

Abstract
Background
Excess storage of lipids in ectopic tissues, such as skeletal muscle, liver, and heart, seems to associate closely with metabolic abnormalities and cardiac disease. Intracellular lipid storage occurs in lipid droplets, which have gained attention as active organelles in cellular metabolism. Recent developments in high-resolution microscopy and microscopic spectroscopy have opened up new avenues to examine the physiology and biochemistry of intracellular lipids.
Scope of review
The aim of this review is to give an overview of recent technical advances in microscopy, and its application for the visualization, identification, and quantification of intracellular lipids, with special focus to lipid droplets. In addition, we attempt to summarize the probes currently available for the visualization of lipids.
Major conclusions
The continuous development of lipid probes in combination with the rapid development of microscopic techniques can provide new insights in the role and dynamics of intracellular lipids. Moreover, in situ identification of intracellular lipids is now possible and promises to add a new dimensionality to analysis of lipid biochemistry, and its relation to (patho)physiology.
Keywords: Metabolic disease, Lipid droplets, Fluorescent lipid probes, Super-resolution, Live-cell imaging, Vibrational microscopy
Abbreviations: BODIPY, Boron-dipyrromethene; CARS, coherent anti-stokes Raman scattering; CLSM, confocal laser scanning microscopy; CLEM, correlative light electron microscopy; DIC, differential interference microscopy; FA, fatty acid; FLIP, fluorescence loss in photobleaching; FRET, fluorescence resonance energy transfer; FRAP, fluorescent recovery after photobleaching; FIB-SEM, focused ion beam scanning electron microscopy; GFP, green fluorescent protein; HCV, hepatitis C virus; LD, lipid droplet; NBD, nitro-benzoxadiazolyl; PALM, photoactivation localization microscopy; SIMS, Secondary Ion Mass Spectrometry; SBEM, serial block face scanning electron microscopy; STED, stimulated emission depletion; SRS, Stimulated Raman Scattering; STORM, stochastic optical reconstruction microscopy; TOF-SIMS, time-of-flight SIMS; TEM, transmission electron microscopy; TAG, triacylglycerol; TPLSM, two-photon laser scanning microscopy
1. Introduction
Lipids are vital to the cell and have key roles in multiple cellular processes. Lipids are structural components of cellular membranes, involved in cellular signaling, and serve as a critical energy source. In current society, where obesity is a global epidemic, partly because of the overconsumption of lipid-dense foods, the role of lipids in human health and disease has taken central stage in biomedical research. In obesity, an overflow of lipids in circulation can lead to excess lipid storage in ectopic tissues, such as skeletal muscle, liver, and heart. This ectopic fat storage is thought to contribute to the development of various metabolic disorders e.g. insulin resistance, non-alcoholic fatty liver disease, and heart failure [1].
More recently, however, a different view has emerged: augmented storage of lipids in peripheral tissues – despite its close association with metabolic and cardiac disorders – does not necessarily detrimentally affect cellular and tissue function. Not only is the amount of intracellular lipid potentially important for the development of metabolic disease but so is the way that lipids are handled and stored [2].
Intracellular lipid storage occurs in lipid droplets (LDs), which are classically viewed as energy depots but now considered to be active organelles in cellular metabolism. The shape and localization of the LDs appear to correlate with the development of metabolic disorders [3], [4]. In addition to shape and size, proteins coating the LD affect LD dynamics as well as the composition of the lipids in the LD [2].
Lipidomic approaches to analyze lipids extracted from tissues have yielded novel information on global lipid composition from many tissues and under a variety of (patho)physiological conditions [5]. However, more detailed information at the individual LD level is warranted to examine the putative differential effects of localization, size, and composition of the LD on cellular function. Recent developments in (high-resolution) microscopy and microscopic spectroscopy open new avenues to examine intracellular lipids. The aim of this review is to give an overview of recent developments in microscopy with a focus on visualization, identification, and quantification of intracellular lipids. We will focus on the practical application of and the information gleaned from different microscopy tools. This review starts with description of probes currently available to visualize lipids and the dynamics of intracellular lipids. Thereafter, we focus on the available microscopy tools to study LD shape, localization, remodeling, and coating proteins. Finally, we discuss new possibilities in the field of microscopy to elucidate intracellular lipid composition in detail.
2. Visualizing lipids
The vast majority of lipids do not possess intrinsic fluorescence. Hence a wide range of probes has been developed that allows visualization of intracellular lipids by microscopy. Many of these probes are suitable for live-cell imaging and therefore allow for detailed study of both the subcellular location of lipids and cellular lipid dynamics. The ongoing development of such probes and the rapid advances in super-resolution microscopy permitting imaging at resolution below the diffraction limit create a powerful combination to further study the dynamics of cellular lipids. Visualization of lipids can be performed via (i) direct labeling of lipids with a fluorophore, (ii) incorporation of (fluorescent) lipid-binding probes, and (iii) the introduction of fluorescent lipid analogs and dyes. The choice for the best probe is dependent not only on the experimental setting (static measurements on histological slices or fixed cells or live imaging on cells, tissues, or organs) but also on the type of microscopy used (given that certain microscopy techniques require probes with specific properties).
2.1. Fluorophore-conjugated lipids
When studying the intracellular localization and mobility of lipid species, covalent linking of a fluorophore to the lipid of interest can be performed. The fluorophore can be linked either to the polar hydrophilic head group, e.g. via polar rhodamine or cyanide dyes, or to the acyl chains or hydrophobic groups. Since large fluorophores like rhodamine and cyanide are not suited to link to acyl chains, frequently used fluorophores include boron-dipyrromethene (BODIPY) and nitro-benzoxadiazolyl (NBD) [6]. Inherent to fluorophore-conjugation is the concern that biophysical properties of the lipids might be altered and/or (yet unknown) functional groups of the lipids are modified or masked [7]. For example, labeling of the acyl chains of phospholipids with NBD can result in looping of the acyl tail to the surface of the lipid bilayer due the moderate polarity of NBD [8]. As a consequence, the intracellular motility and localization of these conjugated lipids can be affected [9], [10], [11]. This could be especially problematic for live-cell imaging but may also have implications in fixed samples, as lipids can retain mobility after fixation [12].
Probe development should aim for minimal functional interaction to allow for optimal investigation of native lipid dynamics. Therefore, lipids have been labeled with an alkyne or azide group, which are most often bound to the terminus of the acyl chains. The advantage of these chemical groups is their small size and bio-orthogonality, meaning that they do not occur in nature and are therefore biologically inert. These lipid probes can subsequently be visualized by ‘clicking’ a fluorophore to the alkyne or azide group via established chemical reactions. This can be done after the lipid probe has reached its cellular destination [13] (Figure 1). Clickable lipid probes have so far been developed and used for various fatty acids, cholesterol, and sphinganine [13]. Besides the clickable group, the lipid probe can have a small photoactivatable group grafted, e.g. in the alkyl chain, which can be used for the investigation of lipid–protein interactions. Activation of this photoactivatable moiety with UV light leads to covalent linking of the lipid to protein binding partners. Covalent cross-linking of the protein–lipid complex before clicking the fluorophore ensures that the fluorophore does not disturb the interaction of lipid and protein [13]. Lipid probes containing both a photoactivatable and clickable group are referred to as bifunctional lipids.
Figure 1.

Example of a bifunctional lipid. Upon activation of a photoactivatable (clickable) fatty acid, the fatty acid can be turned into a variety of bifunctional phospholipids. These functional phospholipids can be linked to proteins e.g., by irradiation with ultraviolet light. Conjugation of the clickable group with reporter molecules (potentially of a wide range) subsequently facilitates imaging of the lipid bound proteins.
2.2. Lipid-binding probes
Lipid-binding probes comprise a group of fluorescently-tagged lipid-binding proteins, toxins, and (conjugated) antibodies. Lipid-binding proteins can especially be used for visualizing the location of lipids and lipid dynamics within living cells. Lipid-binding proteins are often labeled with fluorophores like green fluorescent protein (GFP) or its color variants. Fluorescently-tagged lipid-binding proteins can be directly expressed in the cells of interest, via plasmid transfection or genetic manipulation, or introduced into the cells. The latter requires permeabilization of the plasma membrane to allow entrance of the lipid-binding proteins. A classic example of fluorescently-tagged lipid-binding proteins is the use of the pleckstrin domains of phospholipase Cδ and Akt to visualize the phosphoinositides PI(2,4)P2 and PI(3,4,5)P3, respectively [14]
Several lipid-binding toxins can be used to visualize lipids in cellular lipid bilayers. Again, these molecules, or the lipid-binding domains thereof, can be tagged with fluorescent moieties. Lysenin and cholera toxin subunit B have been used to visualize sphingomyelin [15] and ganglioside GM1 [16], respectively. Pore-forming cytolysins specifically bind to sterols and are used to visualize cholesterol in lipid membranes [17]. It is important to note, however, that the use of the toxin-based lipid probes is mostly restricted to imaging fixed cells, given the cytolytic activity of these probes and that their toxic nature may hamper examination of physiological processes.
Another lipid-binding probe is filipin, which is an intrinsically fluorescent antibiotic that specifically binds to free cholesterol, but not to esterified cholesterol [18]. Because filipin can penetrate through the cell membrane, it can also be easily used for staining of intracellular cholesterol [7]. However, filipin cannot be used in live-cell imaging since it perturbs the lipid membranes. Another disadvantage of filipin is its rapid photobleaching [17].
Although antibodies are being widely used in the visualization of cellular proteins, few antibodies exist that target specific cellular lipids. The main reason for this is the lack of antigenicity of the vast majority of lipids given the endogenous presence of lipids in the animals used [7]. However, some anti-lipid antibodies do exist, such as antibodies against ceramide [19]. Moreover, the use of antibodies is often restricted to fixed and permeabilized samples.
Again, it cannot be excluded that binding of any of the lipid-binding probes affects the functionality of the lipids or disturbs the binding of the lipids with endogenous proteins. Also, sequestering of target lipids in endogenous complexes limits the accessibility of the lipid-binding probes to their targets [12]. As a consequence, it is difficult to exclude that only ‘free’ lipids are detected.
2.3. Lipid analogs and lipid soluble dyes
For visualization of the intracellular localization of cholesterol, two intrinsic fluorescent sterols have been identified, i.e. dehydroergosterol and cholestatrienol. Although these sterols can be used for live-cell imaging, their fluorescence is relatively weak and rapidly photobleached [17]. On the other hand, it is advantageous that these probes have a chemical structure that very closely resembles cholesterol and ergosterol, increasing the likelihood that these are indeed valid cholesterol analogs.
Several lipid dyes are available, specifically used for visualizing and quantifying intracellular lipid stores, each having their advantages and disadvantages. Classical lipid dyes include Oil Red O (ORO) and Sudan Black B [20], which are lipid soluble diazo dyes that are often combined with conventional brightfield microscopy (although modifications to the classical staining protocols also permit Oil red O-based evaluation in fluorescence microscopy [21]). Unfortunately, these dyes can only be used on fixed samples. In addition, these dyes are very sensitive to preparation conditions, requiring fresh solving of powder and filtering, which is time-consuming and leads to less consistent results [22], [23]. Alternatives to these classical dyes have been developed and include the cell-permeable lipophilic fluorophores BODIPY and Nile Red, which can be used in either live or fixed samples.
Besides being conjugated to lipids and lipid-binding probes, BODIPY alone can also be used as a lipophilic fluorescent dye which binds to neutral lipids. The hydrophobicity of BODIPY facilitates rapid uptake by intracellular LDs [23]. While incredibly diverse BODIPY conjugates exist, we have also noticed the binding promiscuity of these conjugates such that mitochondrial, plasma and, nuclear membranes are also labeled, albeit to a lesser extent that neutral lipids. Nile Red is also capable of localizing to hydrophobic intracellular structures such as LDs [24], but it also seems to localize to cellular lysosomal phospholipid inclusions [25]. Interestingly, the spectral properties of Nile Red are dependent on the polarity of the environment and spectral shifts can occur from red (628 nm, polar) to yellow (580 nm, neutral) [22]. LDs storing predominantly neutral lipids are normally visualized in yellow. This spectral shift can yield problems for its application, but the spectral shift of Nile Red can also be used to reveal a change in the composition of cellular LDs [26].
3. Visualizing lipid droplets
As described above, LDs are the main site for intracellular lipid storage. LDs are coated by a phospholipid monolayer with a hydrophobic core containing neutral lipids, mainly triacylglycerols (TAGs) and sterol esters. Moreover, LDs are coated by a range of proteins important in LD dynamics and lipid metabolism in a cell-type and tissue-specific fashion [27]. This dynamic nature of LDs permits tuning of liberation and uptake of neutral fatty acids during fluctuations in demand and supply. Moreover, LD turnover permits uptake and release of bioactive lipids, which interfere with signaling routes in the cell and hence cell and tissue function. Therefore, LDs are emerging as important organelles in cellular (patho)physiology, and a suite of tools to study LDs in physiology and pathology has been emerging.
3.1. LD shape and cellular localization
In order to study LD size, number, and cellular distribution, LDs can be visualized with the previously mentioned lipid soluble dyes. ORO staining in combination with conventional wide-field trans-illumination microscopy has been used to study LD shape [28]. Given the expected diameter of LDs, which ranges from approximately 0.1–2 μm in mammalian cells (with the exception of white adipocytes) [27], the diffracted-limited resolution of wide-field trans-illumination and wide-field fluorescence microscopy (theoretically ∼230 nm in plane, although practically, resolution is often lower) prohibits accurate determination of LD size. In confocal laser scanning microscopy (CLSM), the excitation laser is scanned through the sample in three dimensions. By use of a pinhole in front of the detector, only the fluorescence from the focal point [29] is passed to the detector, while the out-of-focus fluorescent emission is rejected [30]. This improves in plane resolution to ∼180 nm (or lower, at the cost of fluorescence signal) while simultaneously optically sectioning the sample. This allows imaging at various depths (up to ∼100 s of μm) and true three-dimensional (3D) imaging of cells and tissues (Figure 2). Although CLSM allows for a more reliable determination of LD shape, imaging of the smallest LDs may still reside below the resolution limit.
Figure 2.
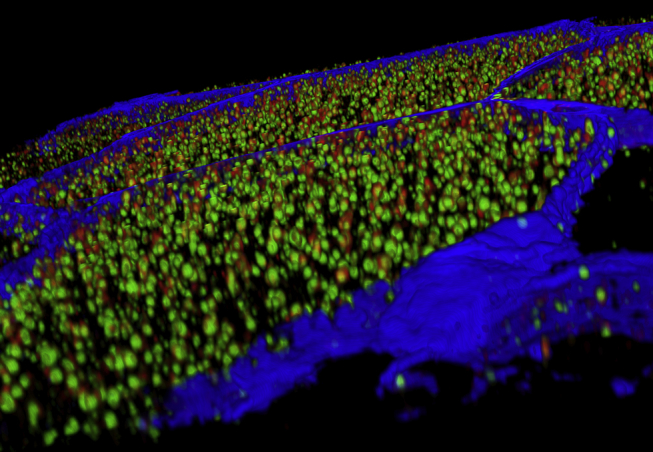
3D reconstruction of the cross-section of human skeletal muscle fibers. 3D reconstruction of the cross-section of human skeletal muscle fibers (cell membrane in blue, stained for laminin), illustrating the well-organized spatial distribution of LDs (green, stained with BODIPY 493/503) and the LD coating protein PLIN5 (red), imaged with CLSM. Image courtesy of Anne Gemmink.
Recently, various super-resolution microscopy techniques – those that produce images with features well beyond the physical diffraction limit showing ∼30 nm features (discussed in detail in Section 3.3) – have gained significant popularity in cell biology. In general, ectopic LDs do not require the technical improvements of nanoscale super-resolution imaging when it comes to LD shape. However, this resolution will certainly be useful for imaging smaller, nascent LDs and for examining LD interaction with proteins or other cellular organelles.
In addition to fluorescence microscopy, two classes of label-free microscopic techniques are currently in use to investigate LD shape and localization, namely differential interference microscopy (DIC) and coherent Raman microscopy (CRM). DIC microscopy is a wide-field technique, with all the previously mentioned limitations in resolution, which uses differences in refractive index between physically neighboring regions of the specimen to produce contrast in the image. DIC microscopy permits visualization of LDs with high contrast, since their refractive index is higher than the surrounding cytosol, and circumvents phototoxicity [31], though this method is largely insensitive to lipid composition. For example, application of DIC microscopy revealed the bidirectional movement of LDs via the dynactin complex on microtubules [32].
CRM is a form of vibrational microscopy that takes advantage of the intrinsic chemical composition of lipids to directly visualize them based on molecular vibrations. LDs consist primarily of TAGs, which contain large numbers of lipid acyl chains with correspondingly abundant CH2 (methylene) groups that collectively generate a strong coherent Raman signal [33]. Two primary modalities for CRM in LD imaging are coherent anti-Stokes Raman scattering (CARS) microscopy and stimulated Raman scattering (SRS) microscopy. These two modalities offer slightly different capabilities but rely on the same underlying physical principle: using coherent excitation of molecular vibrations to generate contrast [34]. Consequently, LDs can be identified with CARS and SRS microscopy without any labeling. For example, quantitative CARS and SRS microscopy have been shown to be useful to visualize intracellular LDs and LD composition in muscle tissue sections [35] and in C. elegans [36].
A clear advantage of these techniques is that they are label-free, enabling the examination of living cells in near physiological settings without photobleaching or phototoxicity [33]. Therefore, samples can be imaged over long periods of time (up to a week) [37]. Real-time and 3D CARS microscopy has been applied to assess the volume of LDs throughout adipogenic differentiation [38]. Moreover, CARS microscopy has been applied in the examination of LD size, density and distribution in a human hepatoma cell line upon hepatitis C virus (HCV) infection [31]. Similarly, we have applied CARS microscopy to examine LD size, density, and distribution in skeletal muscle cells overexpressing the LD coat protein perilipin 5 (PLIN5) [39]. The intracellular movement of LDs has also been studied using CARS microscopy, showing the trafficking of LDs over microtubules [40], [41].
In order to visualize the LDs of all sizes, conventional transmission and scanning electron microscopy (TEM and SEM) has been commonly used to image LDs shape and localization [42], [43]. The advantage of electron microscopy is its excellent resolution (below 10 nm) which allows detailed determination of LD location within the cell and the position relative to other cellular structures and organelles. For example, the use of electron microscopy in skeletal muscle can differentiate between LDs present beneath the sarcolemma and those LDs that lay between the contractile elements of skeletal muscle [42].
Also, conventional electron microscopy initially revealed the close contact between LDs and other organelles such as mitochondria and endoplasmic reticulum (ER) [44], [45]. By merging multiple images one may be able to obtain a high-resolution overview of a larger area of interest (Figure 3). However, this is very laborious and prone to erroneous merging of the images. Another disadvantage of electron microscopy is that it can only be performed in chemically fixed and hence dead material with limited antigenicity. This complicates antibody staining, which, in turn, compromises examination of proteins interacting with LDs.
Figure 3.

Merging of multiple transmission electron microscopy images. Merging of multiple transmission electron microscopy images from a longitudinal section of a muscle fiber located in m. vastus lateralis from a male trained athlete. (Unpublished data MKCH).
High-resolution 3D imaging of cells and tissues has become available through electron tomography, which is based on TEM. To obtain a 3D view, the electron beam is directed through the sample at different angles, after which the images are collected and reconstructed to a 3D image of the entire volume, called a tomogram [46] (Figure 4). Recently, more advanced techniques have been developed that allow 3D imaging with electron microscopy. One of these techniques is focused ion beam scanning electron microscopy (FIB-SEM), using alternating ion and electron beams to etch a thin layer of specimen and subsequently record an image of the newly exposed surface. Besides FIB-SEM, serial block face scanning electron microscopy (SBEM) has been developed, wherein the surface of a resin-embedded sample is imaged via backscattered electron detection using the atomic contract of heavy metal stains. Subsequently, this technique can be used for 3D visualization of subcellular structures. Via 3D segmentation analysis, volumetric data can be obtained.
Figure 4.

Direct membrane contact between ER and LD. (A) Direct membrane contact between ER and LD could be observed in reconstructed tilt-series from high-pressure frozen, freeze-substituted (acetone containing 0.2% uranyl acetate, 2% osmium tetroxide and 1% H2O) and Epon embedded HeLa cells (arrowheads). (B) Model of the reconstructed tomogram showing LDs (light blue), ER membranes (pink), MVB (blue) with internal vesicles (red), mitochondria (green) and the nuclear envelope (purple). While membrane contrast was sufficient to allow for efficient reconstruction of the tilt-series, LDs appeared extracted. er: endoplasmic reticulum, g: Golgi, ld: lipid droplet, m: mitochondria, MVB: multivesicular body, n: nucleus. Images courtesy of Dr. Rob Mesman.
Applications of novel 3D EM based technologies in LD research include studies reporting cell-to-cell heterogeneity of LD volume in individual hepatocytes [47], visualization of the connection of LDs with the ER [48], and quantification of the area of interaction of LDs with the ER [49]. In cardiac tissue of PLIN5 overexpressing mice, FIB-SEM was used to show that PLIN5 overexpression promoted interaction of LDs with mitochondria [46]. Although electron microscopy images revealed close proximity between LDs, ER, and mitochondria, the static nature of electron microscopy cannot resolve if this reflects a functional interaction between the LDs and the other organelles. In general, examination of dynamic processes is complex, if not impossible, to perform with electron microscopy.
3.2. LD biogenesis and remodeling
In most tissues, LDs are highly dynamic organelles, which show growth, remodeling, and degradation as a consequence of lipogenic or lipolytic signals. Detailed investigation of these dynamic processes requires real-time imaging techniques. Real-time microscopic techniques often used for studying LD development and remodeling include CARS microscopy and confocal microscopy. As described above, label-free techniques such as CARS permit long-term imaging of cells without photobleaching in almost native conditions. Therefore, CARS microscopy can be a valuable tool to study the long-term development and growth of LDs. For example, the differentiation of 3T3-L1 adipocytes has been followed for multiple days using CARS microscopy [50]. Paar et al. studied LD growth in both 3T3-L1 adipocytes and human adipose-derived stem cells, induced to differentiate, via CARS imaging for up to 16 h [51]. The researchers also examined the effect of lipolytic stimulation of 3T3-L1 adipocytes on LD540 stained LDs using confocal microscopy. The use of confocal imaging in studying the remodeling of LDs after lipolytic stimulation has also been demonstrated in several reports [49], [52]. Although photobleaching, phototoxicity and abnormality due to staining (e.g. some lipid stains have a tendency to promote LD fusion) must be taken into account when using confocal imaging, it is very useful platform for multiple staining.
Fluorescent recovery after photobleaching (FRAP) and fluorescence loss in photobleaching (FLIP) can be used to study cellular lipid flow and lipid exchange between LDs. FRAP is a quantitative, real-time fluorescent subtechnique that is used to study molecular motility and is often performed using a confocal microscope. With FRAP, a defined region of interest (typically not exceeding 1 μm in radius [53]) is illuminated with high intensity light; the fluorophore within that region being bleached. Subsequently, recovery of fluorescence in the bleached region, by diffusion of non-bleached molecules into the bleached region, can be recorded over time, which yields information on the diffusion coefficients of the non-bleached molecules [54]. FRAP has for example been used to quantify intracellular fatty acid (FA) mobility in adipocytes upon labeling with Bodipy-C16. Subsequently, specific LDs could be photobleached and re-introduction of the label in the LDs was monitored over time [55]. FRAP analysis was also used to examine exchange of neutral lipids between LDs in wild-type adipocytes, a process which was shown to depend on the presence of LD associated protein fat specific protein 27 (Fsp27) in the white adipocytes [56].
Closely related to FRAP is FLIP, which is also used to determine molecular motility in a cell. With this technique, a labeled molecule is photobleached in a specific region and the loss of fluorescence in other regions of the cell can be monitored and related to the diffusion of the photobleached labeled molecule through the cell [54]. Both FRAP and FLIP have several pitfalls that have to be taken into account, including phototoxicity and photo-induced cross-linking, loss in fluorescence, and movement of live cells during the course of the long-lasting experiment [54].
Besides fluorescent excitation and emission, fluorophores are also characterized by their fluorescent lifetime, i.e. the time for the fluorophore to fall back to its ground state after excitation. Moreover, for some probes, the fluorescence lifetime strongly depends on the local environment, e.g., viscosity, pH, [Ca2+]. Fluorescent lifetime imaging microscopy (FLIM) makes use of this probe characteristic and is able to distinguish either between fluorophores with different fluorescent lifetimes or a single probe in different local environments [54]. Thus, FLIM was used in combination with Bodipy-C12 to measure the viscosity of LDs and size of the lipid transfer channel between two LDs [38].
3.3. Proteins at the LD membrane
To determine protein localization and interactions at the LD surface, traditional co-immunofluorescence either in combination with wide-field or confocal imaging is often used. For example, the localization of several proteins (in)directly involved in lipolysis like PLIN2, ATGL, and CGI-58, and their co-localization at the LD surface in human muscle at rest and during exercise was investigated with ORO staining and wide-field trans-illumination microscopy [57]. Given the limited optical resolution, wide-field microscopy, however, cannot determine direct interaction between proteins and the LD. Hashimoto et al. investigated the distribution of LD associated proteins on the LDs and in the cytoplasm during lipolysis using GFP tags and confocal microscopy in 3T3-L1 adipocytes [58]. Other techniques to determine the localization of proteins at the LD surface include CARS in combination with CLSM or TPLSM. Moreover, the techniques of FRAP and FLIP can be used to determine the mobility of proteins associated with LDs. In this respect, it has been shown, with the use of FRAP and FLIP, that in yeast the integral membrane protein DGA1 is able to move between ER and LD and can move freely across the LD membrane [59].
Current, super-resolution microscopy techniques now available allow for significant improvements in plane resolution with fluorescent probes. Such developments include stimulated emission depletion (STED) microscopy and single molecule localization microscopy, namely photoactivation localization microscopy (PALM) and stochastic optical reconstruction microscopy (STORM). Reports using STED and PALM/STORM in relation to LDs and associated proteins are still scarce. In STED microscopy a depletion laser is used in addition to the excitation laser, which depletes all the fluorescence outside the center of the point-spread function by stimulated emission. This technique boosts the lateral resolution down to 30–80 nm [60] (Figure 5). However, it should be noted that not all classically used fluorescent probes can be used in STED microscopy. Probe compatibility, depends, amongst other factors, on exact position (wavelength) of excitation, emission, and depletion bands. Also, the use of multiple probes for dual or more color imaging is less straightforward than in standard CLSM [60]. Wilfling et al. used STED to visualize the localization of endogenous GPAT4 to the LD and to distinguish this from localization of this protein with adjacent ER, using LD and ER marker proteins [61]. In addition, Wilfling et al. showed that GPAT4 moved from ER to LD during the formation of LDs using conventional FRAP.
Figure 5.

Comparative imaging of the mitochondrial outer membrane protein TOMM20. Comparative imaging of the mitochondrial outer membrane protein TOMM20 in cultured human muscle cells with CLSM (A, B) and STED (C, D). Strains of mitochondria surrounding the cell nucleus can be observed with both microscopic techniques (A, C). Whereas identification of individual mitochondria is limited with CLSM (B), the high-resolution of STED is able to differentiate outer membranes of individual mitochondria (D).
Both PALM and STORM are based on the principle of recording large series of images, each of which contains only a few activated fluorophores. This limits the chance of detecting overlapping emitting particles in one image and makes it possible to localize individual fluorophores with nm precision. PALM and STORM became possible by the availability of photoactivatable proteins (e.g. PA-GFP), which were originally used in PALM, and photo switchable dyes (e.g. Cyanine dyes), which STORM originally used. The series of images obtained are reconstructed to a high-resolution image with a resolution down to ∼30 nm [60]. Although 3D PALM/STORM has been shown to be possible, imaging depth is at maximum ∼10 μm [62]. Eggert et al. showed the possibility of imaging LDs with STORM by using the lipophilic dye LipidTox Red [63]. Using this technique, the authors were able to determine the localization of two HCV proteins at the LD and their co-localization at the LD surface in HCV-infected cells. However, this also makes these techniques intrinsically slow, making it less useful for live-cell imaging of rapid processes.
Förster resonance energy transfer (FRET) can be a powerful technique to study co-localization of proteins. This technique relies on the physical proximity of two fluorophores, a so-called donor and an acceptor, wherein energy is transferred between the molecules so that it can only occur when the fluorophores are within Förster distance, typically ∼5 nm, of each other. When two proteins – one labeled with the donor and the other with an acceptor – are co-localized, fluorescence of the donor molecule is quenched; its energy is taken up by the acceptor molecule (without donor fluorescence taking place). The acceptor dye then either emits fluorescent light or the energy is lost as heat [54]. Live FRET imaging was applied to reveal molecular association of PLIN2 and lipids on the LD surface [64]. Next to fluorescent intensity of the acceptor, the fluorescent lifetime of the donor also can be used as donor lifetime decreases upon energy transfer and FRET efficiency can be directly calculated from this change. Lifetime-based FRET appears to be the most reliable FRET technique, as it is insensitive to most of the artifacts that are encountered when using fluorescent intensity of the acceptor as a read out [65]. The fluorescent techniques discussed above are limited by the obvious fact that only fluorescently-tagged structures are visible.
Again, conventional electron microscopy in combination with immunogold labeling of proteins of interest also can provide high-resolution information on protein localization. However, this technique seems to not be suitable for 3D imaging. Because visualization of the protein of interest requires a gold-conjugated primary or secondary antibody, the number of labeled molecules is limited to the number of differently sized gold particles that can be distinctively imaged at the same time. Classically 1, 5, and 10 nm gold particles are used for immunogold electron microscopy. Imaging of the 1 nm gold particles is typically done after silver enhancement, a technique in which the gold particle grows in size upon incubation with a silver solution. This process, however, results in silver enhanced-gold particles of undefined size. Hence, in practice immunogold electron microscopy typically is limited to making the distinction between 2 different proteins of interest.
Correlative light electron microscopy (CLEM) combines the advantages of both fluorescence microscopy and electron microscopy. CLEM allows for the localization of specific proteins with fluorescence in the ultrastructural landscape of EM, also in 3D. In this respect, Romero-Brey et al. used this technique to assess the exact localization of HCV particles within an infected cell [66]. Moreover, CLEM was used to identify the exact location of DAG, labeled with a GFP-tagged probe of PKCε, in subcellular membranes [67].
4. Identifying lipids in situ
Specific research questions that entail a detailed analysis of the intracellular lipid composition such as those related with the so-called athletes paradox or lipid-induced metabolic abnormalities in cells require a different kind of analysis than is typically employed in lipid imaging. Characterization and quantification of specific lipid species is necessary at the tissue, cellular, and perhaps subcellular level. Such a chemical analysis is classically executed via lipid extraction from a sample, which is followed by chromatography, biochemical assays, or conventional mass spectrometry. During these assays, all spatial information is unfortunately lost and the composition of localized cellular structures remains elusive. Recently, several techniques have emerged that can combine chemical identification with spatial information. Promising techniques include hyperspectral CARS microscopy, stimulated Raman scattering (SRS) microscopy and secondary ion mass spectrometry (SIMS).
Both CARS and SRS microscopy (as discussed earlier) make use of the intrinsic vibrations of chemical bonds within molecules in the sample. It is possible to obtain not only images of lipids but also direct biochemistry using these methods. Using a combination of lasers with specific frequencies, these techniques can provide high-speed, label-free chemical fingerprints at each spatial location within a sample with ∼300–500 nm lateral and ∼2–4 μm axial spatial resolution. Both techniques permit distinction between specific classes of lipid molecules based on differences between the fatty acid chains of the lipids. Given that each molecule has a specific combination of chemical bonds, each molecule has a specific vibrational (Raman) spectrum. Single frequency CARS and SRS microscopy, which measure one vibrational signature, have the potential to image and track, in vivo, specific lipid species. In addition, SRS allows direct quantification of the signal comparable to mass spectrometry [33]. Furthermore, these techniques can be combined with fluorescence microscopy, such as CLSM or TPLSM.
The development of hyperspectral CARS and SRS, which provides a whole vibrational spectrum at each spatial location by applying a range of excitation frequencies, has made it possible to elucidate the lipid composition of a biological sample [52], [68]. Individual lipid composition in terms of unsaturation and acyl chain order has been investigated in 3T3-L1 adipocytes and HeLa cells, showing heterogeneity in the composition of LDs within and between individual cells [69], [70]. Billecke et al. studied the effect of overexpression of LD coating protein PLIN5 in both a muscle cell line and rodent muscle on the lipid composition of LDs and identified differences in esterified acyl chain concentration, methylene content, and saturation upon PLIN5 overexpression [39]. We have recently been able to produce vibrational spectra of LDs in human skeletal muscle tissue (Figure 6). In addition, hyperspectral CARS and SRS can better distinguish between specific lipid species given that molecular identification of metabolites depends on subtle changes in multiple vibrational modes. Fu et al. were able to distinguish between specific cholesteryl esters and triacylglycerols, not separable with conventional SRS, at an individual LD level using hyperspectral SRS microscopy [71]. However, especially in the absence of a unique functional group, chemically related lipids have very similar and overlapping Raman spectra, making specific lipid quantification very difficult with CARS and SRS.
Figure 6.

Quantitative imaging of lipid droplet chemistry in muscle using hyperspectral CARS microscopy. One complete hyperspectral dataset contained spectral data from Type I and Type II muscle fibers in a human muscle biopsy of m. vastus lateralis. LDs were identified from the tissue based on image thresholding similar to [39] and additional data segmentation was performed to group LDs by fiber type. This allowed for producing LD specific spectra from each fiber type and from the non-LD area. Average spectra from LDs in Type I (red) and Type II (blue) fibers and the total non-LD area (green) highlight differences in chemical environment between the LDs between different fiber types and from LDs to the sarcoplasm. Images show chemical images of esters (C O) and CH2 symmetric vibrations. LDs in type I fibers contain more esters, yet similar CH2, compared with Type II fibers, which suggests a differential type of neutral lipid storage between fiber types in the same muscle. (Unpublished data, SD, MKCH, and SHP).
An alternative, currently emerging technique combining spatial localization with metabolic profiling is SIMS, an in situ imaging mass spectrometry technique. In SIMS, a primary ion beam is directed at the region of interest in a sample, causing fragmentation of the surface molecules, generating secondary particles. These secondary particles can be identified via mass spectrometry [72]. SIMS has been applied at tissue level in biological samples. For example the use of SIMS in atherosclerotic plaques in the aortic wall regions revealed a heterogeneous distribution of several lipid species [73]. In addition, in human skeletal muscle samples of patients with Duchenne dystrophy, SIMS revealed characteristic lipid distribution patterns [74]. In cerebral cortex of Alzheimer patients, accumulation of cholesterol was observed [75]. Application of this technique in biological samples is limited by the upper mass detection limit (∼ = 1000 m/z), which limits the detection of large complex organic molecules, including lipids. In addition, at present the sensitivity of SIMS is too low to detect low abundance molecules [72].
New instrumentation and technical advances, including the availability of new ion sources for the primary ion beam, which lift the upper mass limit, have increased the application of SIMS in biological samples [68], [72]. Two types of SIMS can be distinguished, namely time-of-flight SIMS (TOF-SIMS) and dynamic SIMS or NanoSIMS. TOF-SIMS generates a whole mass spectrum at each pixel and unknown unlabeled lipid components with distinctive m/z can be identified at specific locations in the samples [72], [76]. On the other hand, NanoSIMS can detect only a few preselected lipid species of interest. Given the complexity of the lipid pool in biological samples, selective labeling of the lipids of interest with distinct stable isotopes is required [72], [76].
TOF-SIMS appears an attractive tool for determining the exact composition of individual cellular LDs. Although an xy-resolution at the 100 nm level is possible, in practice reaching an xy-resolution of less than 1 μm with TOF-SIMS in biological samples seems challenging at the moment. This can limit the application of SIMS in the analysis of the composition of smaller LDs [35], [72]. However, the field of SIMS is rapidly evolving and detailed characterization of individual LD composition is likely to be within reach in the not too distant future. Some examples of the application of NanoSIMS, which has a resolution typically around 100 nm, in relation to LDs can be found [77], [78]. By combining SIMS with stable isotope labeling, Kleinfeld et al. was able to image the levels of 13C-FFA taken up by LDs after culturing adipocytes in the presence of these FFAs [77].
The identification of small organelles is difficult with SIMS. This could be solved by correlating SIMS with another imaging technique, such as fluorescence techniques, as has been shown by Saka et al. They have which has demonstrated the possibility of correlating SIMS to both confocal and STED microscopy [79]. The use of a super-resolution microscopic technique in combination with SIMS could especially be valuable for imaging structures with a size below the diffraction limit [79].
5. Conclusions
Traditionally, the nature of lipids in combination with technical challenges made lipids comparatively less studied than proteins. In recent years, lipids have gained attention in biomedical research as important regulators of (patho)physiologic processes. At the same time, technical advances in both lipid probes and microscopic techniques raised the possibility to visualize intracellular lipids. Especially when it comes to live-cell imaging, further development of lipid probes should aim at minimal functional interaction to enable studying lipids in near-native conditions. Such improvement could, for example, allow answering the question of how subcellular fate of labeled, exogenous fatty acids is affected by differences in chain length and saturation. Similarly, with novel probes, one could potentially delineate if different fatty acids are shuttled towards oxidation for energy provision or preferably stored in LDs. Finally, newly developed probes may also help answer long standing questions such as must fatty acids involved in oxidation be first packaged in LDs or is direct oxidation possible.
LDs are no longer viewed merely as a place for lipid storage but as active organelles interacting with proteins and other organelles and with central roles in cellular metabolism. Recently developed high-resolution microscopic techniques can increase our knowledge on the organization and function of LDs. One of the LD coat proteins (PLIN5) has been hypothesized to tether mitochondria to LDs or chaperone fatty acids from the LD to the mitochondria. PALM/STORM or STED could be potentially used to examine this hypothesis.
Important questions that remain regarding intracellular lipids are how LDs and lipid species are distributed throughout the cell. Moreover, the exact composition of the LD and the putative dynamic changes in LD composition under physiological as well as pathophysiological conditions remain to be elucidated. Where traditionally lipid extractions combined with conventional mass spectroscopy were used to study cellular lipid composition, technical advances in both vibrational microscopy and imaging mass spectrometry allow in situ analysis of cellular lipid distribution. Thus, long standing questions related to the origin and type of insulin desensitizing bioactive lipids can be studied more specifically than ever before. Likewise, in situ information from individual LDs can be linked to the location of these specific LDs (e.g., subsarcolemmal vs. intermyofibrillar). Thus, recent developments undoubtedly open exciting possibilities to address the questions above and could provide conclusive answers to these and other questions in the very near future. Importantly, efforts are being made to combine multiple microscopy platforms. CLEM is an established example of this, but also other combinations have been described, such as SIMS and STED [79]. The combination of these techniques has the potential to provide complete characterization of the organization and metabolism of intracellular lipids.
Acknowledgments
The work of SD is partly supported by Dutch Diabetes Research Foundation (grant DF 2014.00.1756) and by the NUTRIM—School for Nutrition, Toxicology and Metabolism – NWO Graduate Program financially supported from Netherlands Organization for Scientific Research (022.003.011). SHP and MKCH acknowledge financial support from the NanoNextNL, a micro and nanotechnology consortium of the Government of the Netherlands and 130 partners. S.H.P. acknowledges financial support from the Marie Curie Foundation #CIG322284. The authors acknowledge Enabling Technologies BV for their support in purchasing of the STED (Stimulated Emission Depletion).
Contributor Information
Sabine Daemen, Email: sabine.daemen@maastrichtuniversity.nl.
Marc A.M.J. van Zandvoort, Email: mamj.vanzandvoort@maastrichtuniversity.nl.
Sapun H. Parekh, Email: parekh@mpip-mainz.mpg.de.
Matthijs K.C. Hesselink, Email: matthijs.hesselink@maastrichtuniversity.nl.
Conflict of interest
None declared.
References
- 1.Szendroedi J., Roden M. Ectopic lipids and organ function. Current Opinion in Lipidology. 2009;1:50–56. doi: 10.1097/mol.0b013e328321b3a8. [DOI] [PubMed] [Google Scholar]
- 2.Muoio D.M. Revisiting the connection between intramyocellular lipids and insulin resistance: a long and winding road. Diabetologia. 2012;10:2551–2554. doi: 10.1007/s00125-012-2597-y. [DOI] [PubMed] [Google Scholar]
- 3.He J., Goodpaster B.H., Kelley D.E. Effects of weight loss and physical activity on muscle lipid content and droplet size. Obesity Research. 2004;5:761–769. doi: 10.1038/oby.2004.92. [DOI] [PubMed] [Google Scholar]
- 4.Nielsen J., Mogensen M., Vind B.F., Sahlin K., Hojlund K., Schroder H.D., et al. Increased subsarcolemmal lipids in type 2 diabetes: effect of training on localization of lipids, mitochondria, and glycogen in sedentary human skeletal muscle. American Journal of Physiology, Endocrinology and Metabolism. 2010;3:E706–E713. doi: 10.1152/ajpendo.00692.2009. [DOI] [PubMed] [Google Scholar]
- 5.Zhao Y.Y., Miao H., Cheng X.L., Wei F. Lipidomics: novel insight into the biochemical mechanism of lipid metabolism and dysregulation-associated disease. Chemico-Biological Interactions. 2015:220–238. doi: 10.1016/j.cbi.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Klymchenko A.S., Kreder R. Fluorescent probes for lipid rafts: from model membranes to living cells. Chemistry and Biology. 2014;1:97–113. doi: 10.1016/j.chembiol.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Maekawa M., Fairn G.D. Molecular probes to visualize the location, organization and dynamics of lipids. Journal of Cell Science. 2014;22:4801–4812. doi: 10.1242/jcs.150524. [DOI] [PubMed] [Google Scholar]
- 8.Chattopadhyay A., London E. Parallax method for direct measurement of membrane penetration depth utilizing fluorescence quenching by spin-labeled phospholipids. Biochemistry. 1987;1:39–45. doi: 10.1021/bi00375a006. [DOI] [PubMed] [Google Scholar]
- 9.Wang T.Y., Silvius J.R. Different sphingolipids show differential partitioning into sphingolipid/cholesterol-rich domains in lipid bilayers. Biophysical Journal. 2000;3:1478–1489. doi: 10.1016/S0006-3495(00)76399-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Elvington S.M., Nichols J.W. Spontaneous, intervesicular transfer rates of fluorescent, acyl chain-labeled phosphatidylcholine analogs. Biochimica et Biophysica Acta. 2007;3:502–508. doi: 10.1016/j.bbamem.2006.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sezgin E., Levental I., Grzybek M., Schwarzmann G., Mueller V., Honigmann A., et al. Partitioning, diffusion, and ligand binding of raft lipid analogs in model and cellular plasma membranes. Biochimica et Biophysica Acta. 2012;7:1777–1784. doi: 10.1016/j.bbamem.2012.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Takatori S., Mesman R., Fujimoto T. Microscopic methods to observe the distribution of lipids in the cellular membrane. Biochemistry. 2014;4:639–653. doi: 10.1021/bi401598v. [DOI] [PubMed] [Google Scholar]
- 13.Haberkant P., Holthuis J.C. Fat & fabulous: bifunctional lipids in the spotlight. Biochimica et Biophysica Acta. 2014;8:1022–1030. doi: 10.1016/j.bbalip.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 14.Varnai P., Thyagarajan B., Rohacs T., Balla T. Rapidly inducible changes in phosphatidylinositol 4,5-bisphosphate levels influence multiple regulatory functions of the lipid in intact living cells. The Journal of Cell Biology. 2006;3:377–382. doi: 10.1083/jcb.200607116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamaji A., Sekizawa Y., Emoto K., Sakuraba H., Inoue K., Kobayashi H., et al. Lysenin, a novel sphingomyelin-specific binding protein. The Journal of Biological Chemistry. 1998;9:5300–5306. doi: 10.1074/jbc.273.9.5300. [DOI] [PubMed] [Google Scholar]
- 16.Fishman P.H. Role of membrane gangliosides in the binding and action of bacterial toxins. The Journal of Membrane Biology. 1982;2:85–97. doi: 10.1007/BF01872268. [DOI] [PubMed] [Google Scholar]
- 17.Maxfield F.R., Wustner D. Analysis of cholesterol trafficking with fluorescent probes. Methods in Cell Biology. 2012:367–393. doi: 10.1016/B978-0-12-386487-1.00017-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bornig H., Geyer G. Staining of cholesterol with the fluorescent antibiotic “filipin”. Acta Histochemica. 1974;1:110–115. [PubMed] [Google Scholar]
- 19.Krishnamurthy K., Dasgupta S., Bieberich E. Development and characterization of a novel anti-ceramide antibody. Journal of Lipid Research. 2007;4:968–975. doi: 10.1194/jlr.D600043-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Subramaniam H.N., Chaubal K.A. Evaluation of intracellular lipids by standardized staining with a Sudan black B fraction. Journal of Biochemical and Biophysical Methods. 1990;1:9–16. doi: 10.1016/0165-022x(90)90040-j. [DOI] [PubMed] [Google Scholar]
- 21.Koopman R., Schaart G., Hesselink M.K. Optimisation of oil red O staining permits combination with immunofluorescence and automated quantification of lipids. Histochemistry and Cell Biology. 2001;1:63–68. doi: 10.1007/s004180100297. [DOI] [PubMed] [Google Scholar]
- 22.Elle I.C., Olsen L.C., Pultz D., Rodkaer S.V., Faergeman N.J. Something worth dyeing for: molecular tools for the dissection of lipid metabolism in Caenorhabditis elegans. FEBS Letters. 2010;11:2183–2193. doi: 10.1016/j.febslet.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 23.Spangenburg E.E., Pratt S.J., Wohlers L.M., Lovering R.M. Use of BODIPY (493/503) to visualize intramuscular lipid droplets in skeletal muscle. Journal of Biomedicine and Biotechnology. 2011;2011:598358. doi: 10.1155/2011/598358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greenspan P., Mayer E.P., Fowler S.D. Nile red: a selective fluorescent stain for intracellular lipid droplets. The Journal of Cell Biology. 1985;3:965–973. doi: 10.1083/jcb.100.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown W.J., Sullivan T.R., Greenspan P. Nile red staining of lysosomal phospholipid inclusions. Histochemistry. 1992;4:349–354. doi: 10.1007/BF00270037. [DOI] [PubMed] [Google Scholar]
- 26.Diaz G., Melis M., Batetta B., Angius F., Falchi A.M. Hydrophobic characterization of intracellular lipids in situ by Nile Red red/yellow emission ratio. Micron. 2008;7:819–824. doi: 10.1016/j.micron.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Digel M., Ehehalt R., Fullekrug J. Lipid droplets lighting up: insights from live microscopy. FEBS Letters. 2010;11:2168–2175. doi: 10.1016/j.febslet.2010.03.035. [DOI] [PubMed] [Google Scholar]
- 28.van Loon L.J., Koopman R., Manders R., van der Weegen W., van Kranenburg G.P., Keizer H.A. Intramyocellular lipid content in type 2 diabetes patients compared with overweight sedentary men and highly trained endurance athletes. American Journal of Physiology, Endocrinology and Metabolism. 2004;3:E558–E565. doi: 10.1152/ajpendo.00464.2003. [DOI] [PubMed] [Google Scholar]
- 29.Cremer C., Cremer T. Considerations on a laser-scanning-microscope with high resolution and depth of field. Microscopica Acta. 1978;1:31–44. [PubMed] [Google Scholar]
- 30.Sheppard C.J.R., Matthews H.J. Imaging in high-aperture optical systems. Journal of the Optical Society of America A. 1987;8:1354–1360. [Google Scholar]
- 31.Lyn R.K., Kennedy D.C., Stolow A., Ridsdale A., Pezacki J.P. Dynamics of lipid droplets induced by the hepatitis C virus core protein. Biochemical and Biophysical Research Communications. 2010;4:518–524. doi: 10.1016/j.bbrc.2010.07.101. [DOI] [PubMed] [Google Scholar]
- 32.Valetti C., Wetzel D.M., Schrader M., Hasbani M.J., Gill S.R., Kreis T.E., et al. Role of dynactin in endocytic traffic: effects of dynamitin overexpression and colocalization with CLIP-170. Molecular Biology of the Cell. 1999;12:4107–4120. doi: 10.1091/mbc.10.12.4107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y., Ramachandran P.V., Wang M.C. Shedding new light on lipid functions with CARS and SRS microscopy. Biochimica et Biophysica Acta. 2014;8:1120–1129. doi: 10.1016/j.bbalip.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Min W., Freudiger C.W., Lu S., Xie X.S. Coherent nonlinear optical imaging: beyond fluorescence microscopy. Annual Review of Physical Chemistry. 2011:507–530. doi: 10.1146/annurev.physchem.012809.103512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Billecke N., Rago G., Bosma M., Eijkel G., Gemmink A., Leproux P., et al. Chemical imaging of lipid droplets in muscle tissues using hyperspectral coherent Raman microscopy. Histochemistry and Cell Biology. 2014;3:263–273. doi: 10.1007/s00418-013-1161-2. [DOI] [PubMed] [Google Scholar]
- 36.Wang M.C., Min W., Freudiger C.W., Ruvkun G., Xie X.S. RNAi screening for fat regulatory genes with SRS microscopy. Nature Methods. 2011;2:135–138. doi: 10.1038/nmeth.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zumbusch A., Langbein W., Borri P. Nonlinear vibrational microscopy applied to lipid biology. Progress in Lipid Research. 2013;4:615–632. doi: 10.1016/j.plipres.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Jungst C., Klein M., Zumbusch A. Long-term live cell microscopy studies of lipid droplet fusion dynamics in adipocytes. Journal of Lipid Research. 2013;12:3419–3429. doi: 10.1194/jlr.M042515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Billecke N., Bosma M., Rock W., Fleissner F., Best G., Schrauwen P., et al. Perilipin 5 mediated lipid droplet remodelling revealed by coherent Raman imaging. Integrative Biology: Quantitative Biosciences from Nano to Macro. 2015;4:467–476. doi: 10.1039/c4ib00271g. [DOI] [PubMed] [Google Scholar]
- 40.Nan X., Potma E.O., Xie X.S. Nonperturbative chemical imaging of organelle transport in living cells with coherent anti-stokes Raman scattering microscopy. Biophysical Journal. 2006;2:728–735. doi: 10.1529/biophysj.105.074534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jungst C., Winterhalder M.J., Zumbusch A. Fast and long term lipid droplet tracking with CARS microscopy. Journal of Biophotonics. 2011;6:435–441. doi: 10.1002/jbio.201000120. [DOI] [PubMed] [Google Scholar]
- 42.Li Y., Lee S., Langleite T., Norheim F., Pourteymour S., Jensen J., et al. Subsarcolemmal lipid droplet responses to a combined endurance and strength exercise intervention. Physiological Reports. 2014;11 doi: 10.14814/phy2.12187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holloway G.P., Han X.X., Jain S.S., Bonen A., Chabowski A. Chronic muscle stimulation improves insulin sensitivity while increasing subcellular lipid droplets and reducing selected diacylglycerol and ceramide species in obese Zucker rats. Diabetologia. 2014;4:832–840. doi: 10.1007/s00125-014-3169-0. [DOI] [PubMed] [Google Scholar]
- 44.Shaw C.S., Jones D.A., Wagenmakers A.J. Network distribution of mitochondria and lipid droplets in human muscle fibres. Histochemistry and Cell Biology. 2008;1:65–72. doi: 10.1007/s00418-007-0349-8. [DOI] [PubMed] [Google Scholar]
- 45.Cushman S.W. Structure-function relationships in the adipose cell. I. Ultrastructure of the isolated adipose cell. The Journal of Cell Biology. 1970;2:326–341. doi: 10.1083/jcb.46.2.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang H., Lei M., Hsia R.C., Sztalryd C. Analysis of lipid droplets in cardiac muscle. Methods in Cell Biology. 2013:129–149. doi: 10.1016/B978-0-12-408051-5.00008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herms A., Bosch M., Ariotti N., Reddy B.J., Fajardo A., Fernandez-Vidal A., et al. Cell-to-cell heterogeneity in lipid droplets suggests a mechanism to reduce lipotoxicity. Current Biology: CB. 2013;15:1489–1496. doi: 10.1016/j.cub.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kassan A., Herms A., Fernandez-Vidal A., Bosch M., Schieber N.L., Reddy B.J., et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. The Journal of Cell Biology. 2013;6:985–1001. doi: 10.1083/jcb.201305142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ariotti N., Murphy S., Hamilton N.A., Wu L., Green K., Schieber N.L., et al. Postlipolytic insulin-dependent remodeling of micro lipid droplets in adipocytes. Molecular Biology of the Cell. 2012;10:1826–1837. doi: 10.1091/mbc.E11-10-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nan X., Cheng J.X., Xie X.S. Vibrational imaging of lipid droplets in live fibroblast cells with coherent anti-Stokes Raman scattering microscopy. Journal of Lipid Research. 2003;11:2202–2208. doi: 10.1194/jlr.D300022-JLR200. [DOI] [PubMed] [Google Scholar]
- 51.Paar M., Jungst C., Steiner N.A., Magnes C., Sinner F., Kolb D., et al. Remodeling of lipid droplets during lipolysis and growth in adipocytes. The Journal of Biological Chemistry. 2012;14:11164–11173. doi: 10.1074/jbc.M111.316794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barneda D., Frontini A., Cinti S., Christian M. Dynamic changes in lipid droplet-associated proteins in the “browning” of white adipose tissues. Biochimica et Biophysica Acta. 2013;5:924–933. doi: 10.1016/j.bbalip.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 53.Braga J., Desterro J.M., Carmo-Fonseca M. Intracellular macromolecular mobility measured by fluorescence recovery after photobleaching with confocal laser scanning microscopes. Molecular Biology of the Cell. 2004;10:4749–4760. doi: 10.1091/mbc.E04-06-0496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ishikawa-Ankerhold H.C., Ankerhold R., Drummen G.P. Advanced fluorescence microscopy techniques – FRAP, FLIP, FLAP, FRET and FLIM. Molecules. 2012;4:4047–4132. doi: 10.3390/molecules17044047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Somwar R., Roberts C.T., Jr., Varlamov O. Live-cell imaging demonstrates rapid cargo exchange between lipid droplets in adipocytes. FEBS Letters. 2011;12:1946–1950. doi: 10.1016/j.febslet.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 56.Gong J., Sun Z., Wu L., Xu W., Schieber N., Xu D., et al. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. The Journal of Cell Biology. 2011;6:953–963. doi: 10.1083/jcb.201104142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mason R.R., Meex R.C., Russell A.P., Canny B.J., Watt M.J. Cellular localization and associations of the major lipolytic proteins in human skeletal muscle at rest and during exercise. PLoS One. 2014;7:e103062. doi: 10.1371/journal.pone.0103062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hashimoto T., Segawa H., Okuno M., Kano H., Hamaguchi H.O., Haraguchi T., et al. Active involvement of micro-lipid droplets and lipid-droplet-associated proteins in hormone-stimulated lipolysis in adipocytes. Journal of Cell Science. 2012;(Pt 24):6127–6136. doi: 10.1242/jcs.113084. [DOI] [PubMed] [Google Scholar]
- 59.Jacquier N., Choudhary V., Mari M., Toulmay A., Reggiori F., Schneiter R. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae. Journal of Cell Science. 2011;(Pt 14):2424–2437. doi: 10.1242/jcs.076836. [DOI] [PubMed] [Google Scholar]
- 60.Schermelleh L., Heintzmann R., Leonhardt H. A guide to super-resolution fluorescence microscopy. The Journal of Cell Biology. 2010;2:165–175. doi: 10.1083/jcb.201002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilfling F., Wang H., Haas J.T., Krahmer N., Gould T.J., Uchida A., et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Developmental Cell. 2013;4:384–399. doi: 10.1016/j.devcel.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.York A.G., Ghitani A., Vaziri A., Davidson M.W., Shroff H. Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nature Methods. 2011;4:327–333. doi: 10.1038/nmeth.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eggert D., Rosch K., Reimer R., Herker E. Visualization and analysis of hepatitis C virus structural proteins at lipid droplets by super-resolution microscopy. PLoS One. 2014;7:e102511. doi: 10.1371/journal.pone.0102511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McIntosh A.L., Senthivinayagam S., Moon K.C., Gupta S., Lwande J.S., Murphy C.C., et al. Direct interaction of Plin2 with lipids on the surface of lipid droplets: a live cell FRET analysis. American Journal of Physiology Cell Physiology. 2012;7:C728–C742. doi: 10.1152/ajpcell.00448.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sezgin E., Schwille P. Fluorescence techniques to study lipid dynamics. Cold Spring Harbor Perspectives in Biology. 2011;11:a009803. doi: 10.1101/cshperspect.a009803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Romero-Brey I., Merz A., Chiramel A., Lee J.Y., Chlanda P., Haselman U., et al. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathogens. 2012;12:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Peddie C.J., Blight K., Wilson E., Melia C., Marrison J., Carzaniga R., et al. Correlative and integrated light and electron microscopy of in-resin GFP fluorescence, used to localise diacylglycerol in mammalian cells. Ultramicroscopy. 2014:3–14. doi: 10.1016/j.ultramic.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Parekh S.H., Lee Y.J., Aamer K.A., Cicerone M.T. Label-free cellular imaging by broadband coherent anti-Stokes Raman scattering microscopy. Biophysical Journal. 2010;8:2695–2704. doi: 10.1016/j.bpj.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rinia H.A., Burger K.N., Bonn M., Muller M. Quantitative label-free imaging of lipid composition and packing of individual cellular lipid droplets using multiplex CARS microscopy. Biophysical Journal. 2008;10:4908–4914. doi: 10.1529/biophysj.108.137737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bonn M., Muller M., Rinia H.A., Burger K.N.J. Imaging of chemical and physical state of individual cellular lipid droplets using multiplex CARS microscopy. Journal of Raman Spectroscopy. 2009;7:763–769. [Google Scholar]
- 71.Fu D., Yu Y., Folick A., Currie E., Farese R.V., Jr., Tsai T.H., et al. In vivo metabolic fingerprinting of neutral lipids with hyperspectral stimulated Raman scattering microscopy. Journal of the American Chemical Society. 2014;24:8820–8828. doi: 10.1021/ja504199s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boxer S.G., Kraft M.L., Weber P.K. Advances in imaging secondary ion mass spectrometry for biological samples. Annual Review of Biophysics. 2009:53–74. doi: 10.1146/annurev.biophys.050708.133634. [DOI] [PubMed] [Google Scholar]
- 73.Malmberg P., Borner K., Chen Y., Friberg P., Hagenhoff B., Mansson J.E., et al. Localization of lipids in the aortic wall with imaging TOF-SIMS. Biochimica et Biophysica Acta. 2007;2:185–195. doi: 10.1016/j.bbalip.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 74.Tahallah N., Brunelle A., De La Porte S., Laprevote O. Lipid mapping in human dystrophic muscle by cluster-time-of-flight secondary ion mass spectrometry imaging. Journal of Lipid Research. 2008;2:438–454. doi: 10.1194/jlr.M700421-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lazar A.N., Bich C., Panchal M., Desbenoit N., Petit V.W., Touboul D., et al. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathologica. 2013;1:133–144. doi: 10.1007/s00401-012-1041-1. [DOI] [PubMed] [Google Scholar]
- 76.Kraft M.L., Klitzing H.A. Imaging lipids with secondary ion mass spectrometry. Biochimica et Biophysica Acta. 2014;8:1108–1119. doi: 10.1016/j.bbalip.2014.03.003. [DOI] [PubMed] [Google Scholar]
- 77.Kleinfeld A.M., Kampf J.P., Lechene C. Transport of 13C-oleate in adipocytes measured using multi imaging mass spectrometry. Journal of the American Society for Mass Spectrometry. 2004;11:1572–1580. doi: 10.1016/j.jasms.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 78.Jiang H., Goulbourne C.N., Tatar A., Turlo K., Wu D., Beigneux A.P., et al. High-resolution imaging of dietary lipids in cells and tissues by NanoSIMS analysis. Journal of Lipid Research. 2014;10:2156–2166. doi: 10.1194/jlr.M053363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saka S.K., Vogts A., Krohnert K., Hillion F., Rizzoli S.O., Wessels J.T. Correlated optical and isotopic nanoscopy. Nature Communications. 2014;5:3664. doi: 10.1038/ncomms4664. [DOI] [PMC free article] [PubMed] [Google Scholar]