


Abstract
Ku70-binding protein 5 (Kub5)-Hera (K-H)/RPRD1B maintains genetic integrity by concomitantly minimizing persistent R-loops and promoting repair of DNA double strand breaks (DSBs). We used tandem affinity purification-mass spectrometry, co-immunoprecipitation and gel-filtration chromatography to define higher-order protein complexes containing K-H scaffolding protein to gain insight into its cellular functions. We confirmed known protein partners (Ku70, RNA Pol II, p15RS) and discovered several novel associated proteins that function in RNA metabolism (Topoisomerase 1 and RNA helicases), DNA repair/replication processes (PARP1, MSH2, Ku, DNA-PKcs, MCM proteins, PCNA and DNA Pol δ) and in protein metabolic processes, including translation. Notably, this approach directed us to investigate an unpredicted involvement of K-H in DNA mismatch repair (MMR) where K-H depletion led to concomitant MMR deficiency and compromised global microsatellite stability. Mechanistically, MMR deficiency in K-H-depleted cells was a consequence of reduced stability of the core MMR proteins (MLH1 and PMS2) caused by elevated basal caspase-dependent proteolysis. Pan-caspase inhibitor treatment restored MMR protein loss. These findings represent a novel mechanism to acquire MMR deficiency/microsatellite alterations. A significant proportion of colon, endometrial and ovarian cancers exhibit k-h expression/copy number loss and may have severe mutator phenotypes with enhanced malignancies that are currently overlooked based on sporadic MSI+ screening.
INTRODUCTION
Preserving structural and functional integrity of the genome is critical for all living cells. Exogenous and endogenous stresses pose severe threats to genomic stability, creating constant and non-uniform DNA lesions. DNA double-strand breaks (DSBs) are the most potent types of DNA lesions that threaten survival and genomic integrity. If left unrepaired, one DSB can cause lethality (1). If mis-repaired, DSBs can result in mutations and chromosome deletions or rearrangements that compromise the integrity of genome (2). In humans, genomic instability (both at the mutational and chromosomal levels) is considered a leading cause of cancer and cancer progression (3). A relatively unexplored source of genetic instability is the formation of persistent R-loops (DNA-RNA-DNA hybrids) as transcriptional byproducts (4). Several mechanisms were proposed to explain how persistent R-loops may cause genomic instability, including production of complex DSBs (4). A primary source of persistent R-loops is the impaired regulation of RNA Pol II pausing and/or failure to dislodge the enzyme at transcription termination sites (5). Ku70-binding protein 5-Hera (K-H) (also known as RPRD1B (6) or CREPT (7)) is a necessary scaffolding protein that regulates resolution of R-loops at both the transcription termination and DSB repair levels (8).
Emerging data indicate that K-H expression levels must be tightly regulated to maintain genetic stability. Over-expression of K-H promotes tumor growth, potentially by transcriptional promotion (7), whereas, depletion of K-H in normal or cancer cells results in elevated genetic instability (8). Knockout of the k-h gene is lethal, while loss of one k-h allele results in elevated R-loop and DSB formation, ensuring chromosomal aberrations (8). Moreover, copy number variations, single nucleotide polymorphisms (SNPs) and point mutations are present in human k-h gene in a wide variety of cancers (unpublished data).
K-H/RPRD1B is highly conserved across various species, and in yeast its homolog is RTT103 (9,10). The yeast RTT103 protein plays important roles in transcription termination, DNA damage responses and appears to localize at DSB sites (11,12). An Rtt103 deletion strain of yeast is viable, however, double mutants of Rtt103 in combination with condensins (structural maintenance of chromosome (SMC) proteins) or with DNA replication factors, confer growth defects (13,14). These findings suggest that RTT103 may be involved in various cellular processes aside from transcription termination. In contrast to yeast, homozygous deletion of the k-h gene resulted in early embryonic lethality in mice (8).
We recently reported that K-H was important in the physiology of R-loops and subsequent DSB formation and repair by associating with core non-homologous end joining (NHEJ) proteins, particularly Ku70 (8). However, the molecular contributions of K-H remain inadequately understood in diverse cellular processes. Moreover, prior proteomics studies using yeast RTT103 and human K-H proteins reported their association exclusively with proteins involved in RNA metabolism (6,11). Delineating the roles of specific proteins and their related higher-order protein complexes in R-loop clearance and DSB repair are essential to better understand how cells avoid R-loop-induced genetic instability. Thus, a detailed description of proteins associating with K-H/RPRD1B in higher-order protein complexes is required to further elucidate its role in various cellular processes.
We hypothesized that protein–protein association studies for K-H might hold various clues to its molecular functions in several biological processes. These studies represent an important step to further separate and define proteins involved in RNA metabolism and DNA repair, as recently indicated (8). Our goal in this study was to elucidate proteins involved in the K-H/RPRD1B interactome using a combination of proteomics, bioinformatics and biochemical approaches. Collectively, this approach led us to examine an unanticipated involvement of K-H in the regulation of DNA mismatch repair (MMR).
The MMR system performs important proof-reading functions after DNA replication, correcting nucleotide mismatches (15) and triggering G2/M cell cycle checkpoint arrest (16–18) and c-Abl/p73-regulated cell death pathways (19). The MMR system is composed of two complexes: (i) MutS mismatch recognition complex (composed of MutSα and MutSβ complexes) that has functions in repairing nucleotide mismatches and/or loops, respectively (20,21); and (ii) MutL complex (composed of MLH1 and PMS2) that promotes DNA endonucleolytic activity, leading to long-patch excision repair of nucleotide mismatches or small DNA loops (22). The MutSα complex consists of MSH2-MSH6 proteins and the MutSβ complex consists of MSH2-MSH3 proteins. MMR proteins are associated with various DNA damage signaling proteins. The BRCA1 associated genome surveillance complex (BASC) contains MSH2, MSH6 and MLH1 that recognizes and repairs abnormal DNA structures (23). MLH1 and PMS2 (MutLα) also interact with various proteins involved in cell-cycle regulation, signaling and apoptosis (17,19,24), and several studies demonstrated that MMR plays roles in DSB repair (25), either by HR (26–30) or NHEJ (31–33) pathways.
Here, we demonstrate that K-H associates in higher order complexes containing several classes of proteins, including those involved in RNA and/or DNA metabolism. Interactions were confirmed (10/12, >80%) by co-immunoprecipitation (co-IP) validation of key proteins. We show that defects in K-H expression by one allele loss or sh/siRNA-driven knockdown causes a complex DNA repair-deficiency syndrome that includes deficient MMR function, 6-thioguanine (6-TG) damage tolerance and resistance, and global microsatellite content alteration not involving genetic mutation of MHL1 or MSH2 (MSI-H). We show that the MMR deficiency brought on by K-H loss stems from reduced stability of core MMR proteins (MLH1 and PMS2) due to increased basal caspase activation, most likely specific to caspase-3. Treatment of k-h knockdown cells with a pan-caspase inhibitor (z-VAD) restored MLH1-PMS2 levels. Exploring the role(s) of K-H loss in MSI-H colon, endometrial and ovarian cancers may lead to identification of a new subset of MSI+ (with K-H loss) sporadic cancers and open new avenues for patient-specific individualized therapies.
MATERIALS AND METHODS
Cell lines, cell culture and transfection
Human HeLa, MDA-MB-231 (231), HEK 293 and 293T cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM supplemented with 5% FBS and 1 mM L-glutamine in a humidified 5% CO2–95% air atmosphere at 37°C. Triple-negative 231 breast cancer cells and variants were cultured in RPMI medium. Transfections were performed using Lipofectamine®2000 (Life Technologies, Grand Island, NY, USA) according to the manufacturer's recommendation. Puromycin (2 μg/ml) was used 24 h after transfection to select for stable transfectants. Resistant clones derived from pooled populations were established in ∼2 weeks. Individual clones were isolated by limiting dilution assays, and knockdowns or over-expressions of K-H or TAP-K-H protein were tested using Western blotting. A shk-h stable knockdown clone was generated using specific shRNA to target coding region of k-h gene, similarly as described (8). Mouse embryonic fibroblasts (MEFs) from K-H+/+ or K-H+/− mice were generated as described (8).
TAP-K-H cloning, expression and purification
Construction of k-h cDNA was described elsewhere (8). The pIRES-puro-TAP plasmid (CloneTech, Mountain View, CA, USA) was used as a TAP-empty vector control. To clone pIRES-TAP-K-H construct, k-h gene primers (harboring FseI and AscI restriction sites) and k-h cDNA (as a template) were used. Vectors were then used to generate stable cell lines. A modified method (34,35) was used for TAP-K-H protein purification. All incubations and centrifugations were performed at 4°C. HEK 293T cells stably expressing TAP or TAP-K-H were grown in ten 150 mm dishes (up to ∼80% confluency). Cells were trypsinized, harvested, washed with PBS and re-suspended in NP-40 buffer (6 mM Na2HPO4, 4 mM NaH2PO4, 150 mM NaCl, 2 mM EDTA, 50 mM NaF, 100 μM NaVO3, 1 mM benzamidine, 0.5 mM PMSF, 1X protease inhibitor cocktail, 10 mM β-glycerophosphate and 1% Nonidet P-40) and lysed by repeated (3X) freeze/thaw cycles. Lysates were sonicated (4X, 24 s pulses using a Branson Sonifier 450), centrifuged (42 000 g for 15 min) and supernatants were collected. Supernatants were incubated with IgG-Sepharose beads (pre-equilibrated in IgG equilibration buffer (10 mM Tris-HCl (80% cation, pH 8.0), 150 mM NaCl and 0.1% NP-40) overnight with gentle shaking. Beads were then washed with IgG equilibration buffer, then TEV cleavage buffer (10 mM Tris-HCl (80% cation, pH 8.0), 150 mM NaCl and 0.1% NP-40) and incubated with 1 ml TEV cleavage buffer containing 100 units of AcTEV protease with gentle shaking. Eluted proteins were collected and incubated overnight with calmodulin affinity beads (pre-equilibrated in calmodulin binding buffer (10 mM Tris-HCl (80% cation, pH 8.0), 150 mM NaCl, 0.1% NP-40, 10 mM Mg(OAc)2, 1 mM imidazole and 10 mM β-mercaptoethanol)) containing 9 mM CaCl2 with gentle shaking. Beads were then washed (3X) with 1 ml calmodulin binding buffer and TAP or TAP-K-H binding proteins were eluted with EGTA elution buffer (10 mM Tris-HCl (80% cation, pH 8.0), 150 mM NaCl, 0.02% NP-40, 1 mM Mg(OAc)2, 10 mM imidazole, 10 mM β-mercaptoethanol, 20 mM EGTA). Eluted proteins were concentrated using YM-3 centrifugal filters (EMD Millipore, Billerica, MA, USA), quantified, analyzed by SDS-PAGE and used for mass spectrometric analyses.
Mass spectrometry (MS) and bioinformatics analyses
Purified TAP or TAP-K-H complex protein mixtures were separated by 4–20% gradient SDS-PAGE and individual lanes were separately sliced into small pieces and digested overnight with trypsin (Promega, Madison, WI, USA) after reduction and alkylation with DTT and iodoacetamide (Sigma-Aldrich, St Louis, MO, USA). LC-MS/MS analyses were performed using an Ultimate 3000 RSLC-Nano liquid chromatography system (Dionex, Sunnyvale, CA, USA), coupled to a Q Exactive mass spectrometer (Thermo, Bremen). For HPLC, Buffer A consisted of 2% ACN and 0.1% formic acid in water, Buffer B consisted of 80% ACN, 10% TFE and 0.08% formic acid in water. The peptides were separated over 60 min on a 75 μm ID, 50 cm length EasySpray C18 column (Thermo). A data-dependent, top 20 MS method was used with MS scans at 70K resolution followed by up to 20 MS/MS scans at 17.5K resolution after higher-energy CID (HCD) fragmentation. Raw data files were converted to Mascot Generic Format (MGF) using ProteoWizard msconvert (version 3.0.3535) (36) with defaults except that the MS2Denoise filter was employed. Peptide and protein identification was performed within the Central Proteomics Facilities Pipeline (CPFP) version 2.0.3 (37) using the X!Tandem (v 2008.12.01.1) (38) and OMSSA (v 2.1.8) (39) search engines, and iProphet (40) for combination of results. The UniprotKB human whole proteome sequence database (release 2013_03) (41) with common contaminant sequences appended from CRAP (v 2012.01.01 ftp://ftp.thegpm.org/fasta/cRAP) was searched in concatenated target-decoy format (total 175 454 protein sequences). Precursor and fragment Mass tolerances were 20 ppm/0.1 Da, respectively. With tryptic specificity up to 3 missed cleavages were permitted with carbamidomethylation of Cys as a fixed modification, and Oxidation of Met as a variable modification. Protein inference was performed from peptides using ProteinProphet (42), such that ambiguous protein identifications are grouped. Protein-level quantification between samples was performed using the SINQ normalized spectral index tool in CPFP (43). Default parameters were employed such that all proteins at a protein level FDR <1% were quantified using PSMs filtered to a PSM FDR of 1%. Final reported protein IDs were further filtered requiring distinguished peptide identity (i.e. indistinguishable proteins were filtered out), peptide sequences ≥5, PSM ≥5,% coverage ≥5, either exclusively present in the TAP-K-H fraction or enriched in TAP-K-H pull-downs (TAP-K-H/TAP ratio >1.0). Identified proteins were presented by their UniProt accession number along with above-mentioned information (Supplementary Table S1). Proteins fitting all above-mentioned criteria were included in subsequent evaluations. For functional annotation and biological mechanisms analyses, DAVID v6.7 (Database for Annotation, Visualization and Integrated Discovery, (http://david.abcc.ncifcrf.gov/)) bioinformatics tool was used that allowed us to further scrutinize the proteins identified in mass spectrometric screening (44,45).
Protein network building
A protein network was generated using STRING (Search Tool for the Retrieval of Interacting Genes/Proteins) 9.1 (46). The protein–protein interaction prediction methods used were based on neighborhood, gene fusion, co-occurrence, co-expression, experimental data, databases and text mining. The protein interaction layout was created by cytoscape 3.1.0 software (47) using the link from STRING.
Western immunoblotting and antibodies
Proteins were separated by SDS-PAGE and transferred to PVDF membranes. Blots were treated with 1X blocking buffer (Sigma, St. Louis, MO, USA) and incubated with primary and appropriated secondary antibodies conjugated with HRP. Proteins were detected by enhanced chemiluminescent HRP substrates, Super Signal Pico or Dura (Thermo Scientific, Pittsburg, PA, USA). Human antibodies against α-tubulin (DM1A) and K-H (157–170) were purchased from Sigma-Aldrich (St. Louis, MO, USA). The α-Ku70-specific antibody (N3H10) was obtained from Genetex (Irvine, CA, USA). Antibodies against RNA Pol II (8WG16), PARP1 (F-2), p15RS (FF-8), PCNA (PC-10), Ku86 (B-1), SSBP1 (FL-148) and PMS2 (C-20) were obtained from Santa Cruz Biotechnology (Dallas, TX, USA). The antibody against TOP1 (EPR5375) was purchased from Abcam (Cambridge, MA, USA). MSH2 (G219–1129), MLH1 (G168–728) and PMS2 (A16–4) antibodies were purchased from BD Biosciences (Franklin Lakes, NJ, USA) and used for both human and mouse MMR proteins. An antibody against cleaved caspase-3 (5A1E) was obtained from Cell Signaling Technologies Inc. (Beverly, MA, USA).
Co-immunoprecipitations (co-IPs)
HEK 293T cells (∼3 × 106) were seeded onto 150 mm dishes and ∼12 h later co-transfected with 20 nM 3′-UTR-specific siRNA-k-h (to knockdown endogenous K-H) and ∼3.0 μg of pCMV-mycK-H plasmid DNA (to overexpress mycK-H) and incubated for 48 h. Cells were then harvested and whole-cell or nuclear lysates prepared by re-suspending the pellet in 1 ml 1X TNE buffer (100 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.1 mM EDTA and 0.1% NP-40 (v/v) supplemented with 0.5 mM PMSF, 1X protease inhibitor cocktail and 10 mM β-glycerophosphate). For samples treated with nuclease, 100 units of DNase I was added and lysates incubated for 30 min. Cell lysates were centrifuged (10 000 g for 15 min, 4°C), supernatants collected and total protein was quantified using BCA assays (Thermo-Scientific, Waltham, MA, USA). A portion of each sample was saved as input. For co-IP analyses, ∼1 mg protein was incubated with 15 μg mouse normal IgG or anti-myc antibodies. Mixtures were then incubated with A/G plus agarose beads (Santa Cruz Biotechnology, Dallas, TX) overnight at 4°C, collected by centrifugation (1000 g, 5 min, 4°C), washed (3X) with 1X PBS buffer and re-suspended in Laemmli buffer (Bio-Rad, Hercules, CA, USA). Proteins in the supernatants were separated by SDS-PAGE and analyzed by Western immunoblotting using various specific antibodies as indicated.
HeLa whole-cell lysate preparation and gel-filtration chromatography
Procedures for HeLa whole-cell lysate preparation, nuclease treatment (to remove nucleic acid) and gel-filtration chromatography were essentially the same as described (8).
Cell survival assays
A modified cell survival assay that measures DNA content over a 7-day period was employed (48). A stock concentration of 6-TG (Sigma, St. Louis, MO, USA) was prepared in 0.1 M NaOH and stored at −80°C. Cells (20 000 cells/well) were seeded into 48-well plates in 0.5 ml media on day 0. The next day (day 1), media were aspirated and 0.5 ml media containing indicated concentrations of 6-TG (μM) were added. Cells were exposed for 48 h, >2 doublings. Media was then removed, cells washed with PBS and fresh media were added. Cells were then allowed to grow for 7 days. Cells were then lysed in water, freeze-thawed and DNA content (a measure of cell growth) was determined by Hoescht 33258 fluorescence (Sigma, St. Louis, MO, USA) using a Victor X3 plate reader (PerkinElmer, Waltham, MA, USA). Data (means, ±SD) were expressed as treated/control (T/C) values from experiments performed at least three times in triplicate each. P-values were obtained using two-tailed Student's t-tests.
Flow cytometry and cell cycle analyses
Cells (∼1 million) were seeded in 150 mm tissue culture dishes. The next day, cells were treated with 0.5 μM 6-TG for 24, 48 or 72 h and then pulsed-labeled with 10 μM 5-ethynyl-2′-deoxyuridine (EdU) for 1 h. Cells were harvested, fixed in 70% methanol and stored at −20°C until further use. To detect EdU incorporation in DNA, click-iT assays were used according to the manufacturer's protocol (Life Technologies, Grand Island, NY, USA). Cells were analyzed by LSR flow cytometer (BD Bioscience, Franklin Lakes, NJ, USA) using FACSDIVA software and 10 000 events were counted. Representative images are shown and quantification obtained from a minimum of three experiments.
Global microsatellite content measurement
To assess the global microsatellite stability of genomes of control versus shk-h depleted AT cells (characterized previously (8)), we used a custom microarray, which quantifies the total repetitive content for all 1-mer to 6-mer repeat containing loci in a genome. Genomic DNA was prepared using Wizard SV genomic DNA purification system according to manufacturer's protocol (Promega, Madison, WI, USA). Array design, manufacturing, array data processing and statistical analysis were previously described in details (49). Briefly, this array Competitive Genomic Hybridization (aCGH)-like array does not measure any single locus, but instead sums the total content for all loci composed of a given motif, so if there is an instability process in effect that preferentially expands or contracts or replicates or deletes repetitive loci, this effect could be quantified. This array was designed with probes to quantify the total amount of any repetitive motif in the genome. All possible 1-mer to 6-mer microsatellite repeat motifs can be represented by 5356 probes, one for each possible 1-mer to 6-mer, their cyclic permutations and their complements. This array contained 7 copies of all these probes for internal statistical checks. In addition, the array contained a variety of controls, including RepBase probes, ultraconserved region probes, full Arabidopsis/HIV/Lambda genomes and hybridization length control probes. These arrays are 2-color, manufactured by NimbleGen (Madison, WI, USA). Each array is processed to remove background, perform quantile normalization, and only those motif families where the complements and cyclic permutations agree are considered significant. In this experiment, in addition to the internal probe replicates, three arrays with the two samples were co-hybridized. Replicate arrays agreed with an R2 > 0.97.
Protein stability measurement
K-H+/+ and K-H+/− MEFs, or 231 cells stably expressing either a short-hairpin RNA against scramble (shScr) or the coding region or 3′-UTR of the k-h gene (sh/sik-h) were grown in 150 mm dishes (∼80% confluency). For transient knockdown of K-H, shScr 231 cells (∼1 × 106) were seeded onto 10 mm dishes and ∼12 h later co-transfected with 20 nM 3′-UTR-specific siRNA-k-h or siRNA-Scr and incubated for 24, 48 and 72 h. In experiments where the pan-caspase inhibitor z-VAD-fmk was used, the inhibitor was added to a final concentration of 50 μM for 24, 48 or 72 h. For time points greater than 24 h, fresh inhibitor was added every 24 h. Cells were harvested and whole cell lysates prepared by re-suspending pellets in 1X RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1mM EGTA, 1% (v/v) sodium-deoxycholate, 1% (v/v) NP-40, 2.5 mM sodium pyrophosphate supplemented with 0.5 mM PMSF, 1X protease inhibitor cocktail and 10 mM β-glycerophosphate) and incubated on ice for 10 min. Cell lysates were centrifuged (10 000 g for 15 min, 4°C), supernatants collected and total protein content quantified using BCA assays (Thermo-Scientific, Waltham, MA, USA). Proteins (10–40 μg) were separated on 4–20% SDS-PAGE and indicated proteins detected by Western immunoblotting using unique antibodies. Where appropriate, protein band intensities were quantified using NIH ImageJ software and normalized to loading controls.
Quantitative PCR
Quantitative PCR (qPCR) was performed as described (50). Total RNA was isolated from shScr or shk-h cells using RNeasy as directed by the manufacturer (Qiagen, Valencia, CA, USA). Total RNA (∼1–2 μg) was reverse transcribed by high-capacity cDNA reverse transcription according to the manufacturer's (Life Technologies, Grand Island, NY, USA) instructions. Probes for RT PCR (K-H; Hs00221889_m1, MLH1; Hs00179866_m1, PMS2; Hs00241053_m1) were also purchased from Life Technologies.
Statistical analyses
Where appropriate, data are represented as means ±SD from a minimum of three experiments and P-values were obtained using two-tailed Student's t-tests.
RESULTS
Identification, validation and network of novel proteins associated with K-H
The Tandem Affinity Purification (TAP) method is extremely useful to identify protein binding partners (34,35). We utilized this method to purify K-H-associating proteins using an N-terminal TAP tag fused to the K-H protein (Supplementary Figure S1A). Protein complexes were purified with two sequential affinity purification steps, separated by SDS-PAGE and subjected to liquid chromatography-mass spectrometric analyses (LC-MS/MS) as outlined (Supplementary Figure S1B–C). Proteins exclusively associated with TAP-K-H samples, or enriched in these samples (TAP-K-H/TAP ratio >1.0) with distinguished peptide identity, peptide sequences ≥5, PSM ≥5,% coverage ≥5 were considered positive identifications. Identified proteins are summarized in Supplementary Table S1 (see Supplementary Data) with their UniProt accession numbers, protein description, PSMs, peptide sequence numbers,% coverage, spectral indices and enrichment ratios.
We then utilized the Genes Ontology (GO) enrichment analysis tool, DAVID, to further scrutinize and functionally annotate identified proteins. Enriched fourth level of biological process ontology (GOTERM_BP_4) categories with –log10(FDR) >5 were considered significant, and shown in Figure 1A. Proteins co-purified with TAP-K-H were functionally categorized in RNA, DNA and protein metabolism, including translation (Figure 1A, Supplementary Table S2). These analyses were consistent with TAP-MS screening data and further supported K-H's involvement in RNA and DNA metabolic processes. Next, we built an interaction network using proteins of most interest that were involved in RNA and/or DNA metabolism, satisfied the mass spectrometric selection criteria and were supported by bioinformatics analyses through DAVID (Figure 1B). Select proteins listed in Supplementary Table S3 were used as initial input and analyzed by STRING 9.1 (46). The layout in Figure 1B for protein–protein interactions was created by cytoscape 3.1.0 software via the link provided by STRING (47). The STRING tool utilizes protein–protein interaction prediction methods, including neighborhood, gene fusion, co-occurrence, co-expression, experimental data, databases and text mining to build an interaction network (47). The K-H protein is a part of a large protein–protein interaction network (Figure 1B). Each line connecting two given proteins (Figure 1B) represents a link between those two proteins via one of the above-mentioned methods. Multiple lines connecting two proteins signify several connections between them. These analyses strongly suggested that K-H was likely a part of several separately functioning protein complexes in the cell, including processes involved in RNA metabolism, DNA repair/replication, and were consistent with its simultaneous involvement in RNA transcription termination and DSB repair, as recently suggested (8).
Figure 1.

Screening and validation of novel binding partners of K-H. (A) Identification of potential K-H binding partners was performed using TAP-MS screening. Functional annotation of identified proteins was determined by gene ontology (GO) analyses using DAVID v6.7. Bar graph shows the GO terms plotted against negative logarithms to the base 10 of FDR. Functional categories with –log10(FDR) cutoff value of >5 and indicated p-values are shown. (B) Select proteins were used to build a complex network of proteins associating with K-H using STRING 9.1 database and Cytoscape 3.1.0 software as described in ‘Materials and Methods’. Each single line connecting individual proteins represents a connection between given proteins. Proteins represented by green, blue and yellow colors are involved in RNA metabolism, DNA repair and replication, respectively. (C) Whole cell lysates prepared from 293T cells transiently expressing mycK-H were used for co-IP analyses. These cells were simultaneously co-transfected with unique siRNA-k-h oligomers specific for the 3′-UTR sequence of k-h mRNA to knockdown endogenous K-H expression levels. Antibodies to mycK-H or normal mouse IgG were then used to pull-down proteins and then Western blot analyses were performed to detect specific proteins using respective antibodies. Lane 1 represents input for IP and lanes 2 and 3 represent IP by IgG and anti-myc antibodies, respectively. Note that co-IP data validates 10/12 tested interactions for proteins of most interest. (D) Nuclease-treated whole cell lysates were used to fractionate native protein complexes containing K-H using gel-filtration chromatography as described in ‘Materials and Methods’. Indicated proteins were detected by Western blot analyses. Note protein complexes containing K-H in the black box that are separate from RNA Pol II-containing K-H protein complexes.
Within the RNA metabolism category, we found several known protein partners of K-H, such as RNA polymerase II (RNA Pol II), p15RS and PSF (6,8). We also identified novel associating proteins, including RUVBL1, RUVBL2, TOP1, DHX9, DHX15 and DHX30. We discovered a series of novel DNA repair proteins specifically associated with the K-H protein. Within this group, Ku70 was among the known binding partners of K-H, where interaction between them was identified by yeast two-hybrid, co-IP and gel-filtration chromatography (8). Ku86, DNA-PKcs, PARP1, MSH2, RAD23B, SMC1A, SSBP1 and DDB1 were novel K-H binding partners that participate in various overlapping DNA repair processes. Identification of DNA-PKcs and Ku86 was consistent with prior identification of K-H associated with Ku70, suggesting that the entire DNA-PK complex is associated with K-H (8). Another group of proteins co-purified with TAP-K-H was classified as general DNA replication proteins, including PCNA, POLD1 and MCM complex, although many of these proteins (e.g. PCNA) are simultaneously involved in DNA repair. We also found proteins involved in protein metabolic processes in TAP-K-H pull-downs. For all proteins identified above as K-H-associated partners, multiple unique peptides contributed to their identification, suggesting that these interactions were likely specific. Together, mass spectrometric and bioinformatics analyses of proteins co-purified with TAP-K-H revealed numerous associated protein partners that were previously unknown.
To validate interactions of K-H with proteins identified above (Supplementary Table S1, Figure 1A and B), we employed co-IP and gel-filtration chromatography assays. For co-IP experiments, we used an anti-myc antibody to pull-down mycK-H and associated proteins. Anti-IgG and empty vector pull-downs were included as negative controls and final pull-downs, along with corresponding input extracts, were analyzed by Western blotting (Figure 1C and Supplementary Figure S2A–B). Known protein partners of K-H were used as positive controls (6,8), including RNA Pol II and Ku70 (Figure 1C, lane 3). A majority of the current literature points to K-H's involvement in transcription and DNA repair. Given the large number of interactions identified, we focused on validating proteins of most interest related to RNA and DNA metabolic processes, including RNA Pol II, Ku70, p15RS, TOP1, PARP1, MSH2, SMC1, SSBP1, Ku86, DNA-PKcs, DDB1, RAD23B and PCNA. We tested 12 proteins (shown in Figure 1B), and aside from DDB1 and RAD23B (likely due to antibodies interfering with co-IP), we were able to confirm 10 proteins that co-immunoprecipitated with K-H (Figure 1C), represent >80% confirmation.
To further investigate protein complexes (in vivo) containing K-H (without forced over-expression) and to validate our TAP-K-H purification and co-IP data, gel-filtration chromatography was employed as described (8). HeLa cell lysates treated with nuclease (to eliminate nucleic acid) were fractionated by gel-filtration and individual fractions subjected to Western blot analyses (Figure 1D). The elution patterns of K-H protein and its known associating proteins were confirmed (Figure 1D) and similar higher-order complexes were previously noted (8). The molecular weight (MW) of monomeric K-H was ∼37 kDa, consistent with its calculated molecular weight. The presence of K-H protein in fractions (#16–36) was consistent with the notion that this particular protein is part of multiple higher-order protein complexes, ranging from a MW of 2 MDa to as low as 43 kDa (Figure 1D). Fractions containing major peaks of novel K-H protein partners, TOP1 (#24–29), PARP1 (#22–30), MSH2 (#22–28) and PCNA (#28–31) showed substantial overlap with K-H, which peaked around a MW of 158 kDa (black box in Figure 1D). While the gel-filtration data may not provide the exact nature of subunit composition of various novel complexes involving K-H, these data represent an additional validation of specific protein complexes containing K-H and a number of novel protein binding partners. Collectively, our TAP-K-H purification data, bioinformatics analyses, co-IP and gel-filtration data provide multiple corroborating evidence for the novel association of K-H with specific proteins involved in RNA metabolism, DNA repair and replication (Figure 1).
Depletion of K-H protein leads to DNA damage tolerance from 6-TG cytotoxicity
Association of K-H with the MMR protein, MSH2, was particularly interesting and suggested a potential involvement of K-H in regulating MMR function. MSH2 interacts with MSH6 to form the MutSα complex that recognizes nucleotide mismatches (21), and recruits the MutLα complex (MLH1-PMS2) at sites of mismatched DNA for repair processing and G2/M and cell death signaling. To explore possible links between K-H and MMR, we tested the sensitivities of stable K-H knockdown cells to 6-thioguanine (6-TG), which creates 6-TG:T mismatched moieties in DNA after replication (51). MMR competent cells recognize incorporated 6-TG:T moieties in DNA to initiate G2/M cell cycle checkpoint arrest and cell death (apoptosis) signaling responses (52). In contrast, MMR-deficient cells show ‘tolerance’ to this damage and continue to grow (52). Stable shScr and shk-h 231 knockdown cells, as well as genetically matched RKO6 (MMR-deficient) and RKO7 (hMLH1 reconstituted, MMR-proficient) colon cancer cells developed by us (17) were exposed to 6-TG as described (16–18). Stable shk-h knockdown cells showed dramatic tolerance and surviving fractions to 6-TG compared to the much more sensitive MMR+ control shScr 231 cells, which showed significant lethality from ≥0.5 μM 6-TG exposure (Figure 2A). Damage tolerance in shk-h knockdown cells was very similar to that found in hMLH1- RKO6 cells, whereas K-H expressing shScr control 231 cells were sensitive to 6-TG, similar to MMR-proficient (hMLH1+) RKO7 cells (Figure 2A). MMR-, as well as K-H depleted, cells show specific tolerance to 6-TG compared to the higher sensitivities for other DNA damaging agents, such as IR (Table 1 (8,18)). Similarly, MEFs heterozygous for the k-h gene (mk-h+/− cells) showed significant damage tolerance to 6-TG compared to hypersensitive mk-h+/+ cells isolated from wild-type (WT) littermate mice at ≥0.2 μM 6-TG (Figure 2B). These data strongly suggested that loss of K-H protein expression in human or mouse cells (in fact, one allelic loss) resulted in DNA damage tolerance to 6-TG, in a similar manner to that found in MMR-deficient human cells (Figure 2A–B).
Figure 2.
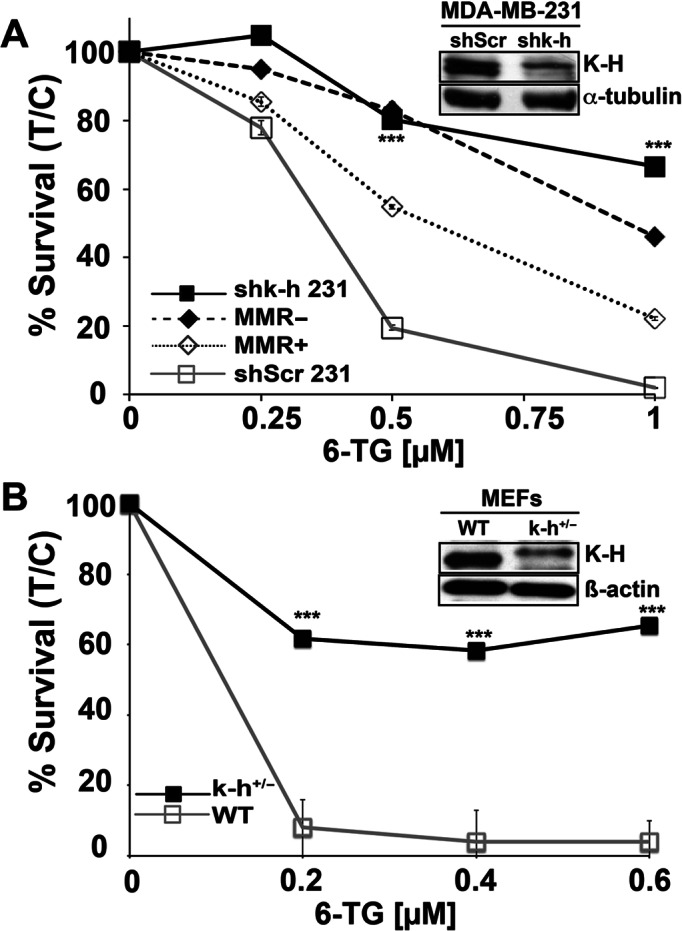
K-H knockdown cells exhibit DNA damage tolerance following 6-TG exposure. (A) shScr or shk-h knockdown 231 TNBC cells and MMR-deficient or MMR-proficient colon cancer cells (as controls) were seeded onto 48-well plates. 6-TG (μM) was added ∼12 h later and cells were exposed for 48 h. After drug removal, cells were grown for 7 days. Relative survival, measured by DNA content, was monitored using Hoechst 33258 dye staining and a Victor X3 plate reader (48). Data were expressed as means, ±SD for treated/control (T/C) cells from experiments performed at least 3 times in triplicate. P-values were obtained via two-tailed Student's t-tests. ***P < 0.001, comparing shScr versus shk-h. K-H and α-tubulin (loading) levels were monitored to compare K-H expression knockdown in shScr versus shk-h cells. (B) Mouse embryonic fibroblasts (MEFs) derived from WT (mk-h+/+) or mk-h+/− heterozygous mice (8) were used to measure sensitivities to 6-TG exposures. Relative cell survival assays were then performed as in Figure 2A. K-H protein levels in MEFs were analyzed by Western blotting using an antibody against human K-H protein. β-Actin was used for loading. K-H levels in these cells were previously confirmed using specific antibodies (8). Antibodies against human K-H also work against mouse K-H due to the conserved nature of the protein (8).
Table 1. K-H knockdown cells show specific DNA damage tolerance to 6-TG treatments.
| Cell Genotype | MSI Status | ∼LD50 valuesb | Reference | |
|---|---|---|---|---|
| IR (Gy) | 6-TG (μM, 48 h) | |||
| shScr 231a | - | 4.0 | 0.3 | (8) and this study |
| shk-h 231a | + | 2.8 | 1.2 | (8) and this study |
| MMR+ HCT116 3–6 | - | 4.0 | 3.0 | (18) |
| MMR- HCT116 | + | 3.8 | 30 | (18) |
a231, Human MDA-MB-231 triple-negative breast cancer cells.
bLD50, dose of agent leading to ∼50% kill.
A comparison of ∼LD50 values after 6-TG or IR treatment of shScr versus shk-h MDA-MB-231 (231) cells. Also included are sensitivity data against 6-TG and IR for known MMR-proficient (MMR+) HCT116 3–6 cells and -deficient (MMR-) HCT116 cells for comparison. Although, genetic background and treatment conditions differ between shScr/shk-h versus MMR-proficient/-deficient cells, the data highlight the unique phenotype of K-H-depleted cells, where they are simultaneously hypersensitive to IR, yet damage-tolerant to 6-TG.
K-H depletion results in deficient G2/M cell cycle checkpoint arrest responses, similar to MMR-deficient cells
MMR-competent cells are capable of recognizing 6-TG:T incorporated in their DNA and activating G2 cell cycle checkpoint arrest responses and cell death after continuous 6-TG exposures (53). We investigated the competence of G2/M cell cycle checkpoint arrest responses in stable shk-h knockdown compared to shScr 231 cells after 6-TG treatment using 5-ethynyl-2′-deoxyuridine (EdU) incorporation assays at 24–72 h exposure as described (54). Upon 6-TG treatment, shScr 231 cells underwent profound G2- cell cycle checkpoint arrest responses after 48 h, whereas genetically matched stable shk-h knockdown 231 cells failed to trigger G2-arrest responses (compare shScr and shk-h to respective mock-treated cells in Figure 3A, B and Supplementary Figure S3 for quantification of G2/M checkpoint arrest responses over time). Similar defective G2/M checkpoint arrest responses were reported in MMR-deficient cells exposed to 6-TG compared to genetically matched MMR-competent cells (16–18). Thus, loss of K-H protein expression mimics DNA damage tolerance, defective G2/M checkpoint arrest activation and lethality after 6-TG exposure in a manner similar to that noted in MMR-compromised cells (16–18).
Figure 3.

K-H depletion results in defective G2 cell cycle checkpoint arrest responses, similar to MMR-deficient cells in response to 6-TG exposure. Stable shScr or shk-h 231 cells were exposed to 0.5 μM 6-TG for 24, 48 or 72 h and analyzed by flow cytometry for changes in cell cycle distributions. (A) Cell populations were gated for G1 (dark blue), S-phase (active EdU incorporation, S+ (orange)) and G2/M (light blue) phases of the cell cycle (represented in top panel) based on PI staining and EdU incorporation. Flow diagrams with cell counts are represented with similar color scheme in the bottom panel. Numbers shown on top of G2 peak represent mean, ±SD from at-least three experiments and asterisk (*) indicates P ≤ 0.05 calculated for shScr versus shk-h. Representative images shown in panel A are for 48 h time point where substantial changes in G2 population of mock versus 6-TG treated shScr cells started to appear. (B) Quantification of cell cycle phase distributions of shScr (left) or shk-h (right) 231 cells after mock (open bars) or 6-TG exposure (closed bars) for 72 h are shown (where most significant changes occurred in mock versus 6-TG treated shScr G2 population) for cells treated as in ‘A’. Shown are means, ±SD from experiments performed at-least three times and P-values determined using two-tailed Student's t-tests. Complete time-course data quantifications are presented in Supplementary Figure S3. ***P ≤ 0.001.
K-H knockdown leads to alteration in global microsatellite stability
A hallmark of MMR-deficient cells is that they exhibit alterations in microsatellite loci, reflecting a ‘mutator’ phenotype (55–61). Using an aCGH-like array, analyses of K-H-proficient versus -depleted fibroblasts cells indicated that there was a significant effect on global microsatellite content stability after K-H depletion. There were 15 motif families (Figure 4) that had increased greater than 1.5-fold, with a P < 0.022 (Fisher's Exact test (two-tailed)). There were 12 motif families (Figure 4) that had decreased in content by 0.5, with a P < 0.0001 (Fisher's Exact test (two-tailed)). Most notably, those repetitive motif families that substantially increased in overall global content consisted mainly of higher GC content probes, while those motif families that significantly decreased in overall genomic content had low GC content, indicating that a MMR deficiency exhibits a GT/AT bias. The microsatellite array analyses that we used is a complement to the Promega microsatellite instability test kit. While the Promega kit measures discrete changes in five homopolymer-A loci, this technology measures all microsatellite loci, but is limited to the summed effect of all loci of a given motif. Importantly, these results strongly support the notion that K-H functions to maintain the stability of microsatellite regions within the genome. Moreover, these data also support a role for K-H depletion in the genesis of MMR-deficiency, and a ‘mutator’ genotype with compromised microsatellite stability.
Figure 4.

Global microsatellite content alteration in K-H-deficient cells. Each dot represents the average of the 7 copies of the 5356 probes. Those probes representing a microsatellite motif family found to be statistically and reproducibly with a fold change of at least 1.5 above 0.5 below are marked in color and P-values (determined by Fisher's Exact test, two-tailed) were <0.022 and <0.0001, respectively. Note that for the K-H-deficient cells, the overall microsatellite content indicates that higher GC-content motifs (15 families) are increased, while low GC-content motifs (12 families) are decreased.
K-H knockdown causes caspase-dependent loss of specific MMR proteins
Previously, we demonstrated that loss of K-H expression influenced the stability and steady-state levels of Artemis protein, even though mRNA levels remained unchanged (8). We subsequently discovered that K-H protein expression was required for Artemis stabilization by protein–protein interactions (8). We, therefore, examined the steady-state protein expression levels of novel K-H-associating proteins in stable shScr versus shk-h knockdown 231 cells. Whole cell lysates were prepared from log-phase cells and levels of different proteins (indicated in Figure 5A) were assessed by Western blot analyses. In contrast, the steady state levels of TOP1, PARP1, MSH2 and PCNA in shScr or shk-h knockdown 231 cells remained unchanged (Figure 5A). These data strongly suggested that knockdown of K-H protein did not affect the stability of TOP1, PARP1, MSH2 or PCNA proteins that are required for a number of important intracellular processes in DNA repair and replication, including MMR (i.e. MSH2). More than 90% of cancers showing MMR deficiencies are the result of losses of MLH1 or MSH2 protein expression (62). However, K-H knockdown cells show equal steady state expression of MSH2 compared to shScr 231 cells (Figure 5A). These data strongly suggest that MSH2 alterations are not the likely mechanism whereby K-H loss induces MMR deficiency, even though K-H associates with MSH2.
Figure 5.

K-H depletion leads to reduced stability of MLH1 and PMS2. (A) Whole cell lysates from shk-h knockdown or shScr 231 cells were used to compare expression of specific proteins by Western blotting as mentioned in ‘Materials and Methods’. (B) MMR protein levels were analyzed in WT mk-h+/+ (lane 1) and k-h heterozygous (mk-h+/−) cells (lane 2) by Western blot analyses. (C) mRNA expression of the specific MMR genes were measured in shk-h knockdown or shScr cells as described in (A). (D) Levels of specific MMR proteins were analyzed by Western blotting using cell lysates from shScr 231 cells transiently expressing siRNA-specific for nonsense (Scr, scramble) or 3′-UTR-specific k-h mRNA sequences (20 nM for 48 h). (E) Activated (cleaved) caspase-3 was detected by Western blot analyses using cell lysates from shScr 231 cells transiently expressing siRNAs specific for Scr (lane 1) or k-h (lanes 2–4) as described above for indicated time points. Flow cytometry-based TUNEL assays were performed to detect apoptotic cells (TUNEL+, subG0/G1) in siScr versus sik-h knockdown 231 cells. (F) MLH1 and PMS2 protein levels were rescued by addition of the pan-caspase inhibitor, z-VAD-fmk. Cells were treated as in ‘E’ in the presence or absence of z-VAD-fmk (50 μM, 72 h) and monitored for MLH1 and PMS2 steady-state levels by Western blot analyses. Lanes 1, 2 and 3 represent cells treated with siRNAs specific to nonsense (Scrambled, Scr), 3′-UTR k-h or 3′-UTR k-h + z-VAD-fmk, respectively. Loading controls for all Western blots were the same as described in Figure 2.
Deregulation of MLH1 protein expression can concomitantly lead to depletion of PMS2 proteins due to loss of protein–protein interactions (63). Although rare, loss of MSH6 can also occur in patients, causing a MMR deficiency (62). Therefore, we examined whether depletion of K-H influenced the steady-state levels of core MLH1, PMS2 or MSH6 MMR proteins. Intriguingly, the stability of both MLH1 and PMS2 proteins were substantially reduced (∼50% and ∼90%, respectively) in stable shk-h knockdown compared to shScr 231 cells (Figure 5A, compare lanes 1 and 2). In contrast, MSH6 levels remained unchanged. We also tested the steady-state levels of MMR proteins in WT (mk-h+/+) compared to mk-h+/- heterozygous MEFs, since these cells also showed DNA damage tolerance to 6-TG exposures (Figure 2B). Consistent with shk-h knockdown 231 cells, we observed significantly lowered (>90%) steady-state levels of MLH1 and PMS2 proteins in mk-h+/- heterozygous versus WT MEFs (Figure 5B, compare lanes 1 and 2). To test whether losses of MLH1 and PMS2 were due to mRNA level changes or to reduced protein stability, we performed quantitative RT-PCR to simultaneously monitor MLH1 and PMS2 mRNA levels. No significant differences were noted in MLH1 or PMS2 mRNA levels in shk-h versus shScr 231 cells (Figure 5C). Similar trends were observed after transient siRNA K-H knockdown in 231 cells (compare lanes 1 to 2, Figure 5D). Together, these data strongly suggest that depletion of K-H protein by sh/siRNA-specific knockdown or by one copy number loss, led to a significant reduction in the overall stability of both MLH1 and PMS2 proteins, resulting in MMR defects.
The MLH1 protein harbors a cleavage site susceptible to proteolysis by caspase-3, and its steady-state levels can be regulated by activated caspase-3 in human cells (64). We therefore investigated whether genetic instability brought about by K-H depletion could cause a general leakiness of caspase activity, leading to degradation of MLH1 in K-H knockdown compared to shScr 231 cells. Western blot analyses for cleaved caspase-3 (activated form, ∼17 kDa) in k-h knockdown 231 cells reveal significant basal level activated caspase-3 in K-H-deficient versus Scr 231 cells (Figure 5E). Since caspase-3 activation could lead to cell death via cellular apoptotic responses (65), we assessed both cells for apoptotic cell populations using TUNEL assays. While we did note an increase in apoptosis in stable shk-h knockdown compared to Scr 231 cells (Figure 5E), <2% of the total cell populations were apoptotic in either growing cell population. These data strongly suggest a basal level increase in activity rather than a caspase-dependent cell death response, consistent with the notion that caspase activation does not always lead to cell death (66). Importantly, inhibition of caspase activity using the small molecule pan-caspase inhibitor, z-VAD-fmk, restored both MLH1 and PMS2 steady-state protein levels in K-H knockdown cells to levels comparable to genetically matched Scr 231 cells (compare lanes 1 and 3 to 2 and 3, Figure 5F). Collectively, these data suggest that low basal-level increases in caspase activation (presumably caspase-3) in K-H knockdown cells likely contributed to MLH1, and consequently PMS2, degradation and steady state protein losses. This increased genetic instability does not appear to be sufficient enough to trigger apoptotic cell death.
DISCUSSION
We initiated the first steps of establishing a detailed interactome of the human Kub5-Hera (K-H)/RPRD1B scaffolding protein to delineate its multiple potential roles in various cellular functions. Using a TAP-tag strategy in conjunction with mass spectrometric and various bioinformatics analyses (including DAVID and STRING), we identified several classes of novel proteins associated with K-H and involved in (i) RNA metabolism, including transcriptional termination and processing events; (ii) DNA metabolism, including DNA repair and replication; and (iii) protein metabolism, including translation. We demonstrated >80% (10/12) co-IP validation of proposed K-H-associated proteins identified under the ‘DNA repair category’, strongly suggesting that the interactome in Figure 1 has established a platform for further hypothesis exploration. Globally, data appear to support a more comprehensive role of K-H in various essential processes in the cell (Figure 1), potentially shedding light on why a mouse does not tolerate K-H gene loss, resulting in embryonic lethality (8).
The current literature supports concomitant roles for K-H in RNA and DNA metabolic processes, with its roles in DNA repair fairly unexplored (8). We, therefore, focused on these two categories. The presence, and validation, of known associating proteins, including RNA Pol II and Ku70, in TAP-K-H pull-down assays shows that the method we employed was effective and valid to define the K-H interactome (Figure 1). The interactions we described are likely protein–protein interactions (albeit not necessarily direct), and not simply bridged by DNA, since we used several different strategies to eliminate any DNA or RNA (see Materials and Methods and Supplementary Data, Figure S2). There were, however, several proteins that we expected to associate with K-H that were not observed. For example, we failed to identify the interaction of K-H with RPAP2, RECQL5, GRINL1A, XRN2, Artemis and BRCA1 (6,8,67–68), even though we previously co-immunoprecipitated some of these proteins from nuclear cell extracts (8). Here, we employed stringent solution conditions during the TAP-K-H purification that could be responsible for disruption of interactions of these proteins with K-H or interactions that have DNA or RNA dependencies. This is particularly true for RPAP2, RECQL5 and GRINL1A, since we were able to detect them in TAP-K-H pull-down under more mild stringency conditions (data not shown).
Below, we discuss the potential involvement of K-H in each of the aforementioned cellular functions defined by the different categories of proteins of most interest (Supplementary Table S3) and indicated in the STRING-generated ‘interactome’ (Figure 1). Certainly, substantially more detailed mechanistic work will be required to delineate the involvement of K-H in each of these cellular processes, including the use of recombinant proteins to detect direct protein–protein interactions.
RNA processing and transcription termination
The existing literature on the functional role(s) of K-H in cells is primarily focused its involvement in transcription, mostly in transcription termination. Data in this paper appear to further confirm K-H's role in transcription termination events by identifying known, as well as novel associating partners that included: RNA Pol II, p15RS (RPRD1A), TOP1, PSF and RNA helicases (Figure 1, Supplementary Table S3). These findings highlight the conserved nature of K-H/RTT103 (yeast homolog of K-H), since in yeast the interaction of RTT103 with RNA Pol II is well established (11). In humans, the interaction of K-H (RPRD1B or CREPT) protein with RNA Pol II was also reported (6–7,68) and further confirmed by our laboratory (Figure 1 and (8)). The interaction of K-H with TOP1 could be direct or mediated through other proteins, such as RNA Pol II and PSF. PSF can form a higher-order complex with TOP1 (69). Notably, PSF binds directly to TOP1 and stimulates its DNA unwinding activity that relieves supercoiling during transcription (69). Several studies demonstrated that TOP1 is capable of binding to the CTD of RNA Pol II (70–72). Thus, it is possible that K-H may function together with TOP1, via protein-protein interactions, to regulate both DNA repair and transcription.
DNA replication
All three experimental approaches described in this study show strong and confirmatory evidence of K-H's association with proliferative cell nuclear antigen (PCNA) in higher-order protein complexes (Figure 1, Supplementary Tables S1–S3), suggesting roles for K-H in DNA damage responses, DSB repair (discussed below) and DNA replication. Of note is K-H's interaction with the majority of the minichromosome maintenance protein complex (MCM2–7 proteins, Figure 1, Supplementary Table S1–S3) involved in initiation and elongation steps of eukaryotic DNA replication, DNA polymerase delta (POLD1) and PCNA. These interactions raise the possibility of K-H's involvement in DNA replication, where the protein may play role(s) in replication fork movement. Indeed, in shk-h knockdown (stable or transient) cells, replication stress (presumably due to replication fork stalling) caused by increased R-loops and DSBs was noted (8). K-H depletion may affect the replication machinery by impeding recruitment of PCNA, as well as TLS polymerases (50). Thus, loss of K-H expression, its downstream loss of MMR, along with greatly enhanced R-loop and DSB formation (8), may result in elevated replication fork stalling. These results further support a role for K-H depletion in cancers as a strong mutator.
DSB repair
Recently our laboratory reported Ku as a novel binding partner of K-H, forming higher-order complexes and confirming an involvement of K-H in DSB repair (8). Previous studies have shown an association between Ku70 and PARP1 (73). Thus, the interaction of K-H with PARP1 could be facilitated through its direct interaction with Ku70. Alternatively, K-H and PARP1 may interact directly to regulate each other's function during DSB repair.
DNA mismatch repair (MMR)
Our studies clearly highlight several links between K-H protein expression loss and defective MMR. Cells deficient in K-H (by si/shRNA-specific knockdown or allelic loss in mouse cells) exhibited DNA damage tolerance and resistant to 6-TG (Figure 2), a functional loss in activation of the G2 cell cycle checkpoint arrest response during 6-TG exposure (Figure 3), compromised microsatellite stability (Figure 4), and concomitant losses of MLH1 and PMS2 (Figure 5). MLH1 is essential for the stability PMS2 (63), and only MLH1 contains a conserved caspase cleavage site, rendering the protein susceptible to caspase-3-dependent degradation (64). Since we demonstrated that MLH1 loss in K-H-deficient cells can be rescued by the pan-caspase inhibitor, z-VAD-fmk (Figure 5F), we assume that low level constitutively-activated caspase activity (presumably caspase-3) is present in cells deficient in K-H, but not active in wild-type cells. Although indirect, data from a recent study seems to support elevated caspase-3 activation in cells down regulated for the Rprd1b (k-h) gene by MiR-1188 (74). Intriguingly, caspase activation required for MLH1 degradation was clearly not sufficient to elicit apoptosis, since <2% of the total cell population was TUNEL+ (Figure 5E). Several studies have suggested a role for non-apoptotic functions of caspases, where activated caspases also do not elicit cell death (66). Thus far, only a limited number of substrates for activated caspase-3 were identified (64), and these are likely to be entirely different than those used under apoptotic conditions. In current study, among the various proteins we tested (Figure 5A), MLH1 seems to be preferred target of caspases-3 under genetically unstable conditions.
We speculate that the source of activated caspase in cells deficient in K-H comes from their inherent genetic instability: increased R-loops and DSBs as recently reported (8). Our proteomics data raised two potential links between K-H and alterations in MMR. K-H formed higher order complexes with MSH2 and Ku. Currently, there is no evidence in our data or in the literature that would support a role for MSH2 functional loss in cells depleted in K-H expression. MSH2 steady state protein levels remain unaltered in K-H-deficient versus WT counterparts. Instead, specific losses in MLH1 and PMS2 were noted and were restored by z-VAD-fmk treatments (Figure 5). We speculate that the K-H-Ku70/86 complex (noted by gel-filtration and co-IPs, Figure 1) binds and regulates a repressor element within the Apaf1 promoter (Supplementary Figure S4), normally suppressing its expression; when stimulated, Apaf1 can activate downstream caspase-3 (75). When K-H is depleted, K-H-Ku-dependent promoter suppression is relieved and Apaf1 expression becomes leaky, which stimulates caspase-3 activity resulting in downstream MLH1 degradation. This notion is supported by several observations, including: (i) K-H associates with Ku (as validated by yeast-two-hybrid screening, co-IP, gel-filtration and TAP-MS analyses ((8) and this study), (ii) K-H physically interacts with Ku (Supplementary Figure S4A–B), (iii) Ku70/86 controls Apaf1 expression (75) and (iv) K-H loss activates caspase-3 (Figure 5). Obviously, further investigation is needed to establish, a link between K-H depletion, Apaf1 expression and caspase-3-induced MLH1 degradation, and the significance of K-H-MSH2 association.
To our knowledge this is the first report describing changes in a transcription termination factor that causes (by indirectly altering caspase activity) the steady-state reduction in MMR core proteins (i.e. MLH1 and PMS2). Gains, as well as losses, of k-h gene copy number and down-regulation of its mRNA are frequent in colon/colorectal cancers (Supplementary Figure S5, panels A–C and D–E, respectively). Similar reports were found for ovarian and endometrial cancers from the Oncomine (http://www.oncomine.org) depository (76–80). Provided that the apoptotic function in these cancers are intact (most likely caspase-3), K-H protein depletion may serve as a prognostic marker to define MSI+ sporadic cancers exhibiting normal MMR genetics and mRNA expression. Furthermore, gains in k-h gene copy number and mRNA over-expression could serve as prognostic markers for radio- and/or chemo-resistance in cancers. We previously showed that yeast Rtt103-/-, human Kub5-Hera (K-H) knockdown or k-h+/− MEF cells were uniformly hypersensitive to various DSB-inducing agents, such as IR, and TOP I and II poisons (8). This is in stark contrast to our current study, where we provide clear evidence that these same human or mouse K-H-deficient cells exhibit DNA damage tolerance against 6-TG (Figure 2, Table 1), a characteristic found fairly exclusively in MMR-deficient cells (18). These findings offer important insights into potential treatments of patients that might have K-H copy number losses or SNPs that compromise K-H function. Such patients would be hypersensitive to DSB-inducing agents, yet resistant to MMR-mediated damage tolerant-inducing agents, such as temozolomide or 5-fluorouracil (81,82).
In summary, we provide a foundation of the K-H scaffold protein interactome using a combination of proteomics (TAP-MS), bioinformatics (DAVID and STRING) and biochemical (co-IP and gel-filtration chromatography) approaches. The number of proteins co-purified with TAP-K-H was very large which is common for TAP-MS analyses (34,35). The complex mixture of proteins co-purified with TAP-K-H is likely to contain direct, as well as indirect, associating proteins. It is imperative to note that the majority of novel interactions presented here required further validation by at-least one alternative method and establishing the biological significance of these novel interaction through in-depth investigation is warranted. However, we were able to validate 10 of 12 (i.e. >80%) tested associations, and we provide evidence for a direct interaction between K-H and Ku70. Overall, the K-H interactome suggests potential involvement of K-H-containing complexes in RNA processing (particularly in transcription termination), various DNA repair pathways (DSB repair and MMR), DNA replication and protein metabolism including translation. Collectively, our findings illustrate the hypomorphic haploinsufficient nature of a deficiency in K-H steady state protein levels, causing a multifaceted phenotype that could clearly contribute to a ‘mutator phenotype’ in cancers at many levels.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are very grateful to help from Dr Wu and the laboratories of Dr Cheng-Ming Chiang, Dr David Chen, Dr Hongtao Yu and Proteomics Core at UT Southwestern Medical Center for providing reagents and instrument support.
Author contribution: P.L.P and D.A.B. designed all experiments, interpreted all data and wrote the manuscript. P.L.P. collected all data. E.A.M. provided technical support on flow cytometry, repeated key experiments and generated TUNEL data, F.J.F. cloned TAP and TAP-K-H constructs, generated stable cell lines harboring these constructs and generated RT-PCR data. Y.Z. and Y.X. performed bioinformatics analyses. J.C.M. generated MEFs and AT cells and H.R.G generated microsatellite data.
FUNDING
National Institutes of Health/National Cancer Institute (NCI/NIH) [R01 CA139217 to D.A.B.]; NCI/NIH [5P30CA142543 to the UT Southwestern Proteomics Core]; NCI/NIH [UT Southwestern CCSG grant 5P30CA142543]; a minority supplement [R01 CA139217–05S1 to E.A.M.]; Cancer Biology Training Grant [T32CA124334–06 (PI: Dr Jerry Shay) to E.A.M.]; Simmons Comprehensive Cancer Center; Cancer Prevention and Research Institute of Texas [CPRIT, RP1206130 to Dr Hamid Mirzaei and UT Southwestern Proteomics Core, in part]. Funding for open access charge: National Institutes of Health/National Cancer Institute (NIH/NCI) [R01 CA139217].
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhou B.B., Elledge S.J. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433–439. doi: 10.1038/35044005. [DOI] [PubMed] [Google Scholar]
- 2.Iyama T., Wilson D.M. 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013;12:620–636. doi: 10.1016/j.dnarep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Negrini S., Gorgoulis V.G., Halazonetis T.D. Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 4.Aguilera A., Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115–124. doi: 10.1016/j.molcel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 5.Skourti-Stathaki K., Proudfoot N.J., Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011;42:794–805. doi: 10.1016/j.molcel.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ni Z., Olsen J.B., Guo X., Zhong G., Ruan E.D., Marcon E., Young P., Guo H., Li J., Moffat J., et al. Control of the RNA polymerase II phosphorylation state in promoter regions by CTD interaction domain-containing proteins RPRD1A and RPRD1B. Transcription. 2011;2:237–242. doi: 10.4161/trns.2.5.17803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu D., Wu Y., Wang Y., Ren F., Wang D., Su F., Zhang Y., Yang X., Jin G., Hao X., et al. CREPT accelerates tumorigenesis by regulating the transcription of cell-cycle-related genes. Cancer Cell. 2012;21:92–104. doi: 10.1016/j.ccr.2011.12.016. [DOI] [PubMed] [Google Scholar]
- 8.Morales J.C., Richard P., Rommel A., Fattah F.J., Motea E.A., Patidar P.L., Xiao L., Leskov K., Wu S.Y., Hittelman W.N., et al. Kub5-Hera, the human Rtt103 homolog, plays dual functional roles in transcription termination and DNA repair. Nucleic Acids Res. 2014;42:4996–5006. doi: 10.1093/nar/gku160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scholes D.T., Banerjee M., Bowen B., Curcio M.J. Multiple regulators of Ty1 transposition in Saccharomyces cerevisiae have conserved roles in genome maintenance. Genetics. 2001;159:1449–1465. doi: 10.1093/genetics/159.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong A.H., Evangelista M., Parsons A.B., Xu H., Bader G.D., Page N., Robinson M., Raghibizadeh S., Hogue C.W., Bussey H., et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- 11.Kim M., Krogan N.J., Vasiljeva L., Rando O.J., Nedea E., Greenblatt J.F., Buratowski S. The yeast Rat1 exonuclease promotes transcription termination by RNA polymerase II. Nature. 2004;432:517–522. doi: 10.1038/nature03041. [DOI] [PubMed] [Google Scholar]
- 12.Srividya I., Tirupataiah S., Mishra K. Yeast transcription termination factor Rtt103 functions in DNA damage response. PLoS One. 2012;7:e31288. doi: 10.1371/journal.pone.0031288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waples W.G., Chahwan C., Ciechonska M., Lavoie B.D. Putting the brake on FEAR: Tof2 promotes the biphasic release of Cdc14 phosphatase during mitotic exit. Mol. Biol. Cell. 2009;20:245–255. doi: 10.1091/mbc.E08-08-0879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suter B., Tong A., Chang M., Yu L., Brown G.W., Boone C., Rine J. The origin recognition complex links replication, sister chromatid cohesion and transcriptional silencing in Saccharomyces cerevisiae. Genetics. 2004;167:579–591. doi: 10.1534/genetics.103.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 16.Meyers M., Wagner M.W., Hwang H.S., Kinsella T.J., Boothman D.A. Role of the hMLH1 DNA mismatch repair protein in fluoropyrimidine-mediated cell death and cell cycle responses. Cancer Res. 2001;61:5193–5201. [PubMed] [Google Scholar]
- 17.Wagner M.W., Li L.S., Morales J.C., Galindo C.L., Garner H.R., Bornmann W.G., Boothman D.A. Role of c-Abl kinase in DNA mismatch repair-dependent G2 cell cycle checkpoint arrest responses. J. Biol. Chem. 2008;283:21382–21393. doi: 10.1074/jbc.M709953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis T.W., Wilson-Van Patten C., Meyers M., Kunugi K.A., Cuthill S., Reznikoff C., Garces C., Boland C.R., Kinsella T.J., Fishel R., et al. Defective expression of the DNA mismatch repair protein, MLH1, alters G2-M cell cycle checkpoint arrest following ionizing radiation. Cancer Res. 1998;58:767–778. [PubMed] [Google Scholar]
- 19.Li L.S., Morales J.C., Hwang A., Wagner M.W., Boothman D.A. DNA mismatch repair-dependent activation of c-Abl/p73alpha/GADD45alpha-mediated apoptosis. J. Biol. Chem. 2008;283:21394–21403. doi: 10.1074/jbc.M709954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Genschel J., Littman S.J., Drummond J.T., Modrich P. Isolation of MutSbeta from human cells and comparison of the mismatch repair specificities of MutSbeta and MutSalpha. J. Biol. Chem. 1998;273:19895–19901. doi: 10.1074/jbc.273.31.19895. [DOI] [PubMed] [Google Scholar]
- 21.Gradia S., Acharya S., Fishel R. The human mismatch recognition complex hMSH2-hMSH6 functions as a novel molecular switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 22.Kadyrov F.A., Dzantiev L., Constantin N., Modrich P. Endonucleolytic function of MutLalpha in human mismatch repair. Cell. 2006;126:297–308. doi: 10.1016/j.cell.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 23.Wang Y., Cortez D., Yazdi P., Neff N., Elledge S.J., Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000;14:927–939. [PMC free article] [PubMed] [Google Scholar]
- 24.Cannavo E., Gerrits B., Marra G., Schlapbach R., Jiricny J. Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 2007;282:2976–2986. doi: 10.1074/jbc.M609989200. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y., Rohde L.H., Wu H. Involvement of nucleotide excision and mismatch repair mechanisms in double strand break repair. Curr. Genomics. 2009;10:250–258. doi: 10.2174/138920209788488544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Elliott B., Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol. Cell Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Villemure J.F., Abaji C., Cousineau I., Belmaaza A. MSH2-deficient human cells exhibit a defect in the accurate termination of homology-directed repair of DNA double-strand breaks. Cancer Res. 2003;63:3334–3339. [PubMed] [Google Scholar]
- 28.Pichierri P., Franchitto A., Piergentili R., Colussi C., Palitti F. Hypersensitivity to camptothecin in MSH2 deficient cells is correlated with a role for MSH2 protein in recombinational repair. Carcinogenesis. 2001;22:1781–1787. doi: 10.1093/carcin/22.11.1781. [DOI] [PubMed] [Google Scholar]
- 29.Franchitto A., Pichierri P., Piergentili R., Crescenzi M., Bignami M., Palitti F. The mammalian mismatch repair protein MSH2 is required for correct MRE11 and RAD51 relocalization and for efficient cell cycle arrest induced by ionizing radiation in G2 phase. Oncogene. 2003;22:2110–2120. doi: 10.1038/sj.onc.1206254. [DOI] [PubMed] [Google Scholar]
- 30.Hong Z., Jiang J., Hashiguchi K., Hoshi M., Lan L., Yasui A. Recruitment of mismatch repair proteins to the site of DNA damage in human cells. J. Cell Sci. 2008;121:3146–3154. doi: 10.1242/jcs.026393. [DOI] [PubMed] [Google Scholar]
- 31.Bannister L.A., Waldman B.C., Waldman A.S. Modulation of error-prone double-strand break repair in mammalian chromosomes by DNA mismatch repair protein Mlh1. DNA Repair (Amst) 2004;3:465–474. doi: 10.1016/j.dnarep.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Smith J.A., Waldman B.C., Waldman A.S. A role for DNA mismatch repair protein Msh2 in error-prone double-strand-break repair in mammalian chromosomes. Genetics. 2005;170:355–363. doi: 10.1534/genetics.104.039362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koh K.H., Kang H.J., Li L.S., Kim N.G., You K.T., Yang E., Kim H., Kim H.J., Yun C.O., Kim K.S., et al. Impaired nonhomologous end-joining in mismatch repair-deficient colon carcinomas. Lab. Invest. 2005;85:1130–1138. doi: 10.1038/labinvest.3700315. [DOI] [PubMed] [Google Scholar]
- 34.Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Seraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- 35.Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Seraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- 36.Kessner D., Chambers M., Burke R., Agusand D., Mallick P. ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics. 2008;24:2534–2536. doi: 10.1093/bioinformatics/btn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trudgian D.C., Thomas B., McGowan S.J., Kessler B.M., Salek M., Acuto O. CPFP: a central proteomics facilities pipeline. Bioinformatics. 2010;26:1131–1132. doi: 10.1093/bioinformatics/btq081. [DOI] [PubMed] [Google Scholar]
- 38.Craig R., Beavis R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- 39.Geer L.Y., Markey S.P., Kowalak J.A., Wagner L., Xu M., Maynard D.M., Yang X., Shi W., Bryant S.H. Open mass spectrometry search algorithm. J. Proteome. Res. 2004;3:958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- 40.Shteynberg D., Deutsch E.W., Lam H., Eng J.K., Sun Z., Tasman N., Mendoza L., Moritz R.L., Aebersold R., Nesvizhskii A.I. iProphet: multi-level integrative analysis of shotgun proteomic data improves peptide and protein identification rates and error estimates. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M111.007690. M111 007690. DOI:10.1074/mcp.M111.007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Consortium U. Reorganizing the protein space at the Universal Protein Resource (UniProt) Nucleic Acids Res. 2012;40:D71–D75. doi: 10.1093/nar/gkr981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 43.Trudgian D.C., Ridlova G., Fischer R., Mackeen M.M., Ternette N., Acuto O., Kessler B.M., Thomas B. Comparative evaluation of label-free SINQ normalized spectral index quantitation in the central proteomics facilities pipeline. Proteomics. 2011;11:2790–2797. doi: 10.1002/pmic.201000800. [DOI] [PubMed] [Google Scholar]
- 44.Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 45.Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szklarczyk D., Franceschini A., Kuhn M., Simonovic M., Roth A., Minguez P., Doerks T., Stark M., Muller J., Bork P., et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cline M.S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Labarca C., Paigen K. A simple, rapid, and sensitive DNA assay procedure. Anal. Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 49.Galindo C.L., McIver L.J., Tae H., McCormick J.F., Skinner M.A., Hoeschele I., Lewis C.M., Minna J.D., Boothman D.A., Garner H.R. Sporadic breast cancer patients’ germline DNA exhibit an AT-rich microsatellite signature. Genes Chromosomes Cancer. 2011;50:275–283. doi: 10.1002/gcc.20853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fattah F.J., Hara K., Fattah K.R., Yang C., Wu N., Warrington R., Chen D.J., Zhou P., Boothman D.A., Yu H. The transcription factor TFII-I promotes DNA translesion synthesis and genomic stability. PLoS Genet. 2014;10:e1004419. doi: 10.1371/journal.pgen.1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bohon J., de los Santos C.R. Structural effect of the anticancer agent 6-thioguanine on duplex DNA. Nucleic Acids Res. 2003;31:1331–1338. doi: 10.1093/nar/gkg203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Swann P.F., Waters T.R., Moulton D.C., Xu Y.Z., Zheng Q., Edwards M., Mace R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science. 1996;273:1109–1111. doi: 10.1126/science.273.5278.1109. [DOI] [PubMed] [Google Scholar]
- 53.Hawn M.T., Umar A., Carethers J.M., Marra G., Kunkel T.A., Boland C.R., Koi M. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- 54.Motea E.A., Lee I., Berdis A.J. A non-natural nucleoside with combined therapeutic and diagnostic activities against leukemia. ACS Chem. Biol. 2012;7:988–998. doi: 10.1021/cb300038f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Akiyama Y., Sato H., Yamada T., Nagasaki H., Tsuchiya A., Abe R., Yuasa Y. Germ-line mutation of the hMSH6/GTBP gene in an atypical hereditary nonpolyposis colorectal cancer kindred. Cancer Res. 1997;57:3920–3923. [PubMed] [Google Scholar]
- 56.Bronner C.E., Baker S.M., Morrison P.T., Warren G., Smith L.G., Lescoe M.K., Kane M., Earabino C., Lipford J., Lindblom A., et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258–261. doi: 10.1038/368258a0. [DOI] [PubMed] [Google Scholar]
- 57.Fishel R., Lescoe M.K., Rao M.R., Copeland N.G., Jenkins N.A., Garber J., Kane M., Kolodner R. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027–1038. doi: 10.1016/0092-8674(93)90546-3. [DOI] [PubMed] [Google Scholar]
- 58.Leach F.S., Nicolaides N.C., Papadopoulos N., Liu B., Jen J., Parsons R., Peltomaki P., Sistonen P., Aaltonen L.A., Nystrom-Lahti M., et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1225. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 59.Miyaki M., Konishi M., Tanaka K., Kikuchi-Yanoshita R., Muraoka M., Yasuno M., Igari T., Koike M., Chiba M., Mori T. Germline mutation of MSH6 as the cause of hereditary nonpolyposis colorectal cancer. Nat. Genet. 1997;17:271–272. doi: 10.1038/ng1197-271. [DOI] [PubMed] [Google Scholar]
- 60.Nicolaides N.C., Papadopoulos N., Liu B., Wei Y.F., Carter K.C., Ruben S.M., Rosen C.A., Haseltine W.A., Fleischmann R.D., Fraser C.M., et al. Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature. 1994;371:75–80. doi: 10.1038/371075a0. [DOI] [PubMed] [Google Scholar]
- 61.Papadopoulos N., Nicolaides N.C., Wei Y.F., Ruben S.M., Carter K.C., Rosen C.A., Haseltine W.A., Fleischmann R.D., Fraser C.M., Adams M.D., et al. Mutation of a mutL homolog in hereditary colon cancer. Science. 1994;263:1625–1629. doi: 10.1126/science.8128251. [DOI] [PubMed] [Google Scholar]
- 62.Peltomaki P. Deficient DNA mismatch repair: a common etiologic factor for colon cancer. Hum. Mol. Genet. 2001;10:735–740. doi: 10.1093/hmg/10.7.735. [DOI] [PubMed] [Google Scholar]
- 63.Chang D.K., Ricciardiello L., Goel A., Chang C.L., Boland C.R. Steady-state regulation of the human DNA mismatch repair system. J. Biol. Chem. 2000;275:18424–18431. doi: 10.1074/jbc.M001140200. [DOI] [PubMed] [Google Scholar]
- 64.Chen F., Arseven O.K., Cryns V.L. Proteolysis of the mismatch repair protein MLH1 by caspase-3 promotes DNA damage-induced apoptosis. J. Biol. Chem. 2004;279:27542–27548. doi: 10.1074/jbc.M400971200. [DOI] [PubMed] [Google Scholar]
- 65.Porter A.G., Janicke R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999;6:99–104. doi: 10.1038/sj.cdd.4400476. [DOI] [PubMed] [Google Scholar]
- 66.Abraham M.C., Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–193. doi: 10.1016/j.tcb.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 67.Woods N.T., Mesquita R.D., Sweet M., Carvalho M.A., Li X., Liu Y., Nguyen H., Thomas C.E., Iversen E.S., Jr, Marsillac S. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci. Signal. 2012;5:rs6. doi: 10.1126/scisignal.2002255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ni Z., Xu C., Guo X., Hunter G.O., Kuznetsova O.V., Tempel W., Marcon E., Zhong G., Guo H., Kuo W.H., et al. RPRD1A and RPRD1B are human RNA polymerase II C-terminal domain scaffolds for Ser5 dephosphorylation. Nat. Struct. Mol. Biol. 2014;21:686–695. doi: 10.1038/nsmb.2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Straub T., Grue P., Uhse A., Lisby M., Knudsen B.R., Tange T.O., Westergaard O., Boege F. The RNA-splicing factor PSF/p54 controls DNA-topoisomerase I activity by a direct interaction. J. Biol. Chem. 1998;273:26261–26264. doi: 10.1074/jbc.273.41.26261. [DOI] [PubMed] [Google Scholar]
- 70.Wu J., Phatnani H.P., Hsieh T.S., Greenleaf A.L. The phosphoCTD-interacting domain of Topoisomerase I. Biochem. Biophys. Res. Commun. 2010;397:117–119. doi: 10.1016/j.bbrc.2010.05.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carty S.M., Greenleaf A.L. Hyperphosphorylated C-terminal repeat domain-associating proteins in the nuclear proteome link transcription to DNA/chromatin modification and RNA processing. Mol. Cell. Proteomics. 2002;1:598–610. doi: 10.1074/mcp.m200029-mcp200. [DOI] [PubMed] [Google Scholar]
- 72.Phatnani H.P., Jones J.C., Greenleaf A.L. Expanding the functional repertoire of CTD kinase I and RNA polymerase II: novel phosphoCTD-associating proteins in the yeast proteome. Biochemistry. 2004;43:15702–15719. doi: 10.1021/bi048364h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Paddock M.N., Bauman A.T., Higdon R., Kolker E., Takeda S., Scharenberg A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair (Amst) 2011;10:338–343. doi: 10.1016/j.dnarep.2010.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Cui W., Huang Z., He H., Gu N., Qin G., Lv J., Zheng T., Sugimoto K., Wu Q. MiR-1188 at the imprinted Dlk1-Dio3 domain acts as a tumor suppressor in hepatoma cells. Mol. Biol. Cell. 2015;26:1416–1427. doi: 10.1091/mbc.E14-11-1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.De Zio D., Bordi M., Tino E., Lanzuolo C., Ferraro E., Mora E., Ciccosanti F., Fimia G.M., Orlando V., Cecconi F. The DNA repair complex Ku70/86 modulates Apaf1 expression upon DNA damage. Cell Death Differ. 2011;18:516–527. doi: 10.1038/cdd.2010.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ramakrishna M., Williams L.H., Boyle S.E., Bearfoot J.L., Sridhar A., Speed T.P., Gorringe K.L., Campbell I.G. Identification of candidate growth promoting genes in ovarian cancer through integrated copy number and expression analysis. PLoS One. 2010;5:e9983. doi: 10.1371/journal.pone.0009983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Reid J.F., Gariboldi M., Sokolova V., Capobianco P., Lampis A., Perrone F., Signoroni S., Costa A., Leo E., Pilotti S., et al. Integrative approach for prioritizing cancer genes in sporadic colon cancer. Genes Chromosomes Cancer. 2009;48:953–962. doi: 10.1002/gcc.20697. [DOI] [PubMed] [Google Scholar]
- 78.Zou T.T., Selaru F.M., Xu Y., Shustova V., Yin J., Mori Y., Shibata D., Sato F., Wang S., Olaru A., et al. Application of cDNA microarrays to generate a molecular taxonomy capable of distinguishing between colon cancer and normal colon. Oncogene. 2002;21:4855–4862. doi: 10.1038/sj.onc.1205613. [DOI] [PubMed] [Google Scholar]
- 79.Schaner M.E., Ross D.T., Ciaravino G., Sorlie T., Troyanskaya O., Diehn M., Wang Y.C., Duran G.E., Sikic T.L., Caldeira S., et al. Gene expression patterns in ovarian carcinomas. Mol. Biol. Cell. 2003;14:4376–4386. doi: 10.1091/mbc.E03-05-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Salvesen H.B., Carter S.L., Mannelqvist M., Dutt A., Getz G., Stefansson I.M., Raeder M.B., Sos M.L., Engelsen I.B., Trovik J., et al. Integrated genomic profiling of endometrial carcinoma associates aggressive tumors with indicators of PI3 kinase activation. Proc. Natl. Acad. Sci. U.S.A. 2009;106:4834–4839. doi: 10.1073/pnas.0806514106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meyers M., Hwang A., Wagner M.W., Bruening A.J., Veigl M.L., Sedwick W.D., Boothman D.A. A role for DNA mismatch repair in sensing and responding to fluoropyrimidine damage. Oncogene. 2003;22:7376–7388. doi: 10.1038/sj.onc.1206941. [DOI] [PubMed] [Google Scholar]
- 82.Meyers M., Wagner M.W., Mazurek A., Schmutte C., Fishel R., Boothman D.A. DNA mismatch repair-dependent response to fluoropyrimidine-generated damage. J. Biol. Chem. 2005;280:5516–5526. doi: 10.1074/jbc.M412105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.