

Abstract
The mainstay of asthma therapy, glucocorticoids (GCs) exert their therapeutic effects through the inhibition of inflammatory signaling and induction of eosinophil apoptosis. However, laboratory and clinical observations of GC-resistant asthma suggest that GCs' effects on eosinophil viability may depend on the state of eosinophil activation. In the present study we demonstrate that eosinophils stimulated with IL-5 show impaired prop-aptoptotic response to GCs. We sought to determine the contribution of GC-mediated transactivating (TA) and transrepressing (TR) pathways in modulation of activated eosinophils' response to GC by comparing their response to the selective GC receptor (GR) agonist Compound A (CpdA) devoid of TA activity to that upon treatment with Dexamethasone (Dex). IL-5-activated eosinophils showed contrasting responses to CpdA and Dex, as IL-5-treated eosinophils showed no increase in apoptosis compared to cells treated with Dex alone, while CpdA elicited an apoptotic response regardless of IL-5 stimulation. Proteomic analysis revealed that both Nuclear Factor IL-3 (NFIL3) and Map Kinase Phosphatase 1 (MKP1) were inducible by IL-5 and enhanced by Dex; however, CpdA had no effect on NFIL3 and MKP1 expression. We found that inhibiting NFIL3 with specific siRNA or by blocking the IL-5-inducible Pim-1 kinase abrogated the protective effect of IL-5 on Dex-induced apoptosis, indicating crosstalk between IL-5 anti-apoptotic pathways and GR-mediated TA signaling occurring via the NFIL3 molecule. Collectively, these results indicate that 1) GCs' TA pathway may support eosinophil viability in IL-5-stimulated cells through synergistic upregulation of NFIL3; and 2) functional inhibition of IL-5 signaling (anti-Pim1) or the use of selective GR agonists that don't upregulate NFIL3 may be effective strategies for the restoring pro-apoptotic effect of GCs on IL-5-activated eosinophils.
Keywords: Eosinophils, Apoptosis, Glucocorticoid Receptor, Signal Transduction
Introduction
Synthetic glucocorticoids (GCs) remain a first-line therapy for eosinophilic inflammatory disorders such as allergy, asthma, eosinophilic esophagitis and hypereosinophilic syndromes (HES) [1-3]. The action of GCs on cells producing eosinophil-active cytokines is considered a major mechanism of reduced eosinophilic inflammation in asthma, as decreased expression of IL-4 and IL-5 in the bronchial mucosa correlates with lower numbers of eosinophils, T cells and tryptase-only positive mast cells in the airways of asthmatic patients during GC therapy [3]. Although the direct, pro-apoptotic of GC on eosinophils is also appreciated, several lines of evidence suggest that it may depend on their state of activation. First, GCs induce eosinophil apoptosis within hours of cell culture in vitro, however, IL-5 at concentrations higher than 23000 fM protects cells from GC-induced cell death [4-6]. Second, rapid eosinopenic effect of GCs is followed by the full anti -eosinophilic clinical response that is delayed for days in patients even with GC-responsive eosinophilic disorders. In the bone marrow, GCs have been shown to stimulate IL-5-mediated eosinopoiesis [7], further arguing against pro-apoptotic effect of GCs on IL-5-activated eosinophils. Finally, recent clinical trials revealed group of severe asthmatics with high levels of IL-5 and eosinophilia in spite of high doses of GCs, an observation consistent with resistance of eosinophils to apoptosis upon exposure to IL-5 at high concentrations [8-10].
The cellular effects of GCs are mediated via the GC receptor (GR). Upon GC binding, the GR translocates to the nucleus, where it regulates gene expression via transactivation (TA) and transrepression (TR). TA requires binding of GR homodimers to palindromic glucocorticoid-responsive elements (GREs)[11], while TR results from inhibiting interactions between GR and other transcription factors, such as NF-κB, AP-1, p53, STATs, IRF-3 and CREB [12,1]. The TR interaction does not require GR dimerization. The role of TA in mediating anti-inflammatory and proapoptotic signaling has been reported in lymphoid cells [13]; however, there is growing evidence that, in parallel with inhibiting cytokine production, TA can mediate anti-apoptotic signaling, as has been shown in some cancer cells that became resistant to chemotherapy and GC treatment [14]. Since IL-5-activated eosinophils show an impaired pro-apoptotic response to GC, it is reasonable to ask whether IL-5-signaling pathways modulate TA-inducible components of GR pathways.
In the present study we sought to determine the contribution of GR-mediated TA and TR in the modulation of eosinophil survival after activation with IL-5. We compared eosinophils' response to the nonselective TR/TA GR agonist Dexamethasone (Dex) and the selective GR agonist Compound A (CpdA). CpdA exhibits the distinct profile of a dissociating GR ligand, preventing GR dimerization and subsequent TA [15]. IL-5-activated eosinophils showed contrasting responses to Dex and CpdA, as IL-5-treated eosinophils were protected from Dex-induced apoptosis, while CpdA elicited an apoptotic response regardless of IL-5 stimulation. We found that MAP Kinase Phosphatase 1 (MKP1) and Nuclear Factor Interleukin-3 (NFIL3) were synergistically upregulated by Dex in IL-5-activated eosinophils, while inhibiting NFIL3 only abrogated the protective effect of IL-5 on GC-induced apoptosis. A similar effect was observed upon inhibition of the IL-5-inducible Pim1 kinase, suggesting crosstalk between IL-5/Pim1 anti-apoptotic pathways and GR-mediated TA signaling occurring via the NFIL-3 molecule. Taken together, we show for the first time that GR-mediated TA may protect IL-5-activated eosinophils from GC-induced apoptosis through synergistic upregulation of NFIL3. This mechanism may explain the steroid- resistant phenotypes of eosinophilic disorders characterized with high level of IL-5. This effect can be interfered with by inhibition IL-5 signaling (anti-Pim1) or by the use of selective GR agonists that don't upregulate NFIL3.
Materials and Methods
Reagents/materials
Recombinant, human IL-5 was purchased from Peprotech (Rocky Hill, NJ). Polyclonal antibodies against NFIL-3, Pim1 and MKP1 were from Santa Cruz Biotechnology (Santa Cruz, CA), and polyclonal antibodies against GR, pGRS11 were from Abcam (Cambridge, MA). Secondary HRP-conjugated anti-rabbit antibody was from Cell Signaling Technology (Danvers, MA). NFIL3 and MKP1 siRNA and the appropriately scrambled control and transfecting reagent were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Dexamethasone and RU 486 were from Sigma-Aldrich (St. Louis, MO), and Compound A was purchased from Enzo Life Sciences (Farmingdale, NY) and dissolved in ethanol. The Pim-1 inhibitor AZD1208 was purchased from SelleckChem (Houston, TX). In experiments involving Pim inhibitor, all control cells were treated with solutions used for dissolving the AZD1208 (DMSO); the final concentration of DMSO never exceeded 0.1% of the culture volume. The chemiluminescence reagent employed was purchased from Millipore Corporation (Bedford, MA).
Eosinophil isolation from peripheral human blood
Peripheral blood (120 ml) was drawn from healthy subjects as we previously reported (27) under a research protocol approved by the IRB at the University of Texas Medical Branch (IRB#04-371). Eosinophils were obtained by sedimentation in 4-6% dextran for 50 min at room temperature (RT), followed by centrifugation in a Ficoll-Hypaque gradient as described previously [16]. After centrifugation at 500 × g, the upper layers of plasma and mononuclear cells were removed and saved for further analysis. Erythrocytes were eliminated by hypotonic lysis, and eosinophils then negatively selected using anti-CD16 immunomagnetic beads to remove neutrophils using the MACS system 9 (Miltenyi Biotec, Sunnyvale, CA). The final eosinophil purity was assessed by microscopic examination using a Wright-stained cytospin preparation. The purity of eosinophil preparations was ≥ 98%.
Human eosinophil cell culture
Although most experiments relating to eosinophil viability were performed on freshly isolated cells in the presence of 10% FBS, in experiments involving inhibition of NFIL3 or MKP1 with siRNA, eosinophils were cultured for the first 8 h in the absence FBS (40). Briefly, eosinophils were suspended in RPMI 1640 medium (GIBCO BRL, Life Technologies, Grand Island, NY) supplemented with 10% FBS (High Clone Laboratories, Inc., Logan, UT), 100 U/ml penicillin G, 100 μg/ml streptomycin, and 0.25 μg/ml amphotericin B (GIBCO BRL/Life Technologies, Grand Island, NY). Cells were cultured at a density of 1 × 106/ml in a humidified atmosphere containing 95% air and 5% CO2. The cultures were maintained in 12-well sterile, flat-bottom plates (Costar Corp., Cambridge, MA).
Western blot analysis
For protein identification after gel electrophoresis, proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MS). After transfer, membranes were blocked with 5% milk Tris-buffered saline (100 mM Tris-HCl, 150 mM NaCl, pH 7.5) containing 0.1% (v/v) Tween 20 for 1 h, and then incubated with the appropriate antibody (1:10000 dilution) overnight at 4˚C. Membranes were washed 4× in Tris-buffered saline with 0.1% (v/v) Tween 20 and then incubated with horseradish peroxidase-conjugated secondary antibody. After washing, immune complexes were detected by reaction with the enhanced-chemiluminescence assay (Millipore) per the manufacturer's protocol.
MKP1 and NFIL3 siRNA transfection
Freshly isolated eosinophils were resuspended (106/ml) in antibiotic-free siRNA Transfection Medium (sc-36868, Santa Cruz, CA). One ml aliquots of eosinophils in transfection medium were added to each well of a 12-well culture plate. An aliquot of 3.6 μl of MKP1 siRNA (sc-35937) or 4.4 μl of NFIL3 siRNA (sc-37821) was added to 40 μl of siRNA Transfection Medium (sc-36868, Santa Cruz, C) and kept at RT for 5 min. In a separate tube, 2.4 μl of siRNA Transfection Reagent (sc-29528) was mixed with 40 μl of siRNA Transfection Medium and kept at RT for 5 min. Subsequently, the contents from both tubes were mixed and incubated at RT for 20 min to form an siRNA-siRNA Transfection Reagent Complex. After 20 min of incubation 0.32 ml of siRNA Transfection Medium was added to each tube containing the siRNA-siRNA Transfection Reagent. Cells were subsequently resuspended in 0.4 ml of resulting siRNA/siRNA Transfection reagent mixture and incubated for 8 h at 37°C (in the absence of FBS). After incubation, cells were resuspended in 1 ml of RPMI 1640 containing 10% of FBS and incubated for additional 36 h. Transfection efficiency was evaluated by Western blotting for each experiment performed on eosinophil viability. Control siRNA (sc-370007) was a scrambled sequence that does not cause degradation of any known cellular mRNA.
Eosinophil viability
An Annexin VPE Apoptosis Detection kit (BD Biosciences) was used to quantitatively determine eosinophils undergoing apoptosisby virtue of their ability to bind annexin V and exclude 7-aminoactinomycin(7-AAD). This assay detected viable cells (Annexin V negative/7-AAD negative) undergoing early apoptosis (Annexin V positive/7-AAD negative), and dead cells (Annexin V positive/7-AAD positive). Eosinophils (2 × 105) were stained with annexin A and 7-ADD per the manufacturer's instructions. Data were acquired on a FACScan instrument (BD Biosciences) and analyzed using CellQuest software (BD Biosciences); 10,000 events per sample were acquired.
Data analysis for eosinophil viability
The results of eosinophil viability measurements are expressed as means ± SD. To determine significant differences between the two groups, a two-tailed Student's t test was performed using a Sigma-Plot software program (SPSS); p< 0.05 was considered significant.
Results
Modulation of eosinophil survival by Dexamethasone (Dex)
When quiescent eosinophils isolated from peripheral blood are cultured in the absence of any modulators of apoptosis, approximately half exhibit morphological features of apoptosis after 36-40 h, depending on donor status and the assays used [17]. We used Annexin V/7-ADD flow cytometry analysis as our major assay due to its high sensitivity and similar performance across different stimuli; however, all experiments were controlled with secondary assessments of cell viability, including morphological examination or staining with Calcein AM (R&D Systems, Minneapolis, MN). Flow cytometric analysis of quiescent eosinophils isolated from non-allergic donors cultured in vitro showed a spontaneous decrease in the number of viable (Annexin V- and 7AAD-negative) cells from 91% at 4 h to 68% at 16 h and 45% at 24 h. Exposure of freshly isolated eosinophils to Dex at a concentration of 1 μM induced a significant decrease in cell viability, as the Annexin V/7ADD-negative cell percentages declined to 81% and 42% at 4 and 16 h of in vitro culture, and decreased to 21% and 18% 2 and 3 days later (Fig. 1A, B). This effect was dose-dependent, as eosinophil apoptosis occurred more slowly when Dex was used at 0.1 and 0.01 μM (data not shown). Similar to previous reports, the GR antagonist RU486 at 1 μM blocked the increase in eosinophil apoptosis when added before Dex, indicating that Dex-induced apoptosis was mediated by GR (Fig. 1A). Upon testing eosinophil viability in the presence of IL-5 we found a significant inhibition of cell apoptosis compared to treatment of quiescent cells with Dex alone as the percentage of Annexin V/7ADD-positive cells did not exceed 10% after the first 24 h of IL-5 stimulation at concentrations 0.01, 0.1 and 1 ng/ml (data not shown). Since the percentage of viable, non-apoptotic eosinophils at 24 h was still above 90% at all used concentrations of IL-5, to assess Dex's effect we tested cell viability after 48 and 72 hours. At a concentration of 0.1 ng/ml IL-5completely inhibited Dex-induced cells death, as there was no significant difference in the number of viable cells between stimulation with Dex/IL-5 and IL-5 alone on day 1 and 3. Surprisingly, the percentage of Annexin/7ADD-negative eosinophils upon IL-5/Dex stimulation was consistently 4-6% higher than the viability of cells stimulated with IL-5 alone for 48 h, reaching statistical significance (Figure 1B). This anti-apoptotic effect was more pronounced at higher concentrations of Dex (0.1 μM > 0.01 μM), suggesting a Dex-mediated effect (not shown); however, at longer incubation times the difference between IL-5/Dex- and IL-5-stimulated cells was not statistically significant. This effect was reproducible using other cell viability assays, including the Calcein AM cell viability assay (not shown). On the other hand, IL-5 at low concentrations (≤ 0.01 ng/ml) had less protective effect from Dex-induced apoptosis, showing a significant decrease in cell viability compared to cells stimulated with IL-5 alone (Figure 1B). However, cells exposed to low concentrations of IL-5 and treated with Dex exhibited higher viability than eosinophils exposed to Dex alone. The observed delay in cell apoptosis observed after 48 h of stimulation with IL-5 and Dex diminished upon pretreatment with RU486, suggesting the involvement of GR signaling pathways in the inhibition of eosinophil apoptosis (Fig. 1C). We also investigated whether the order and time of Dex addition to IL-5 stimulation affects eosinophil viability (Figure 1D). We found that eosinophils pretreated with Dex up to 1 h before stimulation with IL-5 or stimulated simultaneously with Dex and IL-5 showed delayed apoptosis vs. cells cultured with IL-5 alone; however, addition of Dex 3 hours before IL-5 stimulation initiated pro-apoptotic signals that were not fully recoverable by IL-5. Taken together, our findings indicate the crosstalk between IL-5-inducible and GR-inducible pathways resulting in diminished pro-apoptotic response to Dex in IL-5 activated eosinophils.
Figure. 1.
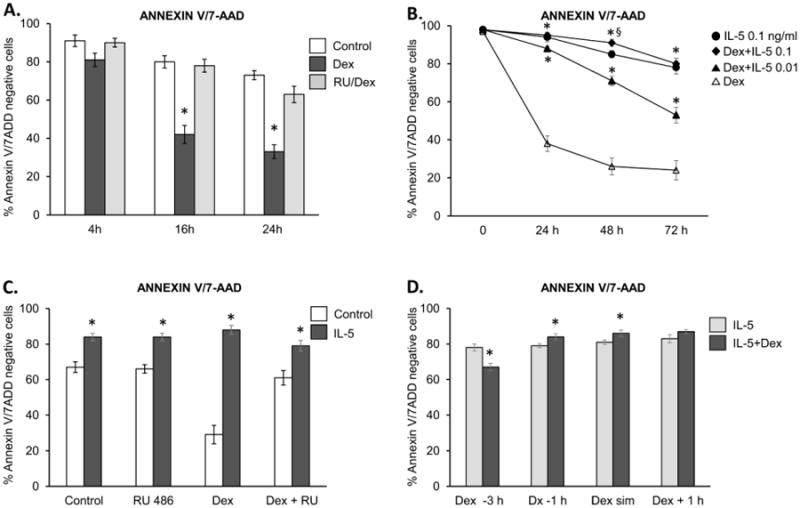
Effect of Dexamethasone and IL-5 on eosinophil viability in vitro. Eosinophil apoptosis was assessed by staining with PE-labelled Annexin V and 7 AAD; viable, non-apoptotic cells are represented as Annexin V/7AAD-negative cells. A). Changes in eosinophil viability upon incubation with Dex at a concentration of 0.1 μM. * = p<0.05 vs. control B). Effect of IL-5 at 0.01 ng/ml and 0.1 ng/ml on eosinophil viability upon exposure to Dex at 0.1 μM. IL-5 was added at the same time as Dex, * = p<0.05 vs. cell stimulated with Dex alone, § = p<0.05 vs cells stimulated with IL-5 alone. C). The GR antagonist RU486 (1μM) inhibits the effect of Dex (1 μM), * = p<0.05 vs. control, D) The effect of preincubating eosinophils with Dex or IL-5 on eosinophil viability. Eosinophils were preincubated with Dex for 3 h or 1 h before stimulation with IL-5, or Dex was added at the same time or 1 h after IL-5, followed by viability assessment at 48 h; * = p<0.05 vs. eosinophils stimulated with IL-5 alone. The error bars represent the standard deviation of at least 3 independent experiments.
Modulation of eosinophil survival by Compound A
To investigate the involvement of transactivating GR activity in the propagation of GR-mediated cell apoptosis, we exposed eosinophils to the selective GR agonist Compound A (CpdA). CpdA was highly effective at inducing rapid apoptosis in quiescent eosinophils, matching the kinetics of eosinophil apoptosis observed when Dex was used at lower concentrations (Fig 2A). Although more than 70% of eosinophils stained positive for both Annexin V and 7AAD at 24 h of culture, the high percentage (>35%) of Annexin V detected 4 h after addition of CpdA suggested apoptosis as a major mechanism of CpdA -induced cell death. Concurrent treatment with RU486 blocked apoptosis induced by CpdA, indicating a GR-dependent mechanism. Since these cells were not stimulated with any proinflammatory or activating cytokines, our results indicated that CpdA prompts apoptotic pathways independently of blocking cytokine-induced anti-apoptotic cascades.
Figure 2.
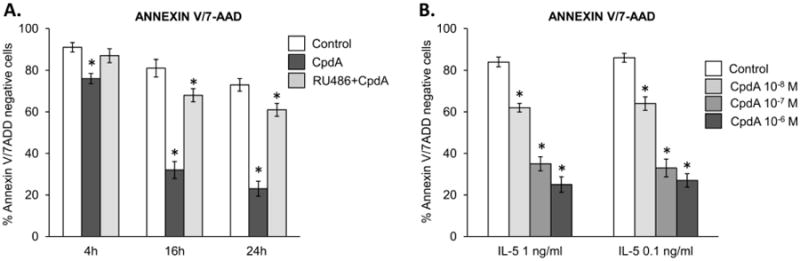
Effect of the selective GR agonist CpdA on the viability of quiescent and IL-5-stimulated eosinophils. A) Induction of apoptosis in quiescent eosinophils stimulated with CpdA (1 μM) and the antagonistic effect of RU486 (1 μM) for 48 h; * p<0.05 vs. control cells. B). The concentration-dependent effect of CpdA on viability of eosinophils stimulated with IL-5 for 48 h. The error bars represent the standard deviation of at least 3 independent experiments.
Importantly, CpdA exerted different effects on IL-5-stimulated eosinophils than Dex, as CpdA effectively induced apoptosis in eosinophils cultured for 4, 16 and 24 h (Figure 2B). The GR antagonist RU486 blocked CpdA's proapoptotic effect on IL-5-activated cells (not shown). Thus, we found for the first time that 1) the selective GR agonist CpdA induces apoptosis in eosinophils, and 2) that IL-5-induced activation does not interfere with this process.
MKP-1 and NFIL3 Expression in Activated Eosinophils
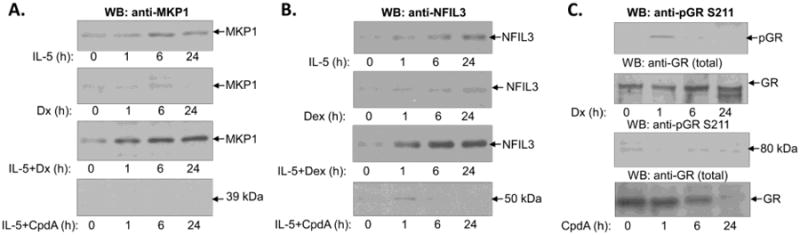
Map Kinase Phosphatase 1 (MKP-1) is upregulated and activated in cytokine-induced granulocytes and in GC-treated cells, and was proposed as one of the major mediators of the anti-inflammatory actions of GCs [18]. Nuclear Factor IL-3 (NFIL3), also known as the basic leucine zipper transcription factor E4BP4, is expressed at low levels in natural killer (NK) cells, B cells, T cells, dendritic cells (DCs) and macrophages, and is upregulated by several cytokines such as IL-3, IL-4, IL-10 and hormones including GCs [19,20]. We tested the expression of MKP-1 and NFIL3 in IL-5-stimulated eosinophils by Western blotting and found low basic expression of both proteins in quiescent eosinophils. MKP1 and NFIL3 levels increased within 1 h of IL-5 stimulation and reached a steady-state level within 6 h. However, the expected upregulation of MKP1 in eosinophils treated with Dex alone was not detected within the first 6 h of incubation, while interpretation of MKP1 expression at the 16 and 24 h time points was affected by the occurrence of apoptosis and protein degradation (Figure. 3A). MKP1 and NFIL3 were clearly upregulated by Dex in IL-5-stimulated eosinophils compared to cells exposed only to IL-5 (Figure 3A, B), suggesting an additive effect of Dex and IL-5 stimulation on MKP1 and NFIL3 expression. CpdA failed to produce a further increase of NFIL3 and MKP1 in IL-5-stimulated cells as assessed after 6 and 24 h of stimulation, suggesting that GR TA was required for the upregulation of these molecules by Dex. We also tested the effects of Dex and CpdA on phosphorylation of GR on Ser 211, as this phosphorylation is critical for GR TA and for MKP1 induction by GC (Fig 3B). We found that Dex upregulated the phosphorylation of GR on Ser 211; however, neither CpdA nor IL-5 had an effect on GR phosphorylation.
Figure 3.
Western blot analysis of MKP1, NFIL3 and GR expression. A), Time-course of MKP1 expression shows upregulation by IL-5 and synergistic upregulation by the combination of IL-5 and Dex. There was no detectable expression of MKP1 in CpdA-treated eosinophils. B), Time-course of NFIL3 expression shows upregulation by IL-5 and synergistic upregulation by the combination of IL-5 and Dex. Treatment with CpdA did not produce upregulation of NFIL3 in either quiescent or activated eosinophils. C) Differential effect of Dex and CpdA on phosphorylation of GR on S211. Analysis of the total GR protein level showed a similar content of GR stimulated and control eosinophils within the first 6h of stimulation, and degradation of GR protein at later times. Blots representative of two independent experiments are shown.
Role of MKP1 and NFIL3 in IL-5-induced inhibition of apoptosis
The inducible nature of MKP1 and NFIL3 expression allowed us to use siRNA to prevent upregulation of these molecules after concurrent stimulation with IL-5 and Dex. Treatment of eosinophils with small interfering RNA against MKP1 or NFIL3 blocked upregulation of MKP1 and NFIL3 proteins, respectively indicating efficient silencing of targeted RNA (Figure 4). Inhibition of MKP1 had no significant effect on expression of GR and did not affect the abundance of F-actin indicating specificity of siRNA-mediated post-transcriptional gene silencing. Similarly, inhibition of NFIL3 did not lead to downregulation of the β chain of IL-5 receptor and did not have significant effect on F-actin expression. We found that the absence of MKP1 had a modest effect on eosinophil viability upon stimulation with GC and IL-5, and did not decrease the viability of cells stimulated with IL-5 alone (Figure 5). Although not significant, a slight prolongation of eosinophil survival was observed in IL-5-stimulated cells with inhibited MKP1, suggesting increased MAP kinase signaling unopposed by MAP kinase phosphatase. Taken together, these observations indicate that GR-mediated transcriptional activation of MKP1 does occur in IL-5-stimulated cells; however, its inhibition has no significant effect on eosinophil viability.
Figure 4.
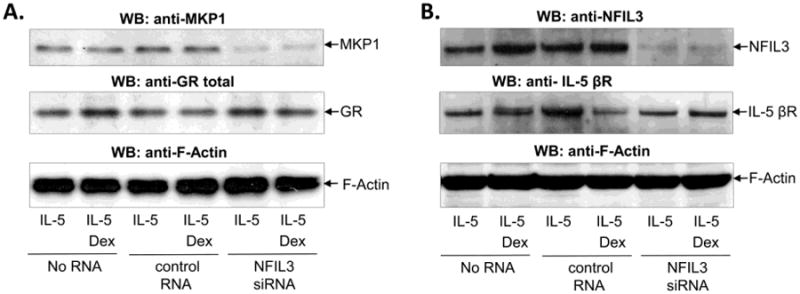
SiRNA-mediated inhibition of MKP1 and NFIL3 expression in IL-5-stimulated eosinophils. Eosinophils were transfected with siRNA against MKP1 (A), against NFIL3 (B), control scrambled RNA (A, B) or no RNA (A, B) for 8 h, followed by stimulation with IL-5 (0.1 ng/ml) and Dex (1 μM) for an additional 40 h in the presence of 10% FBS. A), Western blot of MKP1 showed significant inhibition of MKP1 expression in cells transfected with MKP1 siRNA and no effect of MKP1 siRNA on expression of GR or F-actin B), Western blot of NFIL3 showed inhibition of MKP1 expression in cells transfected with NFIL3 siRNA and no downregulation of F-actin or the β chain of IL-5 receptor. Western blots are representative of at least 2 independent experiments.
Figure 5.
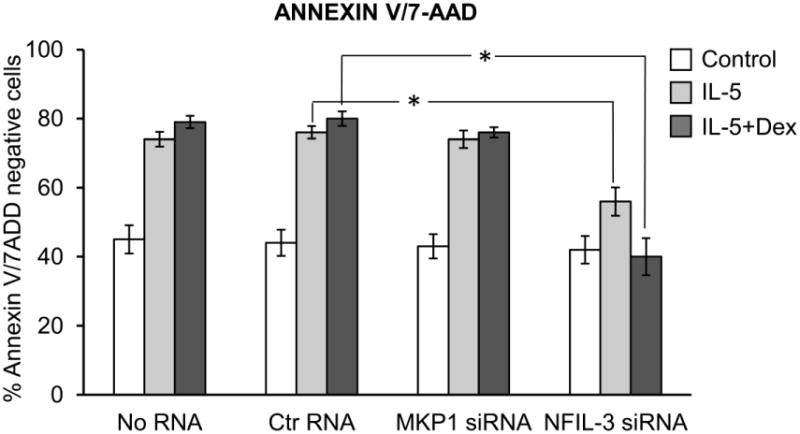
Effect of inhibition of MKP1 and NFIL3 on eosinophil viability in the presence of IL-5 (0.1 ng/ml) and/or Dex (1 μM) for 40 h. Two controls including eosinophils exposed to transfection reagents and transfected with scrambled RNA were used to exclude a potential effect of RNA on eosinophil viability. Inhibition of MKP1 had no effect on eosinophil viability upon stimulation with IL-5 or Dex, while inhibition of NFIL3 decreased the viability of IL-5-stimulated cells and restored the pro-apoptotic effect of Dex. The error bars represent the standard deviation of 3 experiments from 2 independent donors.
On the other hand, inhibition of NFIL3 expression by siRNA had a much stronger effect on eosinophils' response to GC and IL-5 stimulation. In IL-5-stimulated cells, inhibition of NFIL3 decreased the prolongation of survival compared to cells with intact NFIL-3, although the percentage of viable cells was still significantly higher than in quiescent cells. Importantly, Dex was highly effective in inducing apoptosis in IL-5-activated cells with inhibited NFIL3, with apoptosis rates comparable to that of quiescent cells exposed to Dex.
NFIL3 upregulation by eosinophils is Pim1-dependent
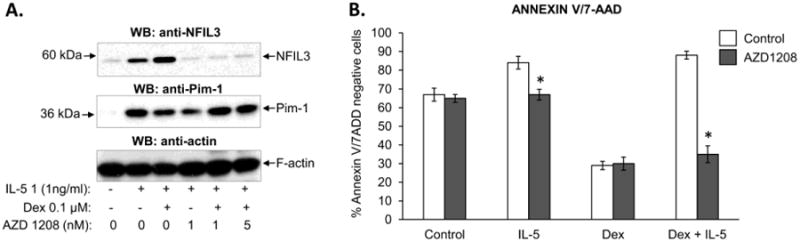
Pim-1 is a constitutively active serine/threonine kinase inducibly expressed in IL-5-stimulated eosinophils [21,22]. It is involved in an anti-apoptotic action of IL-5, and is associated with phosphorylation of Jak2 kinase and increased levels of the anti-apoptotic molecule Mcl-1 [21]. Since NFIL3 has been reported to be regulated in a Jak/STAT-dependent manner, we tested whether inhibition of Pim-1 affects s the expression of NFIL-3 in eosinophils. We used a novel pan-Pim kinase inhibitor AZD1208 that has shown efficacy in models of acute myeloid leukemia [23]. The compound has been shown to preferentially inhibit Pim-1 over Pim-2 and Pim-3 at concentrations lower than 1 nM. At 1 nM, AZD1208 had no effect on the viability of quiescent eosinophils, but it completely blocked the prolongation of eosinophil survival when IL-5 was present (Figure 6B). Further, the addition of IL-5 and Dex to AZD1208-pretreated eosinophils increased cell apoptosis to rates comparable to that observed in quiescent cells treated with GCs. Consistent with previous reports, AZD1208 did not affect the induction of Pim-1 expression, (Figure 6A). Importantly, eosinophils with inhibited activity of Pim-1 kinase failed to upregulate the expression of NFIL3, in both IL-5-activated and IL-5/Dex-treated cells. Taken together, our results indicate a requirement for activated Pim-1 kinase in upregulation of NFIL3 by IL-5 and Dex.
Figure 6.
Effect of the Pim1 inhibitor AZD1208 on the expression of NFIL3 (at 24 h) and viability of eosinophils (at 48 h) stimulated with IL-5 and Dex. A) AZD1208 at a concentration specific for inhibition of Pim1 activity (1ng/ml) blocked upregulation of NFIL3 by IL-5 and Dex/IL-5 (24 h) without a significant effect on Pim1 expression. B) Inhibition of Pim1 activity by AZD1208 blocked the anti-apoptotic effect of IL-5 and restored the proapoptotic effect of Dex on activated eosinophils, * p<0.05 vs. control cells. The error bars represent the standard deviation of 4 experiments from 3 independent donors.
Discussion
GCs are highly effective in inducing apoptosis in quiescent eosinophils; however, the apoptosis of eosinophils is delayed upon IL-5 stimulation at low concentrations, and completely abrogated or reversed in cells exposed to high levels of IL-5. In this paper we present evidence that NFIL3 protects eosinophils from apoptosis and GCs are able to enhance the expression of NFIL-3 via a GR-dependent trans-activation pathway. Importantly, the latter ability is only realized in activated eosinophils in the context of concomitant signaling from proinflammatory cytokines in a Pim-1-dependent manner. Thus we identified a possible molecular basis for the resistance of activated eosinophils to steroid-induced apoptosis.
Inhibition of signaling pathways prolonging eosinophil survival have been considered to be a one of the major mechanisms underlying the anti-inflammatory and anti-eosinophilic action of GC therapy in disorders characterized by eosinophilic inflammation, such as asthma [24]. Our in vitro findings showing that GCs' apoptosis-inducing effect is limited to quiescent eosinophils suggest a perhaps bigger role of inhibition of IL-5 production by other cell types than direct pro-apoptotic action as a major therapeutic mechanism of anti-eosinophilic therapy with GCs. Similar observations of resistance of activated eosinophil to GC-induced apoptosis were reported previously, although no mechanism for such an effect was investigated. An early report by Wallen et al showed that GC inhibited IL-5-induced eosinophil survival in a dose-dependent manner; however, this effect required at least 48 h of GC exposure, and was observed only in eosinophils exposed to IL-5 at concentrations below 23000 fM (∼1 ng/ml) [6]. The next study from the same group extended these observations to 8 different GCs, again confirming their suppressive effects on the prolongation of eosinophil survival induced by low concentrations of IL-5; however, the pro-apoptotic action of even the most potent GCs could be overcome by IL-5 applied at 100 pg/ml [5]. The authors concluded that GCs do inhibit IL-5-mediated eosinophil viability, in spite the observations that survival rate of IL-5 activated eosinophils exposed GC was significantly higher than survival of quiescent cells or cells exposed to GC alone. Interestingly, in the same study fluticasone 17-propionate prolonged survival of quiescent eosinophils, a phenomenon suggesting anti-apoptotic effects of some GCs. Similar observations were reported later by Brode et al showing the potential inhibitory action of IL-5 on GC-induced apoptosis occurring in a dose-dependent manner and reaching maximal effect at 0.1 ng/ml [4].
Our findings of a delayed apoptosis in the first 48 h of incubation of activated eosinophils with Dexamethasone suggest the enhancement of anti-apoptotic signaling by Dex treatment. GCs have been shown to enhance certain IL-5-induced eosinophil functions, as GCs can increase IL-5-induced eosinopoiesis [7], and IL-5 and Dex have been shown to act synergistically in the upregulation of HLA II [25]. Our results suggest that this synergy may depend on the crosstalk between IL-5-induced signaling with the GR-mediated TA pathway. Whether the IL-5-dependent action of GCs observed in our study is related to the therapeutic effectiveness of GCs in vivo is unknown, but the prospects are intriguing. For example, the number of apoptotic eosinophils in the allergic airways is not significantly affected by treatment with inhaled GCs, supporting our in vitro observations on resosistance of activated eosinophils to GCs-induced apoptosis [26,27]. A number of studies reported elevated concentrations of IL-5 in the airways, ranging from 7 pg/ml in the bronchoalveolar lavage fluid (BALF) to 70 pg/ml in the sputum after allergen challenge, the concentration range producing resistance to GC-induced apoptosis in our study [28-30]. Moreover, IL-5 concentrations higher than 1000 pg/ml have been measured in the sera and BALF of patients with eosinophilic pneumonia and hypereosinophilic syndromes (HES)[31,32]. Accordingly, our findings of IL-5-mediated resistance to GC-induced eosinophil apoptosis imply a similar mechanism occurring during exposure to high concentrations of IL-5 in HES, as up to 40% of HES patients show inadequate response to GC therapy [33]. Interestingly, these patients respond favorably to anti-IL-5 therapy and regain responsiveness to GCs [32], another analogy to the restoration of GC-inducible apoptosis observed upon inhibition of IL-5 signaling observed in our study. Along this line, the most efficient clinical response of anti-IL-5 therapy in asthma to date was observed in a selected group of patients characterized by high airway eosinophilia in spite of treatment with high doses of GCs [8,9]. The findings of high eosinophilia, high concentrations of IL-5, aggressive yet inadequate GC therapy, and the effectiveness of anti-IL5 therapy in these patients are consistent with the in vitro conditions tested in the present study.
Here we report for the first time the role of NFIL3 in IL-5 signaling. NFIL3, a basic leucine zipper transcription factor, was first identified as a transcriptional activator of the human IL-3 promoter, and recently has been shown to prevent apoptosis of IL-3-dependent cells during cytokine deprivation [34]. The inducible nature of NFIL-3 expression allowed us to use siRNA to prevent its upregulation; this in turn inhibited the prolongation of eosinophil survival. These observations were complemented with blockade of Pim-1, which prevented upregulation of NFIL3 and inhibited eosinophil survival. Although both Pim-1 and NFIL-3 are thought to depend on Jak and STAT pathways [35,19], the exact mechanism of how Pim-1 regulates NFIL-3 expression is currently not known. Experiments using Pim-1 inhibitors on cancer cells showed significant decrease in NFIL3 expression and phosphorylation of BAD, confirming a functional link between Pim1, NFIL3 and antiapoptotic signaling. Another intriguing question is the mechanism leading to increased expression of NFIL3 via the TA activity of the GR receptor. Our observations that Pim1 is not upregulated by Dex alone, and that a Pim1 inhibitor blocks expression of NFIL-3 expression in IL-5/Dex- stimulated cells, suggest that 1) Dex alone may not upregulate NFIL3, or 2) IL-5 stimulation leads to the expression of additional components necessary for GR transactivation to occur. The other rather trivial possibilities include rapid induction of apoptotic processes induced by Dex in quiescent eosinophils, preventing timely expression of NFIL3. In agreement with this possibility, recently published microarray analysis revealed NFIL3 as one of the several GC-regulated genes associated with the inhibition of apoptosis in breast epithelial cells [14]. Furthermore, although NFIL3 expression was upregulated within 1-2 h of IL-5 stimulation, the possible GR transactivation of NFIL3 may rely on a different mechanism. Previous studies showed the dependence of NFIL-3 expression on new protein synthesis in IL-3-stimulated cells, however, IL-4 and IL-10 induced NFIL3 in the protein synthesis-independent mechanism [36,34,20], supporting the possibility of late, Pim-1-independent transactivation of NFIL3 by Dex.
The observed pro-apoptotic effect of the dissociated GR agonist and delayed apoptosis in response to Dex suggests that the products of GR trans-activated genes protect cells from GC-induced apoptosis. GCs has been shown to activate inhibitors of apoptosis such as the MKP1 (resulting in AP1 inhibition) and the pro-survival serum and GC-inducible protein kinase 1 (SGK1) [37,38]. Signaling from the IL-5 receptor is propagated via a Stat1 and Stat5 mechanism [39,22], and in CD34+ cells a direct GR modulation of STAT5 was suggested to play a role in Dex-induced inhibition of apoptosis [7]. In this regard, GR was shown to act as a co-activator of STAT5 and enhance STAT5-dependent, transcription suggesting possible synergy between GR and STAT in propagating anti-apoptotic signaling [40]. However, since the latter mechanism occurred in a GRE- and thus TA-independent manner, the possible role of GR and STAT5 in enhancement of NFIL3 remains to be investigated.
As a novel piece of information on GR signaling in eosinophils, we found that a non-dimerizing GR agonist with limited TA and preserved TR GR action induced apoptosis in quiescent cells. These results are in contrast to a previous report on T lymphocytes showing that GC-induced apoptosis requires GR TA and is dependent on phosphorylation of GR on S211 [41,13]. How CpdA induces apoptosis in quiescent cells in the absence of apparent pro-inflammatory signals targeted by TR is currently unknown, however, we can't rule out a mechanism of GR-induced apoptosis other than TR and TA occurring through a non-genomic mechanism. In this regard, early activation of JNK kinases and mitochondrial dysfunction occurring during early, Dex-induced apoptosis of eosinophils has been reported [42]; whether the same processes occur upon CpdA treatment remains to be investigated. It was also reported that the aziridine metabolites of CpdA could induce apoptosis of various cell types, including lymphocytes ex vivo, even in the absence of functional GR [43]. Our experiments with the GR antagonist RU486 addressed and excluded this possibility, showing that CpdA induced eosinophil apoptosis in a GR-dependent manner. The molecular target of the TR action of CpdA in IL-5-stimulated eosinophils is currently unknown. Although CpdA has been shown to downregulate AP1, c-fos, and NF-κB signaling in cells stimulated with TH1 and Th17 cytokines [44,45,12,46], only one recent study showed an inhibitory effect on IL-4-mediated STAT-6 nuclear translocation and binding [47]. Our experiments showed no effect of CpdA on IL-5-induced upregulation of Pim1; however, it did inhibit NFIL-3 upregulation in IL-5-stimulated cells, though Dex did not. Thus it is conceivable that signaling molecules repressed by GR are downstream of the Pim-1 kinase.
Although the goal of our use of CpdA was purely mechanistic, to dissociate signaling, the significant implication of our study is that GR-transactivated signals contributing to an IL-5-induced inhibition of apoptosis may ultimately play a role in the resistance to steroid therapy observed in eosinophilic disorders. The goal for the development of selective GR agonists was to diminish multiple metabolic and atrophogenic side effects and overcome resistance to steroid therapy [48]. Our study presents a possible mechanism of resistance to GC-induced apoptosis in activated eosinophils that is driven by Pim-1-induced NFIL-3 and enhanced by GR TA. Blocking of Pim-1/NFIL-3 or selective elimination of GR TA restores GR-mediated apoptosis in IL-5-activated eosinophils. We hope that defining the molecular mechanisms by which GCs regulate NFIL-3 in activated eosinophils will lead to the development of novel approaches for the treatment of eosinophilic disorders resistant to steroid therapy.
Acknowledgments
We thank Dr. David Konkel (Institute for Translational Sciences, UTMB) for his scientific input and for critical editing of the manuscript. We also thank Dr. Mark Griffin (Microbiology and Immunology, UTMB) for his input on the flow cytometry analysis.
Funding: This study was supported by the NIH/NCRR KL2RR029875 (to KP) and NIH/NHLBI Proteomics Initiative NO1-HV-28184 (to AK)
Abbreviations used in this paper
- AAD
aminoactinomycin
- AP1
the Activator Protein 1
- Bcl2
B-cell lymphoma 2
- BclXL
B-cell lymphoma-extra large
- CpdA
Compound A
- IL
interleukin
- HES
hypereosinophilic syndromes
- Jak
Janus tyrosine kinase
- JNK
c-Jun N-terminal kinases
- Mcl-1
myeloid leukemia cell differentiation protein 1
- MKP1
MAP kinase phosphatase
- NFIL3
Nuclear factor Interleuki-3 regulated
- Pim1
Proviral integration site for Moloney murine leukemia virus 1
- Stat
Signal Transducer and Activator of Transcription
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
References
- 1.Barnes PJ. Mechanisms of action of glucocorticoids in asthma. Am J Respir Crit Care Med. 1996;154(2 Pt 2):S21–26. doi: 10.1164/ajrccm/154.2_Pt_2.S21. discussion S26-27. [DOI] [PubMed] [Google Scholar]
- 2.Klion A. Hypereosinophilic syndrome: current approach to diagnosis and treatment. Annu Rev Med. 2009;60:293–306. doi: 10.1146/annurev.med.60.062107.090340. [DOI] [PubMed] [Google Scholar]
- 3.Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol. 2006;117(3):522–543. doi: 10.1016/j.jaci.2006.01.032. [DOI] [PubMed] [Google Scholar]
- 4.Brode S, Farahi N, Cowburn AS, Juss JK, Condliffe AM, Chilvers ER. Interleukin-5 inhibits glucocorticoid-mediated apoptosis in human eosinophils. Thorax. 2010;65(12):1116–1117. doi: 10.1136/thx.2009.124909. [DOI] [PubMed] [Google Scholar]
- 5.Hagan JB, Kita H, Gleich GJ. Inhibition of interleukin-5 mediated eosinophil viability by fluticasone 17-propionate: comparison with other glucocorticoids. Clin Exp Allergy. 1998;28(8):999–1006. doi: 10.1046/j.1365-2222.1998.00363.x. [DOI] [PubMed] [Google Scholar]
- 6.Wallen N, Kita H, Weiler D, Gleich GJ. Glucocorticoids inhibit cytokine-mediated eosinophil survival. J Immunol. 1991;147(10):3490–3495. [PubMed] [Google Scholar]
- 7.Debierre-Grockiego F, Fuentes V, Prin L, Gouilleux F, Gouilleux-Gruart V. Differential effect of dexamethasone on cell death and STAT5 activation during in vitro eosinopoiesis. Br J Haematol. 2003;123(5):933–941. doi: 10.1046/j.1365-2141.2003.04700.x. [DOI] [PubMed] [Google Scholar]
- 8.Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, Marshall RP, Bradding P, Green RH, Wardlaw AJ, Pavord ID. Mepolizumab and exacerbations of refractory eosinophilic asthma. N Engl J Med. 2009;360(10):973–984. doi: 10.1056/NEJMoa0808991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair P, Pizzichini MM, Kjarsgaard M, Inman MD, Efthimiadis A, Pizzichini E, Hargreave FE, O'Byrne PM. Mepolizumab for prednisone-dependent asthma with sputum eosinophilia. N Engl J Med. 2009;360(10):985–993. doi: 10.1056/NEJMoa0805435. [DOI] [PubMed] [Google Scholar]
- 10.Pavord ID, Haldar P, Bradding P, Wardlaw AJ. Mepolizumab in refractory eosinophilic asthma. Thorax. 2010;65(4):370. doi: 10.1136/thx.2009.122697. [DOI] [PubMed] [Google Scholar]
- 11.Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013;154(3):993–1007. doi: 10.1210/en.2012-2045. [DOI] [PubMed] [Google Scholar]
- 12.De Bosscher K, Vanden Berghe W, Vermeulen L, Plaisance S, Boone E, Haegeman G. Glucocorticoids repress NF-kappaB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivator levels in the cell. Proc Natl Acad Sci U S A. 2000;97(8):3919–3924. doi: 10.1073/pnas.97.8.3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller AL, Webb MS, Copik AJ, Wang Y, Johnson BH, Kumar R, Thompson EB. p38 Mitogen-activated protein kinase (MAPK) is a key mediator in glucocorticoid-induced apoptosis of lymphoid cells: correlation between p38 MAPK activation and site-specific phosphorylation of the human glucocorticoid receptor at serine 211. Mol Endocrinol. 2005;19(6):1569–1583. doi: 10.1210/me.2004-0528. [DOI] [PubMed] [Google Scholar]
- 14.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64(5):1757–1764. doi: 10.1158/0008-5472.can-03-2546. [DOI] [PubMed] [Google Scholar]
- 15.Robertson S, Allie-Reid F, Vanden Berghe W, Visser K, Binder A, Africander D, Vismer M, De Bosscher K, Hapgood J, Haegeman G, Louw A. Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound a. J Biol Chem. 2010;285(11):8061–8075. doi: 10.1074/jbc.M109.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Straub C, Pazdrak K, Young TW, Stafford SJ, Wu Z, Wiktorowicz JE, Haag AM, English RD, Soman KV, Kurosky A. Toward the Proteome of the Human Peripheral Blood Eosinophil. Proteomics Clin Appl. 2009;3(10):1151–1173. doi: 10.1002/prca.200900043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walsh GM, Dewson G, Wardlaw AJ, Levi-Schaffer F, Moqbel R. A comparative study of different methods for the assessment of apoptosis and necrosis in human eosinophils. J Immunol Methods. 1998;217(1-2):153–163. doi: 10.1016/s0022-1759(98)00103-3. [DOI] [PubMed] [Google Scholar]
- 18.Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans. 2006;34(Pt 6):1018–1023. doi: 10.1042/BST0341018. [DOI] [PubMed] [Google Scholar]
- 19.Yu YL, Chiang YJ, Yen JJ. GATA factors are essential for transcription of the survival gene E4bp4 and the viability response of interleukin-3 in Ba/F3 hematopoietic cells. J Biol Chem. 2002;277(30):27144–27153. doi: 10.1074/jbc.M200924200. [DOI] [PubMed] [Google Scholar]
- 20.Kashiwada M, Levy DM, McKeag L, Murray K, Schroder AJ, Canfield SM, Traver G, Rothman PB. IL-4-induced transcription factor NFIL3/E4BP4 controls IgE class switching. Proc Natl Acad Sci U S A. 2010;107(2):821–826. doi: 10.1073/pnas.0909235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Andina N, Didichenko S, Schmidt-Mende J, Dahinden CA, Simon HU. Proviral integration site for Moloney murine leukemia virus 1, but not phosphatidylinositol-3 kinase, is essential in the antiapoptotic signaling cascade initiated by IL-5 in eosinophils. J Allergy Clin Immunol. 2009;123(3):603–611. doi: 10.1016/j.jaci.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Stout BA, Bates ME, Liu LY, Farrington NN, Bertics PJ. IL-5 and granulocyte-macrophage colony-stimulating factor activate STAT3 and STAT5 and promote Pim-1 and cyclin D3 protein expression in human eosinophils. J Immunol. 2004;173(10):6409–6417. doi: 10.4049/jimmunol.173.10.6409. [DOI] [PubMed] [Google Scholar]
- 23.Keeton EK, McEachern K, Dillman KS, Palakurthi S, Cao Y, Grondine MR, Kaur S, Wang S, Chen Y, Wu A, Shen M, Gibbons FD, Lamb ML, Zheng X, Stone RM, Deangelo DJ, Platanias LC, Dakin LA, Chen H, Lyne PD, Huszar D. AZD1208, a potent and selective pan-Pim kinase inhibitor, demonstrates efficacy in preclinical models of acute myeloid leukemia. Blood. 2014;123(6):905–913. doi: 10.1182/blood-2013-04-495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilmarinen P, Kankaanranta H. Eosinophil apoptosis as a therapeutic target in allergic asthma. Basic Clin Pharmacol Toxicol. 2014;114(1):109–117. doi: 10.1111/bcpt.12163. [DOI] [PubMed] [Google Scholar]
- 25.Guida L, O'Hehir RE, Hawrylowicz CM. Synergy between dexamethasone and interleukin-5 for the induction of major histocompatibility complex class II expression by human peripheral blood eosinophils. Blood. 1994;84(8):2733–2740. [PubMed] [Google Scholar]
- 26.Duncan CJ, Lawrie A, Blaylock MG, Douglas JG, Walsh GM. Reduced eosinophil apoptosis in induced sputum correlates with asthma severity. Eur Respir J. 2003;22(3):484–490. doi: 10.1183/09031936.03.00109803a. [DOI] [PubMed] [Google Scholar]
- 27.Gibson PG, Saltos N, Fakes K. Acute anti-inflammatory effects of inhaled budesonide in asthma: a randomized controlled trial. Am J Respir Crit Care Med. 2001;163(1):32–36. doi: 10.1164/ajrccm.163.1.9807061. [DOI] [PubMed] [Google Scholar]
- 28.Teran LM, Carroll MP, Shute JK, Holgate ST. Interleukin 5 release into asthmatic airways 4 and 24 hours after endobronchial allergen challenge: its relationship with eosinophil recruitment. Cytokine. 1999;11(7):518–522. doi: 10.1006/cyto.1998.0457. [DOI] [PubMed] [Google Scholar]
- 29.Park SW, Jangm HK, An MH, Min JW, Jang AS, Lee JH, Park CS. Interleukin-13 and interleukin-5 in induced sputum of eosinophilic bronchitis: comparison with asthma. Chest. 2005;128(4):1921–1927. doi: 10.1378/chest.128.4.1921. [DOI] [PubMed] [Google Scholar]
- 30.Sulakvelidze I, Inman MD, Rerecich T, O'Byrne PM. Increases in airway eosinophils and interleukin-5 with minimal bronchoconstriction during repeated low-dose allergen challenge in atopic asthmatics. Eur Respir J. 1998;11(4):821–827. doi: 10.1183/09031936.98.11040821. [DOI] [PubMed] [Google Scholar]
- 31.Ueno T, Miyazaki E, Ando M, Nureki S, Kumamoto T. Osteopontin levels are elevated in patients with eosinophilic pneumonia. Respirology. 2010;15(7):1111–1121. doi: 10.1111/j.1440-1843.2010.01825.x. [DOI] [PubMed] [Google Scholar]
- 32.Kay AB, Klion AD. Anti-interleukin-5 therapy for asthma and hypereosinophilic syndrome. Immunol Allergy Clin North Am. 2004;24(4):645–666, vii. doi: 10.1016/j.iac.2004.06.007. [DOI] [PubMed] [Google Scholar]
- 33.Ogbogu PU, Bochner BS, Butterfield JH, Gleich GJ, Huss-Marp J, Kahn JE, Leiferman KM, Nutman TB, Pfab F, Ring J, Rothenberg ME, Roufosse F, Sajous MH, Sheikh J, Simon D, Simon HU, Stein ML, Wardlaw A, Weller PF, Klion AD. Hypereosinophilic syndrome: a multicenter, retrospective analysis of clinical characteristics and response to therapy. J Allergy Clin Immunol. 2009;124(6):1319–1325 e1313. doi: 10.1016/j.jaci.2009.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ikushima S, Inukai T, Inaba T, Nimer SD, Cleveland JL, Look AT. Pivotal role for the NFIL3/E4BP4 transcription factor in interleukin 3-mediated survival of pro-B lymphocytes. Proc Natl Acad Sci U S A. 1997;94(6):2609–2614. doi: 10.1073/pnas.94.6.2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kirschner AN, Wang J, van der Meer R, Anderson PD, Franco-Coronel OE, Kushner MH, Everett JH, Hameed O, Keeton EK, Ahdesmaki M, Grosskurth SE, Huszar D, Abdulkadir SA. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. J Natl Cancer Inst. 2015;107(2) doi: 10.1093/jnci/dju407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rothman PB. The transcriptional regulator NFIL3 controls IgE production. Trans Am Clin Climatol Assoc. 2010;121:156–171. discussion 171. [PMC free article] [PubMed] [Google Scholar]
- 37.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276(20):16649–16654. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- 38.Kristiansen M, Hughes R, Patel P, Jacques TS, Clark AR, Ham J. Mkp1 is a c-Jun target gene that antagonizes JNK-dependent apoptosis in sympathetic neurons. J Neurosci. 2010;30(32):10820–10832. doi: 10.1523/JNEUROSCI.2824-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pazdrak K, Stafford S, Alam R. The activation of the Jak-STAT 1 signaling pathway by IL-5 in eosinophils. J Immunol. 1995;155(1):397–402. [PubMed] [Google Scholar]
- 40.Stocklin E, Wissler M, Gouilleux F, Groner B. Functional interactions between Stat5 and the glucocorticoid receptor. Nature. 1996;383(6602):726–728. doi: 10.1038/383726a0. [DOI] [PubMed] [Google Scholar]
- 41.Planey SL, Litwack G. Glucocorticoid-induced apoptosis in lymphocytes. Biochem Biophys Res Commun. 2000;279(2):307–312. doi: 10.1006/bbrc.2000.3922. [DOI] [PubMed] [Google Scholar]
- 42.Zhang JP, Wong CK, Lam CW. Role of caspases in dexamethasone-induced apoptosis and activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in human eosinophils. Clin Exp Immunol. 2000;122(1):20–27. doi: 10.1046/j.1365-2249.2000.01344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wust S, Tischner D, John M, Tuckermann JP, Menzfeld C, Hanisch UK, van den Brandt J, Luhder F, Reichardt HM. Therapeutic and adverse effects of a non-steroidal glucocorticoid receptor ligand in a mouse model of multiple sclerosis. PLoS One. 2009;4(12):e8202. doi: 10.1371/journal.pone.0008202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Bosscher K, Vanden Berghe W, Beck IM, Van Molle W, Hennuyer N, Hapgood J, Libert C, Staels B, Louw A, Haegeman G. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci U S A. 2005;102(44):15827–15832. doi: 10.1073/pnas.0505554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.De Bosscher K, Vanden Berghe W, Haegeman G. Glucocorticoid repression of AP-1 is not mediated by competition for nuclear coactivators. Mol Endocrinol. 2001;15(2):219–227. doi: 10.1210/mend.15.2.0591. [DOI] [PubMed] [Google Scholar]
- 46.Yemelyanov A, Czwornog J, Gera L, Joshi S, Chatterton RT, Jr, Budunova I. Novel steroid receptor phyto-modulator compound a inhibits growth and survival of prostate cancer cells. Cancer Res. 2008;68(12):4763–4773. doi: 10.1158/0008-5472.CAN-07-6104. [DOI] [PubMed] [Google Scholar]
- 47.Reber LL, Daubeuf F, Plantinga M, De Cauwer L, Gerlo S, Waelput W, Van Calenbergh S, Tavernier J, Haegeman G, Lambrecht BN, Frossard N, De Bosscher K. A dissociated glucocorticoid receptor modulator reduces airway hyperresponsiveness and inflammation in a mouse model of asthma. J Immunol. 2012;188(7):3478–3487. doi: 10.4049/jimmunol.1004227. [DOI] [PubMed] [Google Scholar]
- 48.Belvisi MG, Wicks SL, Battram CH, Bottoms SE, Redford JE, Woodman P, Brown TJ, Webber SE, Foster ML. Therapeutic benefit of a dissociated glucocorticoid and the relevance of in vitro separation of transrepression from transactivation activity. J Immunol. 2001;166(3):1975–1982. doi: 10.4049/jimmunol.166.3.1975. [DOI] [PubMed] [Google Scholar]