

ABSTRACT
Collagen IV is a major component of basement membranes, and mutations in COL4A1, which encodes collagen IV alpha chain 1, cause a multisystemic disease encompassing cerebrovascular, eye and kidney defects. However, COL4A1 renal disease remains poorly characterized and its pathomolecular mechanisms are unknown. We show that Col4a1 mutations in mice cause hypotension and renal disease, including proteinuria and defects in Bowman's capsule and the glomerular basement membrane, indicating a role for Col4a1 in glomerular filtration. Impaired sodium reabsorption in the loop of Henle and distal nephron despite elevated aldosterone levels indicates that tubular defects contribute to the hypotension, highlighting a novel role for the basement membrane in vascular homeostasis by modulation of the tubular response to aldosterone. Col4a1 mutations also cause diabetes insipidus, whereby the tubular defects lead to polyuria associated with medullary atrophy and a subsequent reduction in the ability to upregulate aquaporin 2 and concentrate urine. Moreover, haematuria, haemorrhage and vascular basement membrane defects confirm an important vascular component. Interestingly, although structural and compositional basement membrane defects occurred in the glomerulus and Bowman's capsule, no tubular basement membrane defects were detected. By contrast, medullary atrophy was associated with chronic ER stress, providing evidence for cell-type-dependent molecular mechanisms of Col4a1 mutations. These data show that both basement membrane defects and ER stress contribute to Col4a1 renal disease, which has important implications for the development of treatment strategies for collagenopathies.
KEY WORDS: Collagen IV, Extracellular matrix, Endoplasmic reticulum stress, Basement membrane, Kidney disease
Summary: Structural and compositional basement membrane defects and ER stress due to Col4a1 mutations cause glomerular and tubular kidney disease, and indicate cell-type-specific disease mechanisms for collagen diseases.
INTRODUCTION
Collagen IV is a major component of the basement membrane (BM), a specialised extracellular matrix structure that provides structural support and influences cell behaviour and signalling. Vertebrates express six collagen IV alpha chains, Col4a1-Col4a6, and in the endoplasmic reticulum (ER) three alpha chains form triple-helical protomers that generate three collagen IV networks: α1α1α2(IV), α3α4α5(IV) and α5α5α6(IV) (Khoshnoodi et al., 2008; Van Agtmael and Bruckner-Tuderman, 2010). During kidney development, all BMs contain α1α1α2(IV), whereas in adult kidney it is mainly expressed in vascular, Bowman's capsule, mesangial and tubular BMs (Hudson et al., 2003). The adult glomerular BM (GBM) predominantly contains α3α4α5(IV), and mutations affecting this network cause Alport syndrome (Hudson et al., 2003).
We have previously identified mouse models with Col4a1 mutations: Col4a1+/Raw and Col4a1+/Svc, containing a lysine (K950E) and a glycine substitution (G1064D), respectively (Van Agtmael et al., 2005). Initial histopathological analysis on a mixed genetic background indicated that within this allelic series the glycine mutations appear to cause a more severe phenotype (Van Agtmael et al., 2005). Col4a1 mouse models are excellent models of the human disease, illustrated by the fact that their analysis led to the identification of humans with COL4A1/COL4A2 mutations (Favor et al., 2007; Gould et al., 2005; Jeanne et al., 2012; Murray et al., 2014; Sibon et al., 2007; Yoneda et al., 2012). Mutations in COL4A1 and COL4A2 cause a multisystemic disorder that leads to cerebrovascular disease, eye defects and muscular dystrophy (Vahedi and Alamowitch, 2011). Some patients present with HANAC (hereditary angiopathy, nephropathy, aneurysms and cramps) syndrome and can develop haematuria, Bowman's capsule defects, large renal cysts and reduced glomerular filtration rate (GFR; Plaisier et al., 2007). HANAC syndrome has been proposed as a clinical sub-entity within COL4A1/COL4A2 disease (Alamowitch et al., 2009), resulting from mutations located in or close to the integrin-binding CB3 region of the collagen protomer predicted to affect integrin signalling (Plaisier et al., 2010). Importantly, COL4A1/COL4A2 variants have also been implicated in sporadic cases of cerebral vascular disease in the general population (Rannikmae et al., 2015; Weng et al., 2012).
COL4A1/COL4A2 mutations are associated with BM defects, ER stress and the unfolded protein response (UPR; Gould et al., 2007; Murray et al., 2014; Van Agtmael et al., 2010). ER stress can be induced by the accumulation of misfolded protein within the ER, and the UPR aims to relieve ER stress by reducing general protein synthesis and increasing the levels of chaperones to promote protein folding (Bateman et al., 2009). Although the UPR is a homeostatic response, chronic ER stress activates pro-apoptotic pathways, in part mediated via activation of the protein CHOP (C/EBP homologous protein; Ron and Walter, 2007), and can become pathogenic. Chronic ER stress has recently been implicated in a variety of matrix diseases (Bateman et al., 2009) in addition to kidney diseases such as uromodulin-associated kidney disease (Williams et al., 2009) and Pierson syndrome, caused by mutations in the basement membrane component laminin beta 2 (Chen et al., 2013).
Our initial analysis in Col4a1 mutant mice revealed a renal component in Col4a1 disease mainly affecting Bowman's capsule (Van Agtmael et al., 2005), whereas analysis of other mouse models indicated mild proteinuria (Favor et al., 2007). Individuals with HANAC syndrome develop similar defects in Bowman's capsule and also a structural phenotype to the tubular BM and the formation of large cysts (Plaisier et al., 2007), although they do not develop a polycystic kidney disease (Plaisier et al., 2010, 2007). However, the role of this essential BM component in renal pathophysiology remains relatively poorly characterized; for example, the potential progression of Col4a1 renal disease and its pathomolecular mechanisms are unknown.
Here, we have uncovered that Col4a1 mutations in mice cause renal glomerular and tubular disease, which becomes more severe with age and leads to proteinuria, polyuria and haematuria. Our data support the suggestion that Col4a1 mutations can display cell-specific pathomolecular mechanisms, because the glomerular and tubular disease components are associated with BM defects and ER-stress-induced apoptosis, respectively. This has important implications for the development of therapeutic approaches.
RESULTS
Col4a1 renal disease includes renal and tubular disease that develops with age
We assessed in vivo renal function in 3- to 4-month-old Col4a1+/Raw (Fig. 1; Table 1) and Col4a1+/Svc (Table 1; Fig. S1) mice, which revealed a reduction in blood pressure of ∼20 mmHg (Fig. 1A; Fig. S1A). Col4a1+/Raw mice display reduced Na+ excretion (Fig. 1B) and GFR as assessed by in vivo inulin clearance assays (Fig. 1C; Table 1). Col4a1 mutant mice have an activated renin-angiotensin system, as indicated by elevated aldosterone levels (Table S1; Van Agtmael et al., 2010). We used diuretic profiling to determine the in vivo activity of the major aldosterone-sensitive sodium transport proteins in Col4a1+/Raw mice (Fig. 1D), whereby responses to furosemide, thiazide and amiloride were measured to assess in vivo activity of the Na+-K+-2Cl− cotransporter (NKCC2), the Na+-Cl− cotransporter (NCC) and the epithelial sodium channel (ENaC), respectively (Bailey et al., 2009; Hunter et al., 2014, 2015). Furosemide-sensitive sodium reabsorption was ∼3.5-fold reduced in Col4a1 mutant mice, but this was a functional downregulation of NKCC2 activity because total protein levels were similar to WT (WT) mice (Fig. S2). Likewise, thiazide-sensitive sodium reabsorption in Col4a1 mutant mice was approximately fourfold reduced, indicating downregulation of NCC. Amiloride-sensitive sodium reabsorption via ENaC was comparable to WT (Fig. 1D), despite the elevated aldosterone levels. The net increase in tubular sodium reabsorption could be explained by the ∼16-fold increased protein levels of active unphosphorylated sodium hydrogen exchanger (NHE3; Fig. 1E,F).
Fig. 1.

Analysis of renal function. (A-D) In vivo renal function analysis of 1-, 3- to 4- and 6- to 8-month-old Col4a1+/Raw (black bars) and WT (white bars) mice. (A) Reduced mean arterial blood pressure in Col4a1+/Raw animals at all ages. (B) Reduced sodium excretion in Col4a1+/Raw animals at all ages. (C) Inulin clearance assays uncovered reduced glomerular filtration rate per gram body weight at 3 months (∼40% reduction), but no further decline with age. (D) Measurement of sodium excretion in the presence of the diuretics furosemide and thiazide remains reduced in mutant animals. Blockade of ENaC by amiloride abolished the difference between WT and mutant mice. (E) Western blotting showed ∼16-fold increased levels of total NHE3 (NHE3) in Col4a1+/Svc (SVC) mice but unaltered phosphorylated NHE3 (p-NHE3). Representative band of total protein stain is given as loading control (entire gel is provided in Fig. S6). (F) ImageJ densitometry analysis of total and p-NHE3. *P<0.05, **P<0.01, ***P<0.001. n=5-7 in A-E; n=3-5 in F.
Table 1.
Renal defects in 3- to 4-month-old female Col4a1+/Raw and Col4a1+/Svc mice
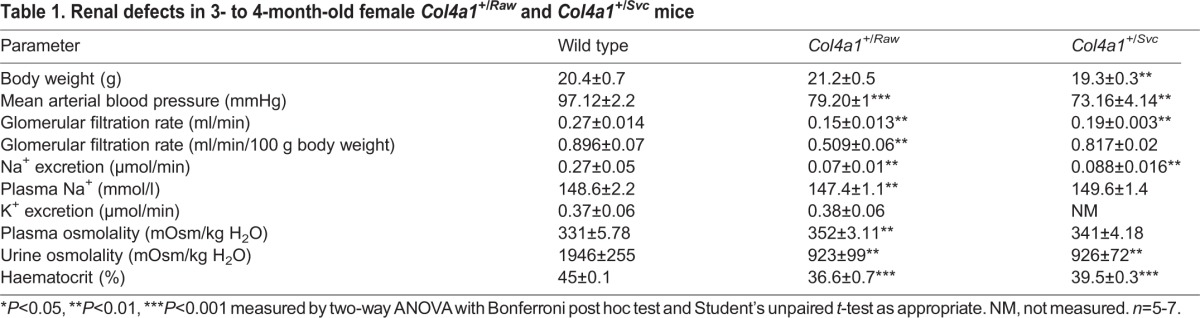
These data indicate that the reduced blood pressure leads to increased sodium reabsorption and activation of the renin-angiotensin-aldosterone system (RAAS) as an attempt to normalize blood pressure. However, the response to the diuretics revealed that Col4a1 mutant mice have aldosterone insensitivity of the distal nephron, thereby reducing the efficacy of the RAAS to increase blood pressure levels, a factor that could contribute to the development of low blood pressure. To compensate for this deficit, mutant mice activate reabsorption in the proximal tubule via NHE3. A reduction in GFR would reduce the filtered sodium load and also help to preserve sodium homeostasis. However, low GFR was not a consistent feature across the age groups; 1-month-old Col4a1+/Raw animals revealed a normal GFR and increased sodium reabsorption (Fig. 1B,C; Table S1). We also failed to detect a decline in GFR in 6- to 8-month-old mice (Fig. 1C; Table S2). This absence of a further decline in GFR indicates that COL4A1 renal disease is unlikely to progress to renal failure, which is supported by a limited reduction of GFR in humans with HANAC syndrome (Plaisier et al., 2007).
Diabetes insipidus resulting from Col4a1 mutations
Polyuria and polydipsia are hallmarks of diabetes insipidus that could be attributable to reduced vasopressin production in the brain (central diabetes insipidus) or inability of the kidney to concentrate urine in response to vasopressin (nephrogenic diabetes insipidus; Babey et al., 2011). Despite the low blood pressure and increased sodium reabsorption, a metabolic cage study revealed polyuria in mutant animals, with Col4a1+/Raw mice displaying a ∼1.8-fold increase in daily urine production (Fig. 2A) and a ∼1.7-fold increase in urine flow rate as measured by in vivo renal clearance assays (Fig. 2B), further confirming the tubular disease. Data from Col4a1+/Svc mice confirmed this (Fig. 2C,D; Table 1; Fig. S1). The polyuria is accompanied by a twofold increase in water consumption (polydipsia; Fig. 2D) and reduced urine osmolality (Fig. 2E; Fig. S1), indicating a defect in concentrating urine that occurs in the distal tubules. Analysis of 1-month-old Col4a1+/Raw animals revealed normal water excretion and urine osmolality (Fig. 2E; Tables S1 and S2). However, there is a further decline in urine osmolality at 6-8 months, indicating that polyuria becomes more severe with age. Polyuria (∼4.7-fold increase; Fig. 2F) and polydipsia (∼1.6-fold increase; Fig. S1E) were also observed in 2.5-month-old Col4a1+/Svc mice.
Fig. 2.

Diabetes insipidus in Col4a1 mutant mice. (A) Metabolic cage study revealed ∼1.8-fold increased 24 h (24H) urine production in Col4a1+/Raw (RAW) mice per gram body weight (g BW; n=6). (B) In vivo urine flow was ∼1.7-fold increased in 3- to 4-month-old mice. Older mutant mice displayed similar trends. (n=4-6). (C) Metabolic cage study revealed increased daily (24 h) urine production in 4- to 5-month-old Col4a1+/Svc (SVC) mice per gram body weight (n=4-6). (D) Metabolic cage study indicates ∼4.2-fold increased daily water consumption in Col4a1+/Svc mice (WT, n=6; Col4a1+/Svc, n=10). (E) Col4a1+/Raw mice develop a reduced urine osmolality at 3 months, which becomes more severe with age. Urine osmolality of 3- to 4-month-old Col4a1+/Svc mice is provided in Fig. S1D (n=5-7). (F) Increased urine production in 2.5-month-old Col4a1+/Svc mice compared with controls (n=3). (G) Effect of water deprivation (WD) on urine production in wild-type and Col4a1+/Svc mice (n=4-5). (H) Percentage weight loss following 24 h water deprivation in 2.5- (2.5M) and 4- to 5-month-old animals (2.5M, n=3; 4-5 months old, n=4-5). (I) Urine production after vasopressin injection. (J) Analysis of Aqp2 protein levels in 4- to 6-month-old Col4a1+/Svc mice. Representative band of total protein stain is given as loading control (entire gel is provided in Fig. S6) (n=3) with densitometry analysis using ImageJ. *P<0.05, **P<0.01, ***P<0.001.
To investigate whether primary polydipsia (increased thirst) causes the apparent diabetes insipidus, mice were deprived of water for 24 h. Although there was an approximately twofold reduction in daily urine output in mutant mice, compared with approximately threefold in controls (Fig. 2G), it remained significantly higher in Col4a1+/Svc mice. This revealed that mutant mice partly retained the ability to concentrate their urine and excludes primary polydipsia as being causative. Mutant mice had increased weight loss when deprived of water, which became more severe with age (Fig. 2H), indicating dehydration, which combined with the elevated urine output during water deprivation (Fig. 2I), further supports the presence of diabetes insipidus. To distinguish between nephrogenic and central diabetes insipidus, urine output was measured following injection (1 µg/kg) with arginine vasopressin (Gabbi et al., 2012). Overall, 4- to 5-month-old mutant mice maintained an elevated urine output, demonstrating a reduced ability of mutant kidneys to respond to vasopression and thus nephrogenic diabetes insipidus (Fig. 2I). However, a dichotomy was observed, because some mutant mice, which were polyuric, responded to vasopressin, suggesting a component of central diabetes insipidus. Analysis of 2.5-month-old mice revealed increased urine output when deprived of water (Fig. S1F). In accordance with nephrogenic diabetes insipidus, untreated 4-month-old Col4a1 mutant mice did not have elevated levels of the water channel aquaporin 2 (Aqp2; Fig. 2J) that plays a crucial role in tubular water reabsorption and urine concentration. The variability observed in Aqp2 expression, supported by the vasopressin data, could reflect differing severity of the phenotype, as is characteristic for Col4a1 mutant mice and patients (Vahedi and Alamowitch, 2011; Van Agtmael et al., 2005), and/or variation in the onset and progression of the defects (see below). Thus, inability to increase Aqp2 expression is in keeping with a generalised homeostatic failure of the distal nephron and, in Col4a1 mutant mice, leads to a failure to concentrate urine and diabetes insipidus.
Erythropoietin treatment rescues the hypotension
Col4a1 mutant mice display reduced blood volume and haematocrit levels but normal plasma volume (Van Agtmael et al., 2010), and recently, anaemia has been described in individuals with a COL4A1 mutation (Tomotaki et al., 2014; Yoneda et al., 2013). The mechanism underlying the reduced red blood cell number remained unexplored, but a defect in erythropoiesis or erythrocyte maturation could be a contributing factor whereby the kidney disease affects renal erythropoietin production. To determine whether the reduced haematocrit in Col4a1 mutant mice can be explained by reduced erythrocyte maturation, Col4a1+/Svc mice were treated with human recombinant erythropoietin (Epo) for 2 weeks. Interestingly, Epo treatment rescued haematocrit levels in 4-month-old Col4a1+/Svc mice (Fig. 3A) and the increased haematocrit levels were associated with an increased in blood pressure in Col4a1 mutant mice (Fig. 3B). These data confirm that reduced renal Epo production and subsequent erythrocyte maturation contribute to the haematological and blood pressure phenotypes.
Fig. 3.

Erythropoietin treatment of Col4a1 mutant mice. (A) Four-month-old Col4a1+/Svc (black bars) and WT (white bars) mice were treated with Epo, and haematological analysis was performed before (−Epo) and afer Epo (+Epo) treatment, which showed that treatment rescued haematocrit values in Col4a1+/Svc mice (WT 44% pretreatment, 44% post-treatment; Col4a1+/Svc 36% pre-treatment, 49% post-treatment; n=4-5). (B) Tail-cuff plethysmography revealed a rescue of blood pressure in Epo-treated mice (WT 122 mmHg pre-treatment, 114 mmHg post-treatment; Col4a1+/Svc 97 mmHg pre-treatment, 110 mmHg post-treatment; n=4-5). *P<0.05, **P<0.01, ***P<0.001, Student's unpaired t-test.
Age-dependent atrophy of the medulla and Bowman's capsule defects contribute to renal dysfunction
To determine any morphological basis to the renal dysfunction, we performed Haematoxylin and Eosin staining on 21- and 40-day-old, 3- to 4- and 6- to 8-month-old Col4a1+/Raw mice (Fig. 4A,B; Table S3). This identified glomerulopathy characterized by hypertrophy and a more cuboidal appearance of the parietal epithelium of Bowman's capsule (Van Agtmael et al., 2005; Fig. 4A), which develops fully by 40 days of age (Table S3). No significant difference in cellularity of the glomerulus or glomerulosclerosis was detected, confirming that the glomerulopathy primarily affects Bowman's capsule. The morphological defects progress with age, because at around 3 months atrophy of the medulla begins to develop and becomes fully penetrant by 6 months (Fig. 4B; Table S3). Hydronephrosis might also contribute to the atrophy of the medulla, because mutant animals develop distended bladders (data not shown). However, mutant mice urinate freely as indicated by our metabolic cage studies, excluding an obstruction in the urethra being the major cause. These data were confirmed in 40-day-old and 3- to 4-month-old Col4a1+/Svc mice (Fig. 4A,B and data not shown). The onset of atrophy of the medulla corresponds with the increased urine flow rate, and further reduced urine osmolality (Fig. 2E). This suggests that the medullary atrophy is likely to prevent the ability to increase Aqp2 levels sufficiently to concentrate the urine, resulting in a nephrogenic component to the diabetes insipidus. Combined with the data showing polyuria in 2.5-month-old animals (Fig. 2F), which is before the medullary atrophy has developed, this provides evidence for the development of an initial central diabetes insipidus with an additional nephrogenic component as animals age.
Fig. 4.

Analysis of kidney histopathology. (A) Bowman's capsule defects in 3- to 4-month-old Col4a1+/Raw (RAW) and Col4a1+/Svc (SVC) mice include thickening of the capsule (arrowhead) and formation of multiple cell layers of parietal epithelial cells, which can develop a more cuboid appearance. (B) Haematoxylin and Eosin staining revealed atrophy of the medulla (indicated by black box) in 8-month-old Col4a1+/Raw and Col4a1+/Svc mice that develops in adulthood (see Table S3). (C) Protein cast (arrowhead) in kidney sections of Col4a1+/Raw and Col4a1+/Svc mice (n=3, 6 months old). (D) Perls' Prussian Blue staining of renal sections identified haem deposits (arrowhead) in 4-month-old Col4a1+/Svc mice as a sign of haemorrhage. Perls' Prussian Blue staining does not detect intact red blood cells (n=3; scale bars: 20 µm). (E) Semi-quantitative analysis of urine samples using a dipstick revealed haematuria in mutant mice. Absence of haematuria was set as a value of 1 (see Fig. S4C for haematuria scale; n=6). ***P<0.001.
To investigate the development of any fibrosis, we performed PicroSirius Red staining and immunostaining against collagen I. Although increased collagen I staining could occasionally be observed in the perivascular tissue of some blood vessels in mutant kidneys, this analysis failed to detect any signs of overt renal fibrosis (Fig. S3A). Finally, absence of renal phenotypes in 3-month-old Col4a1+/Raw mice on a mixed genetic background (Van Agtmael et al., 2005) confirms that genetic modifiers influence Col4a1 renal disease.
Col4a1 mutant mice develop proteinuria and haematuria
Although Col4a1 is not major component of the GBM, histopathological analysis of 3- to 4-month-old mice revealed protein casts within tubules (Fig. 4C), suggesting the development of proteinuria. This is supported by increased total protein levels in Col4a1+/Raw and Col4a1+/Svc urine samples analysed by SDS-PAGE (Fig. S4A). Enzyme-linked immunosorbent assay (ELISA) analysis was performed to measure albumin-to-creatinine ratios of urines samples, which revealed increased albumin-to-creatinine ratios in mutant mice (Fig. S4B), confirming the development of mild proteinuria and highlighting an under-recognised role for α1α1α2(IV) in the filtration apparatus. Besides polyuria and proteinuria, mice also develop haematuria (Fig. 4D,E), similar to humans with HANAC syndrome (Plaisier et al., 2010, 2007), as indicated by dipstick analysis of urine samples collected in metabolic cage studies [Col4a1+/Svc (6/6) Col4a1+/Raw (1/8) and wild-type (0/10); Fig. 4E]. Perls staining of kidney sections revealed haem deposits in renal tissue (Fig. 4D), confirming that renal haemorrhage underlies the haematuria and that vascular defects contribute to renal pathophysiology.
Col4a1 mutations affect BM structure and composition
To shed light on pathomolecular disease mechanisms, electron microscopy was performed on 3- to 4-month-old mice, which revealed structural BM defects predominantly in the BMs of Bowman's capsule and the vasculature (Fig. 5A-C; Fig. S3C). The BM of Bowman's capsule revealed irregular thickening (Fig. 5A) and, in some instances, a basket-weave appearance was observed. Vascular BMs displayed extensive defects, including fragmentation and focal absence (Fig. 5B), likely causing vascular fragility, haemorrhage and haematuria, underscoring the vascular component of Col4a1 renal disease. Analysis of the GBM revealed irregular thickening, although no interruptions or thinning was observed (Fig. 5C). The presence of these defects and proteinuria indicates a role for α1α1α2(IV) in the filtration apparatus. No structural defects were observed in BM of tubules (Fig. S3C).
Fig. 5.
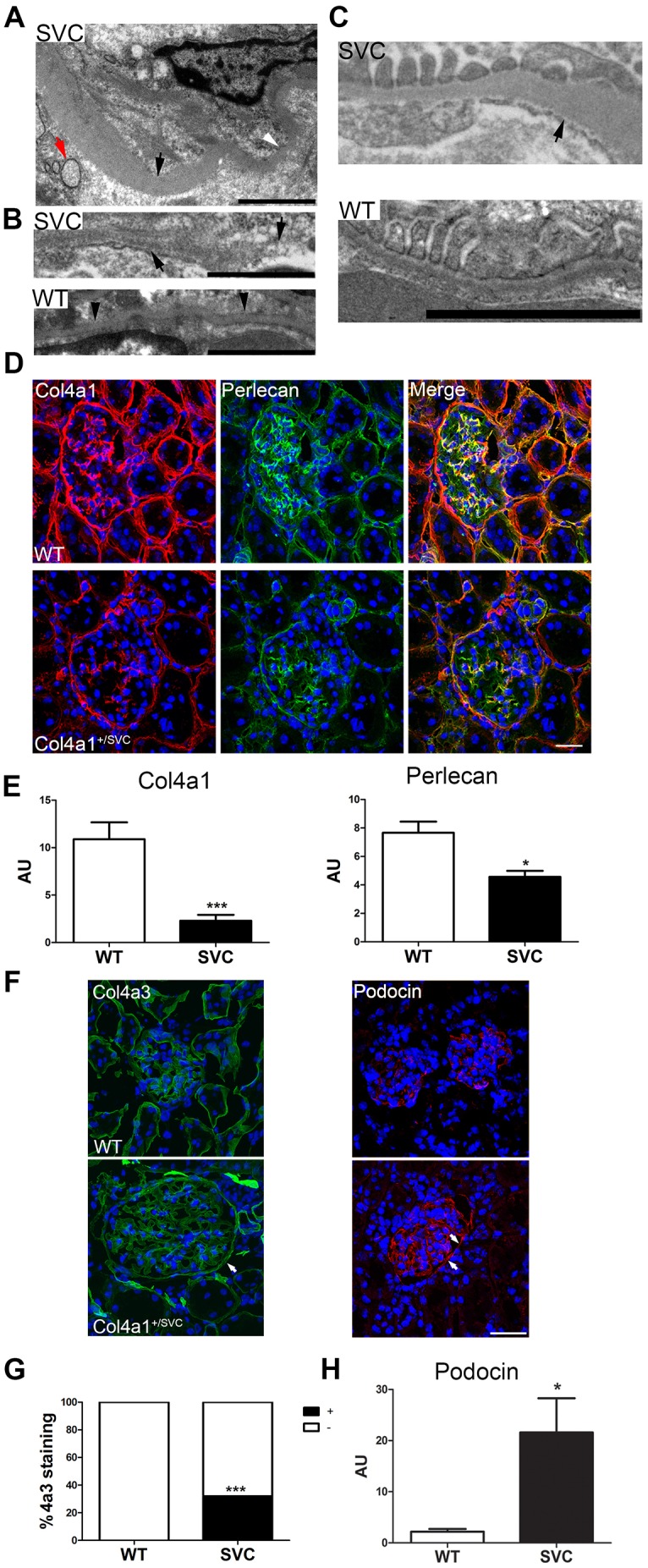
Investigation of mechanisms underlying Col4a1 kidney disease. (A) Transmission electron microscopic analysis revealed irregular thickening of the BM of 4-month-old Col4a1+/Svc Bowman's capsule (indicated by black arrowhead; white arrowhead indicates thinner region). The presence of swollen ER vesicles can also be observed (red arrow). (B) The vascular BM in 4-month-old Col4a1+/Svc mice (SVC) displayed focal interruptions (right black arrow) and an irregular less dense appearance with BM fragments (left arrow). (C) The GBM in 4-month-old Col4a1+/Svc mice showed irregular thickening (black arrow), but no interruptions were detected. Scale bars: 2 µm in A-C (n=3). (D) Immunostaining against Col4a1 and perlecan in 4-month-old Col4a1+/Svc kidney sections followed by confocal microscopy revealed reduced deposition of Col4a1 and perlecan in BMs of Col4a1+/Svc mice (n=3). Scale bar: 20 μm. (E) ImageJ analysis of fluorescence staining in D. (F) Immunostaining against Col4a3 (green) and podocin (red) revealed deposition in 3- to 4-month-old mutant BM of Bowmans capsule (white arrows). Scale bar: 20 µm. (G) Fraction of Bowman's capsules that stain positive (black) and negative (white) for Col4a3 in WT (0% positive) and Col4a1+/Svc mice (32% positive; n=85 glomeruli across three animals, Fisher's exact test). (H) ImageJ analysis of podocin fluorescence staining in H revealed ∼9.8-fold increase in SVC. *P<0.05, ***P<0.001, Student's unpaired t-test in E,H; Fisher's exact test in G.
To investigate whether the BM defects are the result, at least in part, of reduced α1α1α2(IV) incorporation, we performed immunohistochemistry against Col4a1. This revealed a ∼80% reduction in Col4a1 staining in tubular, mesangial and Bowman's capsule BMs (Fig. 5D,E; Fig. S3B). The reduced α1α1α2(IV) deposition was accompanied by ∼40% reduction in perlecan staining in all major BMs (Fig. 5D,E), but no consistent differences were observed for laminin or nidogen 1 and 2 (Fig. S5). Interestingly, ∼32% of mutant Bowman's capsules stained positive for Col4a3, compared with 0% for WT, confirming ectopic deposition of α3α4α5(IV) in damaged BMs of Bowman's capsule (Fig. 5F,G). This ectopic expression could reflect that mutant parietal epithelial cells express different collagen networks and/or that these cells have adopted a more ‘podocyte cell-like’ nature, because Col4a3 is normally produced by podocytes (Abrahamson et al., 2009). Immunostaining against podocin (marker for podocytes) detected podocin staining in Bowman's capsule of mutant mice, suggesting that at least some parietal epithelial cells might have undergone cell reprogramming. Furthermore, a ∼9.8-fold increase in podocin staining was observed within the glomerulus (Fig. 5F,H). These data indicate that Col4a1 mutations not only affect BM structure but also BM composition.
Chronic ER stress in Col4a1 renal disease
Electron microscopic analysis revealed enlarged ER vesicles (Fig. 5A; Fig. 6F) in epithelial cells of Bowman's capsule of mutant mice, suggestive of ER stress. Activation of ER stress was confirmed by western blotting in kidneys of 3- to 4-month-old mutant mice, which had a ∼2.9- and ∼3.6-fold increase in protein levels of the ER stress markers Bip and Atf4, respectively (Fig. 6A,B).
Fig. 6.

ER stress and renal disease. (A) Significantly increased protein levels of ER stress markers Bip (∼2.9-fold increase) and Atf4 (∼4.6-fold increase) in 4-month-old Col4a1+/Svc mice (n=3). Representative band of total protein stain is given as loading control (entire gel is provided in Fig. S6). (B) Densitometric analysis of Bip and Atf4 using ImageJ. (C) Examination of protein levels of Bip and CHOP analysis across Col4a1+/Raw animals of different ages (40 days, 3-4, 8 and 12 months). Increased protein levels of Bip in Col4a1+/Raw animals precedes elevated levels of CHOP, which is a marker for chronic ER-stress-induced apoptosis. Activation of CHOP coincides with development of atrophy of the medulla (see Table S3; n=3). (D) Col4a1+/Svc mice display thinning of the BM of Bowman's capsule at 40 days of age. Scale bar: 2 μm. (E) Average BM thickness as measured by ImageJ (WT ∼257 nm; Col4a1+/Svc ∼121 nm; n=3). (F) Distended ER vesicle (black star) in epithelial cell of Bowman's capsule in 40-day-old Col4a1+/Svc. Scale bar: 2 μm. (G) Analysis of Bip protein levels in 40-day-old Col4a1+/Svc mice (n=3). Representative band of total protein stain is given as loading control (entire gel is provided in Fig. S6). (H) Densitometric analysis of G. (I) Immunostaining against Bip (red) and Col4a1 (green) on kidneys of 3-month-old mice revealed increased expression in epithelial cell of Bowman's capsule (white arrow), and apparent more intense staining in Col4a1+/Svc. Scale bar: 20 µm (n=3).
To shed light on the relative contribution of ER stress and BM defects to the renal pathology, we investigated the association of BM defects and ER stress with the development of renal defects in the Col4a1+/Raw cohorts used to establish phenotype progression (Table S3). Western blot analysis against Bip revealed an apparent transient increase (Fig. 6C) in younger mice, followed by marked elevation of CHOP at time points that appear to correspond to development of medullary atrophy (Fig. 6C). In the UPR, CHOP is downstream of Bip, reflects progression to chronic pathogenic ER stress and is associated with ER-associated apoptosis (Bateman et al., 2009; Ron and Walter, 2007). All 3- to 4-month-old Col4a1+/Raw mice also expressed cleaved caspase 3 (Fig. S6E). Interestingly, although chronic ER stress coincides with development of medullary atrophy, no significant BM abnormalities were observed in tubules (Fig. S3), strongly arguing for a causative role of chronic ER stress in renal tubular disease. By contrast, the glomerulopathy becomes apparent at 21-40 days of age and is associated with BM defects but before the onset of the robust elevation of CHOP levels. Likewise, 40-day-old Col4a1+/Svc kidneys have a ∼50% reduction in thickness of the BM in Bowman's capsule (Fig. 6D,E). Intriguingly, a distended ER could be observed in some parietal epithelial cells of Bowman's capsule (Fig. 6F). However, although this indicates ER retention of collagen IV and suggests ER stress in some cells, this did not translate into higher ER stress levels at a tissue level as we failed to detect elevated Bip protein levels (Fig. 6G). By contrast, significantly elevated Bip levels were detected in protein homogenates of kidneys at 3-4 months (Fig. 6A,B). This was confirmed by immunostaining against Bip on kidney sections of 3-month-old mutant mice, which identified Bip expression in the epithelial cells of Bowman's capsule (Fig. 6I). In some tubules and the mesangium, a more punctate intense staining was also observed (Fig. 6I).
Overall, these data lend support to an adaptive ER stress response in glomeruli and a chronic pathogenic response in tubules. Interestingly and importantly, this differential response has also been observed after pharmacological induction of ER stress (Inagi, 2009). Therefore, our data lend support to a disease mechanism with variable relative contributions of ER stress and BM defects, whereby BM defects play a major role in the glomerulopathy and ER-stress-induced apoptosis in the tubulopathy.
DISCUSSION
Our analysis of the development and progression of renal disease caused by Col4a1 mutations has expanded the spectrum of potential clinical defects that can develop in humans with COL4A1 mutations and identified the importance of α1α1α2(IV) in adult kidney function and pathophysiology. Col4a1 mutations cause a glomerulopathy and tubulopathy with medullary atrophy, leading to proteinuria and diabetes insipidus because of a defect in the ability to concentrate urine. The haematuria attributable to haemorrhage illustrates an important vascular component to the renal disease. The progressive nature of the disease argues for monitoring of renal function with increasing age of patients, thus impacting patient management.
Col4a1 mouse models led to the identification and characterization of mutations in humans and thus the description of clinical phenotypes. Based on this, it is likely that these phenotypes, including the medullary atrophy, diabetes insipidus, polyuria and urine-concentrating defect, proteinuria and mild hypotension, can develop in at least a proportion of these individuals with COL4A1 mutations. The relative small number of families described to date, combined with a high level of clinical heterogeneity between and within families (Kuo et al., 2012; Vahedi and Alamowitch, 2011) and focus on the cerebrovascular disease aspects of Col4a1 disease, can all explain why these phenotypes have yet to emerge and highlight the need for a further detailed clinical analysis of affected individuals. Our data to date strongly support a multifactorial origin of the hypotension in addition to the previously described defects in vascular function (Van Agtmael et al., 2010). Although our previous data suggested an association with the reduced blood volume in the presence of normal plasma volume (Van Agtmael et al., 2010), our Epo experiments described in this article directly establish this contribution. It is interesting in this regard that ER stress through the protein ATF4 can affect Epo production (Chiang et al., 2013). Moreover, we have identified a blunted response of the distal tubules to elevated aldosterone levels and increased compensatory sodium reabsorption in the proximal tubules in response to the hypotension. Given that the experiments with diuretics were performed on young animals, before the onset of medullary atrophy, this indicates a novel role for the basement membrane and the collagen IV network in the regulation of vascular homeostasis in distal tubules by being important for the response to aldosterone. These data show that a compensatory response occurs within the kidney to the reduced blood pressure by activation of RAAS but whereby the reduced response to aldosterone renders this insufficient to normalise blood pressure levels despite increased sodium reabsorption via proximal tubules. Furthermore, it is likely that altered vascular resistance is also an underlying factor, although this remains to be established.
Metabolic cage studies revealed that Col4a1 mutations cause diabetes insipidus, because mice display the classical signs of reduced urine osmolality, polyuria and polydipsia. The diabetes insipidus in 2.5-month-old mice, before the onset of atrophy of the medulla, and reduced urine concentrating in response to vasopressin, suggests the development of diabetes insipidus with a central and nephrogenic origin. The development of chronic ER stress causing medullary atrophy with inability to elevate Aqp2 protein levels represents a molecular basis for the nephrogenic diabetes insipidus. The proteinuria is a consistent feature in Col4a1 mutant mice, and our data indicate that this is not confined to the more severe mutations, such as glycine mutations and deletions (Favor et al., 2007; Gould et al., 2006). However, the origin of the proteinuria remains unclear. Although subtle GBM defects are likely to contribute and indicate the importance of this network for renal function, it has also been proposed that Bowman's capsule, which is severely affected in Col4a1 renal disease, can act as a secondary barrier (Ohse et al., 2009). In addition, reduced tubular protein reabsorption (Nielsen and Christensen, 2010) and medullary atrophy could also contribute to the proteinuria. Although Bowman's capsule defects and haematuria are conserved between our mouse models and HANAC syndrome (Plaisier et al., 2007), we failed to detect any cyst formation or tubular BM defects. These phenotypic differences can reflect a separate disease mechanism, as it has been proposed that HANAC syndrome is caused by a distinct disease mechanism involving altered integrin signalling (Plaisier et al., 2010), supported by data published during the revision of the present paper (Chen et al., 2015). Moreover, our data show that genetic background also influences the development of Col4a1 renal disease, which has also been observed for albuminuria (Long et al., 2013; Tsaih et al., 2009).
Many aspects of the pathomolecular mechanisms of COL4A1 defects remain poorly characterised. Our data reveal that Col4a1 mutations not only affect BM structure but that they also alter BM composition, including reduced incorporation of perlecan. This indicates that α1α1α2(IV) is required for perlecan deposition in the BM, which is supported by data from Drosophila (Pastor-Pareja and Xu, 2011). The ectopic expression of α3α4α5(IV) in Bowman's capsule represents a different mechanism compared with Alport syndrome, in which absence of α3α4α5(IV) can cause continued expression of α1α1α2(IV) in the GBM (Kalluri et al., 1997). To our knowledge, this is the first evidence of collagen IV network ‘switching’ induced by α1α1α2(IV) defects. The more cuboidal appearance of the epithelial cells (Fig. 4; also see Van Agtmael et al., 2005) and podocin expression suggest that this might reflect a cellular reprogramming, perhaps as a compensatory response. This is supported by cell reprogramming because of ER stress in chondrocytes with mutations in Col10a1 (Tsang et al., 2007). Likewise, our analysis also revealed increased podocin expression in the glomerulus. Although reduced levels of podocin are well characterised in kidney disease (Mollet et al., 2009), the significance of increased expression remains unclear but could indicate a response to BM defects while also illustrating altered GBM composition. In conclusion, although their relative importance to the pathophysiology remains to be determined, defects in BM composition are part of the response to, and potentially mechanism of, Col4a1 mutations.
A temporal analysis of ER stress and BM defects revealed that chronic ER stress appears to coincide with the development of tubular disease in the absence of BM defects. By contrast, 40-day-old animals present with BM defects and glomerulopathy, but much lower levels of chronic pathogenic ER stress compared with 3-month-old Col4a1+/Raw mice. This provides evidence for the occurrence of distinct pathomolecular mechanisms reflecting differences in the relative contribution of ER stress and BM defects to phenotype development. This has important consequences for the development of therapeutic avenues, because ER stress can be caused by mutations in other matrix proteins and be targeted by small molecules, including chemical chaperones (Engin and Hotamisligil, 2010; Murray et al., 2014). Rescue of BM defects and their associated phenotypes might require alternative approaches, including regeneration of functional intact BMs and secretion of accurately folded Col4a1. However, it now remains to be determined whether this disease mechanism applies to all Col4a1 mutations, because disease mechanisms and renal phenotypes might be mutation dependent (Alamowitch et al., 2009; Kuo et al., 2014; Plaisier et al., 2010, 2007; Van Agtmael et al., 2005).
In conclusion, Col4a1 mutations cause renal disease that involves cell-specific disease mechanisms, including ER stress and BM defects. These data will inform the development of therapeutic strategies for renal diseases resulting from collagen mutations.
MATERIALS AND METHODS
Animal studies
All animal studies were performed in accordance with the UK Home Office under Project licence 60/4132.
Electron microscopic analysis
Tissue were collected and fixed in 2% glutaraldehyde in phosphate buffer and processed as previously described (Taylor et al., 2011). EM thickness was measured using ImageJ (http://imagej.nih.gov/ij/), whereby thickness was measured on EM images (1900× magnification) using a grid with a measure being taken every ∼800 nm (total of ∼90 measurements).
Histopathology
Kidneys were fixed in 10% neutral buffered formalin or 4% paraformaldehyde and embedded in paraffin wax. Sections were stained with Haematoxylin and Eosin, PicroSirius Red or Perls’ Prussian Blue using standard protocols.
Systolic blood pressure analysis
Systolic blood pressure was measured using tail-cuff plethysmography as previously described (Van Agtmael et al., 2010).
In vivo renal function studies
In vivo renal function studies were performed as described by Craigie et al. (2012). Under anaesthesia, mice were infused intravenously with isotonic saline at a rate of 200 µl/h/10 g body weight. After a 40 min equilibration period, mean arterial blood pressure was recorded for 30-40 min and urine was collected for 1 h. Evans Blue (1 µl/g of a 0.5% solution w:v) was injected for measurement of plasma volume; blood volume was calculated from this and haematocrit. After baseline collections, each mouse received an intravenous injection of furosemide (3 mg/kg), thiazide (2 mg/kg) or amiloride (2 mg/kg). The natriuretic response to these agents was assessed as an index of the in vivo activity of the Na+-K+-2Cl− cotransporter (NKCC2), the Na+-Cl− cotransporter (NCC) and the epithelial sodium channel (ENaC), respectively (Bailey et al., 2009; Hunter et al., 2014, 2015). At the end of the experiment, a 500 µl blood sample was taken for measurements of plasma sodium and potassium. Sodium and potassium concentration in urine and plasma was measured by ISE (Roche 9180, UK).
Metabolic cage studies
Animals were individually housed and allowed to acclimatise for 24 h, after which daily urine samples were collected and water consumption was measured. Water was withdrawn for 24 h. Vasopressin (Sigma) administration occurred via intraperitoneal injection, and animals were subsequently housed in metabolic cages for 6 h. Urine samples were collected 6 h after intraperitoneal injection and via bladder massage/vasopressin. Six microlitres of urine was loaded on an SDS-PAGE gel, followed by silver staining for analysis of proteinuria, whereas ELISA was used to measure albumin-to-creatinine ratios.
Immunoblotting
Protein extracts were prepared in RIPA buffer containing EDTA protease (Roche Applied Science) and phosphatase inhibitors (Phostop Roche). Membranes were blocked with 5% milk or bovine serum albumin before incubation with primary antibodies BIP (1:10,000; BD Transduction 610979), ATF4 (1:2500; Santa Cruz Sc-200), NHE3 (Stressmarq SPC-400D; 1:1000), p-NHE3 (Santa Cruz Sc-53961; 1:200), AQP2 (Santa Cruz Sc-9882; 1:800), NKCC2 (1:1000) and CHOP (Santa Cruz). Membranes were incubated with horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) and developed using chemiluminescense (Millipore). For cleaved caspase 3 (Cell Signaling Technology; 1:1000), primary and secondary antibodies (LI-COR) were incubated using 50% Seablock bluffer (ThermoFisher) and 50% tris-buffered saline-0.1% tween (TBST), and signals were detected using Odyssey Sa imager (LI-COR). Protein levels were corrected for Coomassie staining of total protein gels run in parallel with the western blot gels or by total protein stain on the membrane (Memcode; Pierce). Densitometry was performed using ImageJ software. Statistical analysis (Graphpad Prism) was done using Student's unpaired t-test.
Immunohistochemistry
Immunohistochemistry was performed as described by Taylor et al. (2011). After fixation in acetone and antigen retrieval, sections were incubated with primary antibodies [H11 for Col4a1, H22 for Col4a2 and H31 for Col4a3 (all 1:100), perlecan (1:1000), nidogen 1 (1:1000), nidogen 2 (1:1000), Col1a1 (Abcam Ab292; 1:1000), Bip (Abcam; 1:1000) and laminin (Sigma; 1:50)] overnight at 4°C. Incubation with secondary antibodies (1:400; Jackson ImmunoResearch) was performed and slides were mounted using Vectashield (Vector). Images were captured using a non-inverted fluorescent microscope (Zeiss Axioskop-Axiocam) and captured through AxioVision 4.8 (Zeiss) or as z-stacks using a Zeiss LSM510 Meta confocal microscope and Zeiss LSM software (Zeiss, Germany), which was also used for generating projections. ImageJ analysis was used for quantification of fluorescence as previously described (Murray et al., 2014).
Haematological analysis
Epo was administered via intraperitoneal injection of 150 units (three times per week) of human recombinant erythropoietin (gift from Dr Jo Mountford, University of Glasgow).
Statistical analysis
Statistical analysis (Graphpad Prism) was done by Student's unpaired t-test and by ANOVA and Bonferroni post hoc test for in vivo renal function assays.
Acknowledgements
We would like to thank the following: GlaxoSmithKline for generation of the Col4a1+/Raw and Col4a1+/Svc animal models; Dr J. Mountford and Dr A. Michie (haematological analysis and Epo experiments); T. Sasaki for the perlecan and nidogen antibodies; and Dr F. Howie for performing the ELISAs for the albumin-to-creatinine ratios.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
All authors were involved in writing the paper and gave final approval of the submitted and published versions. F.E.J., M.A.B., L.S.M., Y.L., S.M., U.S.-S., D.G.B. and T.V.A. performed experiments and analysed the data. F.E.J., M.A.B., L.S.M., R.L., D.G.B., J.J.M., K.E.K. and T.V.A. interpreted the data. J.J.M., K.E.K. and T.V.A. supervised the study. M.A.B. and T.V.A. designed the experiments. T.V.A. conceptualised the study.
Funding
This work was supported by the Medical Research Council [G0601268], Kidney Research UK [19/2012], and Research Councils UK Fellowship [EP/E50036611] to T.V.A. and by The Wellcome Trust [091840/Z/10/Z] to K.E.K.
Supplementary information
Supplementary information available online at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.021741/-/DC1
References
- Abrahamson D. R., Hudson B. G., Stroganova L., Borza D.-B. and St. John P. L. (2009). Cellular origins of type IV collagen networks in developing glomeruli. J. Am. Soc. Nephrol. 20, 1471-1479. 10.1681/ASN.2008101086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamowitch S., Plaisier E., Favrole P., Prost C., Chen Z., Van Agtmael T., Marro B. and Ronco P. (2009). Cerebrovascular disease related to COL4A1 mutations in HANAC syndrome. Neurology 73, 1873-1882. 10.1212/WNL.0b013e3181c3fd12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babey M., Kopp P. and Robertson G. L. (2011). Familial forms of diabetes insipidus: clinical and molecular characteristics. Nat. Rev. Endocrinol. 7, 701-714. 10.1038/nrendo.2011.100 [DOI] [PubMed] [Google Scholar]
- Bailey M. A., Mullins J. J. and Kenyon C. J. (2009). Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of Cushing syndrome. Hypertension 54, 890-896. 10.1161/HYPERTENSIONAHA.109.134973 [DOI] [PubMed] [Google Scholar]
- Bateman J. F., Boot-Handford R. P. and Lamandé S. R. (2009). Genetic diseases of connective tissues: cellular and extracellular effects of ECM mutations. Nat. Rev. Genet. 10, 173-183. 10.1038/nrg2520 [DOI] [PubMed] [Google Scholar]
- Chen Y. M., Zhou Y., Go G., Marmerstein J. T., Kikkawa Y. and Miner J. H. (2013). Laminin beta2 gene missense mutation produces endoplasmic reticulum stress in podocytes. J. Am. Soc. Nephrol. 24, 1223-1233. 10.1681/ASN.2012121149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z., Migeon T., Verpont M.-C., Zaidan M., Sado Y., Kerjaschki D., Ronco P. and Plaisier E. (2015). HANAC syndrome Col4a1 mutation causes neonate glomerular hyperpermeability and adult glomerulocystic kidney disease. J. Am. Soc. Nephrol. [E-pub ahead of print]. 10.1681/ASN.2014121217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C.-K., Nangaku M., Tanaka T., Iwawaki T. and Inagi R. (2013). Endoplasmic reticulum stress signal impairs erythropoietin production: a role for ATF4. Am. J. Physiol. Cell Physiol. 304, C342-C353. 10.1152/ajpcell.00153.2012 [DOI] [PubMed] [Google Scholar]
- Craigie E., Evans L. C., Mullins J. J. and Bailey M. A. (2012). Failure to downregulate the epithelial sodium channel causes salt sensitivity in Hsd11b2 heterozygote mice. Hypertension 60, 684-690. 10.1161/HYPERTENSIONAHA.112.196410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin F. and Hotamisligil G. S. (2010). Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes. Metab. 12 Suppl. 2, 108-115. 10.1111/j.1463-1326.2010.01282.x [DOI] [PubMed] [Google Scholar]
- Favor J., Gloeckner C. J., Janik D., Klempt M., Neuhauser-Klaus A., Pretsch W., Schmahl W. and Quintanilla-Fend L. (2007). Type IV procollagen missense mutations associated with defects of the eye, vascular stability, the brain, kidney function and embryonic or postnatal viability in the mouse, Mus musculus: an extension of the Col4a1 allelic series and the identification of the first two Col4a2 mutant alleles. Genetics 175, 725-736. 10.1534/genetics.106.064733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbi C., Kong X., Suzuki H., Kim H.-J., Gao M., Jia X., Ohnishi H., Ueta Y., Warner M., Guan Y. et al. (2012). Central diabetes insipidus associated with impaired renal aquaporin-1 expression in mice lacking liver X receptor beta. Proc. Natl. Acad. Sci. USA 109, 3030-3034. 10.1073/pnas.1200588109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould D. B., Phalan F. C., Breedveld G. J., van Mil S. E., Smith R. S., Schimenti J. C., Aguglia U., van der Knaap M. S., Heutink P. and John S. W. M. (2005). Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 308, 1167-1171. 10.1126/science.1109418 [DOI] [PubMed] [Google Scholar]
- Gould D. B., Phalan F. C., van Mil S. E., Sundberg J. P., Vahedi K., Massin P., Bousser M. G., Heutink P., Miner J. H., Tournier-Lasserve E. et al. (2006). Role of COL4A1 in small-vessel disease and hemorrhagic stroke. N. Engl. J. Med. 354, 1489-1496. 10.1056/NEJMoa053727 [DOI] [PubMed] [Google Scholar]
- Gould D. B., Marchant J. K., Savinova O. V., Smith R. S. and John S. W. M. (2007). Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum. Mol. Genet. 16, 798-807. 10.1093/hmg/ddm024 [DOI] [PubMed] [Google Scholar]
- Hudson B. G., Tryggvason K., Sundaramoorthy M. and Neilson E. G. (2003). Alport's syndrome, Goodpasture's syndrome, and type IV collagen. N. Engl. J. Med. 348, 2543-2556. 10.1056/NEJMra022296 [DOI] [PubMed] [Google Scholar]
- Hunter R. W., Craigie E., Homer N. Z. M., Mullins J. J. and Bailey M. A. (2014). Acute inhibition of NCC does not activate distal electrogenic Na+ reabsorption or kaliuresis. Am. J. Physiol. Renal. Physiol. 306, F457-F467. 10.1152/ajprenal.00339.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter R. W., Ivy J. R., Flatman P. W., Kenyon C. J., Craigie E., Mullins L. J., Bailey M. A. and Mullins J. J. (2015). Hypertrophy in the distal convoluted tubule of an 11β-hydroxysteroid dehydrogenase type 2 knockout model. J. Am. Soc. Nephrol. 27, 1537-1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagi R. (2009). Endoplasmic reticulum stress in the kidney as a novel mediator of kidney injury. Nephron Exp. Nephrol. 112, e1-e9. 10.1159/000210573 [DOI] [PubMed] [Google Scholar]
- Jeanne M., Labelle-Dumais C., Jorgensen J., Kauffman W. B., Mancini G. M., Favor J., Valant V., Greenberg S. M., Rosand J. and Gould D. B. (2012). COL4A2 mutations impair COL4A1 and COL4A2 secretion and cause hemorrhagic stroke. Am. J. Hum. Genet. 90, 91-101. 10.1016/j.ajhg.2011.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalluri R., Shield C. F., Todd P., Hudson B. G. and Neilson E. G. (1997). Isoform switching of type IV collagen is developmentally arrested in X-linked Alport syndrome leading to increased susceptibility of renal basement membranes to endoproteolysis. J. Clin. Invest. 99, 2470-2478. 10.1172/JCI119431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnoodi J., Pedchenko V. and Hudson B. G. (2008). Mammalian collagen IV. Microsc. Res. Tech. 71, 357-370. 10.1002/jemt.20564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D. S., Labelle-Dumais C. and Gould D. B. (2012). COL4A1 and COL4A2 mutations and disease: insights into pathogenic mechanisms and potential therapeutic targets. Hum. Mol. Genet. 21, R97-R110. 10.1093/hmg/dds346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo D. S., Labelle-Dumais C., Mao M., Jeanne M., Kauffman W. B., Allen J., Favor J. and Gould D. B. (2014). Allelic heterogeneity contributes to variability in ocular dysgenesis, myopathy and brain malformations caused by Col4a1 and Col4a2 mutations. Hum. Mol. Genet. 23, 1709-1722. 10.1093/hmg/ddt560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D. A., Kolatsi-Joannou M., Price K. L., Dessapt-Baradez C., Huang J. L., Papakrivopoulou E., Hubank M., Korstanje R., Gnudi L. and Woolf A. S. (2013). Albuminuria is associated with too few glomeruli and too much testosterone. Kidney Int. 83, 1118-1129. 10.1038/ki.2013.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollet G., Ratelade J., Boyer O., Muda A. O., Morisset L., Lavin T. A., Kitzis D., Dallman M. J., Bugeon L., Hubner N. et al. (2009). Podocin inactivation in mature kidneys causes focal segmental glomerulosclerosis and nephrotic syndrome. J. Am. Soc. Nephrol. 20, 2181-2189. 10.1681/ASN.2009040379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray L. S., Lu Y., Taggart A., Van Regemorter N., Vilain C., Abramowicz M., Kadler K. E. and Van Agtmael T. (2014). Chemical chaperone treatment reduces intracellular accumulation of mutant collagen IV and ameliorates the cellular phenotype of a COL4A2 mutation that causes haemorrhagic stroke. Hum. Mol. Genet. 23, 283-292. 10.1093/hmg/ddt418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. and Christensen E. I. (2010). Proteinuria and events beyond the slit. Pediatr. Nephrol. 25, 813-822. 10.1007/s00467-009-1381-9 [DOI] [PubMed] [Google Scholar]
- Ohse T., Chang A. M., Pippin J. W., Jarad G., Hudkins K. L., Alpers C. E., Miner J. H. and Shankland S. J. (2009). A new function for parietal epithelial cells: a second glomerular barrier. Am. J. Physiol. Renal Physiol. 297, F1566-F1574. 10.1152/ajprenal.00214.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastor-Pareja J. C. and Xu T. (2011). Shaping cells and organs in Drosophila by opposing roles of fat body-secreted Collagen IV and perlecan. Dev. Cell 21, 245-256. 10.1016/j.devcel.2011.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plaisier E., Gribouval O., Alamowitch S., Mougenot B., Prost C., Verpont M. C., Marro B., Desmettre T., Cohen S. Y., Roullet E. et al. (2007). COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 357, 2687-2695. 10.1056/NEJMoa071906 [DOI] [PubMed] [Google Scholar]
- Plaisier E., Chen Z., Gekeler F., Benhassine S., Dahan K., Marro B., Alamowitch S., Paques M. and Ronco P. (2010). Novel COL4A1 mutations associated with HANAC syndrome: a role for the triple helical CB3[IV] domain. Am. J. Med. Genet A 152A, 2550-2555. 10.1002/ajmg.a.33659 [DOI] [PubMed] [Google Scholar]
- Rannikmae K., Davies G., Thomson P. A., Bevan S., Devan W. J., Falcone G. J., Traylor M., Anderson C. D., Battey T. W. K., Radmanesh F. et al. (2015). Common variation in COL4A1/COL4A2 is associated with sporadic cerebral small vessel disease. Neurology 84, 918-926. 10.1212/WNL.0000000000001309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D. and Walter P. (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519-529. 10.1038/nrm2199 [DOI] [PubMed] [Google Scholar]
- Sibon I., Coupry I., Menegon P., Bouchet J.-P., Gorry P., Burgelin I., Calvas P., Orignac I., Dousset V., Lacombe D. et al. (2007). COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann. Neurol. 62, 177-184. 10.1002/ana.21191 [DOI] [PubMed] [Google Scholar]
- Taylor S. H., Al-Youha S., Van Agtmael T., Lu Y., Wong J., McGrouther D. A. and Kadler K. E. (2011). Tendon is covered by a basement membrane epithelium that is required for cell retention and the prevention of adhesion formation. PLoS ONE 6, e16337 10.1371/journal.pone.0016337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomotaki S., Mizumoto H., Hamabata T., Kumakura A., Shiota M., Arai H., Haginoya K. and Hata D. (2014). Severe hemolytic jaundice in a neonate with a novel COL4A1 mutation. Pediatr. Neonatol. [E-pub ahead of print]. 10.1016/j.pedneo.2014.04.001 [DOI] [PubMed] [Google Scholar]
- Tsaih S.-W., Pezzolesi M. G., Yuan R., Warram J. H., Krolewski A. S. and Korstanje R. (2009). Genetic analysis of albuminuria in aging mice and concordance with loci for human diabetic nephropathy found in a genome-wide association scan. Kidney Int. 77, 201-210. 10.1038/ki.2009.434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang K. Y., Chan D., Cheslett D., Chan W. C. W., So C. L., Melhado I. G., Chan T. W. Y., Kwan K. M., Hunziker E. B., Yamada Y. et al. (2007). Surviving endoplasmic reticulum stress is coupled to altered chondrocyte differentiation and function. PLoS Biol. 5, e44 10.1371/journal.pbio.0050044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahedi K. and Alamowitch S. (2011). Clinical spectrum of type IV collagen (COL4A1) mutations: a novel genetic multisystem disease. Curr. Opin. Neurol. 24, 63-68. 10.1097/WCO.0b013e32834232c6 [DOI] [PubMed] [Google Scholar]
- Van Agtmael T. and Bruckner-Tuderman L. (2010). Basement membranes and human disease. Cell Tissue Res. 339, 167-188. 10.1007/s00441-009-0866-y [DOI] [PubMed] [Google Scholar]
- Van Agtmael T., Schlotzer-Schrehardt U., McKie L., Brownstein D. G., Lee A. W., Cross S. H., Sado Y., Mullins J. J., Poschl E. and Jackson I. J. (2005). Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum. Mol. Genet. 14, 3161-3168. 10.1093/hmg/ddi348 [DOI] [PubMed] [Google Scholar]
- Van Agtmael T., Bailey M. A., Schlotzer-Schrehardt U., Craigie E., Jackson I. J., Brownstein D. G., Megson I. L. and Mullins J. J. (2010). Col4a1 mutation in mice causes defects in vascular function and low blood pressure associated with reduced red blood cell volume. Hum. Mol. Genet. 19, 1119-1128. 10.1093/hmg/ddp584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y.-C., Sonni A., Labelle-Dumais C., de Leau M., Kauffman W. B., Jeanne M., Biffi A., Greenberg S. M., Rosand J. and Gould D. B. (2012). COL4A1 mutations in patients with sporadic late-onset intracerebral hemorrhage. Ann. Neurol. 71, 470-477. 10.1002/ana.22682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams S. E., Reed A. A. C., Galvanovskis J., Antignac C., Goodship T., Karet F. E., Kotanko P., Lhotta K., Morinière V., Williams P. et al. (2009). Uromodulin mutations causing familial juvenile hyperuricaemic nephropathy lead to protein maturation defects and retention in the endoplasmic reticulum. Hum. Mol. Genet. 18, 2963-2974. 10.1093/hmg/ddp235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda Y., Haginoya K., Arai H., Yamaoka S., Tsurusaki Y., Doi H., Miyake N., Yokochi K., Osaka H., Kato M. et al. (2012). De novo and inherited mutations in COL4A2, encoding the type IV collagen alpha2 chain cause porencephaly. Am. J. Hum. Genet. 90, 86-90. 10.1016/j.ajhg.2011.11.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda Y., Haginoya K., Kato M., Osaka H., Yokochi K., Arai H., Kakita A., Yamamoto T., Otsuki Y., Shimizu S.-I. et al. (2013). Phenotypic spectrum of COL4A1 mutations: porencephaly to schizencephaly. Ann. Neurol. 73, 48-57. 10.1002/ana.23736 [DOI] [PubMed] [Google Scholar]