

Abstract
Aortic valve stenosis is a heart disease prevalent in the elderly characterized by valvular calcification, fibrosis, and inflammation, but its exact pathogenesis remains unclear. Previously, aortic valve stenosis was thought to be caused by chronic passive and degenerative changes associated with aging. However, recent studies have demonstrated that atherosclerotic processes and inflammation can induce valvular calcification and bone deposition, leading to valvular stenosis. In particular, the most abundant cell type in cardiac valves, valvular interstitial cells, can differentiate into myofibroblasts and osteoblast-like cells, leading to valvular calcification and stenosis. Differentiation of valvular interstitial cells can be trigged by inflammatory stimuli from several immune cell types, including macrophages, dendritic cells, T cells, B cells, and mast cells. This review indicates that crosstalk between immune cells and valvular interstitial cells plays an important role in the development of aortic valve stenosis.
Keywords: Valve, Stenosis, VIC, VEC, Immune cells
INTRODUCTION
Aortic valve stenosis is a degenerative valvular heart disease characterized by narrowing of the aortic valve orifice by severe calcification, fibrosis, and lipid deposition, leading to valve sclerosis. Narrowing of the orifice increases the pressure burden on the left ventricle (1,2). This is a common disease in the elderly, with approximately 21 to 26% of people over 65 years of age with degenerative valve disease, and around 2.8% of those over 75 years of age showing aortic valve stenosis (3,4). The most adverse aspect of aortic valve stenosis is the low survival rate with which it is associated. Without aortic valve replacement, patients with severe aortic stenosis demonstrate poor prognoses. For instance, following detection of symptoms, two- and five-year survival rates of 50 and 25%, respectively, have been reported (5). Another important factor is the limited number of therapy options currently available for aortic valve stenosis patients. Some trials have indicated that statins are effective in slowing the progression of this disease (6,7), but more recently, larger randomized trials have reported negative statin therapy results (8,9). It is thought that inflammation initiates degenerative valve disease and valvular calcification (10,11). Therefore, to develop a novel therapeutic drug, it is important to understand which immune cells are involved in aortic valve stenosis. In this review, we focus on immune and non-immune cells of the aortic valve and their functions in the progression of this condition.
VALVULAR INTERSTITIAL CELLS (VICs)
VICs, the most numerous cell type in cardiac valve, are located under the valvular endothelium, and together with valvular endothelial cells (VECs) are important in maintaining cardiac valve tissue homeostasis (12,13). VICs are highly involved in the progression of aortic valve stenosis, being capable of differentiating into myofibroblasts and osteoblast-like cells, which cause fibrosis and valve calcification, respectively (14,15). Such differentiation is dependent on TGF-β (16,17,18). In aortic valve stenosis, myofibroblasts produce extracellular collagen and tenascin-C, causing changes to components of the extracellular matrix and tissue fibrosis (19). Osteoblast-like VICs induce calcification through a mechanism similar to osteogenesis (20). These cells produce bone morphogenetic protein 2 and osteopontin, which are important for bone formation (21), and express runt-related transcription factor 2 and osterix—transcription factors involved in osteoblast differentiation (22). VICs can also take up lipids, but not to the same extent as macrophages (23). A recent study by Syvaranta and colleagues showed that, in the stenotic state, myofibroblasts upregulate the scavenger receptors CD36 and lectin-like oxidized low density lipoprotein receptor-1, which are able to bind to oxidized low-density lipoprotein (LDL). In response to oxidized LDL, myofibroblasts produce inflammatory cytokines and chemokines, including MCP-1, IL-6, IL-8, and M-CSF (24). These findings indicate that VIC-derived myofibroblasts are able to take up lipids, and that lipid accumulation is associated with pro-inflammatory processes in aortic valve stenosis. In 2008, Meng and colleagues showed that VICs express TLR2 and TLR4. Treatment with agonists of these TLRs induces NF-κB activation and upregulation of ICAM-1, bone morphogenetic protein 2, and runt-related transcription factor 2 in VICs (25). TLR2- and TLR4-stimulation in these cells also promotes alkaline phosphatase activity and increases calcified nodule formation (26). These findings indicate that the TLR-activated pro-inflammatory process in VICs closely correlates with valvular calcification. In conclusion, VICs participate in various processes, including calcification, fibrosis, lipid uptake, and inflammation in aortic valve stenosis.
VALVULAR ENDOTHELIAL CELLS (VECs)
In the normal state, VECs and VICs are important for the maintenance of homeostasis in the cardiac valve (12,13). VECs can differentiate into VICs through endothelial-mesenchymal transition to preserve valve homeostasis (27). Mechanical shearing force or other types of stress can reduce the integrity of valvular endothelium, leading to accumulation of lipoproteins in the subendothelial space. The accumulated LDL molecules are modified into oxidized LDL, and trigger inflammation of cardiac valves by upregulating cell adhesion molecules, such as ICAM-1 and VCAM-1, which allow T cells and monocyte-derived macrophages to infiltrate the tissue (28,29,30,31). These infiltrating T cells and macrophages release pro-inflammatory cytokines, including IL-1β and TNF-α (1,32,33). Inflammatory cytokines such as IL-6 and TNF-α induce the differentiation of VECs to VICs via endothelial-mesenchymal transition (34). These observations indicate that VECs play important roles in the initiation and progression of aortic valve disease. Interestingly, in hypercholesterolemia, the peroxisome proliferator-activated receptor gamma (PPARγ) pathway is activated, and related genes, including ATP-binding cassette subfamily A member 1 and fatty acid-binding protein 1, are upregulated in the aortic valve endothelium (35). PPARγ activation in endothelial cells is known to inhibit inflammation of the endothelium (36,37). Therefore, PPARγ may constitute a drug target in valvular endothelium for the treatment of aortic valve stenosis.
MACROPHAGES
In cardiac valve disease, monocytes infiltrate valve tissues and differentiate into macrophages (38,39). Macrophages are the principal immune cell population in various valvular heart diseases. Endothelial damage to cardiac valves is known to upregulate endothelial cell adhesion molecules such as ICAM-1 and VCAM-1 that subsequently induce the recruitment of monocytes and other leukocytes (31,38). In addition, a gene expression profiling study by Bosse and colleagues showed that various chemokines and chemokine receptors are upregulated in aortic valve stenosis (40). This indicates that chemotaxis is also involved in the pathogenesis of this disease. After having infiltrated the tissue, macrophages release inflammatory cytokines such as TNF-α, IL-1β, and TGF-β leading to cardiac valve inflammation. Moreover, TNF-α and TGF-β promote VIC activation and induce alkaline phosphatase expression, eventually leading to valvular calcification (16,32,41,42,43). Macrophages also release matrix metalloproteinases and cathepsins, which alter extracellular matrix components in valvular disease (38). Inflammatory macrophages promote VEC and VIC calcification in a cathepsin S-dependent manner in calcific aortic valve disease and aortic valve stenosis (44). In atherosclerosis, the uptake of LDLs by macrophages results in their becoming foam cells (45,46). Likewise, in hypercholesterolemia-induced valvular sclerosis, recruited macrophages engulf lipids and develop a foamy appearance (23). This suggests that macrophages play important roles in valvular disease, as well as in atherosclerosis. However, further studies are required for a more detailed understanding of macrophage functions and to validate them as therapeutic targets in aortic valve stenosis.
DENDRITIC CELLS (DCs)
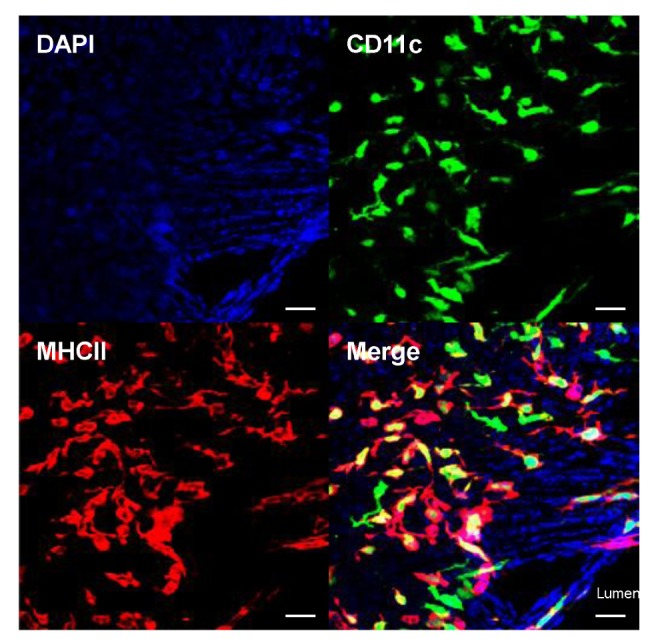
DCs, present in normal cardiac valves (Fig. 1), are another important immune cell type. In the aortic valve, DCs are particularly located beneath the aortic-side endothelium, a site exposed to turbulent flow (47). In a normal state, these cells are thought to be classical DCs; however, their role in cardiac valves remains unknown. Considering the atheroprotective role of FLT3-dependent DCs (48), valvular DCs may play regulatory functions in aortic valve stenosis, but additional investigation is needed. In hyperlipidemia, lipids accumulate in the aortic side of the aortic valve, where the majority of valvular DCs are located (47). It has been confirmed that a large number of monocyte-derived DCs amass during atherosclerosis (49), and these same cells have been proposed to accumulate in sclerotic cardiac valves, potentially playing a significant role in aortic valve stenosis progression. However, the relationship between lipid accumulation and DCs and their roles in aortic valve stenosis remains to be elucidated.
Figure 1. Presence of dendritic cells (DCs) in mouse cardiac valve. Mitral valve from a CD11c-EYFP transgenic mouse was whole-mount immunostained with MHCII antibody. The CD11c+ MHCII+ DCs were then visualized (green, CD11c; red, MHCII). CD11c+ MHCII+ DCs are also present in other cardiac valves (aortic, tricuspid, and pulmonary; data not shown). Scale bars, 20 µm.
T CELLS
T cells also play a role in cardiac valve disease, gathering in valvular tissue during the progression of conditions such as aortic valve stenosis (50). Infiltrating T cells secrete various pro-inflammatory cytokines, including TNF-α, IL-1β, and TGF-β (1,33). In a previous study, multiple oligoclonal CD4+ and CD8+ T cells were observed to accumulate in stenotic aortic valves, suggesting that clonally expanded T cells are highly involved in the pathogenesis of aortic valve stenosis (51). Moreover, the number of T cells, especially of the CD8+ and CD8+ CD28null memory-effector subsets, is increased in the stenotic aortic valve and peripheral blood of calcific aortic valve stenosis patients (52). T cell proliferation may occur not only in the lymphoid organs but also in the stenotic aortic valve itself, although more investigations are needed. To conclude, the T cell-mediated adaptive immune system is involved in aortic valve stenosis. Considering their abundancy, valvular DCs, powerful antigen-presenting cells that can actively interact with T cells (47), may have an important role in regulating valvular T cell activation and proliferation.
B CELLS
In the normal state, B cells are not present in cardiac valves, while in the stenotic aortic valve tissue, CD20+ B cells and CD138+ plasma cells accumulate (50). A recent study by Natorska and colleagues showed that B cells infiltrate the aortic side of human stenotic aortic valve (53). It has been demonstrated that B cells can be activated by TLR signaling (54) or macrophage-secreted cytokines, such as B cell-activating factor belonging to the TNF family (BAFF). The binding of BAFF to its receptor on B cells results in signaling that promotes their survival, maturation, and proliferation (55,56,57). Importantly, B cells in the stenotic valve also express BAFF receptors, and a positive correlation is evident between the number of these cells and the degree of valve calcification. In addition, correlations exist between the number of B cells and macrophages, and between the number of BAFF receptor-positive B cells and macrophages (53). These findings indicate that, in collaboration with macrophages, B cells also contribute to the progression of aortic valve stenosis via BAFF/BAFF receptor signaling. Monocyte-derived DCs are also known to secrete BAFF (55,58). Therefore, it is plausible that these cells may play a role in B cell activation in aortic valve stenosis.
MAST CELLS
Mast cells also participate in the progression of aortic valve stenosis. Under normal conditions, a small number of mast cells are located in the aortic valve. However, their numbers are markedly increased in the subendothelial space of the aortic side of stenotic valve, in the vicinity of valvular macrophages (59,60). During disease, mast cells are activated and they produce cathepsin G, which causes elastin degradation in the stenotic valve (59). These cells appear to induce angiogenesis during the aortic valve stenosis process, and a positive correlation between mast cell and neovessel density has been documented in this condition. Mast cells can produce vascular endothelial growth factor (VEGF) and they are closely localized to neovessels in the stenotic valve (61). Tryptase, which is released by activated mast cells, negatively regulates levels of the endogenous angiogenesis inhibitor endostatin (61,62). Thus, these cells promote angiogenesis in aortic valve stenosis by VEGF secretion and downregulation of endostatin. In contrast, mast cells play a regulatory function in lymphangiogenesis by suppressing VEGF-C secreted by valvular myofibroblasts (63). These dual functions of mast cells indicate their versatility in regulating neovascularization in aortic valve disease.
CONCLUSION
Various cellular components participate in the progression of aortic valve stenosis. Damage to VECs triggers the onset of disease and upregulates cell adhesion molecules, promoting inflammatory immune cell infiltration of the aortic valve. Infiltrating inflammatory cells secrete cytokines, and induce endothelial-mesenchymal transition of VECs to VICs. These events trigger the differentiation of VICs into myofibroblasts and osteoblast-like cells, causing fibrosis and calcification. Macrophages and mast cells directly affect the disease progression and cause lipid accumulation, angiogenesis, and extracellular matrix remodeling. The adaptive immune response effected by T cells and B cells also affects the progress of this disease. In addition, DCs may participate in aortic valve stenosis by regulating T cell immunity. However, the pathogenesis of this condition remains largely unknown. Additional studies are required to understand valvular immune cell networks and their crosstalk with VICs and VECs, which will provide new therapeutic options for aortic valve stenosis.
ACKNOWLEDGEMENTS
This work was supported by the research fund of Hanyang University (HY-2012-N).
Abbreviations
- VIC
valvular interstitial cell
- VEC
valvular endothelial cell
- BMP2
bone morphogenetic protein 2
- Runx2
runt-related transcription factor 2
- LDL
low-density lipoprotein
- PPARγ
peroxisome proliferator-activated receptor gamma
- DC
dendritic cell
- BAFF
B cell-activating factor belonging to the TNF family
- VEGF
vascular endothelial growth factor
Footnotes
CONFLICTS OF INTEREST: The authors have no financial conflict of interest.
References
- 1.Dweck MR, Boon NA, Newby NA. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. 2012;60:1854–1863. doi: 10.1016/j.jacc.2012.02.093. [DOI] [PubMed] [Google Scholar]
- 2.Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. 2013;113:198–208. doi: 10.1161/CIRCRESAHA.113.300155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nkomo VT, Gardin JM, Skelton TN, Gottdiener JS, Scott CG, Enriquez-Sarano M. Burden of valvular heart diseases: a population-based study. Lancet. 2006;368:1005–1011. doi: 10.1016/S0140-6736(06)69208-8. [DOI] [PubMed] [Google Scholar]
- 4.Otto CM, Lind BK, Kitzman DW, Gersh BJ, Siscovick DS. Association of aortic-valve sclerosis with cardiovascular mortality and morbidity in the elderly. N Engl J Med. 1999;341:142–147. doi: 10.1056/NEJM199907153410302. [DOI] [PubMed] [Google Scholar]
- 5.Otto CM. Timing of aortic valve surgery. Heart. 2000;84:211–218. doi: 10.1136/heart.84.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moura LM, Ramos SF, Zamorano JL, Barros IM, Azevedo LF, Rocha-Goncalves F, Rajamannan NM. Rosuvastatin affecting aortic valve endothelium to slow the progression of aortic stenosis. J Am Coll Cardiol. 2007;49:554–561. doi: 10.1016/j.jacc.2006.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenhek R, Rader F, Loho N, Gabriel H, Heger M, Klaar U, Schemper M, Binder T, Maurer G, Baumgartner H. Statins but not angiotensin-converting enzyme inhibitors delay progression of aortic stenosis. Circulation. 2004;110:1291–1295. doi: 10.1161/01.CIR.0000140723.15274.53. [DOI] [PubMed] [Google Scholar]
- 8.Cowell SJ, Newby DE, Prescott RJ, Bloomfield P, Reid J, Northridge DB, Boon NA. A randomized trial of intensive lipid-lowering therapy in calcific aortic stenosis. N Engl J Med. 2005;352:2389–2397. doi: 10.1056/NEJMoa043876. [DOI] [PubMed] [Google Scholar]
- 9.Rossebo AB, Pedersen TR, Boman K, Brudi P, Chambers JB, Egstrup K, Gerdts E, Gohlke-Barwolf C, Holme I, Kesaniemi YA, Malbecq W, Nienaber CA, Ray S, Skjaerpe T, Wachtell K, Willenheimer R. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–1356. doi: 10.1056/NEJMoa0804602. [DOI] [PubMed] [Google Scholar]
- 10.Aikawa E, Otto CM. Look more closely at the valve: imaging calcific aortic valve disease. Circulation. 2012;125:9–11. doi: 10.1161/CIRCULATIONAHA.111.073452. [DOI] [PubMed] [Google Scholar]
- 11.New SE, Aikawa E. Molecular imaging insights into early inflammatory stages of arterial and aortic valve calcification. Circ Res. 2011;108:1381–1391. doi: 10.1161/CIRCRESAHA.110.234146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Combs MD, Yutzey KE. Heart valve development: regulatory networks in development and disease. Circ Res. 2009;105:408–421. doi: 10.1161/CIRCRESAHA.109.201566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Leinwand LA, Anseth KS. Cardiac valve cells and their microenvironment--insights from in vitro studies. Nat Rev Cardiol. 2014;11:715–727. doi: 10.1038/nrcardio.2014.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu AC, Joag VR, Gotlieb AI. The emerging role of valve interstitial cell phenotypes in regulating heart valve pathobiology. Am J Pathol. 2007;171:1407–1418. doi: 10.2353/ajpath.2007.070251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajamannan NM, Evans FJ, Aikawa E, Grande-Allen KJ, Demer LL, Heistad DD, Simmons CA, Masters KS, Mathieu P, O'Brien KD, Schoen FJ, Towler DA, Yoganathan AP, Otto CM. Calcific aortic valve disease: not simply a degenerative process: A review and agenda for research from the National Heart and Lung and Blood Institute Aortic Stenosis Working Group. Executive summary: Calcific aortic valve disease-2011 update. Circulation. 2011;124:1783–1791. doi: 10.1161/CIRCULATIONAHA.110.006767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jian B, Narula N, Li QY, Mohler ER, III, Levy RJ. Progression of aortic valve stenosis: TGF-beta1 is present in calcified aortic valve cusps and promotes aortic valve interstitial cell calcification via apoptosis. Ann Thorac Surg. 2003;75:457–465. doi: 10.1016/s0003-4975(02)04312-6. [DOI] [PubMed] [Google Scholar]
- 17.Li C, Gotlieb AI. Transforming growth factor-beta regulates the growth of valve interstitial cells in vitro. Am J Pathol. 2011;179:1746–1755. doi: 10.1016/j.ajpath.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Osman L, Yacoub MH, Latif N, Amrani M, Chester AH. Role of human valve interstitial cells in valve calcification and their response to atorvastatin. Circulation. 2006;114:I547–I552. doi: 10.1161/CIRCULATIONAHA.105.001115. [DOI] [PubMed] [Google Scholar]
- 19.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–537. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 20.Rajamannan NM, Subramaniam M, Rickard D, Stock SR, Donovan J, Springett M, Orszulak T, Fullerton DA, Tajik AJ, Bonow RO, Spelsberg T. Human aortic valve calcification is associated with an osteoblast phenotype. Circulation. 2003;107:2181–2184. doi: 10.1161/01.CIR.0000070591.21548.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang X, Meng X, Su X, Mauchley DC, Ao L, Cleveland JC, Jr, Fullerton DA. Bone morphogenic protein 2 induces Runx2 and osteopontin expression in human aortic valve interstitial cells: role of Smad1 and extracellular signal-regulated kinase 1/2. J Thorac Cardiovasc Surg. 2009;138:1008–1015. doi: 10.1016/j.jtcvs.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Alexopoulos A, Bravou V, Peroukides S, Kaklamanis L, Varakis J, Alexopoulos D, Papadaki H. Bone regulatory factors NFATc1 and Osterix in human calcific aortic valves. Int J Cardiol. 2010;139:142–149. doi: 10.1016/j.ijcard.2008.10.014. [DOI] [PubMed] [Google Scholar]
- 23.Filip DA, Nistor A, Bulla A, Radu A, Lupu F, Simionescu M. Cellular events in the development of valvular atherosclerotic lesions induced by experimental hypercholesterolemia. Atherosclerosis. 1987;67:199–214. doi: 10.1016/0021-9150(87)90280-2. [DOI] [PubMed] [Google Scholar]
- 24.Syväranta S, anne-Kinnunen M, Oorni K, Oksjoki R, Kupari M, Kovanen PT, Helske-Suihko S. Potential pathological roles for oxidized low-density lipoprotein and scavenger receptors SR-AI, CD36, and LOX-1 in aortic valve stenosis. Atherosclerosis. 2014;235:398–407. doi: 10.1016/j.atherosclerosis.2014.05.933. [DOI] [PubMed] [Google Scholar]
- 25.Meng X, Ao L, Song Y, Babu A, Yang X, Wang M, Weyant MJ, Dinarello CA, Cleveland JC, Jr, Fullerton DA. Expression of functional Toll-like receptors 2 and 4 in human aortic valve interstitial cells: potential roles in aortic valve inflammation and stenosis. Am J Physiol Cell Physiol. 2008;294:C29–C35. doi: 10.1152/ajpcell.00137.2007. [DOI] [PubMed] [Google Scholar]
- 26.Yang X, Fullerton DA, Su X, Ao L, Cleveland JC, Jr, Meng X. Pro-osteogenic phenotype of human aortic valve interstitial cells is associated with higher levels of Toll-like receptors 2 and 4 and enhanced expression of bone morphogenetic protein 2. J Am Coll Cardiol. 2009;53:491–500. doi: 10.1016/j.jacc.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 27.Bischoff J, Aikawa E. Progenitor cells confer plasticity to cardiac valve endothelium. J Cardiovasc Transl Res. 2011;4:710–719. doi: 10.1007/s12265-011-9312-0. [DOI] [PubMed] [Google Scholar]
- 28.Olsson M, Thyberg J, Nilsson J. Presence of oxidized low density lipoprotein in nonrheumatic stenotic aortic valves. Arterioscler Thromb Vasc Biol. 1999;19:1218–1222. doi: 10.1161/01.atv.19.5.1218. [DOI] [PubMed] [Google Scholar]
- 29.Mohty D, Pibarot P, Despres JP, Cote C, Arsenault B, Cartier A, Cosnay P, Couture C, Mathieu P. Association between plasma LDL particle size, valvular accumulation of oxidized LDL, and inflammation in patients with aortic stenosis. Arterioscler Thromb Vasc Biol. 2008;28:187–193. doi: 10.1161/ATVBAHA.107.154989. [DOI] [PubMed] [Google Scholar]
- 30.O'Brien KD, Reichenbach DD, Marcovina SM, Kuusisto J, Alpers CE, Otto CM. Apolipoproteins B, (a), and E accumulate in the morphologically early lesion of 'degenerative' valvular aortic stenosis. Arterioscler Thromb Vasc Biol. 1996;16:523–532. doi: 10.1161/01.atv.16.4.523. [DOI] [PubMed] [Google Scholar]
- 31.Sucosky P, Balachandran K, Elhammali A, Jo H, Yoganathan AP. Altered shear stress stimulates upregulation of endothelial VCAM-1 and ICAM-1 in a BMP-4- and TGF-beta1-dependent pathway. Arterioscler Thromb Vasc Biol. 2009;29:254–260. doi: 10.1161/ATVBAHA.108.176347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaden JJ, Dempfle CE, Grobholz R, Tran HT, Kilic R, Sarikoc A, Brueckmann M, Vahl C, Hagl S, Haase KK, Borggrefe M. Interleukin-1 beta promotes matrix metalloproteinase expression and cell proliferation in calcific aortic valve stenosis. Atherosclerosis. 2003;170:205–211. doi: 10.1016/s0021-9150(03)00284-3. [DOI] [PubMed] [Google Scholar]
- 33.Weiss RM, Miller JD, Heistad DD. Fibrocalcific aortic valve disease: opportunity to understand disease mechanisms using mouse models. Circ Res. 2013;113:209–222. doi: 10.1161/CIRCRESAHA.113.300153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahler GJ, Farrar EJ, Butcher JT. Inflammatory cytokines promote mesenchymal transformation in embryonic and adult valve endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33:121–130. doi: 10.1161/ATVBAHA.112.300504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guerraty MA, Grant GR, Karanian JW, Chiesa OA, Pritchard WF, Davies PF. Hypercholesterolemia induces side-specific phenotypic changes and peroxisome proliferator-activated receptor-gamma pathway activation in swine aortic valve endothelium. Arterioscler Thromb Vasc Biol. 2010;30:225–231. doi: 10.1161/ATVBAHA.109.198549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan SZ, Usher MG, Mortensen RM. Peroxisome proliferator-activated receptor-gamma-mediated effects in the vasculature. Circ Res. 2008;102:283–294. doi: 10.1161/CIRCRESAHA.107.164384. [DOI] [PubMed] [Google Scholar]
- 37.Jackson SM, Parhami F, Xi XP, Berliner JA, Hsueh WA, Law RE, Demer LL. Peroxisome proliferator-activated receptor activators target human endothelial cells to inhibit leukocyte-endothelial cell interaction. Arterioscler Thromb Vasc Biol. 1999;19:2094–2104. doi: 10.1161/01.atv.19.9.2094. [DOI] [PubMed] [Google Scholar]
- 38.Aikawa E, Nahrendorf M, Sosnovik D, Lok VM, Jaffer FA, Aikawa M, Weissleder R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation. 2007;115:377–386. doi: 10.1161/CIRCULATIONAHA.106.654913. [DOI] [PubMed] [Google Scholar]
- 39.Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis Histological and immunohistochemical studies. Circulation. 1994;90:844–853. doi: 10.1161/01.cir.90.2.844. [DOI] [PubMed] [Google Scholar]
- 40.Bossé Y, Miqdad A, Fournier D, Pepin A, Pibarot P, Mathieu P. Refining molecular pathways leading to calcific aortic valve stenosis by studying gene expression profile of normal and calcified stenotic human aortic valves. Circ Cardiovasc Genet. 2009;2:489–498. doi: 10.1161/CIRCGENETICS.108.820795. [DOI] [PubMed] [Google Scholar]
- 41.Kaden JJ, Kilic R, Sarikoc A, Hagl S, Lang S, Hoffmann U, Brueckmann M, Borggrefe M. Tumor necrosis factor alpha promotes an osteoblast-like phenotype in human aortic valve myofibroblasts: a potential regulatory mechanism of valvular calcification. Int J Mol Med. 2005;16:869–872. [PubMed] [Google Scholar]
- 42.Khan R, Sheppard R. Fibrosis in heart disease: understanding the role of transforming growth factor-beta in cardiomyopathy, valvular disease and arrhythmia. Immunology. 2006;118:10–24. doi: 10.1111/j.1365-2567.2006.02336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parameswaran N, Patial S. Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr. 2010;20:87–103. doi: 10.1615/critreveukargeneexpr.v20.i2.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron. 2006;37:208–222. doi: 10.1016/j.micron.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 46.Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. 1983;52:223–261. doi: 10.1146/annurev.bi.52.070183.001255. [DOI] [PubMed] [Google Scholar]
- 47.Choi JH, Do Y, Cheong C, Koh H, Boscardin SB, Oh YS, Bozzacco L, Trumpfheller C, Park CG, Steinman RM. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J Exp Med. 2009;206:497–505. doi: 10.1084/jem.20082129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi JH, Cheong C, Dandamudi DB, Park CG, Rodriguez A, Mehandru S, Velinzon K, Jung IH, Yoo JY, Oh GT, Steinman RM. Flt3 signaling-dependent dendritic cells protect against atherosclerosis. Immunity. 2011;35:819–831. doi: 10.1016/j.immuni.2011.09.014. [DOI] [PubMed] [Google Scholar]
- 49.Koltsova EK, Ley K. How dendritic cells shape atherosclerosis. Trends Immunol. 2011;32:540–547. doi: 10.1016/j.it.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steiner I, Krbal L, Rozkos T, Harrer J, Laco J. Calcific aortic valve stenosis: Immunohistochemical analysis of inflammatory infiltrate. Pathol Res Pract. 2012;208:231–234. doi: 10.1016/j.prp.2012.02.009. [DOI] [PubMed] [Google Scholar]
- 51.Wu HD, Maurer MS, Friedman RA, Marboe CC, Ruiz-Vazquez EM, Ramakrishnan R, Schwartz A, Tilson MD, Stewart AS, Winchester R. The lymphocytic infiltration in calcific aortic stenosis predominantly consists of clonally expanded T cells. J Immunol. 2007;178:5329–5339. doi: 10.4049/jimmunol.178.8.5329. [DOI] [PubMed] [Google Scholar]
- 52.Winchester R, Wiesendanger M, O'Brien W, Zhang HZ, Maurer MS, Gillam LD, Schwartz A, Marboe C, Stewart AS. Circulating activated and effector memory T cells are associated with calcification and clonal expansions in bicuspid and tricuspid valves of calcific aortic stenosis. J Immunol. 2011;187:1006–1014. doi: 10.4049/jimmunol.1003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Natorska J, Marek G, Sadowski J, Undas A. Presence of B cells within aortic valves in patients with aortic stenosis: Relation to severity of the disease. J Cardiol. 2016;67:80–85. doi: 10.1016/j.jjcc.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 54.Han SB, Yoon YD, Ahn HJ, Lee HS, Lee CW, Yoon WK, Park SK, Kim HM. Toll-like receptor-mediated activation of B cells and macrophages by polysaccharide isolated from cell culture of Acanthopanax senticosus. Int Immunopharmacol. 2003;3:1301–1312. doi: 10.1016/S1567-5769(03)00118-8. [DOI] [PubMed] [Google Scholar]
- 55.Mackay F, Browning JL. BAFF: a fundamental survival factor for B cells. Nat Rev Immunol. 2002;2:465–475. doi: 10.1038/nri844. [DOI] [PubMed] [Google Scholar]
- 56.Mackay F, Schneider P, Rennert P, Browning J. BAFF AND APRIL: a tutorial on B cell survival. Annu Rev Immunol. 2003;21:231–264. doi: 10.1146/annurev.immunol.21.120601.141152. [DOI] [PubMed] [Google Scholar]
- 57.Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, cha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J. BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med. 1999;189:1747–1756. doi: 10.1084/jem.189.11.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nardelli B, Belvedere O, Roschke V, Moore PA, Olsen HS, Migone TS, Sosnovtseva S, Carrell JA, Feng P, Giri JG, Hilbert DM. Synthesis and release of B-lymphocyte stimulator from myeloid cells. Blood. 2001;97:198–204. doi: 10.1182/blood.v97.1.198. [DOI] [PubMed] [Google Scholar]
- 59.Helske S, Syvaranta S, Kupari M, Lappalainen J, Laine M, Lommi J, Turto H, Mayranpaa M, Werkkala K, Kovanen PT, Lindstedt KA. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006;27:1495–1504. doi: 10.1093/eurheartj/ehi706. [DOI] [PubMed] [Google Scholar]
- 60.Wypasek E, Natorska J, Grudzien G, Filip G, Sadowski J, Undas A. Mast cells in human stenotic aortic valves are associated with the severity of stenosis. Inflammation. 2013;36:449–456. doi: 10.1007/s10753-012-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Syväranta S, Helske S, Laine M, Lappalainen J, Kupari M, Mayranpaa MI, Lindstedt KA, Kovanen PT. Vascular endothelial growth factor-secreting mast cells and myofibroblasts: a novel self-perpetuating angiogenic pathway in aortic valve stenosis. Arterioscler Thromb Vasc Biol. 2010;30:1220–1227. doi: 10.1161/ATVBAHA.109.198267. [DOI] [PubMed] [Google Scholar]
- 62.O'Reilly MS, Boehm T, Shing Y, Fukai N, Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR, Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 63.Syväranta S, Helske S, Lappalainen J, Kupari M, Kovanen PT. Lymphangiogenesis in aortic valve stenosis--novel regulatory roles for valvular myofibroblasts and mast cells. Atherosclerosis. 2012;221:366–374. doi: 10.1016/j.atherosclerosis.2011.12.034. [DOI] [PubMed] [Google Scholar]