

Abstract
Ampicillin, a β-lactam antibiotic, dose-dependently protects neurons against ischemic brain injury. The present study was performed to investigate the neuroprotective mechanism of ampicillin in a mouse model of transient global forebrain ischemia. Male C57BL/6 mice were anesthetized with halothane and subjected to bilateral common carotid artery occlusion for 40 min. Before transient forebrain ischemia, ampicillin (200 mg/kg, intraperitoneally [i.p.]) or penicillin G (6,000 U/kg or 20,000 U/kg, i.p.) was administered daily for 5 days. The pretreatment with ampicillin but not with penicillin G signifi cantly attenuated neuronal damage in the hippocampal CA1 subfield. Mechanistically, the increased activity of matrix metalloproteinases (MMPs) following forebrain ischemia was also attenuated by ampicillin treatment. In addition, the ampicillin treatment reversed increased immunoreactivities to glial fibrillary acidic protein and isolectin B4, markers of astrocytes and microglia, respectively. Furthermore, the ampicillin treatment significantly increased the level of glutamate transporter-1, and dihydrokainic acid (DHK, 10 mg/kg, i.p.), an inhibitor of glutamate transporter-1 (GLT-1), reversed the neuroprotective effect of ampicillin. Taken together, these data indicate that ampicillin provides neuroprotection against ischemia-reperfusion brain injury, possibly through inducing the GLT-1 protein and inhibiting the activity of MMP in the mouse hippocampus.
Keywords: Ampicillin, Dihydrokainic acid, Glutamate transporter-1, Matrix metalloproteinase, Transient global forebrain ischemia
INTRODUCTION
Transient global forebrain ischemia induces delayed neuronal death in the brain, especially in the hippocampus [1]. Neuronal death following ischemia-reperfusion injury is mediated by several mechanisms, including glutamate excitotoxicity, oxidative stress, inflammation, and apoptosis [2,3]. As the transient release of glutamate from synapses during ischemia and the early period of reperfusion has been shown to trigger the cascade of neuronal cell death, many studies have focused on identifying therapeutic tools to effectively reduce the excitotoxicity of glutamate. For example, selective blockade of the NMDA receptor or the adenosine receptor attenuated ischemic insults [4,5]. In addition, accumulating evidence indicates that modulation of the activity of the glial glutamate transporter (GLT-1) provides neuroprotection against brain ischemic insults and even pilocarpine-induced status epilepticus and Alzheimer's disease [6,7,8].
Glutamate transporters, also known as excitatory amino acid transporters (EAATs), modulate the concentration of synaptic glutamate by clearing glutamate from the extracellular space [9]. Thus far, five EAAT isoforms have been identified: glutamate/aspartate transporter-1 (GLAST-1, EAAT1), glutamate transporter-1 (GLT-1, EAAT2), excitatory amino acid carrier-1 (EAAC-1, EAAT3), EAAT4 and EAAT5 [10]. Interestingly, about 80% of glutamate transporters expressed in the hippocampus were found to be GLT-1. Thus, most of the glutamate released in the hippocampus is cleared by this subtype [11,12,13]. Although GLT-1 has also been observed on neuronal axon terminals, it is predominantly expressed in astrocytes and plays a crucial role in glutamate uptake from the synaptic cleft. Thus, GLT-1 is believed to ameliorate glutamate-mediated excitotoxicity [14]. Supporting this idea, reduced activity of GLT-1 was reported in several kinds of neurodegenerative diseases and pharmacological interventions, such as ceftriaxone, successfully reduced neuronal cell death by increasing GLT-1 expression [15,16,17,18].
Recently, we reported that ampicillin played a functional role in chemical preconditioning [19]. Although its mechanism of action was not clear, ampicillin pretreatment protected hippocampal neurons against severe ischemic insults [19]. Ampicillin is a well-known β-lactam antibiotic. Interestingly, in common with ceftriaxone, ampicillin was reported to induce the expression of GLT-1 in vitro [20]. Considering the recent evidence supporting the role of GLT-1 in neurodegenerative diseases and its potential as a therapeutic candidate, it is important to elucidate the mechanistic link between β-lactam antibiotics and neuronal protection in ischemic insults. The present study explored the neuroprotective capacity of ampicillin and its mechanism of action in global forebrain ischemia in mice.
METHODS
Animals and induction of transient global forebrain ischemia
All animal procedures were approved by the Ethics Committee of the Catholic University of Korea and were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23). Male C57BL/6 mice (Koatec, Kyungki-do, Korea), weighing 20~25 g, were kept in cages under light-controlled conditions (lights on from 08:00 to 20:00 h), with access to food and water ad libitum. The induction of global forebrain ischemia was performed as described previously [21]. Briefly, the bilateral common carotid arteries were occluded for 40 min. Then, animals satisfying the inclusion criterion of regional cerebral blood flow of less than 15% of the baseline during bilateral carotid artery occlusion were selected for the experiments. Rectal temperature was maintained at 37.5±0.5℃ by using a heating pad during the surgery. In sham-manipulated animals, the bilateral common carotid arteries were not occluded and only isolated from the adjacent vagus nerve. After perfusion, animals satisfying the exclusion criterion of a posterior communicating artery less than one-third of the diameter of the basilar artery were selected for further processing and data analysis.
Drug treatment schedule
Ampicillin (200 mg/kg/day, Yungjin Pharmaceutical Co. Ltd, Hwa Sung, Korea) and penicillin G sodium salt (6,000 U/kg or 20,000 U/kg, Sigma Chemical Co., MO, U.S.A.) were dissolved in normal saline and administered intraperitoneally (i.p.) for 5 days. Forebrain ischemia was induced as described earlier 24 h after the last injection [19]. To elucidate the molecular mechanism of ampicillin, dihydrokainic acid (DHK) (10 mg/kg, Tocris, Missouri, U.S.A.), a GLT-1 inhibitor [22], was administrated i.p. 30 min before the onset of ischemia. In the control animals, saline was administered instead of ampicillin or penicillin G, at the same volume and time schedule.
Cresyl violet staining and analysis of hippocampal damage
Three days after transient forebrain ischemia, the animals were transcardially perfused with 4% paraformaldehyde in 0.1 M phosphate buffer for 30 min, followed by 1.5 ml of latex solution to evaluate the plasticity of the posterior communicating artery. After the mice had been cooled on ice for 1 day, the brains were isolated and photographed with a digital camera (EOS 300D, Canon, Tokyo, Japan). The brains were then postfixed in 4% paraformaldehyde for 24 h and cut using a cryotome (20 µm thickness) after cryoprotection with 30% sucrose in 0.1 M phosphate buffer.
For cresyl violet staining, the sections were mounted on gelatin-coated slides and allowed to air-dry overnight. The mounted sections were rehydrated in a graded series of alcohol and distilled water and submerged in 0.1% cresyl violet solution for 10 min. The sections were rinsed in 70% ethanol and dehydrated in graded series of ethanol, immersed in xylene, mounted with Canada balsam (Kanto Chemical Co., Tokyo, Japan), and cover slipped.
The severity of the ischemic neuronal damage in the medial CA1 area of the hippocampus was evaluated by an examiner blinded to the study groups. The severity was semiquantitatively assessed according to the method described previously [21,23]. Ischemic neuronal damage was measured on the following scale: no ischemic neuronal damage, grade 0; less than 30% ischemic neuronal damage, grade 1; 31–64% ischemic neuronal damage, grade 2; 65~100% ischemic neuronal damage, grade 3.
Western blotting
To evaluate the expression level of the GLT-1 protein in the hippocampus, A Western blot analysis was employed. First, ampicillin was administered (100 mg/kg, twice a day for 5 days, i.p.). Hippocampi were then isolated 24 h after the last injection. After homogenization in isolation buffer containing 10 mM triethanolamine, 250 mM sucrose, 1% sodium dodecyl sulfate (SDS), and a complete protease inhibitor cocktail (Roche, Mannheim, Germany), the samples were centrifuged (13,000 rpm, 40 min), and the supernatant was collected. The protein concentration was determined using a Bicinchoninic acid assay. For gel electrophoresis, the samples (6 µg for each sample) were denatured by heating at 95℃ for 5 min and separated on a 8% SDS gel. After transferring the proteins to nitrocellulose transfer membranes, they were incubated in a blocking buffer [5% nonfat milk with 0.1% Tween-20 in phosphate buffered saline (PBST)] for 1 h at room temperature and then incubated overnight with anti-GLT-1 IgG (1:15000; Millipore, Billerica, MA, U.S.A.) or anti-beta-tubulin IgG (1:200; Santa Cruz, California, U.S.A.). After washing membranes for 10 min, five times with PBST, the membranes were incubated for 1 h with peroxidase-conjugated anti-guinea pig IgG (1:5000, Santa Cruz, California, U.S.A.) or peroxidase-conjugated anti-mouse IgG (1:5000, Vector Laboratories, California, U.S.A.). The signals were visualized with enhanced chemiluminiscence detection reagents (SuperSignal West Femto Maximum Sensitivity Substrate; Thermo Fisher Scientific, Rockford, IL, U.S.A.). The band intensity was measured using Multi Guage V2.2 LAS 3000 software (Fujifilm, Tokyo, Japan).
In situ zymography
In situ zymography was performed as described previously [24]. To assess the activity of gelatinases, including matrix metalloproteinase-2 (MMP-2) and MMP-9, the mice were transcardially perfused with ice-cold PBS (pH 7.4) 3 days after ischemia (control group: n=6; ampicillin-pretreated group: n=6). Thawed frozen, nonfixed coronal brain sections (20 um thick) were incubated for 18 h at 37℃ in a humid dark chamber in reaction buffer containing 50 µg/ml of FITC-labeled DQ-gelatin (Invitrogen Molecular Probes, Eugene, OR, U.S.A.) that was quenched intramolecularly. The gelatin with a fluorescent tag remained caged (no fluorescence) until the gelatin was cleaved by gelatinase activity. Fresh brain slices (20 µm) from each experimental group were incubated with reaction solution containing FITC-labeled gelatin (Invitrogen Molecular Probes, Eugene, OR, U.S.A.).
Immunohistochemistry
For immunohistochemistry, the sections were washed with PBS. After normal serum blocking for 1 h, the sections were incubated with anti-glial fibrillary acidic protein (GFAP) IgG (1:200; Chemicon, Billerca, MA, U.S.A.) or anti-isolectin-B4 IgG (1:200; Vector Laboratories, Burlingame, CA, U.S.A.) overnight. After several washes, the sections were incubated with peroxidase-conjugated IgG (Jackson Immunoresearch, West Grove, PA, U.S.A.) for 2 h, and diaminobenzidine and hydrogen peroxide were used to visualize the signal.
Statistical analysis
Quantitative measurements of the neuronal damage were analyzed by an χ2 test. The Western blot data for evaluating the GLT-1 protein expression level were analyzed using a t-test with GraphPad Prism (GraphPad Software, San Diego, CA, U.S.A.). A p-value of <0.05 was considered significant.
RESULTS
Effect of the ampicillin pretreatment
In our previous study, ampicillin showed neuroprotective effects against transient forebrain ischemia in the mouse hippocampus in a dose-dependent manner [19]. In this study, ampicillin (200 mg/kg) or vehicle was administered for 5 days before the induction of transient global forebrain ischemia, and neuronal cell death was examined 3 days after reperfusion. As shown in Fig. 1, marked pyramidal cell death was observed in the hippocampal CA1 area of the saline-treated ischemic group. However, pyramidal cell death was markedly reduced in the ampicillin-treated group (Fig. 1A). The semiquantitative analysis of the data showed a significant reduction in pyramidal cell death in the medial hippocampus of the ampicillin-treated group compared to that of the saline-treatment group (Fig. 1B). These data indicate that ampicillin has a marked protective effect against ischemic hippocampal neuronal cell death in mice.
Fig. 1. Effect of the ampicillin pretreatment (200 mg/kg for 5 days) on delayed neuronal death in the hippocampus of mice after transient global forebrain ischemia.
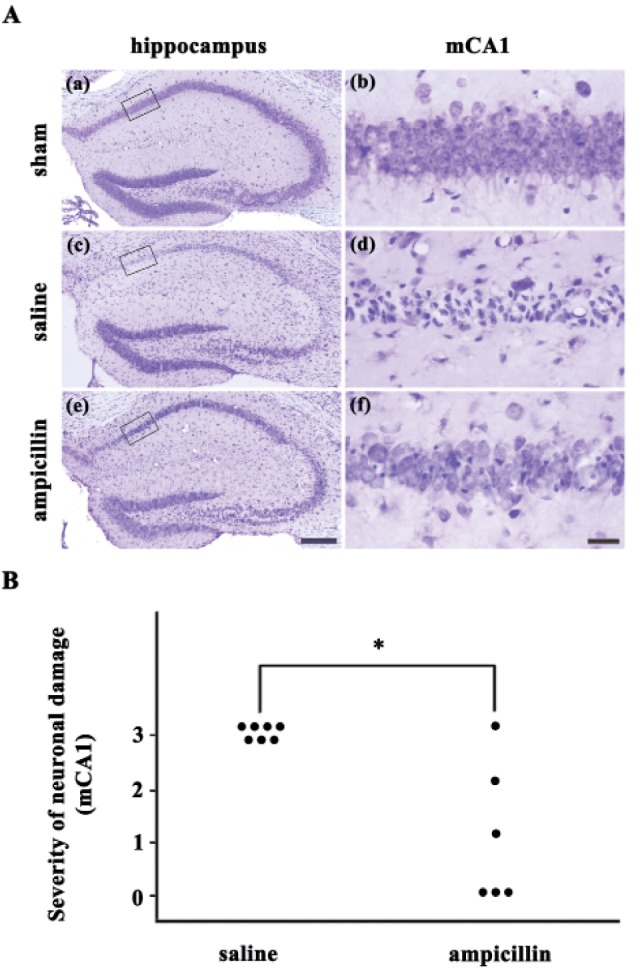
(A) Representative images of cresyl violet-stained brain coronal sections 3 days after transient forebrain ischemia or sham manipulation. Daily treatment with ampicillin protected the medial CA1 pyramidal cells of the hippocampus 3 days after forebrain ischemia. The scale bars in e and f indicate 200 µm and 20 µm, respectively. (B) Quantitative analysis of the neuronal damage in the saline- and ampicillin-treated groups. *p<0.05.
Effect of the penicillin G sodium salt
Next, we tested the effect of penicillin G, which is one of the most commonly used β-lactam antibiotic, and its distribution in the central nervous system is very low. In this study, penicillin G (6,000 U/kg or 20,000 U/kg, i.p.) was administered for 5 days before the induction of transient forebrain ischemia. Neuronal cell death was then examined in the medial CA1 region (Fig. 2). However, there was no significant difference in the neuronal cell death in the hippocampus, indicating that the neuroprotective effect of ampicillin was dependent on the accumulation in the central nervous system.
Fig. 2. Effect of the penicillin G sodium salt pretreatment (6,000, 20,000 U/kg, for 5 days) on delayed hippocampal neuronal death after transient global forebrain ischemia in mice.
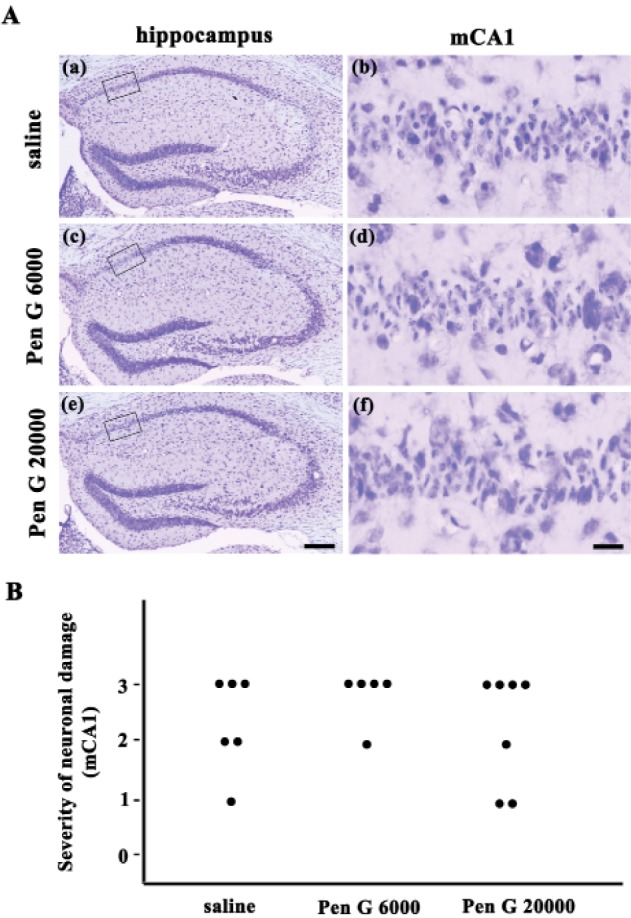
(A) Representative images of cresyl violet-stained brain coronal sections 3 days after transient forebrain ischemia or sham manipulation. The penicillin G sodium salt did not protect the medial CA1 pyramidal cells of the hippocampus 3 days after ischemia/reperfusion. The scale bars in e and f indicate 200 µm and 20 µm, respectively. (B) Quantitative analysis of the neuronal damage in the saline- and ampicillin-treated groups. There was no significant difference in neuronal damage between the saline- and penicillin G-treated groups.
Effect of ampicillin on glial cells and gelatinase reactivation
To examine the activities of astrocyte and microglial cells, immunohistochemistry with GFAP and the isolectin B4 antibody was performed. As shown in Fig. 3, the reactivity of both GFAP and isolectin-B4 in the ampicillin-treated animals seemed to be reduced compared to that of the control animals. Next, it has been known that MMPs, including MMP-2 and MMP-9, are activated during cerebral ischemia and play a role in the degradation of the extracellular matrix, and reportedly inhibition of the activities of MMPs reduced ischemic lesions in cerebral ischemia [25,26]. In the present study, in situ zymography was used to examine whether ampicillin administration reduce the activities of MMPs. As shown in Fig. 4, ampicillin administration distinctly reduced the activities of MMPs in the hippocampus.
Fig. 3. Glial reaction following transient forebrain ischemia and the effect of ampicillin.

Representative images show that the ampicillin treatment apparently reduced the immunoreactivities of isolectin-B4 (A and B) and GFAP (C and D) in the hippocampal CA1 area. The animals were administered saline (A and C) or ampicillin (B and D) and sacrificed 3 days after transient forebrain ischemia. The scale bars indicate 50 µm in B and D.
Fig. 4. Gelatinase activity in the hippocampus.
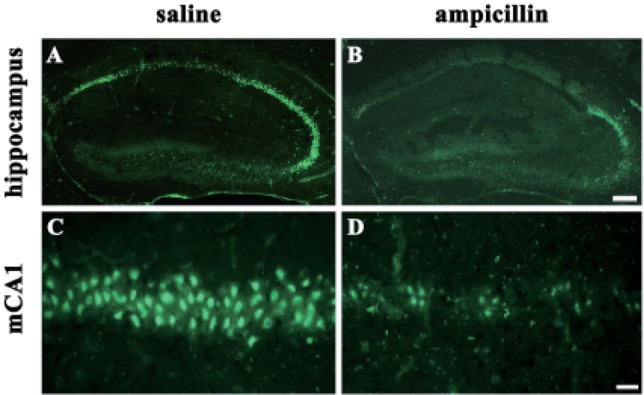
In the saline-treated group, transient forebrain ischemia induced gelatinase activity in the hippocampus. However, ampicillin treatment reversed this activity. In this study, freshly cut hippocampal sections were used for in situ zymography. Gelatinolytic activity was detected as green fluorescence. The scale bars indicate 200 µm and 50 µm in B and D, respectively.
Ampicillin-induced induction of the GLT-1 protein
Next, to identify the role of ampicillin in the induction of GLT-1, a Western blot analysis was employed. Ampicillin or saline was administered to normal mice for 5 days, and the hippocampi were isolated. As shown in Fig. 5, the expression level of GLT-1 increased significantly (~70%) in response to the administration of ampicillin. These data suggest that ampicillin may protect hippocampal neurons by inducing GLT-1 in the hippocampus.
Fig. 5. Expression level of the GLT-1 protein in the hippocampus.

(A) Representative image of a western blot from the control and ampicillin-treated mice. In this study, normal mice were intraperitoneally administered ampicillin or saline for 5 days. (B) Quantitative data of GLT-1 expression in the hippocampus. The data are presented as the mean±SEM. *p<0.05.
DHK reversed the neuroprotective effects of ampicillin
Finally, to identify the molecular mechanism of ampicillin, we tested whether co-administration of DHK, a GLT-1 antagonist, reversed the protective effect of ampicillin. DHK (10 mg/kg) was administered to the mice 30 min before the onset of ischemia, and neuronal cell death was then examined in the hippocampus. As shown in Fig. 6, the ampicillin treatment significantly reduced neuronal damage in the medial CA1 area. However, the neuroprotective effect of ampicillin disappeared following co-administration of the DHK. These data clearly indicate that ampicillin protects pyramidal neurons in the hippocampus against global forebrain ischemia by increasing the expression level and activity of GLT-1.
Fig. 6. Effect of dihydrokainic acid (DHK), a selective antagonist of the glutamate transporter GLT-1, on hippocampal neuronal death following global forebrain ischemia.
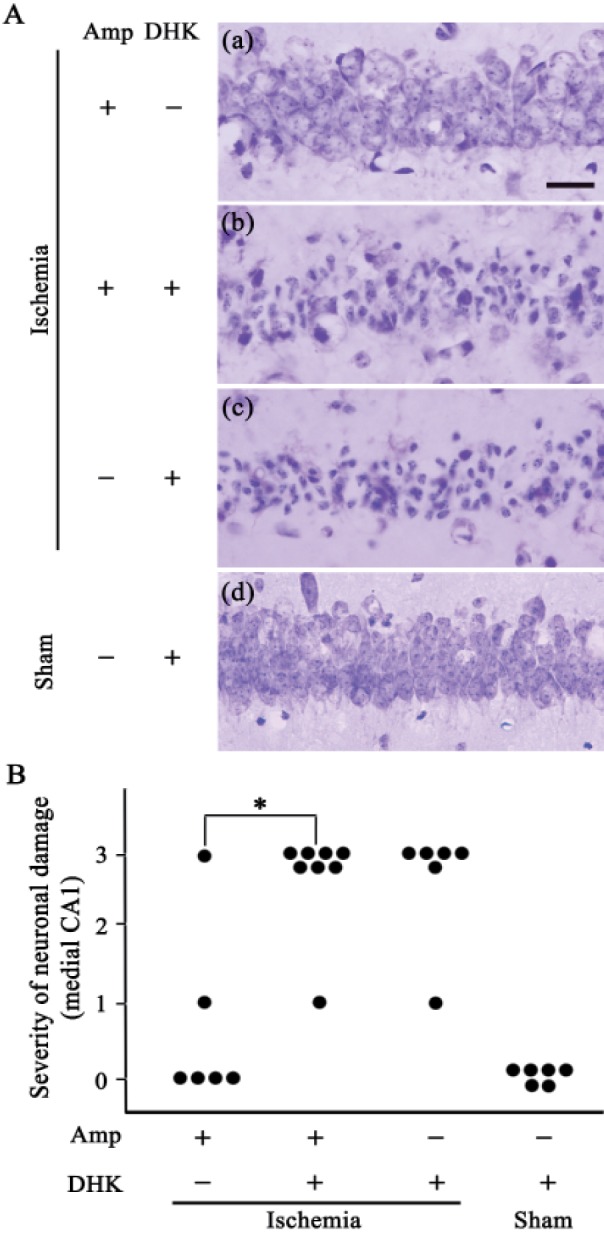
(A) Representative images from each group indicate that the ampicillin treatment (200 mg/kg for 5 days) reduced neuronal cell death in the medial CA1 of the hippocampus. In addition, the protective effect of ampicillin was reversed by co-administration of DHK (10 mg/kg). The scale bar indicates 20 µm. (B) The quantitative analysis of the neuronal damage indicated that the ampicillin treatment significantly reduced neuronal cell death and that DHK reversed its neuroprotective effect. Animal numbers are 6, 8, 6, and 6 for ampicillin treated-, ampicillin+DHK treated-, DHK treated-, and sham-manipulated groups, respectively. *p<0.05 by χ2 test.
DISCUSSION
In the present study, ampicillin reduced the activities of MMPs and increased the expression level of GLT-1. In addition, pretreatment with ampicillin significantly reduced medial hippocampal cell death following global forebrain ischemia, and its protective effect disappeared following co-administration of DHK, a GLT-1 antagonist. Combined, the data indicate that ampicillin-induced neuroprotection against transient forebrain ischemia is mediated by the induction of GLT-1.
Glutamate transporters regulate the synaptic concentration of glutamate by clearing glutamate released into the synapses [13]. The accumulation of synaptic glutamate during ischemia and the early period of reperfusion triggers a cascade of neuronal cell death. Pharmacological tools and genetic approaches have been used to minimize glutamate signaling and confer neuroprotection against neurological diseases, such as cerebral ischemia and epilepsy. Interestingly, among the five subtypes of glutamate transporters, GLT-1 accounts for about 80% of those in the hippocampus. Many pharmacological approaches to inhibit the activity of GLT-1 have been developed. Several candidates, such as ceftriaxone, successfully provided therapeutic evidence in brain ischemia [17]. This study explored the therapeutic efficacy of the β-lactam antibiotics ampicillin and penicillin G, both of which, like ceftriaxone, have been reported to induce GLT-1 expression [20]. In the present study, the pretreatment with ampicillin, but not with penicillin G, significantly attenuated neuronal damage in the hippocampal CA1 subfield. These findings are supported by the following observation. Unlike ampicillin, penicillin G, administered systemically, does not accumulate in the central nervous system because the outward clearance of penicillin G from the cerebrospinal fluid is ~14 times faster than the inward clearance [27]. As a result, in both animal and human studies, the concentration of penicillin G in the cerebrospinal fluid was 1~2% that of the plasma concentration. In addition, the present study demonstrated that the pretreatment with ampicillin significantly increased the expression level of GLT-1 in the hippocampus. Therefore, the findings of the present study suggest that ampicillin provides protection against global forebrain ischemia, possibly by the increased expression of GLT-1 in the hippocampus. The neuroprotective mechanism of ampicillin was further studied by co-administration with DHK, a selective inhibitor of GLT-1. DHK reportedly reversed the protective effect of ceftriaxone against global forebrain ischemia and Parkinson's disease in rodents [28,29]. As expected, DHK effectively reversed the neuroprotective effect of ampicillin. Taken together, the present results indicate that β-lactam antibiotics including ampicillin which can increase the activity of GLT-1 in the brain potentially protect the hippocampus from ischemic insults.
MMPs are involved in the degradation of the extracellular matrix. They are thought to inhibit interactions of neurons with the extracellular matrix and induce neuronal cell death. In accordance with this idea, MMP inhibitors, such as resveratrol and melatonin, provided neuronal protection against ischemic injuries [30,31]. The mechanisms by which MMPs mediate neuronal damage have not been fully elucidated. However, they have been shown to degrade the vascular matrix, including collagen and laminin. In addition, MMP-9 was reported to degrade ZO-1, a blood-brain barrier protein, thereby disrupting the integrity of the blood-brain barrier [32]. In recent studies, neuronal damage was markedly reduced in MMP-9 knockouts following traumatic brain injury and focal ischemia compared to wild-type littermates [26,33]. On the other hand, MMP-2 knockouts were not neuroprotective in an animal model of focal cerebral ischemia [34]. Therefore, MMP-9 is thought to be a critical mediator of the induction of neuronal cell death following cerebral ischemia. In the present study, we examined the activity of gelatinases including MMP-9 and MMP-2. Their activity was increased in the hippocampus following global forebrain ischemia, whereas the pretreatment with ampicillin apparently reduced their activity. Although it is not clear whether ampicillin directly inhibits the activity of MMPs, such as MMP-9 or MMP-2, the data from this study suggest that the neuroprotective effects of ampicillin are mediated, at least in part, by its attenuation of the increased activity of MMPs following brain ischemia. Further studies are needed to elucidate the mechanism of action of ampicillin related to the inhibition of the activities of MMP.
In summary, transiently induced global forebrain ischemia resulted in neuronal cell death in discrete regions of the hippocampus, and ampicillin conferred neuroprotection both by increasing the activity of GLT-1 and by decreasing the activities of MMPs. Although further studies are needed to fully understand the mechanism of neuroprotective action of ampicillin, the present data clearly indicate that ampicillin may be a potential candidate as a therapeutic tool against ischemic brain insults.
Footnotes
Author contributions: K.-E.L. and S.Y.K. designed the study. K.-E.L., K.-O.C. and Y.-S.C. performed experiments and analyzed and interpreted the data. K.-O.C. and S.Y.K. supervised and coordinated the study. K.-E.L., Y.-S.C. and S.Y.K. wrote the manuscript.
References
- 1.Kirino T. Delayed neuronal death in the gerbil hippocampus following ischemia. Brain Res. 1982;239:57–69. doi: 10.1016/0006-8993(82)90833-2. [DOI] [PubMed] [Google Scholar]
- 2.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 3.Chong W, Kim SN, Han SK, Lee SY, Ryu PD. Low non-NMDA receptor current density as possible protection mechanism from neurotoxicity of circulating glutamate on subfornical organ neurons in rats. Korean J Physiol Pharmacol. 2015;19:177–181. doi: 10.4196/kjpp.2015.19.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melani A, Dettori I, Corti F, Cellai L, Pedata F. Time-course of protection by the selective A2A receptor antagonist SCH58261 after transient focal cerebral ischemia. Neurol Sci. 2015;36:1441–1448. doi: 10.1007/s10072-015-2160-y. [DOI] [PubMed] [Google Scholar]
- 5.Yu G, Wu F, Wang ES. BQ-869, a novel NMDA receptor antagonist, protects against excitotoxicity and attenuates cerebral ischemic injury in stroke. Int J Clin Exp Pathol. 2015;8:1213–1225. [PMC free article] [PubMed] [Google Scholar]
- 6.Kong Q, Takahashi K, Schulte D, Stouffer N, Lin Y, Lin CL. Increased glial glutamate transporter EAAT2 expression reduces epileptogenic processes following pilocarpine-induced status epilepticus. Neurobiol Dis. 2012;47:145–154. doi: 10.1016/j.nbd.2012.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zumkehr J, Rodriguez-Ortiz CJ, Cheng D, Kieu Z, Wai T, Hawkins C, Kilian J, Lim SL, Medeiros R, Kitazawa M. Ceftriaxone ameliorates tau pathology and cognitive decline via restoration of glial glutamate transporter in a mouse model of Alzheimer's disease. Neurobiol Aging. 2015;36:2260–2271. doi: 10.1016/j.neurobiolaging.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Zhang M, Li WB, Liu YX, Liang CJ, Liu LZ, Cui X, Gong JX, Gong SJ, Hu YY, Xian XH. High expression of GLT-1 in hippocampal CA3 and dentate gyrus subfields contributes to their inherent resistance to ischemia in rats. Neurochem Int. 2011;59:1019–1028. doi: 10.1016/j.neuint.2011.08.023. [DOI] [PubMed] [Google Scholar]
- 9.Fontana AC. Current approaches to enhance glutamate transporter function and expression. J Neurochem. 2015;134:982–1007. doi: 10.1111/jnc.13200. [DOI] [PubMed] [Google Scholar]
- 10.Massie A, Cnops L, Smolders I, McCullumsmith R, Kooijman R, Kwak S, Arckens L, Michotte Y. High-affinity Na+/K+-dependent glutamate transporter EAAT4 is expressed throughout the rat fore- and midbrain. J Comp Neurol. 2008;511:155–172. doi: 10.1002/cne.21823. [DOI] [PubMed] [Google Scholar]
- 11.Scofield MD, Kalivas PW. Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist. 2014;20:610–622. doi: 10.1177/1073858413520347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mookherjee P, Green PS, Watson GS, Marques MA, Tanaka K, Meeker KD, Meabon JS, Li N, Zhu P, Olson VG, Cook DG. GLT-1 loss accelerates cognitive deficit onset in an Alzheimer's disease animal model. J Alzheimers Dis. 2011;26:447–455. doi: 10.3233/JAD-2011-110503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soni N, Reddy BV, Kumar P. GLT-1 transporter: an effective pharmacological target for various neurological disorders. Pharmacol Biochem Behav. 2014;127:70–81. doi: 10.1016/j.pbb.2014.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J, Sutherland ML. Glutamate transporter cluster formation in astrocytic processes regulates glutamate uptake activity. J Neurosci. 2004;24:6301–6306. doi: 10.1523/JNEUROSCI.1404-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meeker KD, Meabon JS, Cook DG. Partial loss of the glutamate transporter GLT-1 alters brain Akt and insulin signaling in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2015;45:509–520. doi: 10.3233/JAD-142304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA. Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci. 2015;35:5187–5201. doi: 10.1523/JNEUROSCI.4255-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu YY, Xu J, Zhang M, Wang D, Li L, Li WB. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem. 2015;132:194–205. doi: 10.1111/jnc.12958. [DOI] [PubMed] [Google Scholar]
- 18.Chu K, Lee ST, Sinn DI, Ko SY, Kim EH, Kim JM, Kim SJ, Park DK, Jung KH, Song EC, Lee SK, Kim M, Roh JK. Pharmacological induction of ischemic tolerance by glutamate transporter-1 (EAAT2) upregulation. Stroke. 2007;38:177–182. doi: 10.1161/01.STR.0000252091.36912.65. [DOI] [PubMed] [Google Scholar]
- 19.Lee KE, Kim SK, Cho KO, Kim SY. Pre-ischemic treatment with ampicillin reduces neuronal damage in the mouse hippocampus and neostriatum after transient forebrain ischemia. Korean J Physiol Pharmacol. 2008;12:287–291. doi: 10.4196/kjpp.2008.12.6.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Dykes Hoberg M, Vidensky S, Chung DS, Toan SV, Bruijn LI, Su ZZ, Gupta P, Fisher PB. Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–77. doi: 10.1038/nature03180. [DOI] [PubMed] [Google Scholar]
- 21.Cho KO, Kim SK, Cho YJ, Sung KW, Kim SY. A simple method for predicting hippocampal neurodegeneration in a mouse model of transient global forebrain ischemia. Korean J Physiol Pharmacol. 2006;10:167–172. [Google Scholar]
- 22.Zhang M, Li WB, Geng JX, Li QJ, Sun XC, Xian XH, Qi J, Li SQ. The upregulation of glial glutamate transporter-1 participates in the induction of brain ischemic tolerance in rats. J Cereb Blood Flow Metab. 2007;27:1352–1368. doi: 10.1038/sj.jcbfm.9600441. [DOI] [PubMed] [Google Scholar]
- 23.Murakami K, Kondo T, Kawase M, Chan PH. The development of a new mouse model of global ischemia: focus on the relationships between ischemia duration, anesthesia, cerebral vasculature, and neuronal injury following global ischemia in mice. Brain Res. 1998;780:304–310. doi: 10.1016/s0006-8993(97)01217-1. [DOI] [PubMed] [Google Scholar]
- 24.Lee SR, Tsuji K, Lee SR, Lo EH. Role of matrix metalloproteinases in delayed neuronal damage after transient global cerebral ischemia. J Neurosci. 2004;24:671–678. doi: 10.1523/JNEUROSCI.4243-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pandey AK, Bhattacharya P, Shukla SC, Paul S, Patnaik R. Resveratrol inhibits matrix metalloproteinases to attenuate neuronal damage in cerebral ischemia: a molecular docking study exploring possible neuroprotection. Neural Regen Res. 2015;10:568–575. doi: 10.4103/1673-5374.155429. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000;20:1681–1689. doi: 10.1097/00004647-200012000-00007. [DOI] [PubMed] [Google Scholar]
- 27.Spector R. Nature and consequences of mammalian brain and CSF efflux transporters: four decades of progress. J Neurochem. 2010;112:13–23. doi: 10.1111/j.1471-4159.2009.06451.x. [DOI] [PubMed] [Google Scholar]
- 28.Hu YY, Xu J, Zhang M, Wang D, Li L, Li WB. Ceftriaxone modulates uptake activity of glial glutamate transporter-1 against global brain ischemia in rats. J Neurochem. 2015;132:194–205. doi: 10.1111/jnc.12958. [DOI] [PubMed] [Google Scholar]
- 29.Kelsey JE, Neville C. The effects of the β-lactam antibiotic, ceftriaxone, on forepaw stepping and L-DOPA-induced dyskinesia in a rodent model of Parkinson's disease. . Psychopharmacology (Berl) 2014;231:2405–2415. doi: 10.1007/s00213-013-3400-6. [DOI] [PubMed] [Google Scholar]
- 30.Gao D, Huang T, Jiang X, Hu S, Zhang L, Fei Z. Resveratrol protects primary cortical neuron cultures from transient oxygen-glucose deprivation by inhibiting MMP-9. Mol Med Rep. 2014;9:2197–2204. doi: 10.3892/mmr.2014.2086. [DOI] [PubMed] [Google Scholar]
- 31.Kim SJ, Lee SR. Protective effect of melatonin against transient global cerebral ischemia-induced neuronal cell damage via inhibition of matrix metalloproteinase-9. Life Sci. 2014;94:8–16. doi: 10.1016/j.lfs.2013.11.013. [DOI] [PubMed] [Google Scholar]
- 32.Harkness KA, Adamson P, Sussman JD, Davies-Jones GA, Greenwood J, Woodroofe MN. Dexamethasone regulation of matrix metalloproteinase expression in CNS vascular endothelium. Brain. 2000;123:698–709. doi: 10.1093/brain/123.4.698. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Jung J, Asahi M, Chwang W, Russo L, Moskowitz MA, Dixon CE, Fini ME, Lo EH. Effects of matrix metalloproteinase-9 gene knock-out on morphological and motor outcomes after traumatic brain injury. J Neurosci. 2000;20:7037–7042. doi: 10.1523/JNEUROSCI.20-18-07037.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asahi M, Sumii T, Fini ME, Itohara S, Lo EH. Matrix metalloproteinase 2 gene knockout has no effect on acute brain injury after focal ischemia. Neuroreport. 2001;12:3003–3007. doi: 10.1097/00001756-200109170-00050. [DOI] [PubMed] [Google Scholar]