


Abstract
HIV-1 efficiently hijacks host cellular machinery and exploits a plethora of host–viral interactions for its successful survival. Identifying host factors that affect susceptibility or resistance to HIV-1 may offer a promising therapeutic strategy against HIV-1. Previously, we have reported that heat shock proteins, HSP40 and HSP70 reciprocally regulate HIV-1 gene-expression and replication. In the present study, we have identified HSP70 binding protein 1 (HspBP1) as a host-intrinsic inhibitor of HIV-1. HspBP1 level was found to be significantly down modulated during HIV-1 infection and virus production inversely co-related with HspBP1 expression. Our results further demonstrate that HspBP1 inhibits HIV-1 long terminal repeat (LTR) promoter activity. Gel shift and chromatin immunoprecipitation assays revealed that HspBP1 was recruited on HIV-1 LTR at NF-κB enhancer region (κB sites). The binding of HspBP1 to κB sites obliterates the binding of NF-κB hetero-dimer (p50/p65) to the same region, leading to repression in NF-κB mediated activation of LTR-driven gene-expression. HspBP1 also plays an inhibitory role in the reactivation of latently infected cells, corroborating its repressive effect on NF-κB pathway. Thus, our results clearly show that HspBP1 acts as an endogenous negative regulator of HIV-1 gene-expression and replication by suppressing NF-κB-mediated activation of viral transcription.
INTRODUCTION
Human immunodeficiency virus-1 (HIV-1) continues to be a successful pathogen for the last three decades, owing to its ability to undergo frequent mutations and the capability to manipulate host cell micro-environment to its advantage. The virus employs multiple strategies to escape the host immune system including latency (1), inhibition of antigen processing and presentation (2) and high rate of mutations (3) to avoid recognition by immune molecules (4). Moreover, highly evolved accessory proteins add to its pathogenicity (5). It has a relatively small genome, approximately 9.8 kb in length that encodes 15 proteins. Therefore, in addition to its own proteins, HIV-1 exploits various host cellular proteins for successful completion of its life cycle. Genome-wide studies including siRNA and shRNA screens (6–10), protein-protein interactions (11–14), bio-informatic analysis with patient samples (15–18) and meta-analysis of genome-wide studies (19) have revealed the significance of over a thousand cellular proteins in HIV-1 replication and gene-expression.
In past 30 years, viral enzymes (including reverse transcriptase, integrase and protease) have been extensively targeted to develop therapeutics against HIV. Although the combination therapy of these anti-retrovirals have worked well for the management of the disease but issues related to drug resistance and cellular toxicity have induced researchers to look for novel therapeutic targets (20). As mentioned above, in recent years, a large number of cellular factors have been shown to be absolutely essential for HIV-1 life cycle. Thus, targeting such host cellular factors required for successful infection and propagation of the virus in addition to currently available anti-retrovirals might offer a better therapeutic strategy.
The gene expression of HIV-1 is tightly regulated by interaction of numerous host cellular proteins with cis-acting DNA sequences present on the LTR promoter of the provirus (21). HIV-1 LTR is divided into three regions- U3, R and U5. The major regulatory sites in LTR promoter are ‘core promoter’ and ‘core enhancer’ sites, positioned in the ‘U3’ region. The ‘core promoter’ contains a TATA box and three binding sites for SP1, whereas the ‘core enhancer’ has two NF-κB binding sites (22,23). Binding of the NF-κB heterodimer (p50/p65) to LTR promoter is one of the strongest triggers for HIV-1 transcription. p50/p65 facilitates the accessibility and processivity of RNA Pol II on the promoter by recruiting various cellular factors including p300 and P-TEFb complex (24,25). Hence, this association of NF-κB proteins to the LTR promoter is very critical for efficient viral transcription.
Among the different host factors that have been characterized as pro- or anti-viral regulators, heat shock proteins (HSPs) have been shown to play an important role in the life cycle of different viruses. HSPs are chaperone molecules, which participate in housekeeping functions and also confer protection against stress conditions. HSP70 has been found to be incorporated into the HIV-1 virion along with HSP60 (26). HSP70 is also involved in the nuclear import of HIV-1 pre-integration complex (PIC) and in the inhibition of Vpr-mediated cell-cycle arrest and apoptosis (27). Furthermore, HSP70 and HSP90 have been reported to regulate Tat-mediated viral transcription by stabilizing the CDK9/Cyclin T1 complex (28). HSP40 has been demonstrated as an essential factor for Nef-mediated up-regulation of HIV-1 gene-expression (29). Previously, we have reported that HSP70 and HSP40 reciprocally regulate HIV-1 gene-expression and replication (30). A recent study has revealed that even slight increase in temperature stimulates HIV-1 replication in HSP90-dependent manner (31). HSP90 has also been shown to reactivate HIV-1 latency (32), highlighting the significance of HSPs in regulation of HIV-1 gene-expression.
In the present study, we have focused on deciphering the role of a co-chaperone, HSP70 binding protein1 (HspBP1), in HIV-1 pathogenesis. It has been reported that anti-HspBP1 IgG levels increase in the serum of HIV-1 infected subjects (33) but the role of HspBP1 in HIV-1 infection has not been established. HspBP1 was identified in 1998 as a HSP70 interacting protein by yeast-two-hybrid screening of a heart cDNA library (34). HspBP1 binds to the ATPase domain of HSP70 and inhibits its refolding activity (34,35). HspBP1 negatively affects the binding of substrates to HSP70 by accelerating nucleotide exchange of ATP domain (36). Also, HspBP1 regulates protein degradation activities of molecular chaperones (37). HspBP1 can regulate HSP70 activity and the levels of antibody against it are altered in infected individuals, suggesting that HspBP1 may play a role in HIV-1 pathogenesis by regulating the function of HSP70, an important determinant of HIV-1 life cycle. In the present study, we demonstrate that HspBP1 acts as an intrinsic inhibitor of HIV-1 replication. We also show that it physically interacts with κB sites in the LTR promoter, thereby obstructing the binding of p50/p65 at those sites; consequently leading to inhibition of LTR driven gene-expression. Thus, HspBP1 restricts HIV-1 replication by suppressing NF-κB-mediated activation of HIV-1 transcription.
MATERIALS AND METHODS
Cell lines, plasmids and reagents
Jurkat (CD4+ human T cell line) and HEK293T (human embryonic kidney cell line) were obtained from the NCCS Cell Repository, Pune, India. CEM-GFP (cat # 3655), a GFP expressing CD4+ reporter human T-cell line (38) was obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, USA. ACH2 (cat # 349), a HIV-1 latently infected T-cell line with one copy of the provirus integrated in the genome and U1 (cat # 165), a sub-clone of U937 (monocyte), chronically infected with HIV-1 and harbouring two copies of provirus DNA integrated in the genome were obtained from NIH AIDS Reagent program (39–41). Both the cell lines can be induced with phorbol myristate acetate (PMA) or TNF-α to secrete high levels of infectious HIV-1. HEK293T was grown in DMEM medium (Invitrogen, USA) whereas Jurkat, CEM-GFP, U1 and ACH2 cells were grown in RPMI 1640 medium (Invitrogen, USA), both supplemented with 10% fetal bovine serum (Invitrogen, USA) and penicillin-streptomycin (Invitrogen, USA) at 37°C with 5% CO2, in a humidified incubator. For CEM-GFP cells, growth media was supplemented with 500 μg/ml G418 (Invitrogen, USA).
The HIV-1 molecular clone, pNL4-3 (cat # 114) was obtained from the National Institutes of Health AIDS repository (42). pcDNA-HspBP1 and its deletion mutant construct have been described earlier (35,43). The plasmids, pcDNA-Hsp70 and pcDNA-Hsp40 (pcDNA3 expressing human Hsp70 and Hsp40, respectively) were obtained from Dr Margarida D. Amaral (44). The HIV-1 LTR reporter vector, pLTR-luc was sub-cloned from pU3RIII (45,46) into pGL3basic (47). HIV-1 LTR and its deletion mutant constructs of CD series were a kind gift from Dr. T. Okamoto (48). CD12 luc is wild type LTR promoter luciferase construct. CD23, CD52 and CD54 luc are deletion mutants of LTR promoter. CD23 luc lacks upstream regulatory regions of LTR promoter but includes enhancer (2 NF-κB binding sites) and core regions (3 SP1 binding sites). CD52 contains 2 SP1 binding sites of core promoter. CD54 lacks all SP1 binding sites and contains small region of core promoter from −48 to +80. LTR del NF-κB luc, the NF-κB enhancer region deletion mutant of LTR promoter was constructed by primer extension PCR strategy (49) using pLTR-luc as a template and the primers listed in Table 1. Briefly, primers A and D are the forward and reverser primers respectively to amplify the full-length fragment and primers B and C are located on either side of the sequence to be deleted and contain sequence from both sides of the deletion. The first round of PCR was performed using primer pairs A/B and C/D (Table 1). The two resulting PCR products were mixed together with the primer pair A/D for a second round of PCR, which resulted in generation of final, full-length product with the desired area deleted. LTR NF-κB mut luc, a point mutant of LTR promoter having mutations in both NF-κB sites was generated using QuikChange II Site-Directed Mutagenesis kit, according to the manufacturer's protocol (Agilent technologies, USA). The mutation was generated using pLTR-luc as a template and the primers listed in Table 1. Both the series of mutants described above are also schematically represented in Figure 4E and G. pNF-κB-Luc, a reporter vector expressing luciferase under the control of five tandem copies of NF-κB enhancer was obtained from Stratagene (USA). pcDNA-Tat was previously cloned in our lab (50). p50 was sub-cloned from pBluescript-NF-κB1 (p50) (NIH AIDS Reagent Program, cat. # 2627) into pcDNA3.1(+) (Invitrogen) in HindIII and BamH1 cloning sites. p65 was sub-cloned from pBluescript-RelA (p65) (NIH AIDS Reagent Program, cat. # 2625) into pcDNA3.1(+) (Invitrogen) in HindIII and Not1 cloning sites.
Table 1. List of the primers used for generating NF-κB mutant constructs of LTR promoter.
| Construct | Primer sequence 5′ to 3′ |
|---|---|
| LTR del NF-κB luc | A (F) CGGGCTCGAGACCTGGAAAAACATGG |
| B (R) CCGAAGCTTTATTGAGGCTTAAGCAGTGGG | |
| C (F) CTTGTTACAAAGGGAGGCGTGGCCTGGG | |
| D (R) CGCCTCCCTTTGTAACAAGCTCGATGTC | |
| LTR NF-κB mut luc | F CGAGCTTGTTACAACTCACTTTCCGCTGCTCACTTTCCAGGGAGGCG |
| R CGCCTCCCTGGAAAGTGAGCAGCGGAAAGTGAGTTGTAACAAGCTCG |
F- Forward primer; R-Reverse primer.
Figure 4.

HspBP1 inhibits HIV-1 LTR promoter activity through NF-κB enhancer region. (A) Luciferase reporter assays showing inhibition of LTR-driven gene-expression by HspBP1 in dose-dependent manner in HEK293T cells. Cells were co-transfected with increasing amounts of HspBP1 construct (0.25–1.5 ug) and the HIV-1 LTR luciferase reporter construct (LTR-luc) followed by luciferase assay after 36 h of transfection. (B) HspBP1 over-expression inhibits LTR promoter activity in dose dependent manner in Jurkat cells. Jurkat cells were co-transfected with 0.5 and 1 ug of HspBP1-construct along with LTR-luc. Luciferase assays were performed 36 h post transfection. (C) HspBP1 silencing enhances HIV-1 LTR driven gene expression in dose-dependent manner. Control siRNA and siRNA against HspBP1 were transfected in HEK293T cells. LTR-luc was also transfected 24 h after siRNA-transfection. Luciferase activities were determined 24 h post second transfection. Efficiency of gene silencing was assessed by immunoblotting. (D) HspBP1 inhibits LTR-driven gene-expression in presence of Tat. HEK293T cells were co-transfected with indicated plasmids and luciferase assays were performed 36 h post transfection. (E) Schematic representation of HIV-1 LTR and LTR-luciferase mutant constructs (48). (F) Luciferase reporter assays depicting inhibition of LTR promoter activity by HspBP1 through NF-κB and SP1 binding sites. HEK293T cells were co-transfected with various LTR luc constructs and HspBP1. Luciferase assays was performed 36 h post-transfection. (G) Schematic representation of NF-κB mutants of HIV-1 LTR promoter. (H) Effect of HspBP1 on HIV-1 LTR promoter is specifically through NF-κB binding sites. HEK293T cells were co-transfected with indicated plasmids and reporter assays were performed 36 h post-transfection. (I) HspBP1 inhibits NF-κB driven gene expression in dose-dependent manner. HEK293T cells were co-transfected with increasing concentrations of HspBP1 vector (0.25–1.5 ug) and luciferase reporter construct under the control of five NF-κB binding sites. Luciferase activities were determined 36 h post-transfection. Results shown in (A), (C), (D) and (I) are representative of three experiments. (B), (F) and (H) represent data from two experiments. Error bars represent the mean ± SD values and significance is defined as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
p24 antiserum (cat. # 4250) and anti-Tat monoclonal antibody (cat. # 4138) were obtained from the National Institutes of Health AIDS repository. HspBP1 antibody was used for immunoblotting as reported earlier (51). Antibodies against GAPDH (sc-32233), HSP40 (sc-1800), HSP70 (sc-59571), p50 (sc-7178-X), p65 (sc-372-X) and RNA Pol II (sc-899) were procured from Santa Cruz Biotechnology, USA. Tubulin antibody was obtained from Sigma, USA. HspBP1 antibody (NBP201061) used in EMSA was obtained from Novus biological, USA. Immunoprecipitation and ChIP were performed using HspBP1 antibody (SAB1401597) from Sigma, USA. Control and HspBP1 siGENOME SMARTpool siRNAs were obtained from Dharmacon, USA. siRNAs against HSP40 and HSP70 were obtained from Santa Cruz Biotechnology, USA.
Transient transfection and luciferase assay
HEK293T cells were co-transfected with reporter vectors along with other expression vectors or molecular clones using Lipofectamine 2000 (Invitrogen, USA) and harvested 36 h post-transfection for luciferase assay. The cells were lysed in cell lysis reagent (Promega, USA) and luciferase assays were performed using Steady-Glo substrate (Promega, USA) as described earlier (30). Jurkat cells were transfected using x-treme gene HP DNA transfection reagent (Roche Applied Bioscience, Germany).
For silencing studies, cells were first transfected with siRNA using Lipofectamine 2000 as per the manufacturer's instructions, followed by second transfection (reporter plasmid/ expression vectors) or infection as described earlier (52). Cells were harvested 48 h post-transfection/infection. Knockdown was confirmed by immunoblotting with respective antibodies. Immunoblotting with GAPDH served as the loading control.
Immunoblotting and immuno-precipitation assays
Cells were lysed in lysis buffer (50 mM Tris–HCl pH 7.4, 5 mM EDTA, 0.12 M NaCl, 0.5% NP40, 0.5 mM NaF, 1 mM DTT, 0.5 mM PMSF) supplemented with protease inhibitor cocktail (Roche Applied Bioscience, Germany) on ice for 45 min with intermittent mixing using vortex. Protein concentration was determined using Bradford assay reagent (Biorad, USA) and equal amounts of protein were examined on a 10–12% SDS-PAGE gel. Proteins were transferred to a PVDF membrane (GE Healthcare, USA), which was then blocked with 5% non-fat dry milk or BSA, and probed with respective antibodies. The blots were developed using the ECL Prime system (GE Healthcare, USA).
For co-immunoprecipitation assays, clarified lysates were incubated with indicated antibodies and the antigen–antibody complex was pulled down by an equal mixture of protein A and G agarose beads (Invitrogen, USA), followed by resolution on 10–12% SDS–PAGE. Proteins were then transferred to PVDF membrane and probed with indicated antibodies.
HIV-1 infection and virus quantitation
Jurkat and CEM-GFP cells were infected with HIV-1NL4-3 virus at various multiplicities of infection (MOI) in the presence of polybrene (1 μg/ml) as described previously (53). Peripheral blood mononuclear cells (PBMCs) were isolated from the blood of normal seronegative donors obtained from local blood bank using Ficoll-Hypaque (Amersham Biosciences, USA) gradient centrifugation. Cells were activated with 5 μg/ml phytohemagglutinin (PHA) (Sigma, USA) for 36–48 h. Activated PBMCs were infected with 0.1 MOI of HIV-1NL4-3 virus as described earlier (53). Cells were washed and plated in complete medium supplemented with human interleukin-2 (Roche Applied Bioscience, Germany) at 20 units/ml and incubated at 37°C in a humidified CO2 incubator. The culture supernatants of infected and pNL4-3 transfected cells were collected at various time points and were used to determine virus production by p24gag antigen capture ELISA (Perkin Elmer Life Sciences, USA).
Quantitative real-time PCR
RNA was prepared from 2 × 106 Jurkat (uninfected and infected) and 1 × 106 PBMCs (uninfected and infected) using TRIzol Reagent (Invitrogen, USA). The cDNA was prepared using Moloney Murine Leukemia Virus reverse transcriptase (MMLV-RT) (Invitrogen, USA). Quantitative real time PCR (qRT-PCR) was used to analyze the expression of HspBP1 and p24. 10 μl reaction mixtures containing cDNA template, SYBR Green IQ supermix (Bio-Rad, USA) and 10 pmol of each of hGAPDH or respective gene-specific oligonucleotide primer pairs (Table 2) were run on a Realplex4 Mastercycler (Eppendorf, Germany) using the following program: initial denaturation at 95°C for 5 min and 40 cycles of 95°C for 30 s, 55°C for 30 s and 72°C for 30 s followed by the melt curve analysis. The fold change in the target gene relative to housekeeping gene (GAPDH) was calculated as:
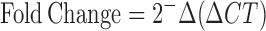 |
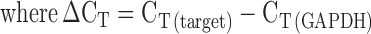 |
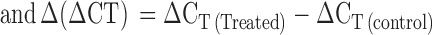 |
Table 2. List of the primers used for qRT-PCR analysis.
| Primer | Primer sequence 5′ to 3′ |
|---|---|
| GAPDH | F GAAGGTGAAGGTCGGAGTC |
| R GAAGATGGTGATGGGATTTC | |
| p24 | F ATAATCCACCTATCCCAGTAGGAGAAAT |
| R TTTGGTCCTTGTCTTATGTCCAGAATGC | |
| HspBP1 | F CGTGCAGTCAGAACGTGG |
| R TCTCGGACCAGACAGGAGAT |
Electrophoretic mobility shift assay (EMSA)
Equimolar amounts of complementary oligonucleotide probes spanning the NF-κB consensus region of the HIV-1 LTR were annealed to generate double stranded oligonucleotide. The annealed oligo was then labeled with [γ-32P]-dATP (BRIT, India) using T4 polynucleotide kinase (New England Biolabs, USA) by incubating at 37°C for 30 min and purified by Illustra ProbeQuant G-50 micro columns (GE Healthcare Life Sciences, USA). Binding reaction with the labeled probe was performed with either purified recombinant HspBP1 protein or nuclear extract from HEK293T cells in buffer containing 10 mM Tris (pH 7.5), 1 mM EDTA, 50 mM KCl, 0.1 mM DTT, 5% glycerol, 0.01 mg/ml BSA, 200 ng poly dI/dC at 25°C for 20 min and resolved on 6% polyacrylamide gel (acrylamide:bis-acryamide 39:1). Nuclear extract was prepared from HEK293T cells using NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce, USA) as per the manufacturer's protocol. Recombinant His-HspBP1 protein was purified from BL21 (DE3) strain of E. coli cells using Ni-NTA beads as reported earlier (34). Mutated NF-κB region containing oligo was used as non-specific competitor. For the competition assay, 10X, 50X and 100X excess of the specific cold oligo was used. The nucleotide sequences of probe ‘P’ and mutated probe are listed in Table 3.
Table 3. List of the primers used in EMSA.
| Specific Oligo, P | F 5′ GGGACTTTCCGCTGGGGACTTTCC 3′ |
| R 5′ GGAAAGTCCCCAGCGGAAAGTCCC 3′ | |
| Mutant Oligo | F 5′ TTTCCAAAGGAGGGAGGTGTGGC 3′ |
| R 5′ GCCACACCTCCCTCCTTTGGAAA 3′ |
F, forward primer; R, reverse primer.
Chromatin-immunoprecipitation (ChIP)
CEM-GFP cells (1 × 107) were infected with HIV-1NL4-3 at 0.1 MOI. On Day 5 post-infection, cells were cross-linked by addition of 0.75% formaldehyde to the media and neutralized with 125 mM glycine. HEK293T cells were also cross-linked similarly. The fixed cells were then lysed in lysis buffer containing 50 mM HEPES–KOH pH 7.5, 140 mM NaCl, 1 mM EDTA, 1% Triton-X100, 0.1% Na deoxycholate, 0.1% SDS and protease inhibitor cocktail and subjected to sonication using Bioruptor (Diagenode, Belgium) to shear the chromatin into fragments of 200–500 bp (for HEK293T: total time 8 min with 30 s ‘on’ and 30 s ‘off’ at high settings; CEM-GFP: total time 7 min with 30 s ‘on’ and 30 s ‘off’ at high settings). The lysate was pre-cleared for 2 h with protein A/G beads (Pierce, USA). The pre-cleared lysate was immunoprecipitated with specific antibody or normal IgG as isotype control (Santacruz, USA). The immune complexes were then pulled down by protein A/G beads and washed once with RIPA wash buffer [50 mM Tris–HCl pH 8, 0.1% SDS, 0.5% sodium deoxycholate, 150 mM NaCl, 2 mM EDTA and 1% NP-40] followed by three washes with Wash Buffer containing 20 mM Tris–HCl pH 8, 2 mM EDTA, 0.1% SDS, 1% Triton-X100 and 150 mM NaCl and finally one wash with final wash buffer [20 mM Tris-HCl pH 8, 2 mM EDTA, 0.1% SDS, 1% Triton-X100 and 500 mM NaCl]. The complexes were then eluted in 1% SDS and 100 mM NaHCO3. Antibody-bound chromatin complexes were reverse cross-linked with NaCl for 6 h at 65°C, treated with RNAse with 1 h at 37°C and proteinase K for 2 h at 45°C. DNA was extracted using phenol:chloroform extraction and subjected to PCR amplification to check the occupancy of HspBP1 and p65 on HIV-1 LTR promoter, using primers listed in Table 4. F1/R1 encompasses NF-κB-SP1 (−143 to +14) enhancer region whereas F2/R2 and F3/R3 cover −453 to −349 and +75 to +180 regions of the LTR promoter, respectively.
Table 4. List of the primers used in ChIP analysis.
| Primer | Location on LTR | Sequence (5′ to 3′) |
|---|---|---|
| F1 | −143 to +14 | GGAGTACTACAAAGACTGCT |
| R1 | TAACCAGAGAGACCCAGTA | |
| F2 | −453 to −349 | TGGAAGGGCTAATTTGGTC |
| R2 | CTGGCCCTGGTGTGTAGTTC | |
| F3 | +75 to +180 | TAAAGCTTGCCTTGAGTGCT |
| R3 | TGCTAGAGATTTTCCACACTGA |
F, forward primer; R, reverse primer.
Nucleofection and stimulation of ACH2 and U1 cells
HspBP1 was either over-expressed or silenced using siRNA in U1 and ACH2 cells by nucleofection using Amaxa Biosystem Nucleofector II (Lonza, Switzerland) with programs V-001 and T-014, respectively. 24 h post-nucleofection, cells were activated with PMA (50 ng/ml) or TNFα (10 ng/ml). Twenty-four hours post-treatment, the culture supernatants were used to determine virus production by p24gag antigen capture ELISA (Perkin Elmer Life Sciences, USA).
Statistical analysis
The statistical analysis of the experimental data was performed using Student's t-test. The results are plotted as mean ± SD. The level of significance is shown in the figures with asterisks and defined as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
RESULTS
HspBP1 interacts with both HSP40 and HSP70
HSP40 and HSP70 are known to interact with each other and play an important role in regulating HIV-1 transcription (30). HspBP1 has also been shown to interact with HSP70 and regulates its activity (34). Taking these observations into consideration, we investigated whether HspBP1 also interacts with HSP40. To accomplish this, we performed co-immunoprecipitation assays with lysate from HEK293T cells, over-expressing HspBP1 and HSP40 or HSP70. Immunoprecipitation was carried out with either HSP40 or HSP70 antibodies with respective lysates, followed by immunoblotting with HspBP1 antibody and vice versa. We observed that HspBP1 was pulled down with both HSP70 and HSP40 antibodies (Figure 1A). Likewise, HSP40 and HSP70 were also pulled down with HspBP1 antibody (Figure 1A), suggesting that in addition to HSP70, HspBP1 also interacts with HSP40. We further confirmed this observation at endogenous level in HIV-1NL4-3 infected CEM-GFP cells, where immunoprecipitation was performed with HspBP1 antibody and presence of HSP40/HSP70 was examined in the immunoprecipitated lysate by immunoblotting (Figure 1B).
Figure 1.
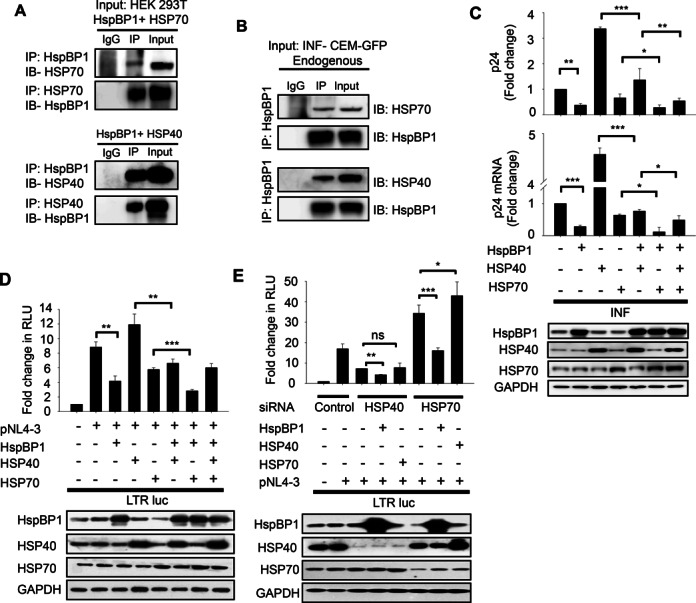
HspBP1 interacts with both HSP40 and HSP70 and interferes with HIV-1 gene-expression and replication. (A) Co-immunoprecipitation showing interaction of HspBP1 with HSP70 and HSP40. HEK293T cells were co-transfected with indicated plasmids. Forty-eight hours post-transfection, immunoprecipitation was performed with indicated antibodies followed by immunoblotting for specified proteins. (B) Co-immunoprecipitation showing interaction of HspBP1 with HSP70 and HSP40 at endogenous level in infected CEM-GFP cells. Immunoprecipitation was performed with HspBP1 antibody with day-5 0.1 MOI HIV-1NL4-3 infected CEM-GFP cells followed by immunoblotting with either HSP40 or HSP70 antibodies. (C) Upper panel: p24 ELISA showing the effect of over-expression of different HSPs on virus production. Jurkat cells were transfected with indicated plasmids. Twenty-four hours post-transfection, cells were infected with HIV-1NL4-3. Twenty-four hours post-infection, cells were harvested and culture supernatants were collected. Culture supernatants were examined for amount of virus produced using p24 antigen capture ELISA. Lower panel: Quantitative real time PCR analysis showing effect of over-expression of different HSPs on viral gene-expression. Cells were utilized to isolate RNA and cDNA was prepared. The expression of p24 mRNA was analyzed using qRT-PCR. (D) Luciferase reporter assays showing effect of over-expression of different HSPs on LTR driven gene-expression. HEK293T cells were co-transfected with the indicated plasmids and luciferase assay was performed 36 h post transfection. (E) Luciferase reporter assay indicating that the inhibitory function of HspBP1 on viral transcription is independent of HSP40 and HSP70. HEK293T cells were depleted of HSP40 and HSP70 using indicated siRNA. 24 h post siRNA transfection, HIV-1 LTR luciferase reporter construct (LTR-luc) was co-transfected along with indicated plasmids. Luciferase activity was determined 24 h post second transfection. Silencing using siRNA or over-expression of various plasmids was confirmed by immunoblotting in each experiment. (A) and (B) are one of the representative images of two independent experiments. (C), (D) and (E) represent data from three independent experiments. The error bars are presented as the mean ± SD values and significance is defined as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
HspBP1 interferes with HIV-1 replication
HSP70 and HSP40 have been reported to play a significant role in HIV-1 replication (30) and we have observed that HspBP1 also interacts with both HSP70 and HSP40. Therefore it is reasonable to investigate the relevance of this interaction during HIV-1 infection, if any. In order to pursue this, we assessed both individual role of HspBP1 and cumulative role of these three proteins during HIV-1 infection. First, we examined the effect of these stress family proteins on virus production by p24gag antigen capture ELISA. Jurkat cells were co-transfected with different HSP constructs, followed by HIV-1NL4-3 infection and supernatant was assessed for p24 levels. Significant inhibition in virus production was observed with HspBP1 over-expression. Moreover, HspBP1 was found to suppress HSP40-mediated increase in virus production. Further, HspBP1, along with HSP70 completely abolished the positive effect of HSP40 on virus production (Figure 1C upper panel). Effect of these chaperones on viral mRNA expression (p24 mRNA) was also analyzed using qRT-PCR and almost identical profile was observed (Figure 1C lower panel), suggesting that these HSPs affect viral gene-expression probably at transcription level.
HSP70 and HSP40 have been previously shown to alter LTR-driven gene-expression (30). We next wanted to assess the effect of HspBP1 alone and in combination with HSP70/40 on the LTR promoter activity. HEK293T cells were co-transfected with LTR-luc construct and pNL4-3 molecular clone with different HSP constructs. As shown in Figure 1D, HspBP1 over-expression resulted in inhibition of LTR driven gene-expression. Furthermore, HspBP1 inhibited HSP40-mediated increase in LTR promoter activity and potentiated HSP70-mediated inhibition in LTR driven gene-expression (Figure 1D). These results suggest a possible collaborative function between HspBP1 and HSP70 that inhibits HSP40-mediated increase in viral transcription. Taking all these observations into consideration, we hypothesized that these proteins are involved in regulation of HIV-1 transcription by similar mechanism and are part of the same pathway. So, in order to validate this hypothesis, we next determined if the inhibitory activity of HspBP1 on LTR-driven gene-expression is HSP70/HSP40-dependent. HEK293T cells were depleted of HSP40 or HSP70 by siRNA knockdown and the role of HspBP1 on LTR promoter was analyzed by luciferase assays. Surprisingly, our results showed that HspBP1 inhibits LTR driven gene-expression even when HSP40 and HSP70 are greatly reduced, suggesting an independent role of HspBP1 in suppression of LTR activity (Figure 1E). These results demonstrate that although HSP40, HSP70 and HspBP1 seem to interact with each other but they have different effects on HIV-1 replication probably utilizing different mechanistic pathways. Also, HspBP1 potently inhibits LTR-driven gene-expression and virus production and thus may act as an important molecule regulating HIV-1 gene-expression and replication. This observation intrigued us to further investigate the role of HspBP1 in HIV-1 infection and decipher the mechanism involved in regulation of viral gene-expression.
HspBP1 is down-modulated during HIV-1 infection
To explore the biological relevance of inhibitory effect of HspBP1 on HIV-1 replication and gene-expression, we next examined HspBP1 expression levels during HIV-1 infection. As it is evident from Figure 1 that HspBP1 inhibits HIV-1 replication and virus production, we were curious to know that what happens to HspBP1 levels during infection. Kinetic expression profile studies for HspBP1 were performed following HIV-1 infection in T-cells (Jurkat) and primary cells (PBMCs). Jurkat cells were infected with HIV-1NL4-3 virus and expression of HspBP1 was analyzed at mRNA and protein level at various time points. HIV-1 infection resulted in down-modulation of HspBP1 mRNA and protein expression in T-cells (Figure 2A–C). This finding was confirmed by analyzing the expression of HspBP1 in HIV-1 infected human PBMCs isolated from the blood of three normal seronegative donors as described in Materials and Methods. These results clearly indicate that HspBP1 mRNA and protein levels are significantly down-modulated following HIV-1 infection in PBMCs also (Figure 2D and E).
Figure 2.
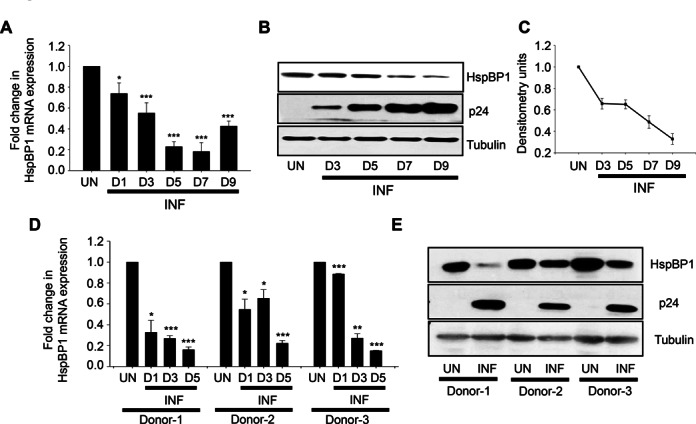
HspBP1 is down modulated during HIV-1 infection in both T cells and PBMCs. (A) mRNA expression profiling of HspBP1 during HIV-1 infection. Jurkat cells were infected with 0.1 MOI HIV-1NL4-3 and were harvested on day 1 (D1), day 3 (D3), day 5 (D5), day 7 (D7) and day 9 (D9), post infection for quantitative real time PCR analysis. (B) Representative western blot showing expression profile of HspBP1 in HIV-1NL4-3 infected Jurkat cells at different time points. (C) Densitometry analysis of HspBP1 expression in western blot shown in (B). (D) qRT-PCR analysis of HspBP1 mRNA expression in HIV-1NL4-3 (0.1MOI) infected PBMCs on day 1 (D1), day 3 (D3) and day 5 (D5) post infection. (E) HspBP1 protein expression analysis in HIV-1NL4-3 infected PBMCs on day 5 (D5) post-infection using immunoblotting. PBMCs were isolated from blood of three healthy donors obtained from local blood bank and were infected with HIV-1NL4-3. The expression of viral capsid protein p24 was measured to monitor the progression of infection. The results shown in (A) and (D) are represented as fold change in expression as compared to uninfected cells (UN). The results shown in (A) and (C) are representative of three independent experiments. (D) represents data from two experiments. The error bars are presented as the mean ± SD values and significance is defined as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
HspBP1 expression inversely correlates with HIV-1 production
To decipher the role of endogenous HspBP1 in HIV-1 infection, virus production was examined following HspBP1 depletion. We performed single cycle replication studies in HEK293T cells and infection studies in Jurkat cells to assess the effect of knockdown of HspBP1 on virus production. HEK293T cells were first transfected with increasing doses of siRNA and 24 h post-siRNA transfection, cells were transfected with pNL4-3 molecular clone. 48 h following the second transfection, cells were harvested and supernatant was collected. p24 ELISA of the culture supernatant revealed that knockdown of HspBP1 resulted in enhanced virus production in a dose dependent manner (Figure 3A). A more biologically relevant scenario would be to infect T-cells with HIV-1NL4-3 and monitor the effect of HspBP1 depletion. Jurkat cells were transfected with HspBP1 siRNA followed by HIV-1NL4-3 infection 24 h post-transfection. It was observed that silencing of HspBP1 leads to higher virus production from infected Jurkat cells as compared to control siRNA-transfected cells (Figure 3B).
Figure 3.
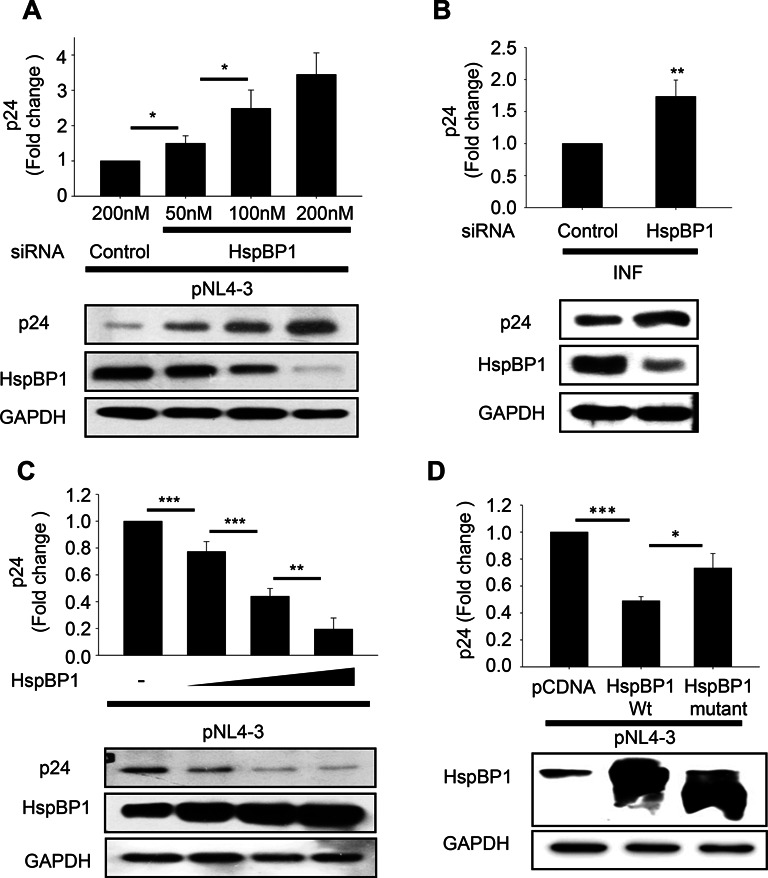
HspBP1 levels inversely correlate with HIV-1 virus production. (A) HspBP1 knockdown results in increased HIV-1 virus production in a dose dependent manner in HEK293T cells. Virus production was analyzed by p24 ELISA with culture supernatants from HEK293T cells co-transfected with pNL4-3 and increasing doses of HspBP1 siRNA. Supernatant was collected 48 h post-transfection. (B) HspBP1 silencing enhances virus production in HIV-1 infected Jurkat cells. Jurkat cells were transfected with HspBP1 siRNA followed by infection with HIV-1NL4-3 virus after 24 h. p24 ELISA was performed on the supernatant collected after 48 h of infection. (C) HspBP1 over-expression inhibits virus production in dose dependent manner in HEK293T cells. Culture supernatants of HEK293T cells transfected with pNL4-3 and increasing doses of HspBP1 vector were analyzed for virus production using the p24 antigen capture ELISA. (D) HIV-1 inhibition by HspBP1 is a specific occurrence as mutant failed to inhibit virus production. HEK293T cells were co-transfected with either wild type or mutant HspBP1 and pNL4-3. Cells were harvested 48 h post-transfection. Virus production was determined by p24 ELISA on culture supernatant. Silencing using siRNA or over-expression of HspBP1 was confirmed by immunoblotting in each experiment. The results are representative of three experiments and presented as the mean ± SD (*P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001).
To further validate the inverse correlation of HspBP1 expression with HIV-1, over-expression studies were performed in both HEK293T and Jurkat cells. Increasing concentration of HspBP1 expression vector and pNL4-3 were co-transfected in HEK293T cells and virus production was examined in the supernatant using p24 antigen capture ELISA assay. We observed that over-expression of HspBP1 reduces virus production in a dose-dependent manner (Figure 3C). As demonstrated earlier, HspBP1 over-expression also inhibits virus production in T-cells (Figure 1C). In order to determine whether inhibitory effect of HspBP1 on HIV-1 is a specific occurrence, we have utilized an N-terminal deletion mutant of HspBP1 (84–359), which lacks the first domain of HspBP1 but can still bind HSP70 and has some HSP70 inhibitory activity. We observed that mutant failed to inhibit virus production as efficiently as wild type (Figure 3D), suggesting that the Domain I of HspBP1 is required for the inhibition of HIV-1 replication. Collectively, these results clearly suggest that HspBP1 acts as an intrinsic inhibitor for the virus.
HspBP1 inhibits HIV-1 LTR promoter activity
The luciferase assay results presented earlier in Figure 1D and E demonstrated that HspBP1 inhibits HIV-1NL4-3 virus induced LTR driven gene-expression. In order to understand the mechanistic details of the inhibitory effect of HspBP1 on LTR-mediated gene-expression, we used bioinformatic analysis to predict the DNA binding activity of HspBP1. Both DNAbinder (54) and iDNA-Prot (55) suggested that HspBP1 might have DNA binding activity. Therefore, it was plausible to hypothesize that HspBP1 might have a direct influence on the LTR promoter. The direct role of HspBP1 on LTR driven gene-expression was examined by performing luciferase reporter assays in HEK293T and Jurkat cells. It was observed that HspBP1 inhibits basal LTR driven gene-expression in dose-dependent manner in both HEK293T (Figure 4A) and Jurkat cells (Figure 4B). This was confirmed by HspBP1 siRNA knockdown studies. A dose dependent increase in basal LTR-driven gene-expression was observed upon gradual depletion of HspBP1, suggesting a repressive role of HspBP1 on LTR promoter activity (Figure 4C). We have also performed these experiments in a more biologically relevant scenario, where we checked the effect of HspBP1 on LTR promoter activity in the presence of Tat as successful HIV-1 transcription and productive infection is not possible in the absence of Tat (Figure 4D). Increasing amount of HspBP1 were transfected in HEK293T cells along with LTR luc reporter construct and Tat expressing vector. Our results demonstrate that HspBP1 also inhibits HIV-1 LTR driven gene expression in presence of Tat.
HspBP1 inhibits HIV-1 LTR promoter activity through NF-κB enhancer region
Various cellular proteins have been shown to bind specific regions of HIV-1 LTR sequence and thereby exert physiological effects (56). Therefore, we attempted to identify the region of the LTR, which was targeted by HspBP1. Different LTR promoter deletion mutant constructs as schematically represented in Figure 4E (48) were used to perform luciferase reporter assays. The inhibition of promoter activity by HspBP1 was observed with full-length LTR construct, CD12 luc and deletion mutant CD23 luc. These results suggest that the upstream region (upstream to CD23 luc) of LTR does not play a role in HspBP1 mediated inhibition. However, CD52 luc and CD54 luc demonstrated no significant decrease in reporter activity with HspBP1, suggesting that the region between −117 and −65 contains HspBP1 regulated site. This region harbours binding sites for NF-κB and Sp1, suggesting that the NF-κB-SP1 (−117 to −65) enhancer region is the target site for HspBP1 (Figure 4F). NF-κB-Sp1 enhancer region harbours two NF-κB sites and one of the three SP1 binding sites. To further define the specific target site, we constructed two NF-κB mutants of LTR promoter- LTR del-NF-κB luc (deletion mutant in which both NF-κB binding sites are deleted) and LTR NF-κB mut luc (point mutant that has mutations in both NF-κB binding sites) (Figure 4G). These mutants were then used to perform luciferase assays to see if effect of HspBP1 on HIV-1 LTR promoter is specifically through NF-κB binding sites. HspBP1 lost its inhibitory effect on LTR promoter activity with both NF-κB deletion and point mutants of LTR in the presence and absence of Tat, indicating that suppression of LTR activity by HspBP1 is mediated by NF-κB cis-acting elements (Figure 4H). Furthermore, we examined the effect of HspBP1 on NF-κB enhancer driven luciferase construct containing 5 copies of the NF-κB response elements. We found that HspBP1 suppresses NF-κB driven gene-expression in a dose-dependent manner (Figure 4I), suggesting a potential role of HspBP1 in regulation of NF-κB enhancer regulated pathways. Thus, overall, our results demonstrate that HspBP1 targets NF-κB-mediated activation of LTR-driven gene-expression.
HspBP1 interacts with NF-κB consensus region on LTR promoter
The result above indicates a probable role of NF-κB enhancer region in HspBP1 mediated inhibition of LTR activity and therefore potential binding of HspBP1 on NF-κB enhancer. To test this possibility, electrophoretic mobility shift assays (EMSA) were performed using a 24 bp probe (probe P, Figure 5A) containing both NF-κB binding sites of the LTR promoter with purified recombinant HspBP1 protein. We observed a dose-dependent increase in binding of His-HspBP1 to NF-κB enhancer region of LTR promoter with increasing amounts of HspBP1 protein (Figure 5B). We also tested the specificity of this interaction by using cold and mutated oligo. The loss in binding was only observed by competition with unlabeled specific oligo and not with non-specific oligo (Figure 5C), demonstrating specificity of the interaction. In order to check the binding of cellular HspBP1, we used nuclear extract of HEK293T cells. Binding of the probe to nuclear extract was inhibited when the nuclear extract was pre-incubated with HspBP1 antibody while binding remained unaffected by post-incubation with HspBP1 antibody (Figure 5D). This data suggests that HspBP1 present in the nuclear extract binds to the NF-κB enhancer region on the LTR promoter and its binding site to NF-κB enhancer region is also the site for antibody binding. Complex formation was also inhibited by HspBP1 antibody in dose-dependent manner whereas the IgG control did not have any effect on complex formation (Figure 5E). Specificity of the interaction was also confirmed using cold and mutant oligo. Excess of cold oligo completely inhibited the complex formation, which was not observed with mutant oligo (Figure 5F).
Figure 5.

HspBP1 interacts with NF-κB consensus region on LTR promoter. (A) Schematic illustration of HIV-1 LTR promoter showing the position of oligonucleotide probe (P) and primers (F1/R1, F2/R2 and F3/R3) used in EMSA and ChIP assays, respectively. (B) His-HspBP1 binds to probe in dose-dependent manner. (C) His-HspBP1 binding to probe is inhibited by cold excess oligo but not by mutant oligo. (D) Probe binds to HEK293T nuclear extract, which is inhibited by pre-incubation with HspBP1 antibody while binding remains unaffected during post-incubation with HspBP1 antibody. (E) Binding of probe to nuclear extract is inhibited by HspBP1 antibody in dose-dependent manner while IgG control does not have any effect. (F) Specificity of probe binding to nuclear extract is analyzed by competition with specific (cold) and non-specific (mutant) oligos. (G) HspBP1 is recruited on LTR promoter at NF-κB-SP1 enhancer region in HIV-1NL4-3 infected CEM-GFP cells. Fixed and sonicated chromatin from day 5 infected CEM-GFP cells were immunoprecipitated with HspBP1 antibody followed by qRT-PCR analysis with indicated primers spanning three different regions on LTR promoter. (B), (C), (D), (E) and (F) are one of the representative figure of at least two independent experiments. Results shown in (G) represent data from three experiments. Results are presented as the mean ± SD and significance is defined as *P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001.
We then also investigated in-vivo binding of HspBP1 to LTR promoter by chromatin immunoprecipitation assay. Cross-linked chromatin from HIV-1NL4-3 virus infected CEM-GFP cells was immuno-precipitated with HspBP1 antibody. Quantitative-RT-PCR was carried out with primers- F1/R1, F2/R2 and F3/R3 to determine the region occupied by HspBP1 on the LTR promoter. F1/R1 encompasses NF-κB-SP1 (−143 to +14) in the enhancer region. F2/R2 and F3/R3 include −453 to −349 and +75 to +180 regions on the LTR promoter, respectively (Table 4). A significant enrichment using HspBP1 antibody over IgG was observed with primers F1/R1 (Figure 5G) indicating that HspBP1 is recruited on NF-κB-SP1 enhancer region. No enrichment was observed with primers F2/R2 and F3/R3, indicating HspBP1 is not recruited on the regions covered by these primer sets (Figure 5G).
HspBP1 inhibits NF-κB mediated activation of HIV-1 transcription by blocking the recruitment of p65 on LTR promoter
Our observation of HspBP1 recruitment on the LTR promoter at NF-κB consensus region led us to further investigate whether HspBP1 competes with p50/p65 hetero-dimer for occupancy on the LTR promoter. Competition assays between p50/p65 and HspBP1 were performed for LTR-driven gene-expression. Luciferase reporter assays revealed that HspBP1 competes with p65 for regulation of LTR promoter activity, which was not visible with HspBP1 mutant (Figure 6A). Furthermore, our data shows that HspBP1 inhibits p50/p65-mediated activation of LTR promoter in dose dependent manner (Figure 6B), suggesting an active competition between HspBP1 and p65 for controlling LTR promoter activity. We also performed ChIP assays to determine whether over-expression of HspBP1 causes any change in the recruitment of p65 on LTR promoter. A significant reduction in the occupancy of p65 at NF-κB-SP1 enhancer region [with F1/R1 (−143 to +14)] on LTR was observed in HspBP1 and pNL4-3 co-transfected HEK293T cells compared to pNL4-3 alone transfected cells (Figure 6C). However, there was no difference observed with F2/R2 and F3/R3 encompassing regions. Thus, our results clearly indicate that HspBP1 inhibits p65 recruitment to NF-κB enhancer region on the LTR promoter. We also observed in Figure 6A and B that HspBP1 over-expression causes decrease in p50/p65 protein levels. Thus, it became necessary to confirm whether decrease in LTR activity by HspBP1 over-expression is only due to an HspBP1-mediated decrease in p50/p65 levels or competition between these proteins for LTR occupancy. To confirm this, we checked the effect of HspBP1 depletion on p65-mediated induction of LTR promoter. A significant increase in promoter activity was observed in HspBP1 depleted cells compared to control siRNA-transfected cells with no change in expression levels of p65 upon HspBP1 knockdown, indicating that increased availability of LTR to p65 and not its expression level (remains same) is responsible for higher LTR induction in HspBP1 depleted condition (Figure 6D). Thus, our results indicate that inhibition of LTR activity by HspBP1 is primarily due to lesser accessibility of LTR to p50/p65 in presence of HspBP1.
Figure 6.
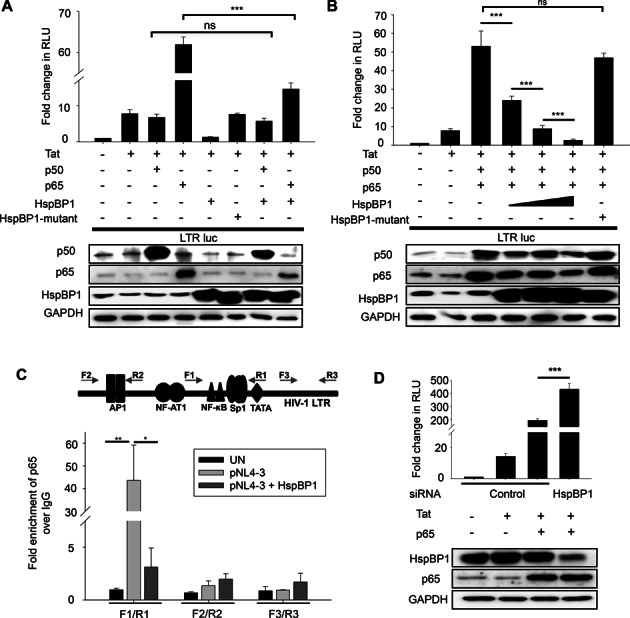
HspBP1 inhibits NF-κB mediated activation of HIV-1 transcription by obstructing p65-recruitment on LTR promoter. (A) HspBP1 competes with p65 for regulation of LTR driven gene-expression. HEK293T cells were transfected with indicated plasmids. Luciferase assays were performed 36 h post-transfection. (B) HspBP1 inhibits p50/p65-mediated activation of LTR promoter in a dose-dependent manner. HEK293T cells were co-transfected with the indicated plasmids. HspBP1-construct was transfected in three doses (0.25–0.75 ug). Luciferase assays were performed 36 h post-transfection. Expression of various constructs used in the assay was confirmed by immunoblotting. (C) HspBP1 over-expression diminishes the occupancy of p65 at LTR promoter. HEK293T cells were co-transfected with either control DNA and pNL4-3 or HspBP1 and pNL4-3. 48 h post-transfection, cells were fixed and chromatin was prepared followed by immunoprecipitation with p65 antibody or IgG control antibody. qRT-PCR analysis was performed with indicated primers spanning three different regions of LTR promoter. (D) HspBP1 knockdown causes an increase in p65-mediated induction of LTR promoter. HEK293T cells were transfected with either control siRNA or HspBP1 siRNA. 24 h post transfection, LTR promoter, Tat and p65 were over-expressed as indicated and luciferase assay was performed after 24 h of second transfection. Results represent data from three experiments and presented as the mean ± SD (*P ≤ 0.05, **P ≤ 0.01 and ***P ≤ 0.001).
HspBP1 inhibits reactivation of latently infected cells
NF-κB signaling forms an important part of reactivation of HIV-1 from latency. In order to understand the functional significance of HspBP1-mediated-transcriptional repression of HIV-1 by targeting NF-κB pathway, we examined the expression levels of HspBP1 in unstimulated and stimulated cells that are latently infected with HIV-1 (ACH2 and U1). ACH2 is a T cell-line and has a single copy of the provirus integrated in its genome. U1 is a latent monocytic cell-line harbouring two copies of integrated proviral DNA. Both the cell lines can be stimulated by phorbol myristate acetate (PMA) or TNF-α to produce high levels of infectious HIV-1. We observed that HspBP1 levels were high during latent phase and upon activation/stimulation (via PMA or TNFα), HspBP1 was depleted, suggesting that the physiological levels of HspBP1 might be relevant for HIV-1 latency (Figure 7A and B). Furthermore, we also assessed the effect of HspBP1 on NF-κB mediated activation of latently infected cells. We analyzed the activation status of latently infected cells via PMA and TNFα in HspBP1 over-expressed or HspBP1 depleted ACH2 (Figure 7C) and U1 (Figure 7D) cells. The activation of latent cells was determined by measuring the released viral p24 antigen in the supernatant. PMA and TNFα induced significant HIV reactivation, which was evident from increased p24 levels in the supernatant (Figure 7C and D). HspBP1 over-expression inhibited this reactivation of latently infected cells by PMA in both ACH2 (Figure 7C left panel) and U1 (Figure 7D left panel) cells. Furthermore, knockdown of HspBP1 causes an increase in reactivation of HIV-1 in both ACH2 (Figure 7C right panel) and U1 (Figure 7D right panel) cells, suggesting the active involvement of HspBP1 in suppressing NF-κB-mediated activation of HIV-1 in chronically infected cells.
Figure 7.
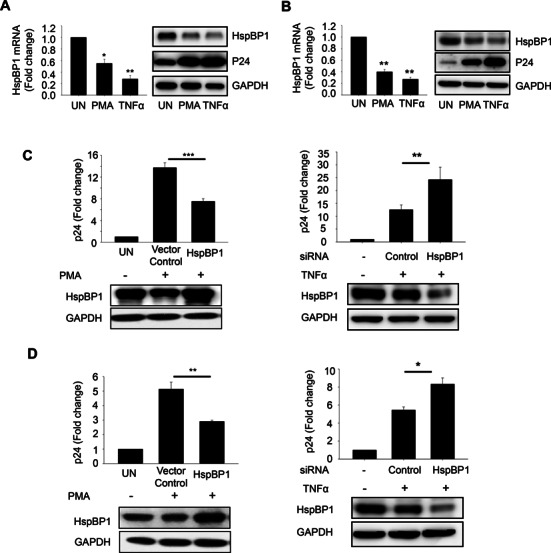
HspBP1 inhibits activation of latently infected cells. Activation of latently infected cells [(A) ACH2 and (B) U1] leads to depletion in HspBP1 levels. ACH2 and U1 were stimulated with PMA (50ng/ml) and TNFα (10 ng/ml). 36 h post-treatment, cells were harvested for RNA and protein lysate preparation. Expression of HspBP1 was analyzed by qRT-PCR and immunoblotting. (C) HspBP1 over-expression inhibits virus production whereas knockdown causes an increase in virus production in ACH2 cells. Nucleofection was performed to over-express or knockdown HspBP1 in these cell lines. 24 h post-nucleofection, cells were stimulated with PMA (50 ng/ml) or TNFα (10 ng/ml). 24 h post-activation, p24 ELISA was performed with the supernatant to determine the amount of virus produced. HspBP1 over-expression and knockdown was confirmed by immunoblotting. (D) HspBP1 over-expression inhibits virus production whereas knockdown causes an increase in virus production in U1 cells. The results shown in (A), (B) and (C) are representative of three experiments. (D) represents data from two experiments. Error bars represent the mean ± SD values and significance is defined as **P ≤ 0.01 and ***P ≤ 0.001.
Thus, overall, our results demonstrate that HspBP1 is an important regulator of HIV-1 gene-expression, which restricts HIV-1 by suppressing NF-κB-mediated activation of viral transcription.
DISCUSSION
Despite substantial advancements in our understanding of HIV/AIDS pathogenesis, the disease still remains incurable. More than 39 million people have already died because of AIDS-related opportunistic infections and at present 35 million people are living with AIDS (57). HIV manipulates host cellular gene-expression for its benefit (8,58) and simultaneously its life cycle is also influenced by different host factors (6,7,10). Multiple cellular proteins have been identified which either facilitate or inhibit various steps of viral replication (59,60).
In the present study, we report that HSP70 binding protein 1 (HspBP1), one of the HSP70 interacting proteins, acts as an intrinsic inhibitor of HIV-1 and restricts its replication. Previously, it has been reported that HspBP1 IgG levels increase in the serum of HIV-1 infected subjects (33). However, no clear function has been attributed to HspBP1 in HIV pathogenesis to date. This protein has also been associated with other diseases like cancer and its expression is inversely related to tumor aggressiveness (61,62).
HspBP1 is a HSP70 co-chaperone and binds to ATPase domain of HSP70, which inhibits its refolding activity (34,35). Several reports have demonstrated the significance of HSP70 in HIV-1 life cycle. It was shown to regulate viral transcription by stabilizing CDK9/Cyclin T1 complex (28), elicit cellular and humoral immune responses against HIV-1 (63) and exhibit neutralizing activity against the virus (64). HSP70 also protects cells from HIV induced cell cycle arrest and cell death (27). Because HspBP1 can regulate HSP70 activity, it is reasonable to speculate the association of HspBP1 with HIV-1 pathogenesis.
It has been well established that HSP40 and HSP70 interact with each other and play an important role in HIV-1 life cycle (30). Previous studies from our laboratory have shown that HSP40 and HSP70 reciprocally regulate HIV-1 gene-expression and replication, where HSP40 activates viral transcription and HSP70 blocks it by interfering with phosphorylation of C-terminal domain of RNA pol II (29,30). HspBP1 has also been reported to be independently associated with HSP70. In the present study, we investigated whether HspBP1 also interacts with HSP40. The results of co-immunoprecipitation assays revealed that indeed, HspBP1 interacts with both HSP40 and HSP70. To understand the relevance of this association of chaperones during HIV-1 infection, we investigated the individual and cumulative effect of HspBP1, HSP40 and HSP70 on viral gene-expression and virus production. Our results show that HspBP1 inhibits virus production as well as LTR driven gene-expression. Along with HSP70, HspBP1 completely abolished the positive effect of HSP40 on the virus, suggesting a possible collaborative interaction between HspBP1 and HSP70. These observations led us to hypothesize that HspBP1 acts in concert with HSP70 and HSP40 and regulates HIV-1 transcription. Contrary to our expectation that HspBP1 would act on HIV-1 transcription by a similar mechanism and be part of the same pathway as Hsp40 and Hsp70, we found that the inhibitory activity of HspBP1 on HIV-1 can be observed in absence of both HSP40 and HSP70, implying that HspBP1 employs a distinct mechanism for regulation of viral gene-expression. Overall, our results suggest that although these three proteins interact but they exert independent effects on HIV-1 replication, manifested via different mechanisms, however, intricate interplay between these proteins may not be ruled out.
Earlier, we have studied the mechanism of HSP40 and HSP70-mediated regulation of HIV-1 gene-expression (30). As it seems that HspBP1 employs a different mechanism, so, here, in the present study we intended to understand the role of HspBP1 in HIV-1 infection and decipher the involved mechanism. In our attempt to decipher the significance of HspBP1 in HIV-1 pathogenesis, we have examined the expression levels of HspBP1 during HIV-1 infection. HspBP1 was down modulated upon HIV-1 infection both in T-cells and primary cells. This suggested the presence of a plausible viral defence mechanism to down-regulate the levels of a protein, which might interfere with its replication. It will be interesting to study the mechanism of HspBP1 down regulation during HIV infection. Altered expression of cellular proteins has been demonstrated to have an impact on viral replication and gene expression (58). Knockdown and over-expression of HspBP1 in T-cells confirmed the inverse co-relation between HspBP1 and virus production. These results strongly suggest that HspBP1 act as a negative factor for HIV-1.
Having established the negative role of HspBP1 for HIV-1 replication, we next characterized the molecular mechanism of this inhibition. HIV-1 LTR promoter regulates the transcription of HIV-1 provirus and can be activated by several stimuli. Many cellular proteins/transcription factors have been shown to regulate LTR driven gene expression (56). Using bioinformatic analysis, we predicted DNA binding activity for HspBP1. We investigated the effect of HspBP1 on basal HIV-1 LTR promoter activity. The results of the luciferase reporter assays clearly showed that HspBP1 inhibits LTR driven gene-expression in dose dependent manner through the NF-κB enhancer region. In addition, gel-shift and chromatin immunoprecipitation assays revealed that HspBP1 is recruited on the LTR promoter at the NF-κB enhancer region, both in-vitro and in-vivo.
NF-κB pathway plays a critical role in host immune response against viruses. But many viruses, including HIV-1 exploit this pathway for their replication, spread and pathogenic functions (65). Activation of the NF-κB pathway involves proteasomal degradation of its inhibitor IκB, leading to nuclear translocation of NF-κB proteins and subsequent binding to LTR promoter. Binding of NF-κB p50/p65 hetero-dimer to the κB-sites present in LTR promoter is very critical for efficient HIV-1 transcription (65). p50/p65 associates with p300, which in turn increases the accessibility of LTR for RNA pol II and hence facilitates transcription initiation (24). Moreover, this hetero-dimer has also been reported to recruit the P-TEFb complex, which increases the processivity of RNA Pol II and hence facilitates transcription elongation (66).
Our results indicated that HspBP1 is also recruited on LTR promoter at NF-κB enhancer region, therefore we investigated whether there is competition between HspBP1 and p50/p65 hetero-dimer to occupy the viral promoter and hence, in the regulation of LTR-driven gene-expression. Luciferase reporter assays indicated that HspBP1 inhibits p50/p65-mediated activation of LTR promoter in dose-dependent manner. The ChIP assays demonstrated that the over-expression of HspBP1 leads to inhibition in recruitment of p65 on LTR promoter suggesting that HspBP1 and p65 compete for viral LTR promoter and the binding of HspBP1 to the κB-sites on viral promoter leads to obstruction in binding of NF-κB proteins, consequently resulting in suppression of viral transcription.
NF-κB signaling is one of the key players involved in reactivation of HIV-1 from latency, as latency can be broken by multiple pathways, including NF-κB signaling (66). Our results demonstrate that the activation of latently infected cells via PMA or TNFα leads to depletion in HspBP1 levels, suggesting the significance of physiological levels of HspBP1 for HIV-1 latency. We also checked the effect of HspBP1 on the activation status of latently infected cells by over-expression and knockdown approaches. Strikingly, our results show that HspBP1 inhibits NF-κB-mediated reactivation of latently infected cells. Hence, our results corroborated that HspBP1 inhibits HIV-1 transcription by suppressing NF-κB-mediated activation of HIV-1 gene-expression.
To summarize, we have identified a novel role for HspBP1 as a host intrinsic inhibitor of HIV-1, which negatively regulates HIV-1 gene-expression and replication. It was observed that the expression of HspBP1 is reduced upon infection. We show that HspBP1 competes with p50/p65 for recruitment at κB-sites on HIV-1 LTR promoter and binding of HspBP1 to these sites obliterates the binding of NF-κB proteins leading to repression in HIV-1 transcription (Figure 8). Hence, HspBP1 restricts HIV-1 replication by inhibiting NF-κB mediated activation of viral gene expression. In conclusion, our study establishes that HspBP1 is an endogenous negative regulator of HIV-1 gene-expression and replication and the information generated in the present study might be useful for developing novel therapeutic strategy against HIV-1.
Figure 8.
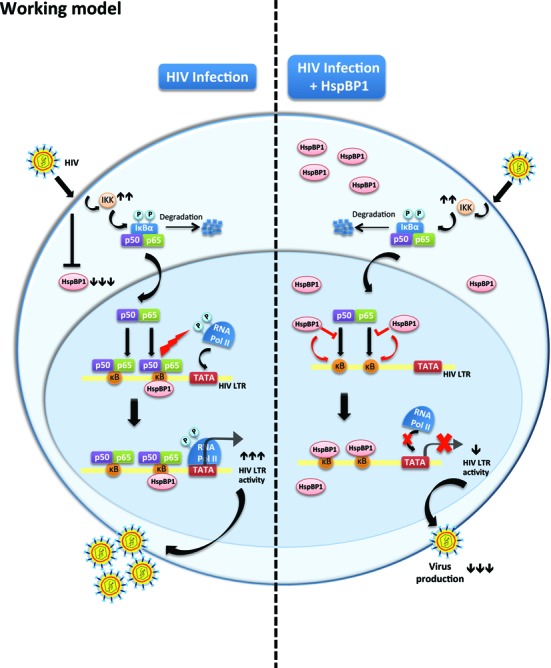
Proposed mechanistic model showing restriction of HIV-1 by HspBP1. Left, HIV-1 infection causes down-modulation of HspBP1 expression. Therefore, HspBP1 cannot inhibit HIV-1 production, consequently leading to efficient virus production. Right, when HspBP1 is over-expressed in an infected cell, HspBP1 interacts with NF-κB enhancer region (κB sites) on the HIV-1 LTR promoter and inhibits LTR driven gene-expression. The binding of HspBP1 to κB sites obliterates the binding of NF-κB hetero-dimer (p50/p65) to the same region, leading to repression in HIV-1 transcription.
Acknowledgments
We are thankful to NIH AIDS Reagent Program, Division of AIDS, NIAID, USA for providing cell lines (CEM-GFP, U1 and ACH2), plasmid constructs (pNL4-3, p50 and p65) and antibodies (p24 and Tat). We thank Dr T. Okamoto and Dr Margarida D. Amaral for providing different plasmid constructs as described in the text. We also thank Dr Neeru Dhamija for the technical help.
Footnotes
Present addresses:
Sohrab Zafar Khan, Center for Cancer and Immunology Research, Children's Research Institute, Children's National Medical Center, Washington, DC 20010, USA.
Pratima Rawat, Department of Pediatrics, Division of Infectious Diseases, 9500 Gilman Dr, University of California San Diego, La Jolla, CA 92093, USA.
FUNDING
Department of Biotechnology (DBT) grant number [BT/PR14226/MED/29/195/2010]; Government of India; intramural support from National Centre for Cell Science, Pune, India; Tata innovation Fellowship to DM, Department of Biotechnology, Ministry of Science and Technology, Government of India; Council of Industrial and Scientific Research fellowship (to P.C.), Government of India; Indian Council of Medical Research fellowship (to T.A.), Government of India. Funding for open access charge: National Centre for Cell Science, Pune, India.
Conflict of interest statement. None declared.
REFERENCES
- 1.Archin N.M., Sung J.M., Garrido C., Soriano-Sarabia N., Margolis D.M. Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014;12:750–764. doi: 10.1038/nrmicro3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chien P.C., Jr, Cohen S., Tuen M., Arthos J., Chen P.D., Patel S., Hioe C.E. Human immunodeficiency virus type 1 evades T-helper responses by exploiting antibodies that suppress antigen processing. J. Virol. 2004;78:7645–7652. doi: 10.1128/JVI.78.14.7645-7652.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Preston B.D., Poiesz B.J., Loeb L.A. Fidelity of HIV-1 reverse transcriptase. Science. 1988;242:1168–1171. doi: 10.1126/science.2460924. [DOI] [PubMed] [Google Scholar]
- 4.Guha D., Ayyavoo V. Innate immune evasion strategies by human immunodeficiency virus type 1. ISMn Aids. 2013;2013:954806. doi: 10.1155/2013/954806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kirchhoff F. Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe. 2010;8:55–67. doi: 10.1016/j.chom.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Brass A.L., Dykxhoorn D.M., Benita Y., Yan N., Engelman A., Xavier R.J., Lieberman J., Elledge S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 7.Konig R., Zhou Y., Elleder D., Diamond T.L., Bonamy G.M., Irelan J.T., Chiang C.Y., Tu B.P., De Jesus P.D., Lilley C.E., et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landi A., Vermeire J., Iannucci V., Vanderstraeten H., Naessens E., Bentahir M., Verhasselt B. Genome-wide shRNA screening identifies host factors involved in early endocytic events for HIV-1-induced CD4 down-regulation. Retrovirology. 2014;11:118. doi: 10.1186/s12977-014-0118-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yeung M.L., Houzet L., Yedavalli V.S., Jeang K.T. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J. Biol. Chem. 2009;284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou H., Xu M., Huang Q., Gates A.T., Zhang X.D., Castle J.C., Stec E., Ferrer M., Strulovici B., Hazuda D.J., et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Gautier V.W., Gu L., O'Donoghue N., Pennington S., Sheehy N., Hall W.W. In vitro nuclear interactome of the HIV-1 Tat protein. Retrovirology. 2009;6:47. doi: 10.1186/1742-4690-6-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jager S., Cimermancic P., Gulbahce N., Johnson J.R., McGovern K.E., Clarke S.C., Shales M., Mercenne G., Pache L., Li K., et al. Global landscape of HIV-human protein complexes. Nature. 2012;481:365–370. doi: 10.1038/nature10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kramer G., Moerland P.D., Jeeninga R.E., Vlietstra W.J., Ringrose J.H., Byrman C., Berkhout B., Speijer D. Proteomic analysis of HIV-T cell interaction: an update. Front. Microbiol. 2012;3:240. doi: 10.3389/fmicb.2012.00240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L., Zhang X., Ma Q., Zhou H. Host proteome research in HIV infection. Genomics Proteomics Bioinformatics. 2010;8:1–9. doi: 10.1016/S1672-0229(10)60001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borner K., Hermle J., Sommer C., Brown N.P., Knapp B., Glass B., Kunkel J., Torralba G., Reymann J., Beil N., et al. From experimental setup to bioinformatics: an RNAi screening platform to identify host factors involved in HIV-1 replication. Biotechnol. J. 2010;5:39–49. doi: 10.1002/biot.200900226. [DOI] [PubMed] [Google Scholar]
- 16.Luque M.C., Santos C.C., Mairena E.C., Wilkinson P., Boucher G., Segurado A.C., Fonseca L.A., Sabino E., Kalil J.E., Cunha-Neto E. Gene expression profile in long-term non progressor HIV infected patients: in search of potential resistance factors. Mol. Immunol. 2014;62:63–70. doi: 10.1016/j.molimm.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 17.Peretz Y., Cameron C., Sekaly R.P. Dissecting the HIV-specific immune response: a systems biology approach. Curr. Opin. HIV AIDS. 2012;7:17–23. doi: 10.1097/COH.0b013e32834ddb0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu J.Q., Sasse T.R., Wolkenstein G., Conceicao V., Saksena M.M., Soedjono M., Perera S.S., Wang B., Dwyer D.E., Saksena N.K. Transcriptome analysis of primary monocytes shows global down-regulation of genetic networks in HIV viremic patients versus long-term non-progressors. Virology. 2013;435:308–319. doi: 10.1016/j.virol.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 19.Bushman F.D., Malani N., Fernandes J., D'Orso I., Cagney G., Diamond T.L., Zhou H., Hazuda D.J., Espeseth A.S., Konig R., et al. Host cell factors in HIV replication: meta-analysis of genome-wide studies. PLoS Pathogens. 2009;5:e1000437. doi: 10.1371/journal.ppat.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clavel F., Hance A.J. HIV drug resistance. N. Engl. J. Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- 21.Brown P.H., Tiley L.S., Cullen B.R. Efficient polyadenylation within the human immunodeficiency virus type 1 long terminal repeat requires flanking U3-specific sequences. J. Virol. 1991;65:3340–3343. doi: 10.1128/jvi.65.6.3340-3343.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaynor R. Cellular transcription factors involved in the regulation of HIV-1 gene expression. Aids. 1992;6:347–363. doi: 10.1097/00002030-199204000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Nabel G., Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- 24.Williams S.A., Chen L.F., Kwon H., Ruiz-Jarabo C.M., Verdin E., Greene W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006;25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams S.A., Greene W.C. Regulation of HIV-1 latency by T-cell activation. Cytokine. 2007;39:63–74. doi: 10.1016/j.cyto.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurer C., Cimarelli A., Luban J. Specific incorporation of heat shock protein 70 family members into primate lentiviral virions. J. Virol. 2002;76:4666–4670. doi: 10.1128/JVI.76.9.4666-4670.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iordanskiy S., Zhao Y., Dubrovsky L., Iordanskaya T., Chen M., Liang D., Bukrinsky M. Heat shock protein 70 protects cells from cell cycle arrest and apoptosis induced by human immunodeficiency virus type 1 viral protein R. J. Virol. 2004;78:9697–9704. doi: 10.1128/JVI.78.18.9697-9704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Keeffe B., Fong Y., Chen D., Zhou S., Zhou Q. Requirement for a kinase-specific chaperone pathway in the production of a Cdk9/cyclin T1 heterodimer responsible for P-TEFb-mediated tat stimulation of HIV-1 transcription. J. Biol. Chem. 2000;275:279–287. doi: 10.1074/jbc.275.1.279. [DOI] [PubMed] [Google Scholar]
- 29.Kumar M., Mitra D. Heat shock protein 40 is necessary for human immunodeficiency virus-1 Nef-mediated enhancement of viral gene expression and replication. J. Biol. Chem. 2005;280:40041–40050. doi: 10.1074/jbc.M508904200. [DOI] [PubMed] [Google Scholar]
- 30.Kumar M., Rawat P., Khan S.Z., Dhamija N., Chaudhary P., Ravi D.S., Mitra D. Reciprocal regulation of human immunodeficiency virus-1 gene expression and replication by heat shock proteins 40 and 70. J. Mol. Biol. 2011;410:944–958. doi: 10.1016/j.jmb.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 31.Roesch F., Meziane O., Kula A., Nisole S., Porrot F., Anderson I., Mammano F., Fassati A., Marcello A., Benkirane M., et al. Hyperthermia stimulates HIV-1 replication. PLoS Pathogens. 2012;8:e1002792. doi: 10.1371/journal.ppat.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson I., Low J.S., Weston S., Weinberger M., Zhyvoloup A., Labokha A.A., Corazza G., Kitson R.A., Moody C.J., Marcello A., et al. Heat shock protein 90 controls HIV-1 reactivation from latency. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1528–1537. doi: 10.1073/pnas.1320178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papp D., Prohaszka Z., Kocsis J., Fust G., Banhegyi D., Raynes D.A., Guerriero V. Development of a sensitive assay for the measurement of antibodies against heat shock protein binding protein 1 (HspBP1): increased levels of anti-HspBP1 IgG are prevalent in HIV infected subjects. J. Med. Virol. 2005;76:464–469. doi: 10.1002/jmv.20384. [DOI] [PubMed] [Google Scholar]
- 34.Raynes D.A., Guerriero V., Jr Inhibition of Hsp70 ATPase activity and protein renaturation by a novel Hsp70-binding protein. J. Biol. Chem. 1998;273:32883–32888. doi: 10.1074/jbc.273.49.32883. [DOI] [PubMed] [Google Scholar]
- 35.McLellan C.A., Raynes D.A., Guerriero V. HspBP1, an Hsp70 cochaperone, has two structural domains and is capable of altering the conformation of the Hsp70 ATPase domain. J. Biol. Chem. 2003;278:19017–19022. doi: 10.1074/jbc.M301109200. [DOI] [PubMed] [Google Scholar]
- 36.Shomura Y., Dragovic Z., Chang H.C., Tzvetkov N., Young J.C., Brodsky J.L., Guerriero V., Hartl F.U., Bracher A. Regulation of Hsp70 function by HspBP1: structural analysis reveals an alternate mechanism for Hsp70 nucleotide exchange. Mol. Cell. 2005;17:367–379. doi: 10.1016/j.molcel.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 37.Alberti S., Bohse K., Arndt V., Schmitz A., Hohfeld J. The cochaperone HspBP1 inhibits the CHIP ubiquitin ligase and stimulates the maturation of the cystic fibrosis transmembrane conductance regulator. Mol. Biol. Cell. 2004;15:4003–4010. doi: 10.1091/mbc.E04-04-0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gervaix A., West D., Leoni L.M., Richman D.D., Wong-Staal F., Corbeil J. A new reporter cell line to monitor HIV infection and drug susceptibility in vitro. Proc. Natl. Acad. Sci. U.S.A. 1997;94:4653–4658. doi: 10.1073/pnas.94.9.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Clouse K.A., Powell D., Washington I., Poli G., Strebel K., Farrar W., Barstad P., Kovacs J., Fauci A.S., Folks T.M. Monokine regulation of human immunodeficiency virus-1 expression in a chronically infected human T cell clone. J. Immunol. 1989;142:431–438. [PubMed] [Google Scholar]
- 40.Folks T.M., Clouse K.A., Justement J., Rabson A., Duh E., Kehrl J.H., Fauci A.S. Tumor necrosis factor alpha induces expression of human immunodeficiency virus in a chronically infected T-cell clone. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2365–2368. doi: 10.1073/pnas.86.7.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Folks T.M., Justement J., Kinter A., Dinarello C.A., Fauci A.S. Cytokine-induced expression of HIV-1 in a chronically infected promonocyte cell line. Science. 1987;238:800–802. doi: 10.1126/science.3313729. [DOI] [PubMed] [Google Scholar]
- 42.Adachi A., Gendelman H.E., Koenig S., Folks T., Willey R., Rabson A., Martin M.A. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gottwald E., Herschbach M., Lahni B., Miesfeld R.L., Kunz S., Raynes D.A., Guerriero V. Expression of the cochaperone HspBP1 is not coordinately regulated with Hsp70 expression. Cell Biol. Int. 2006;30:553–558. doi: 10.1016/j.cellbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 44.Farinha C.M., Nogueira P., Mendes F., Penque D., Amaral M.D. The human DnaJ homologue (Hdj)-1/heat-shock protein (Hsp) 40 co-chaperone is required for the in vivo stabilization of the cystic fibrosis transmembrane conductance regulator by Hsp70. Biochem. J. 2002;366:797–806. doi: 10.1042/BJ20011717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosen C.A., Sodroski J.G., Kettman R., Haseltine W.A. Activation of enhancer sequences in type II human T-cell leukemia virus and bovine leukemia virus long terminal repeats by virus-associated trans-acting regulatory factors. J. Virol. 1986;57:738–744. doi: 10.1128/jvi.57.3.738-744.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sodroski J., Rosen C., Goh W.C., Haseltine W. A transcriptional activator protein encoded by the x-lor region of the human T-cell leukemia virus. Science. 1985;228:1430–1434. doi: 10.1126/science.2990028. [DOI] [PubMed] [Google Scholar]
- 47.Dandekar D.H., Kumar M., Ladha J.S., Ganesh K.N., Mitra D. A quantitative method for normalization of transfection efficiency using enhanced green fluorescent protein. Anal. Biochem. 2005;342:341–344. doi: 10.1016/j.ab.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Takada N., Sanda T., Okamoto H., Yang J.P., Asamitsu K., Sarol L., Kimura G., Uranishi H., Tetsuka T., Okamoto T. RelA-associated inhibitor blocks transcription of human immunodeficiency virus type 1 by inhibiting NF-kappaB and Sp1 actions. J. Virol. 2002;76:8019–8030. doi: 10.1128/JVI.76.16.8019-8030.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho S.N., Hunt H.D., Horton R.M., Pullen J.K., Pease L.R. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 50.Joseph A.M., Ladha J.S., Mojamdar M., Mitra D. Human immunodeficiency virus-1 Nef protein interacts with Tat and enhances HIV-1 gene expression. FEBS Lett. 2003;548:37–42. doi: 10.1016/s0014-5793(03)00725-7. [DOI] [PubMed] [Google Scholar]
- 51.Raynes D.A., Thomson C.A., Stroster J., Newton T., Cuneo P., Guerriero V. Human serum contains detectable levels of the Hsp70 cochaperone HspBP1 and antibodies bound to HspBP1. J. Immunoassay Immunochem. 2006;27:251–264. doi: 10.1080/15321810600734935. [DOI] [PubMed] [Google Scholar]
- 52.Rawat P., Mitra D. Cellular heat shock factor 1 positively regulates human immunodeficiency virus-1 gene expression and replication by two distinct pathways. Nucleic Acids Res. 2011;39:5879–5892. doi: 10.1093/nar/gkr198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu D., Donegan J., Nuovo G., Mitra D., Laurence J. Stable human immunodeficiency virus type 1 (HIV-1) resistance in transformed CD4+ monocytic cells treated with multitargeting HIV-1 antisense sequences incorporated into U1 snRNA. J. Virol. 1997;71:4079–4085. doi: 10.1128/jvi.71.5.4079-4085.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar M., Gromiha M.M., Raghava G.P. Identification of DNA-binding proteins using support vector machines and evolutionary profiles. BMC Bioinformatics. 2007;8:463. doi: 10.1186/1471-2105-8-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lin W.Z., Fang J.A., Xiao X., Chou K.C. iDNA-Prot: identification of DNA binding proteins using random forest with grey model. PLoS One. 2011;6:e24756. doi: 10.1371/journal.pone.0024756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pereira L.A., Bentley K., Peeters A., Churchill M.J., Deacon N.J. A compilation of cellular transcription factor interactions with the HIV-1 LTR promoter. Nucleic Acids Res. 2000;28:663–668. doi: 10.1093/nar/28.3.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.UNAIDS. Geneva, Switzerland: UNAIDS; 2014. pp. 1–422. [Google Scholar]
- 58.Serrao E., Wang C.H., Frederick T., Lee C.L., Anthony P., Arribas-Layton D., Baker K., Millstein J., Kovacs A., Neamati N. Alteration of select gene expression patterns in individuals infected with HIV-1. J. Med. Virol. 2014;86:678–686. doi: 10.1002/jmv.23872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arhel N., Kirchhoff F. Host proteins involved in HIV infection: new therapeutic targets. Biochim. Biophys. Acta. 2010;1802:313–321. doi: 10.1016/j.bbadis.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 60.Friedrich B.M., Dziuba N., Li G., Endsley M.A., Murray J.L., Ferguson M.R. Host factors mediating HIV-1 replication. Virus Res. 2011;161:101–114. doi: 10.1016/j.virusres.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 61.Raynes D.A., Graner M.W., Bagatell R., McLellan C., Guerriero V. Increased expression of the Hsp70 cochaperone HspBP1 in tumors. Tumour Biol. 2003;24:281–285. doi: 10.1159/000076459. [DOI] [PubMed] [Google Scholar]
- 62.Souza A.P., Albuquerque C., Torronteguy C., Frasson A., Maito F., Pereira L., Duval da Silva V., Zerwes F., Raynes D., Guerriero V., et al. HspBP1 levels are elevated in breast tumor tissue and inversely related to tumor aggressiveness. Cell Stress Chaperones. 2009;14:301–310. doi: 10.1007/s12192-008-0085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suzue K., Young R.A. Adjuvant-free hsp70 fusion protein system elicits humoral and cellular immune responses to HIV-1 p24. J. Immunol. 1996;156:873–879. [PubMed] [Google Scholar]
- 64.Babaahmady K., Oehlmann W., Singh M., Lehner T. Inhibition of human immunodeficiency virus type 1 infection of human CD4+ T cells by microbial HSP70 and the peptide epitope 407–426. J. Virol. 2007;81:3354–3360. doi: 10.1128/JVI.02320-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chan J.K., Greene W.C. Dynamic roles for NF-kappaB in HTLV-I and HIV-1 retroviral pathogenesis. Immunol. Rev. 2012;246:286–310. doi: 10.1111/j.1600-065X.2012.01094.x. [DOI] [PubMed] [Google Scholar]
- 66.Williams S.A., Kwon H., Chen L.F., Greene W.C. Sustained induction of NF-kappa B is required for efficient expression of latent human immunodeficiency virus type 1. J. Virol. 2007;81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]