated NO release of (A) RG 503H PLGA-SNAP microspheres in PBSE (black) and in PBSACu (red) release medium and (B) as a dry powder at room temperature. Dashed lines show the estimated release in percent.
The encapsulated microcrystalline SNAP showed excellent stability also at elevated temperature, which is in agreement with our previous findings for SNAP in certain polymeric materials [39]. Indeed, the SNAP loading did not decrease below 90% after a one-month storage period in the dark (Figure 8) at room temperature, and even when stored at an elevated temperature of 37 °C, there was still ca. 70% of the active NO donor present in the dry microparticles after one year.
Figure 8.
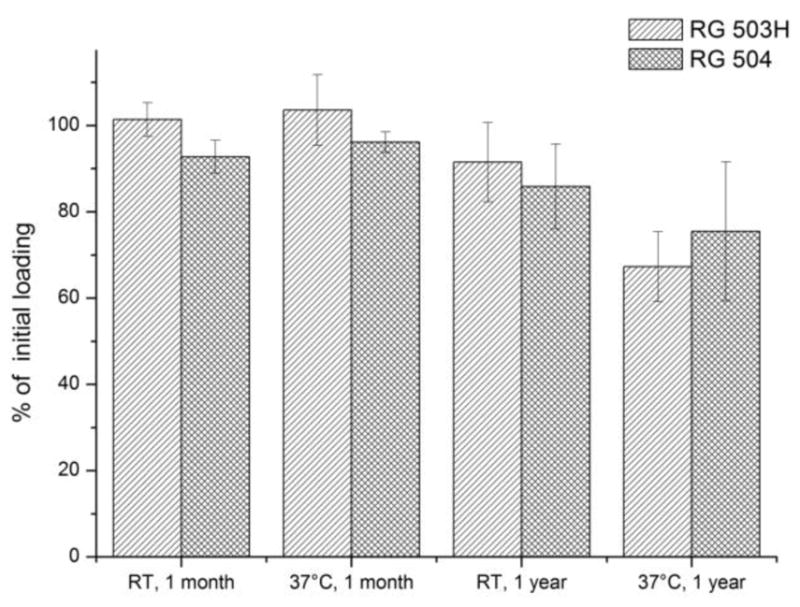
One month and one year storage stability of RG 504 and RG 503H PLGA-SNAP microspheres at room temperature (RT) and at 37°C, averages and standard deviations are calculated from the loadings of the three formulations for each polymer.
Conclusion
In this study it has been shown, for the first time, that the NO donor SNAP can be encapsulated into PLGA microspheres using S/O/W method with high efficiency for long-term controlled release of NO. These microspheres release SNAP in a controlled manner for up to four weeks and in the presence of copper(II) and ascorbate the released SNAP is instantaneously decomposed to generate localized NO. The PLGA encapsulated SNAP is highly stable when stored at room temperature for up to 1 year, demonstrating an enhanced shelf-life over many other NO donor systems. Given that the products produced from the SNAP decomposition are non-toxic, the microparticles reported here could ultimately be injected in numerous locations within in the body or incorporated into creams or hydrogels to create wound healing patches that may be useful in treating various types of wounds, including ulcers related to diabetes. Research in this direction is currently in progress using appropriate animal models.
Supplementary Material
Acknowledgments
We gratefully acknowledge Ms. Yaqi Wo for developing and helping with the LC-MS method that was used to determine the levels of SNAP, NAP, and (NAP)2 dimer emitted by the PLGA microspheres into the soaking media. Support for this research was provided by NIH (EB000783) and The University of Michigan MCubed program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tare M, Parkington HC, Coleman Ha, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- 3.Nathan CF, Hibbs JB. Role of nitric oxide synthesis in macrophage antimicrobial activity. Current Opinion in Immunology. 1991;3:65–70. doi: 10.1016/0952-7915(91)90079-G. [DOI] [PubMed] [Google Scholar]
- 4.Bogdan C. Nitric oxide and the immune response. Nature Immunology. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 5.De Groote MA, Fang FC. NO inhibitions: Antimicrobial properties of nitric oxide. Clinical Infectious Diseases. 1995;21:S162–S165. doi: 10.1093/clinids/21.Supplement_2.S162. [DOI] [PubMed] [Google Scholar]
- 6.Davis KL, Martin E, Turko IV, Murad F. Novel effects of nitric oxide. Annual Review of Pharmacology and Toxicology. 2001;41:203–236. doi: 10.1146/annurev.pharmtox.41.1.203. [DOI] [PubMed] [Google Scholar]
- 7.Vaughn MW, Kuo L, Liao JC. Estimation of nitric oxide production and reaction rates in tissue by use of a mathematical model. The American Journal of Physiology. 1998;274:H2163–H2176. doi: 10.1152/ajpheart.1998.274.6.H2163. http://www.ncbi.nlm.nih.gov/pubmed/9134920. [DOI] [PubMed] [Google Scholar]
- 8.Lancaster JR. A tutorial on the diffusibility and reactivity of free nitric oxide. Nitric Oxide: Biology and Chemistry / Official Journal of the Nitric Oxide Society. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 9.Vaughn MW, Kuo L, Liao JC. Effective diffusion distance of nitric oxide in the microcirculation. The American Journal of Physiology. 1998;274:H1705–H1714. doi: 10.1152/ajpheart.1998.274.5.H1705. [DOI] [PubMed] [Google Scholar]
- 10.Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. New England Journal of Medicine. 2000;342:469–474. doi: 10.1056/NEJM200002173420704. [DOI] [PubMed] [Google Scholar]
- 11.Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. British Journal of Pharmacology. 2007;151:305–21. doi: 10.1038/sj.bjp.0707224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller MR, Wadsworth RM. Understanding organic nitrates - a vein hope? British Journal of Pharmacology. 2009;157:565–567. doi: 10.1111/j.1476-5381.2009.00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Agvald P, Adding LC, Gustafsson LE, Persson MG. Nitric oxide generation, tachyphylaxis and cross-tachyphylaxis from nitrovasodilators in vivo. European Journal of Pharmacology. 1999;385:137–145. doi: 10.1016/S0014-2999(99)00720-7. [DOI] [PubMed] [Google Scholar]
- 14.Núñez C, Víctor VM, Tur R, Alvarez-Barrientos A, Moncada S, Esplugues JV, et al. Discrepancies between nitroglycerin and NO-releasing drugs on mitochondrial oxygen consumption, vasoactivity, and the release of NO. Circulation Research. 2005;97:1063–1069. doi: 10.1161/01.RES.0000190588.84680.34. [DOI] [PubMed] [Google Scholar]
- 15.Wang PG, Cai TB, Taniguchi N. Nitric oxide donors. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, FRG: 2005. [DOI] [Google Scholar]
- 16.Carpenter AW, Schoenfisch MH. Nitric oxide release: part II. Therapeutic applications. Chemical Society Reviews. 2012;41:3742–52. doi: 10.1039/c2cs15273h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Broniowska KA, Diers AR, Hogg N. S-Nitrosoglutathione. Biochimica et Biophysica Acta (BBA) - General Subjects. 2013;1830:3173–3181. doi: 10.1016/j.bbagen.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Al-Sa’doni H, Ferro A. S-Nitrosothiols: a class of nitric oxide-donor drugs. Clinical Science (London, England: 1979) 2000;98:507–520. doi: 10.1042/CS19990267. [DOI] [PubMed] [Google Scholar]
- 19.Pulfer SK, Ott D, Smith DJ. Incorporation of nitric oxide-releasing crosslinked polyethyleneimine microspheres into vascular grafts. Journal of Biomedical Materials Research. 1997;37:182–189. doi: 10.1002/(SICI)1097-4636(199711)37:2<182::AID-JBM6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 20.Jeh HS, Lu S, George SC. Encapsulation of PROLI/NO in biodegradable microparticles. Journal of Microencapsulation. 2004;21:3–13. doi: 10.1080/02652040310001619767. [DOI] [PubMed] [Google Scholar]
- 21.Zhang H, Annich GM, Miskulin J, Stankiewicz K, Osterholzer K, Merz SI, et al. Nitric oxide-releasing fumed silica particles: synthesis, characterization, and biomedical application. Journal of the American Chemical Society. 2003;125:5015–5024. doi: 10.1021/ja0291538. [DOI] [PubMed] [Google Scholar]
- 22.Rothrock AR, Donkers RL, Schoenfisch MH. Synthesis of nitric oxide-releasing gold nanoparticles. Journal of the American Chemical Society. 2005;127:9362–3. doi: 10.1021/ja052027u. [DOI] [PubMed] [Google Scholar]
- 23.Shin JH, Metzger SK, Schoenfisch MH. Synthesis of nitric oxide-releasing silica nanoparticles. Journal of the American Chemical Society. 2007;129:4612–9. doi: 10.1021/ja0674338. [DOI] [PubMed] [Google Scholar]
- 24.Stasko NA, Schoenfisch MH. Dendrimers as a scaffold for nitric oxide release. Journal of the American Chemical Society. 2006;128:8265–8271. doi: 10.1021/ja060875z. [DOI] [PubMed] [Google Scholar]
- 25.Carpenter AW, Reighard KP, Saavedra JE, Schoenfisch MH. O (2)-Protected diazeniumdiolate-modified silica nanoparticles for extended nitric oxide release from dental composites. Biomaterials Science. 2013;1:456–459. doi: 10.1039/C3BM00153A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yoo JW, Lee JS, Lee CH. Characterization of nitric oxide-releasing microparticles for the mucosal delivery. Journal of Biomedical Materials Research Part A. 2010;92:1233–43. doi: 10.1002/jbm.a.32434. [DOI] [PubMed] [Google Scholar]
- 27.Coneski PN, Schoenfisch MH. Competitive formation of N-diazeniumdiolates and N-nitrosamines via anaerobic reactions of polyamines with nitric oxide. Organic Letters. 2009;11:5462–5465. doi: 10.1021/ol902282y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frost MC, Meyerhoff ME. Synthesis, characterization, and controlled nitric oxide release from S-nitrosothiol-derivatized fumed silica polymer filler particles. Journal of Biomedical Materials Research - Part A. 2005;72:409–419. doi: 10.1002/jbm.a.30275. [DOI] [PubMed] [Google Scholar]
- 29.Stasko NA, Fischer TH, Schoenfisch MH. S-Nitrosothiol-modified dendrimers as nitric oxide delivery vehicles. Biomacromolecules. 2008;9:834–841. doi: 10.1021/bm7011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Babich H, Zuckerbraun HL, Hirsch ST, Blau L. In vitro cytotoxicity of the nitric oxide donor, S-nitroso-N-acetyl-penicillamine, towards cells from human oral tissue. Pharmacology & Toxicology. 1999;84:218–225. doi: 10.1111/j.1600-0773.1999.tb01486.x. [DOI] [PubMed] [Google Scholar]
- 31.Levitskaia TG, Creim Ja, Curry TL, Luders T, Morris JE, Woodstock AD, et al. Evaluation of Cuprimine® and Syprine® for decorporation of 60Co and 210Po. Health Physics. 2010;98:471–479. doi: 10.1097/HP.0b013e3181bcdf4f. [DOI] [PubMed] [Google Scholar]
- 32.Wischke C, Schwendeman SPS. Principles of encapsulating hydrophobic drugs in PLA/PLGA microparticles. International Journal of Pharmaceutics. 2008;364:298–327. doi: 10.1016/j.ijpharm.2008.04.042. [DOI] [PubMed] [Google Scholar]
- 33.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nature Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fontijn A, Sabadell AJ, Ronco RJ. Homogeneous chemiluminescent measurement of nitric oxide with ozone. Implications for continuous selective monitoring of gaseous air pollutants. Analytical Chemistry. 1970;42:575–579. doi: 10.1021/ac60288a034. [DOI] [Google Scholar]
- 35.Antus B, Horvath I, Barta I. Assessment of exhaled nitric oxide by a new hand-held device. Respiratory Medicine. 2010;104:1377–1380. doi: 10.1016/j.rmed.2010.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Dicks AP, Beloso PH, Williams DLH. Decomposition of S-nitrosothiols: the effects of added thiols, Journal of the Chemical Society. Perkin Transactions. 1997;2:1429–1434. doi: 10.1039/a701594a. [DOI] [Google Scholar]
- 37.Williams DLH. The chemistry of S-nitrosothiols. Accounts of Chemical Research. 1999;32:869–876. doi: 10.1021/ar9800439. [DOI] [Google Scholar]
- 38.Williams DLH. Nitric oxide release from S-nitrosothiols (RSNO) - the role of copper ions. Transition Metal Chemistry. 1996;21:189–191. doi: 10.1007/BF00136555. [DOI] [Google Scholar]
- 39.Wo Y, Li Z, Brisbois EJ, Colletta A, Wu J, Major TC, et al. Origin of long-term storage stability and nitric oxide release behavior of CarboSil polymer doped with S-Nitroso-N-acetyl-D-penicillamine. ACS Applied Materials & Interfaces. 2015;7:22218–22227. doi: 10.1021/acsami.5b07501. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.