

In this prospective phase III trial, afatinib combined with paclitaxel improved progression-free survival and objective response, compared with single-agent chemotherapy, in patients with NSCLC who were clinically enriched for ErbB dependency having failed platinum-based chemotherapy, gefitinib/erlotinib and afatinib monotherapy after initial benefit on each tyrosine kinase inhibitor.
Keywords: afatinib, paclitaxel, NSCLC, squamous cell
Abstract
Background
Afatinib has demonstrated clinical benefit in patients with non-small-cell lung cancer progressing after treatment with erlotinib/gefitinib. This phase III trial prospectively assessed whether continued irreversible ErbB-family blockade with afatinib plus paclitaxel has superior outcomes versus switching to chemotherapy alone in patients acquiring resistance to erlotinib/gefitinib and afatinib monotherapy.
Patients and methods
Patients with relapsed/refractory disease following ≥1 line of chemotherapy, and whose tumors had progressed following initial disease control (≥12 weeks) with erlotinib/gefitinib and thereafter afatinib (50 mg/day), were randomized 2:1 to receive afatinib plus paclitaxel (40 mg/day; 80 mg/m2/week) or investigator's choice of single-agent chemotherapy. The primary end point was progression-free survival (PFS). Other end points included objective response rate (ORR), overall survival (OS), safety and patient-reported outcomes.
Results
Two hundred and two patients with progressive disease following clinical benefit from afatinib were randomized to afatinib plus paclitaxel (n = 134) or single-agent chemotherapy (n = 68). PFS (median 5.6 versus 2.8 months, hazard ratio 0.60, P = 0.003) and ORR (32.1% versus 13.2%, P = 0.005) significantly improved with afatinib plus paclitaxel. There was no difference in OS. Global health status/quality of life was maintained with afatinib plus paclitaxel over the entire treatment period. The median treatment duration was 133 and 51 days with afatinib plus paclitaxel and single-agent chemotherapy, respectively; 48.5% of patients receiving afatinib plus paclitaxel and 30.0% of patients receiving single-agent chemotherapy experienced drug-related grade 3/4 adverse events. Treatment-related adverse events were consistent with those previously reported with each agent.
Conclusion
Afatinib plus paclitaxel improved PFS and ORR compared with single-agent chemotherapy in patients who acquired resistance to erlotinib/gefitinib and progressed on afatinib after initial benefit. LUX-Lung 5 is the first prospective trial to demonstrate the benefit of continued ErbB targeting post-progression, versus switching to single-agent chemotherapy.
Trial registration number
NCT01085136 (clinicaltrials.gov).
introduction
Patients with non-small-cell lung cancer (NSCLC) harboring activating epidermal growth factor receptor (EGFR) mutations are exquisitely sensitive to first-line treatment with EGFR tyrosine kinase inhibitors (TKIs) [1]. However, almost all patients acquire resistance, leading to disease progression [2]. It has been hypothesized that, at the time of progression, NSCLC tumors comprise a heterogeneous mix of cells, some of which remain sensitive to EGFR inhibition [3]. Clinical observations are consistent with this hypothesis; withdrawal of an EGFR TKI at the onset of resistance can lead to rapid tumor growth [4, 5]. Therefore, there may be rationale for continued exposure to EGFR TKIs in combination with post-progression therapy. Indeed, recent retrospective [6] and single-arm [7] studies in patients with confirmed EGFR mutations suggested that continuation of EGFR TKIs beyond progression may improve outcomes in NSCLC patients.
Afatinib is a potent, irreversible ErbB family blocker that inhibits tyrosine kinase activity of EGFR, human epidermal growth factor 2 (HER2, ErbB2), ErbB4 and all relevant ErbB family dimers [8, 9]. Afatinib monotherapy has demonstrated progression-free survival (PFS) and overall survival (OS) benefits versus platinum-based doublet chemotherapy in previously untreated patients with NSCLC with activating EGFR mutations [10–12]. It has also demonstrated modest clinical activity in patients who had progressed following gefitinib and/or erlotinib [13, 14]. In order to prospectively evaluate the benefit of continued ErbB targeting beyond progression, this study was conducted in two parts. Part A enrolled patients who had failed ≥1 line of chemotherapy and progressed following ≥12 weeks' clinical benefit on erlotinib/gefitinib. All patients received single-agent afatinib (50 mg daily) to identify patients who derive clinical benefit from ErbB blockade, from a uniformly treated ‘late line’ population. The experimental part of the study (Part B) compared afatinib plus paclitaxel versus single-agent chemotherapy in patients who progressed following ≥12 weeks' clinical benefit from afatinib given as third- or higher-line treatment. Paclitaxel was chosen as a combination partner based on preclinical evidence for synergism [15]. The safety and maximum tolerated dose (MTD) of the combination has been previously established [16]. As there is no established biomarker to predict clinical benefit from afatinib in a greater than or equal to third-line setting, ≥12 weeks of disease control from afatinib monotherapy were chosen as the primary selection criterion for enrollment in the randomized study.
methods
Patients had stage IIIb (wet) or IV NSCLC with measurable disease and had failed treatment with ≥1 line of chemotherapy (including platinum and pemetrexed) and erlotinib/gefitinib after ≥12 weeks of treatment, and must have attained ≥12 weeks' clinical benefit on afatinib monotherapy [complete response (CR), partial response (PR) or stable disease (SD)] with subsequent progression according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Documentation of EGFR mutation status was not mandatory as this was not universal clinical practice at the time of study planning and conduct. Patients were ≥18 years old, had Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 and life expectancy of ≥12 weeks. Exclusion criteria included absence of clinical benefit from afatinib monotherapy; abnormal hepatic, renal or hematologic function. Patients provided written informed consent before afatinib monotherapy and before randomization. This trial is registered at clinicaltrials.gov (NCT01085136).
study design
Patients were randomized 2:1 to afatinib plus paclitaxel or investigator's choice of single-agent chemotherapy. Randomization was stratified based on duration of benefit on prior gefitinib/afatinib (≥6 versus <6 months) and sex. Patients were treated until disease progression, unacceptable tolerability or withdrawal of consent.
The primary end point was PFS. Secondary end points included OS, and objective response (OR). Other end points included health-related quality of life (QoL; defined as time-to-deterioration for cough, dyspnea and pain (supplementary Methods S1, available at Annals of Oncology online) and safety.
The study protocol, designed in accordance with the Declaration of Helsinki, was approved by institutional review boards and the responsible regulatory authorities of each participating country. An independent Data and Safety Monitoring Committee (DMC) monitored study progress.
treatments
Patients randomized to afatinib plus paclitaxel received 40 mg daily and 80 mg/m2 weekly, respectively (the MTD) [16]. Patients who had two dose reductions on afatinib monotherapy during Part A (i.e. eventually received 30 mg daily) were permitted to start on 30 mg daily in Part B. Investigator's choice of chemotherapy was administered according to the package insert. Up to two 10 mg dose reductions of afatinib were permitted if patients encountered any grade ≥3 drug-related adverse events [AEs; assessed according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 3.0], or grade 2 diarrhea lasting ≥2 days or nausea or vomiting for ≥7 consecutive days despite best supportive care. Paclitaxel dose reductions were conducted according to the Summary of Product Characteristics.
assessments
Tumor assessments were carried out by computed tomography or magnetic resonance imaging of less than or equal to five target lesions at baseline and every 8 weeks thereafter. Response was evaluated by the investigator. AE assessments were carried out weekly. Patients were assessed for the likelihood of being EGFR mutation-positive based on a modification of published clinical enrichment criteria (higher clinical enrichment criteria: CR/PR or ≥48 weeks of benefit on erlotinib/gefitinib) [2].
statistical plan
PFS and OS were assessed using a stratified log-rank test. A stratified Cox proportional hazards model was used to derive the hazard ratios (HRs) and 95% confidence intervals (CIs). The Kaplan–Meier analysis was used to estimate the median survival and 95% CIs at landmark time points. Differences in objective response rate (ORR; CR plus PR) and disease control rate (DCR; ORR plus SD) were assessed by logistic regression.
For the primary analysis, 351 patients (279 PFS events) would be required to provide 90% power at a two-sided 5% significance level for the log-rank test, assuming an HR of 0.67 in favor of afatinib plus paclitaxel. Assuming an exponentially distributed median PFS of 8 weeks with afatinib monotherapy, 35% of patients would be alive and progression free at 12 weeks. Therefore, ∼1100 patients needed to be treated with afatinib in Part A, assuming an ineligibility rate of 10%. Enrollment to Part B was lower than anticipated due to several unforeseen factors, including: patients' desire for a break in treatment, patients' refusal to proceed due to non-disease-related reasons and the requirement for screening to be within 14 days of progression under afatinib monotherapy. Consequently, the calculated number of 351 eligible patients (279 PFS events) was considered unachievable, and the protocol was amended following discussion with the DMC on 18 January 2013. The planned time points for the primary analysis of PFS and OS were amended to be undertaken once the final randomized patients had the chance to be followed for at least 6 months. Primary analysis was thus undertaken on 10 December 2013 after 163 PFS events had occurred.
results
patients
This trial was conducted in 115 enrolling centers in 23 countries. Between April 2010 and May 2011, 1154 patients were treated with afatinib monotherapy (supplementary Table S1, available at Annals of Oncology online). Of those, 223 patients with clinical benefit of at least 12 weeks were screened and 202 patients were randomized (134 to afatinib plus paclitaxel and 68 to chemotherapy; supplementary Figure S1, available at Annals of Oncology online). Reasons for not being randomized following afatinib monotherapy included <12 weeks of clinical benefit (n = 625) and exclusion at screening (n = 21). At the time of database lock, seven patients were still on treatment in Part A. Of the remaining 299 patients who were not randomized following progression on afatinib monotherapy, most declined participation due to general health deterioration, treatment-related, or other reasons (supplementary Table S1, available at Annals of Oncology online). The demographics and clinical characteristics of patients treated with afatinib monotherapy are shown in supplementary Table S2, available at Annals of Oncology online. There were no discernable differences in baseline characteristics between the 202 patients randomized to Part B and the 299 patients who were not randomized (supplementary Table S3, available at Annals of Oncology online).
Of the 202 patients randomized, 132 patients received afatinib plus paclitaxel and 60 received single-agent chemotherapy (Table 1). Few patients (13%) in the chemotherapy arm had received the same agent previously; six paclitaxel, one docetaxel and one pemetrexed.
Table 1.
Patient demographics and clinical characteristics
| Characteristic |
n = 202 |
|
|---|---|---|
| Afatinib plus paclitaxel (n = 134) | Chemotherapya (n = 68) | |
| Female, n (%) | 65 (48.5) | 34 (50.0) |
| Median age (years) | 60.0 | 60.5 |
| Baseline ECOG status, n (%) | ||
| 0 | 47 (35.1) | 14 (20.6) |
| 1 | 77 (57.5) | 46 (67.6) |
| 2 | 10 (7.5) | 8 (11.8) |
| Race, n (%) | ||
| East Asianb | 52 (38.8) | 30 (44.1) |
| Caucasian | 53 (39.6) | 24 (35.3) |
| Other | 3 (2.2) | 2 (2.9) |
| Unknownc | 26 (19.4) | 12 (17.6) |
| Smoking status, n (%) | ||
| Never smoked | 71 (53.0) | 37 (54.4) |
| <15 pack-years, stopped | 14 (10.4) | 10 (14.7) |
| >1 year before diagnosis | ||
| Current/other ex-smoker | 49 (36.6) | 21 (30.9) |
| Clinical stage at screening, n (%) | ||
| IIIb | 1 (0.7) | 3 (4.4) |
| IV | 133 (99.3) | 65 (95.6) |
| Tumor histology, n (%) | ||
| Adenocarcinoma | 113 (84.3) | 61 (89.7) |
| Squamous | 11 (8.2) | 6 (8.8) |
| Other | 10 (7.5) | 1 (1.5) |
| Centrally confirmed activating EGFR mutation status, n (%) | ||
| Positive | 6 (4.5) | 3 (4.4) |
| Negative | 2 (1.5) | 3 (4.4) |
| Higher clinical enrichment criteria,d n (%) | ||
| Yes | 78 (58.2) | 42 (61.8) |
| No | 56 (41.8) | 26 (38.2) |
| Prior EGFR TKI therapy, n (%) | ||
| Erlotinib | 96 (71.6) | 47 (69.1) |
| Gefitinib | 32 (23.9) | 16 (23.5) |
| Both | 6 (4.5) | 5 (7.4) |
| Lines of prior chemotherapy, n (%) | ||
| 0 | 5 (3.7) | 2 (2.9) |
| 1 | 41 (30.6) | 12 (17.6) |
| 2 | 39 (29.1) | 28 (41.2) |
| >2 | 49 (36.6) | 26 (38.2) |
| Previous pemetrexed, n (%) | ||
| Yes | 72 (53.7) | 39 (57.4) |
| No | 62 (46.3) | 29 (42.6) |
| Previous taxane, n (%) | ||
| Yes | 67 (50.0) | 38 (55.9) |
| No | 67 (50.0) | 30 (44.1) |
CR, complete response; ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor; PR, partial response; TKI, tyrosine kinase inhibitor.
aPaclitaxel (35.0%), docetaxel (15.0%), pemetrexed (26.7%), vinorelbine (8.3%), gemcitabine (6.7%), carboplatin (1.7%), non-protocol defined chemotherapy (6.7%).
bChina, Taiwan or Korea.
cDue to local regulations that did not allow the collection of such information.
dDefined as CR or PR to prior EGFR TKI or ≥48 weeks' treatment with prior EGFR TKI.
primary efficacy end point
The median PFS was significantly longer with afatinib plus paclitaxel versus chemotherapy alone (5.6 versus 2.8 months, HR 0.60, 95% CI 0.43–0.85, P = 0.003, Figure 1A). PFS benefit was consistent across predefined subgroups (supplementary Figure S2, available at Annals of Oncology online) and was apparent versus individual chemotherapy regimens, although patient numbers in these analyses were small (supplementary Table S4, available at Annals of Oncology online). The median PFS attained in patients treated with paclitaxel monotherapy, pemetrexed monotherapy or other cytotoxic monotherapies was 3.8 (n = 21), 2.9 (n = 16) and 2.1 (n = 23) months, respectively.
Figure 1.
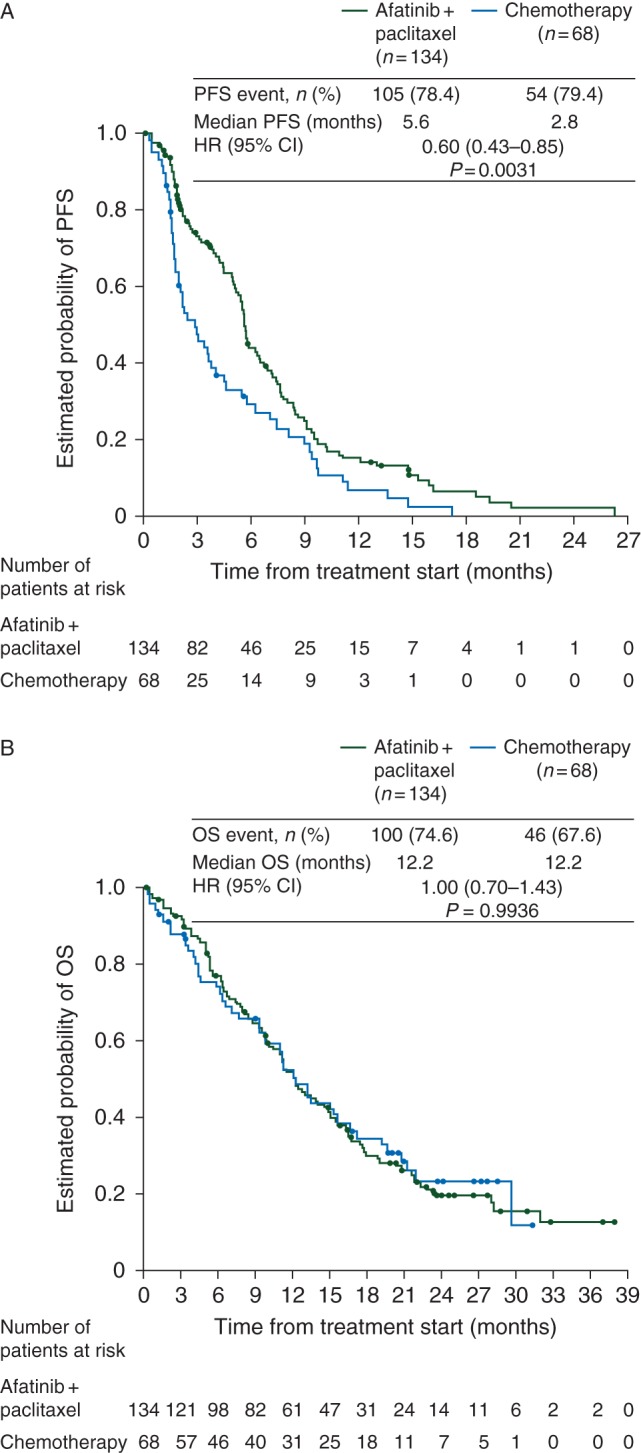
Kaplan–Meier curves for (A) PFS and (B) OS. CI, confidence interval; HR, hazard ratio; OS, overall survival; PFS, progression-free survival.
secondary efficacy end points
Clinical benefit rate (74.5 versus 45.6%, OR 3.41, 95% CI 1.85–6.26, P < 0.0001) and ORR (32.1 versus 13.2%, OR 3.41, 95% CI 1.41–6.79, P = 0.005) were superior with afatinib plus paclitaxel versus chemotherapy (Table 2). The median maximum percentage decrease from baseline sum of target lesion diameters was 15.1% and 1.2%, respectively (supplementary Figure S3, available at Annals of Oncology online). Tumor response and PFS for patients treated with afatinib monotherapy are shown in supplementary Table S5 and Figure S4, available at Annals of Oncology online.
Table 2.
Tumor response
| Outcome |
n = 202 |
|
|---|---|---|
| Afatinib + paclitaxel (n = 134) | Chemotherapy (n = 68) | |
| Disease control, n (%)a | 100 (74.6) | 31 (45.6) |
| Objective response, n (%)b | 43 (32.1) | 9 (13.2) |
| CR | 1 (0.7) | 0 (0.0) |
| PR | 42 (31.3) | 9 (13.2) |
| SD | 57 (42.5) | 22 (32.4) |
| PD | 19 (14.2) | 22 (32.4) |
| NE | 15 (11.2) | 15 (22.1) |
| Median duration of objective response, months | 4.2 | 3.3 |
CI, confidence interval; CR, complete response; NE, not evaluable; PD, progressive disease; PR, partial response; SD, stable disease.
aOdds ratio 3.41, 95% CI 1.85–6.26, P < 0.0001.
bOdds ratio 3.41, 95% CI 1.41–6.79, P = 0.005.
There was no difference in the median OS between treatment arms (12.2 versus 12.2 months, HR 1.00, 95% CI 0.70–1.43, P = 0.994, Figure 1B). Approximately 60% of patients received greater than or equal to one post-progression therapy. More patients in the chemotherapy arm received two additional lines of therapy than in the afatinib plus paclitaxel arm (36% versus 15%, supplementary Figure S5 and Table S6, available at Annals of Oncology online). Time to deterioration of global health status/quality of life and predefined symptoms was not adversely affected by treatment with afatinib plus paclitaxel versus single-agent chemotherapy (supplementary Table S7, available at Annals of Oncology online).
safety
The median exposure to afatinib plus paclitaxel was longer than for chemotherapy alone (133 versus 51 days). Eighty-two percent of patients started on the 40 mg afatinib dose. A total of 27.3% and 4.5% of patients required one or two afatinib dose reductions, respectively; 23.5% and 35.6% of patients required one or two paclitaxel dose reductions, respectively.
The most common treatment-related AEs in the combination arm were diarrhea (53.8%), alopecia (32.6%), asthenia (27.3%), decreased appetite (22.0%) and rash (20.5%, Table 3). The incidence of treatment-related peripheral neuropathy was 9.1% versus 8.3% with chemotherapy. Serious treatment-related AEs were reported in 11.4% and 3.3% of patients in the combination and chemotherapy arms, respectively. Dose-reduction rates due to AEs were 32.6% in the combination arm and 11.7% with chemotherapy. Discontinuation rates due to drug-related AEs were 18.9% in the combination arm and 6.7% with chemotherapy. One patient experienced a fatal AE of pneumonia that was considered treatment-related (attributed to paclitaxel).
Table 3.
Drug-related AEs reported in >10% of patients (ordered by rate of occurrence in afatinib + paclitaxel arm)
| Clinical characteristic | Afatinib + paclitaxel (n = 132) |
Chemotherapy (n = 60) |
||
|---|---|---|---|---|
| All grades, n (%) | Grade ≥3, n (%) | All grades, n (%) | Grade ≥3, n (%) | |
| Patients with any drug-related AE | 117 (88.6) | 64 (48.5) | 42 (70.0) | 18 (30.0) |
| Leading to discontinuation | 25 (18.9) | 13 (9.8) | 4 (6.7) | 2 (3.3) |
| Diarrhea | 71 (53.8) | 16 (12.1) | 4 (6.7) | 0 (0) |
| Alopecia | 43 (32.6) | 1 (0.8) | 9 (15.0) | 3 (5.0) |
| Asthenia | 36 (27.3) | 11 (8.3) | 17 (28.3) | 2 (3.3) |
| Decreased appetite | 29 (22.0) | 2 (1.5) | 10 (16.7) | 1 (1.7) |
| Fatigue | 27 (20.5) | 6 (4.5) | 9 (15.0) | 3 (5.0) |
| Rash | 27 (20.5) | 2 (1.5) | 6 (10.0) | 0 (0) |
| Neutropenia | 24 (18.2) | 15 (11.3) | 8 (13.3) | 5 (8.3) |
| Nausea | 23 (17.4) | 2 (1.5) | 10 (16.7) | 1 (1.7) |
| Paronychia | 23 (17.4) | 3 (2.3) | 0 (0) | 0 (0) |
| Vomiting | 21 (15.9) | 3 (2.3) | 4 (6.7) | 0 (0) |
| Anemia | 20 (15.2) | 5 (3.8) | 3 (5.0) | 0 (0) |
| Leukopenia | 20 (15.2) | 6 (4.5) | 7 (11.7) | 3 (5.0) |
| Epistaxis | 16 (12.1) | 0 (0) | 1 (1.7) | 0 (0) |
| Stomatitis | 13 (9.8) | 2 (1.5) | 2 (3.3) | 0 (0) |
| Mucosal inflammation | 12 (9.1) | 1 (0.8) | 0 (0) | 0 (0) |
| Pruritus | 10 (7.6) | 0 (0) | 3 (5.0) | 1 (1.7) |
| Dry skin | 6 (4.5) | 0 (0) | 0 (0) | 0 (0) |
AEs, adverse events.
discussion
In this study, afatinib plus paclitaxel significantly improved PFS and ORR versus single-agent chemotherapy in patients who had failed platinum-based chemotherapy, erlotinib/gefitinib and afatinib, after achieving an initial benefit on each TKI. Randomized patients were clinically enriched based on their response to afatinib monotherapy and had previously achieved a median PFS of 5.6 months from initiation of afatinib treatment. Thus, by clinically confirming continued sensitivity to EGFR inhibition and ErbB-family blockade, over multiple lines of therapy, this study identified a selected population of patients who achieved a median of nearly 1 year of clinical benefit following failure of chemotherapy and gefitinib or erlotinib.
Owing to the lack of precedent for prospective trials in the greater than or equal to fourth-line setting, we acknowledge a number of dilemmas in design and conduct with LUX-Lung 5. One of these was the use of physician's choice of single-agent chemotherapy. This was a pragmatic decision, reflecting the absence of evidence-based, standard late-line treatment options in NSCLC. However, subset analysis (albeit with small patient numbers) indicated that there was a trend toward improved PFS with afatinib/paclitaxel versus individual chemotherapy comparators. It was also a pragmatic decision for response to be assessed locally by investigators. We wanted treating oncologists to decide the point at which patients moved from one line of treatment to the next, thus reflecting clinical practice and medical need in a late-line setting. In support, recent studies indicate that local site evaluation has a high correlation to blinded independent central review and does not bias trial outcomes [17, 18]. Another limitation was our failure to enroll the planned number of 351 patients in the randomized study. Since the randomized patients were recruited from the afatinib-treated pool in Part A which was fully recruited before the high attrition was recognized, a decision was made in discussion with the independent DMC to analyze the randomized part of the trial without going back to increase recruitment in Part A. Based on this experience, future trials of fourth-line therapies will have to expect such high attrition rates. Also, EGFR mutation testing was not mandated in this study, reflecting community practice at the time of patient recruitment (2010–2011). Instead, we clinically enriched patients for continued ErbB dependency before randomization.
Given the likelihood of several lines of post-progression therapy after study discontinuation, PFS was chosen as the primary end point. Randomized patients had received (and responded to) greater than or equal to two prior TKIs and most subsequently received greater than or equal to one line of post-study treatment. Moreover, there was an imbalance in the number of post-progression therapies between treatment arms. Hence, it was not surprising that no difference in OS was observed. The absence of OS benefit necessitated that the clinical meaningfulness of PFS was validated by patient-reported outcomes and safety. Notably, global health status/QoL was not significantly different with afatinib plus paclitaxel, despite a doubling of median treatment time versus chemotherapy. The AE profile of afatinib (±paclitaxel) was consistent with previous studies [10, 11, 13, 14]. The frequency and severity of AEs was higher in the afatinib plus paclitaxel arm versus chemotherapy (possibly reflecting prolonged treatment exposure) and discontinuation was higher in the afatinib plus paclitaxel arm. However, differences in AEs between arms did not seem to impact on global health status/QoL.
In previous studies, modest activity has been demonstrated with afatinib monotherapy in some patients with acquired resistance to first-generation EGFR TKIs [13, 14, 19]. Recently, emerging third-generation EGFR inhibitors, such as AZD-9291, rociletinib and HM-61713, have shown much greater response rates (>50%) in patients with EGFRT790M-positive NSCLC [20–22]. Consequently, the role of afatinib monotherapy in this setting may, ultimately, be limited. However, third-generation EGFR inhibitors are less active in patients with EGFRT790M-independent resistance mechanisms. This leaves up to 50% of patients with acquired resistance to first-generation EGFR TKIs underserved, thus constituting a major unmet medical need. Therefore, novel afatinib-based combination regimens, including afatinib plus paclitaxel, warrant consideration in these patients, as well as in patients who are unsuitable for tumor rebiopsy. Indeed, recent data have demonstrated that afatinib plus cetuximab may be particularly effective post-erlotinib/gefitinib in both EGFRT790M-positive and -negative tumors [23].
In conclusion, this study demonstrates that, in a clinically selected patient population, continuous ErbB blockade with afatinib in patients with NSCLC who had failed platinum-based chemotherapy, gefitinib/erlotinib and afatinib monotherapy improves PFS and ORR, but not OS, when combined with paclitaxel. This is the first prospective evidence to support the concept for maintaining target inhibition beyond formal disease progression in oncogene-addicted lung cancer.
funding
This work was supported by Boehringer Ingelheim. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Lynn Pritchard of GeoMed, an Ashfield company, part of UDG Healthcare plc, during the preparation of this article. There is no applicable grant number.
disclosure
MS has received grants from Boehringer Ingelheim during the conduct of the study; grants and personal fees from Novartis Pharma; and personal fees from AstraZeneca, Pfizer, GlaxoSmithKline and Lilly, outside the submitted work. JC-HY has received personal fees for honoraria from Boehringer Ingelheim, Pfizer, AstraZeneca, Roche and Bayer, and advisory and consultation fees from AstraZeneca, Pfizer, Roche, Clovis Oncology, Bayer, Merck Serono, Takeda, Innopharma and TTY. KP is an advisor to Boehringer Ingelheim. JB has received honoraria from Roche, Boehringer Ingelheim, Novartis and Pierre Fabre and has consulting/advisory roles for Roche, Boehringer Ingelheim, Novartis and Pierre Fabre Medicament. CC has received personal fees from AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Roche, Lilly, Sanofi, Novartis, Amgen and Pfizer. FG has received grants from Boehringer Ingelheim during the conduct of the study and personal fees for lectures from Boehringer Ingelheim, Roche and AstraZeneca, outside the submitted work. D-WK has received personal fees for honoraria from Lilly, Pfizer and Novartis, outside the submitted work. RW's institution has received fees for conduct of the study and during the conduct of the study from Boehringer Ingelheim; grants, personal fees for lectures and advisory fees from Roche and Eli Lilly; and personal fees for lectures and advisory fees from Novartis, Boehringer Ingelheim, AstraZeneca, Intermune and Grifolis, outside the submitted work. VKC is an employee of Boehringer Ingelheim. DP reports personal fees from Boehringer Ingelheim for advisory boards, outside the submitted work. All remaining authors have declared no conflicts of interest.
Supplementary Material
acknowledgements
We thank the patients, their families, the investigators and staff who participated in the study. The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development and have approved the final version.
appendix
Members of the LUX-Lung 5 study group were as follows
Publication steering committee: Sai Hong Ignatius Ou, David Planchard, Keunchil Park, Martin Schuler, James Yang, Vikram Chand, Klaus Rohr.
Investigators: Argentina: Claudia Bagnes, Claudio Marcelo Martin, Gonzalo Recondo, Juan Jose Zarba, Cesar Blajman, Martín Richardet; Australia: Sue-Anne McLachlan, Phillip Parente, Craig Underhill, Catherine Crombie, Paul Mainwaring; Austria: Richard Greil; Belgium: Yves Humblet, Frédérique Bustin, Luciano Carestia, Danny Galdermans, Marc Lambrechts, Laetitia Delval, Piet Vercauter; China: Caicun Zhou, Jin Wang, Cheng Huang, Xiaoyan Lin, Yilong Wu, Xiaoqing Liu, Ying Cheng, Shukui Qin, Jifeng Feng, Jianjin Huang, Yiping Zhang, Shun Lu; Brazil: Manuela Zereu, Bernardo Garicochea, Cyntia Albuquerque Zadra; Finland: Henrik Riska, Tuomo Alanko; France: Jacques Cadranel, Christos Chouaid, Gérard Zalcman, Denis Moro Sibilot, Maurice Perol, David Planchard, Jaafar Bennouna, Pierre Fournel, Radj Gervais, Maciej Rotarski, Bruno Coudert; Germany: Martin Schuler, Michael Thomas, Thomas Wehler, Martin Faehling, Ulrich Keilholz, Eckart Laack, Joachim von Pawel, Rudolf Huber, Nicolas Dickgreber, Rainer Wiewrodt; Hungary: Zsuzsanna Mark, Sandor Tehenes, Janos Strausz, Veronika Sarosi; India: Kumar Prabhash, Minish Jain, Srinivasan Venkatesan, Lalit Sharma, Hemant Dadhich, Rajnish Vasant Nagarkar; Israel: Amir Onn, Maya Gottfried, Solomon Stemmer; Italy: Maria Rita Migliorino, Francesco Grossi, Paolo Bidoli, Alessandra Bearz, Cesare Gridelli, Carlo Milandri, Marco Platania, Giovanni Luca Ceresoli, Giorgio Cruciani; Mexico: Francisco Gutierrez Delgado, José Luis Gonzalez Perez, Gabriela Alvarado Luna, Othon Padilla Baca; The Netherlands: Joachim Aerts, Jos Stigt, Anne-Marie Dingemans, Gerarda Herder, Steven Gans; Peru: Jorge Fernando Salas Sánchez, Renzo Luzgardo Alvarez Barreda, Wilbert Rodriguez Pantigoso, Osbert Luis Mejia Palomino; Poland: Piotr Jaskiewicz, Andrzej Kazarnowicz, Piotr Serwatowski, Aleksandra Szczesna, Jacek Jassem; Russia: Vladimir Lubennikov, Nina Karaseva, Sergey Orlov, Yuri Ragulin; Spain: Pilar Garrido, José Luis González Larriba, Carlos Camps, Rosario García Campelo, Pilar Lianes, Manuel Cobo, Enriqueta Felip; South Korea: Dong-Wan Kim, Sang-We Kim, Keunchil Park, Joo-Hang Kim, Ji-Youn Han, Young-Chul Kim; Taiwan: Chih-Hsin Yang, Te-Chun Hsia, Yuh-Min Chen, Ying-Huang Tsai, Gee-Chen Chang, Thomas Chang-Yao Tsao, Wu-Chou Su, Ming-Shyan Huang, Ching-Liang Ho, Ruey-Kuen Hsieh; Ukraine: Yuriy Vinnyk, Oleksandr Popovych, Olga Ponomarova, Igor Bondarenko, Iryna Polishchuk; United Kingdom: Riyaz Shah, Sanka Mitra, Sanjaykumar Popat, James Spicer, Elizabeth Toy, Sanjaykumar Popat, Toby Talbot, Emma Brown, Sunil Upadhyay, Yvonne Summers; United States: Jayne Gurtler, Luis Meza, John Thropay.
Contributor Information
Collaborators: for the LUX-Lung 5 Investigators, Sai Hong Ignatius Ou, David Planchard, Keunchil Park, Martin Schuler, James Yang, Vikram Chand, Klaus Rohr, Claudia Bagnes, Claudio Marcelo Martin, Gonzalo Recondo, Juan Jose Zarba, Cesar Blajman, Martín Richardet, Sue-Anne McLachlan, Phillip Parente, Craig Underhill, Catherine Crombie, Paul Mainwaring, Richard Greil, Yves Humblet, Frédérique Bustin, Luciano Carestia, Danny Galdermans, Marc Lambrechts, Laetitia Delval, Piet Vercauter, Caicun Zhou, Jin Wang, Cheng Huang, Xiaoyan Lin, Yilong Wu, Xiaoqing Liu, Ying Cheng, Shukui Qin, Jifeng Feng, Jianjin Huang, Yiping Zhang, Shun Lu, Manuela Zereu, Bernardo Garicochea, Cyntia Albuquerque Zadra, Henrik Riska, Tuomo Alanko, Jacques Cadranel, Christos Chouaid, Gérard Zalcman, Denis Moro Sibilot, Maurice Perol, David Planchard, Jaafar Bennouna, Pierre Fournel, Radj Gervais, Maciej Rotarski, Bruno Coudert, Martin Schuler, Michael Thomas, Thomas Wehler, Martin Faehling, Ulrich Keilholz, Eckart Laack, Joachim von Pawel, Rudolf Huber, Nicolas Dickgreber, Rainer Wiewrodt, Zsuzsanna Mark, Sandor Tehenes, Janos Strausz, Veronika Sarosi, Kumar Prabhash, Minish Jain, Srinivasan Venkatesan, Lalit Sharma, Hemant Dadhich, Rajnish Vasant Nagarkar, Amir Onn, Maya Gottfried, Solomon Stemmer, Maria Rita Migliorino, Francesco Grossi, Paolo Bidoli, Alessandra Bearz, Cesare Gridelli, Carlo Milandri, Marco Platania, Giovanni Luca Ceresoli, Giorgio Cruciani, Francisco Gutierrez Delgado, José Luis Gonzalez Perez, Gabriela Alvarado Luna, Othon Padilla Baca, J.G.J.V. Aerts, J.A. Stigt, A.M.C. Dingemans, G.J.M. Herder, S. J. M. Gans, Jorge Fernando Salas Sánchez, Renzo Luzgardo Alvarez Barreda, Wilbert Rodriguez Pantigoso, Osbert Luis Mejia Palomino, Piotr Jaskiewicz, Andrzej Kazarnowicz, Piotr Serwatowski, Aleksandra Szczesna, Jacek Jassem, Vladimir Lubennikov, Nina Karaseva, Sergey Orlov, Yuri Ragulin, Pilar Garrido, José Luis González Larriba, Carlos Camps, Rosario García Campelo, Pilar Lianes, Manuel Cobo, Enriqueta Felip, Dong-Wan Kim, Sang-We Kim, Keunchil Park, Joo-Hang Kim, Ji-Youn Han, Young-Chul Kim, Chih-Hsin Yang, Te-Chun Hsia, Yuh-Min Chen, Ying-Huang Tsai, Gee-Chen Chang, Thomas Chang-Yao Tsao, Wu-Chou Su, Ming-Shyan Huang, Ching-Liang Ho, Ruey-Kuen Hsieh, Yuriy Vinnyk, Oleksandr Popovych, Olga Ponomarova, Igor Bondarenko, Iryna Polishchuk, Riyaz Shah, Sanka Mitra, Sanjaykumar Popat, James Spicer, Elizabeth Toy, Sanjaykumar Popat, Toby Talbot, Emma Brown, Sunil Upadhyay, Yvonne Summers, Jayne Gurtler, Luis Meza, and John Thropay
references
- 1.Lee CK, Brown C, Gralla RJ et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst 2013; 105: 595–605. [DOI] [PubMed] [Google Scholar]
- 2.Jackman D, Pao W, Riely GJ et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol 2010; 28: 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chmielecki J, Foo J, Oxnard GR et al. Optimization of dosing for EGFR-mutant non-small cell lung cancer with evolutionary cancer modeling. Sci Transl Med 2011; 3: 90ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaft JE, Oxnard GR, Sima CS et al. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res 2011; 17: 6298–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Riely GJ, Kris MG, Zhao B et al. Prospective assessment of discontinuation and reinitiation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of everolimus. Clin Cancer Res 2007; 13: 5150–5155. [DOI] [PubMed] [Google Scholar]
- 6.Goldberg SB, Oxnard GR, Digumarthy S et al. Chemotherapy with erlotinib or chemotherapy alone in advanced non-small cell lung cancer with acquired resistance to EGFR tyrosine kinase inhibitors. Oncologist 2013; 18: 1214–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshimura N, Okishio K, Mitsuoka S et al. Prospective assessment of continuation of erlotinib or gefitinib in patients with acquired resistance to erlotinib or gefitinib followed by the addition of pemetrexed. J Thorac Oncol 2013; 8: 96–101. [DOI] [PubMed] [Google Scholar]
- 8.Li D, Ambrogio L, Shimamura T et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008; 27: 4702–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Solca F, Dahl G, Zoephel A et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther 2012; 343: 342–350. [DOI] [PubMed] [Google Scholar]
- 10.Sequist LV, Yang JC, Yamamoto N et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol 2013; 31: 3327–3334. [DOI] [PubMed] [Google Scholar]
- 11.Wu YL, Zhou C, Hu CP et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol 2014; 15: 213–222. [DOI] [PubMed] [Google Scholar]
- 12.Yang JC, Wu YL, Schuler M et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol 2015; 16: 141–151. [DOI] [PubMed] [Google Scholar]
- 13.Katakami N, Atagi S, Goto K et al. LUX-Lung 4: a phase II trial of afatinib in patients with advanced non-small-cell lung cancer who progressed during prior treatment with erlotinib, gefitinib, or both. J Clin Oncol 2013; 31: 3335–3341. [DOI] [PubMed] [Google Scholar]
- 14.Miller VA, Hirsh V, Cadranel J et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): a phase 2b/3 randomised trial. Lancet Oncol 2012; 13: 528–538. [DOI] [PubMed] [Google Scholar]
- 15.Solca F, Baum A, Himmelsbach F et al. Efficacy of BIBW 2992, an irreversible dual EGFR/HER2 receptor tyrosine kinase inhibitor, in combination with cytotoxic agents. Eur J Cancer Suppl 2006; 4: 172. [Google Scholar]
- 16.Spicer JF, Harris D, Ang J et al. A phase I study of daily BIBW 2992, an irreversible EGFR/HER2 dual kinase inhibitor, in combination with weekly paclitaxel. Ann Oncol 2008; 19(Suppl. 8): viii157–viii158. [Google Scholar]
- 17.Dodd LE, Korn EL, Freidlin B et al. Blinded independent central review of progression-free survival in Phase III clinical trials: important design element or unnecessary expense? J Clin Oncol 2008; 26: 3791–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang JJ, Chen H, He K et al. Evaluation of blinded independent central review of tumor progression in oncology clinical trials: a meta-analysis. Ther Innov Regul Sci 2013; 47: 167–174. [DOI] [PubMed] [Google Scholar]
- 19.Choi MK, Lee JY, Hong JY et al. An open label compassionate use programme of BIBW 2992/afatinib in advanced non-small cell lung cancer patients pre-treated with erlotinib or gefitinib in Korea. J Thorac Oncol 2013; 8(Suppl. 2): S1204. [DOI] [PubMed] [Google Scholar]
- 20.Janne PA, Yang JC, Kim DW et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med 2015; 372: 1689–1699. [DOI] [PubMed] [Google Scholar]
- 21.Kim DW, Lee DH, Kang JH et al. Clinical activity and safety of HM61713, an EGFR-mutant selective inhibitor, in advanced non-small cell lung cancer (NSCLC) patients (pts) with EGFR mutations who had received EGFR tyrosine kinase inhibitors (TKIs). J Clin Oncol 2014; 32(Suppl. 15): 8011. [Google Scholar]
- 22.Sequist LV, Soria JC, Goldman JW et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med 2015; 372: 1700–1709. [DOI] [PubMed] [Google Scholar]
- 23.Janjigian YY, Smit EF, Groen HJ et al. Dual inhibition of EGFR with afatinib and cetuximab in kinase inhibitor-resistant EGFR-mutant lung cancer with and without T790M mutations. Cancer Discov 2014; 4: 1036–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.