

Abstract
Here we present a combined experimental and theoretical study on the secondary structure of isolated proteins as a function of charge state. In infrared spectra of the proteins ubiquitin and cytochrome c, amide I (C=O stretch) and amide II (N–H bend) bands can be found at positions that are typical for condensed‐phase proteins. For high charge states a new band appears, substantially red‐shifted from the amide II band observed at lower charge states. The observations are interpreted in terms of Coulomb‐driven transitions in secondary structures from mostly helical to extended C5‐type hydrogen‐bonded structures. Support for this interpretation comes from simple energy considerations as well as from quantum chemical calculations on model peptides. This transition in secondary structure is most likely universal for isolated proteins that occur in mass spectrometric experiments.
Keywords: gas-phase reactions, IR spectroscopy, low-temperature physics, mass spectrometry, protein structures
The structure and dynamics of proteins is determined by a delicate balance between various attractive and repulsive interactions. Aside from covalent bonds, various noncovalent interactions such as Pauli repulsion, dispersive interactions, hydrogen bonding, and Coulomb interactions shape the potential energy surface and dictate the resulting structural and dynamical preferences. In the condensed phase, interactions with the surroundings play an important role. Those are explicitly absent when, either for analytical purposes or in order to study intrinsic properties, experiments are performed on gas‐phase species. Under those conditions, the interplay between hydrogen bonding and Coulomb repulsion plays a crucial role in structure formation. In a hallmark experiment, gas‐phase ion‐mobility methods were used to observe the Coulomb driven transition from folded, compact shapes to more extended conformations.1 By determining the size (collision cross‐section) of the ions via measuring their gas‐phase mobility as function of charge for the proteins cytochrome c1, 2 and ubiquitin,2b, 3 these experiments showed that a variety of structures can be populated. For high charge states, presumably helical structures undergo a gradual increase in collision cross‐section, which is an indication for the possible unzipping of helices to even more‐extended structures.1b, 2b However, while ion‐mobility spectrometry provides information on the overall size and shape of the protein, it is not directly sensitive to structural details, such as the secondary structure. This gap is filled by infrared (IR) spectroscopy, where the position and shape of absorption bands can be used to gain information on the more local secondary structure. The C=O stretching modes (amide I) and N–H bending modes (amide II) are of particular importance in IR spectroscopy because their positions and shapes are sensitive to the local hydrogen bonding. This is why IR spectroscopy is a an established tool for investigating the secondary structures of peptides and proteins in the condensed phase.4 Here, we use IR radiation to investigate the change in secondary structure of the proteins ubiquitin and cytochrome c as a function of charge state in the ultracold environment provided by helium droplets. Our data indicate a smooth transition from helical to extended structures that can be traced by distinct bands within the amide II region.
The experimental setup is shown schematically in Figure 1. It consists of a newly built apparatus that follows the same operational principle as the experimental setup employed in the past.5 Ions, brought into the gas phase via electrospray ionization, are mass‐to‐charge selected with a quadrupole mass filter and accumulated inside a hexapole ion trap for several seconds. Once the trap is filled, the ions can be picked up by a pulsed beam of helium droplets that traverses the trap; the helium droplets employed in this work consist on average of ca. 106 helium atoms. After pick‐up takes place, the ion cools down to the ca. 0.37 K equilibrium temperature of the droplet and the doped droplets can leave the trap, because their kinetic energy is much larger than the longitudinal trapping potential. Further downstream, the ion‐doped helium droplets are then probed using the Fritz Haber Institute IR free‐electron laser (FHI FEL6). Upon resonant absorption of (multiple) photons, the ion can be ejected from the droplet.5b, 7 Although this process is not yet fully understood, ejection can be used as a marker for photon absorption, and when the ejection yield is monitored while the IR frequency is scanned, an IR spectrum is obtained.7b, 8
Figure 1.
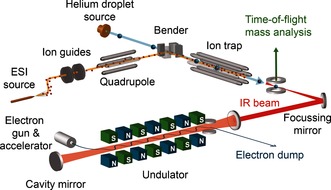
Schematic diagram of the experimental setup. Ions are brought into the gas phase, mass‐to‐charge selected and accumulated in the ion trap. Helium droplets can pick up those ions and doped droplets are interrogated using the free‐electron laser.
IR spectra of the proteins ubiquitin and cytochrome c in the wavenumber range from 1400 cm−1 to 1800 cm−1 are shown in Figures 2 a and 2 b. For each protein the IR spectra are classified and labeled according to the charge state of the ions as low (L), intermediate (I), and high (H). With the exception of cytochrome c 24+, all spectra show a band near 1650 cm−1. For proteins, a band at this position usually dominates the mid‐IR spectrum and stems from backbone C=O stretch (amide I) vibrations. In the spectra shown here, the position of this band is observed to shift to slightly higher wavenumbers with increasing charge state, concomitant with a decrease in relative intensity. On the lower wavenumber side, two additional bands are observed. They occur in the region where one characteristic band from backbone N–H bending (amide II) vibrations is expected in protein IR spectra. Here, however, two bands are observed: one near 1480 cm−1 (labeled amide IIa) and the other one near 1550 cm−1 (labeled amide IIb). The amide IIb band is observed only for low and intermediate charge states. In contrast, the amide IIa band appears for intermediate charge states and grows in relative intensity with increasing charge, while the amide IIb band disappears. Besides the spectra shown in Figure 2 a and 2 b, spectra for most in‐between charge states were measured, showing the same trends. The relative intensities of the amide IIa and IIb bands as a function of charge state for the two proteins are shown in Figure 2 c. Clearly, the curves show very similar behavior, with the difference being that the crossover from amide IIb to IIa is observed to occur around charge state 11+ for ubiquitin and around 14+ for the larger protein cytochrome c.
Figure 2.
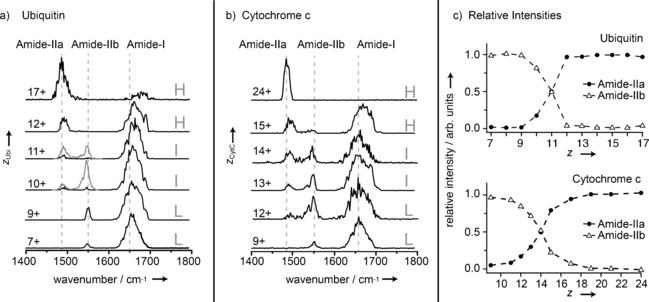
IR spectra of a) ubiquitin and b) cytochrome c for various charge states. Gray trace segments for ubiquitin 10+ and 11+ denote a higher photon density. Three types of bands can be identified which are labeled amide IIa, amide IIb, and amide I. c) Relative intensities of the amide IIa and IIb bands as a function of charge state.
It is also important to note that the obtained spectra result from the absorption of multiple photons. Although bands in the spectra will scale nonlinearly with absorption cross‐section and laser power, here, as a first‐order approximation, a linear correction has been performed by dividing the signal by the laser power. Therefore, relative intensities in the present experiment should only be regarded as a general guide. Peak positions, on the other hand, are not expected to be significantly influenced by the multiple‐photon process in the present experiment. Due to the time structure of the IR light pulse, consecutive absorption events will take place over a period of up to several microseconds. As the ion is embedded in the helium droplet, it will most likely have cooled down in between absorption events and, as a result, each absorption will take place from the ion's vibrational ground state. Therefore, in contrast to the experiments using IR multiple photon dissociation (IRMPD),9 anharmonicities or cross‐anharmonicities are not expected to play a role here. This is confirmed by recent IR spectra of peptides in helium droplets.7b
In the condensed phase, amide I bands generally show a red‐shift with increasing hydrogen bonding and are typically found in a range between 1654 cm−1 for mostly helical or random‐coil species to 1633 cm−1 for β‐sheet‐rich structures.4 The position of the amide I band observed here (ca. 1650 cm−1) falls in this range and its position could be considered as an indicator of a mostly helical structure. The amide II band is usually found around 1550 cm−1 and its exact position is not as directly correlated to the secondary structure.4 The position of the band observed here near 1550 cm−1, labeled amide IIb, is thus in good agreement with typical amide II band positions of proteins in the condensed phase. So what is the origin of the band found to the red of the amide IIb band—the amide IIa band?
For a smaller range of charge states, IR spectra of cytochrome c at room temperature have been measured previously using the IRMPD technique.11 There, qualitatively similar but much broader spectra were observed, which confirms that the (unknown) dynamics of the ejection process or vibrational anharmonicities do not have a significant effect on the spectra shown here. Interestingly, in those spectra, an additional band at around 1483 cm−1 was observed as well; however, at the time, a satisfying assignment of this band could not be given.11 Ion‐mobility experiments have clearly shown that proteins can undergo a transition from compact, possibly native‐like structures to more extended, helical structures when the charge of the ion increases.1b For cytochrome c, this initial unfolding transition manifests itself as a sharp increase in collision cross‐section (size), which occurs in a charge‐state range of 7+ to 8+1b and for ubiquitin around 6+ to 7+.3a When the charge on the molecule increases further, a gradual increase in collision cross‐section is observed. The predicted cross‐section limit of a fully extended string‐like structure is, however, not reached, and, using a model, such structures are calculated to occur only at very high charge states.1b
The transition from the amide IIb to IIa band observed here might indicate a drastic change in structure, which occurs over a small range of charge states. Helical structures allow for a stable hydrogen‐bonding pattern and, at the same time, provide for a relatively large distance between charges in order to minimize Coulomb repulsion. As has been recognized earlier,1b, 2b increasing the number of charges and, hence Coulomb repulsion further, will at one point cause an unzipping of the helix, thereby allowing charges to be at even larger distances. In such an unzipped, completely extended structure, which resembles the backbone orientation of a single‐strand β‐sheet, C5‐type hydrogen bonds between adjacent C=O and N–H groups serve as stabilizing elements.
To estimate the energy difference between a helical and fully extended structure, the corresponding Coulomb potential energy (E C) of N charges separated by distances rij can be calculated as:
where ɛ 0 and ɛ r are the vacuum and relative permittivities, respectively. If one assumes N equal charges being distributed equidistant over a length L, the above equation can be expressed as:
The calculated difference between the Coulomb energy per amino acid residue (ΔE C) of an extended and a helical structure as a function of the charge state of each protein is depicted in Figure 3 a. The length of an α‐helix is taken as L Helix=1.5 n A (Å) and that of an extended structure by L Ext.=3.5 n A (Å) (n A: the number of amino acids) and ɛ r=1.
Figure 3.
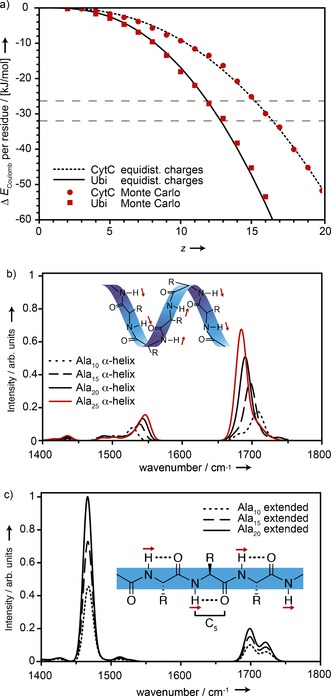
a) Difference in Coulomb energy (ΔE C) per amino acid residue between helical and extended conformations of ubiquitin and cytochrome c. The dashed horizontal lines indicate helix stabilization energies obtained from quantum chemistry calculations.10 b,c) Calculated IR spectra for neutral polyalanine in α‐helical (b) and extended (c) conformations. Shown schematically as red arrows are the directions of the amide II transition dipoles. C5 hydrogen bonds for extended structures are shown schematically as dotted lines.
The black lines in Figure 3 a represent the values of ΔE C obtained for equally spaced charges. In a more realistic model, the molecular charge distribution has to be based on individual amino acid basicities and the Coulomb potential energy. In order to calculate such a charge distribution, we use a Monte Carlo simulated annealing scheme, similar to the approach taken by Schnier et al.12 In this model, the well‐known amino acid sequences of ubiquitin and cytochrome c (the heme group is ignored) are used and, both the helical as well as extended conformations are approximated as linear chains with a total length of L Helix=1.5 n A (Å) and L Ext.=3.5 n A (Å), respectively, and equidistant spacing between amino acids. The initial charge distributions are assigned randomly and the annealing parameters are chosen such that the same lowest energy charge distribution is reliably reached for repeated calculations. The resulting ΔE C values are shown in Figure 3 a as red squares and circles and are in good agreement with the simple equidistant model.
Depending on the method, quantum theory gives values for the helix stabilization energy per amino acid with reference to a fully extended structure of −26.4 kJ mol−1[10a] and −32.6 kJ mol−1.10b Those values are shown as gray dashed lines in Figure 3 a. When the charge on the molecule increases, the extended structure becomes more stable once the difference in Coulomb energy ΔE C falls below the helix stabilization energy. The model thus predicts that for ubiquitin, an extended structure is favored starting at around 12+ or 13+ and for cytochrome c at around 15+ and 16+. This roughly coincides with the charge‐state regions where the experimental transition between amide Ilb to amide IIa bands occurs. Therefore, the unzipping of a helix to form a C5‐type secondary structure seems a viable explanation from an energetic point of view.
To test whether such a change in secondary structure is supported by the observed spectral signatures, IR spectra of neutral polyalanine peptides were calculated for extended C5‐type as well as for helical secondary structures. The calculations were performed at the B3LYP level of theory, using the def2‐SVP basis set and adding dispersion corrections using the Grimme D3 method,13 as implemented in Gaussian 09.14 All vibrational frequencies are scaled by 0.965. Figure 3 b shows calculated spectra for Ala10 to Ala25 in α‐helical conformation. Due to an increase in hydrogen bonding and macro‐dipole, the amide I band position shifts from 1708 cm−1 for the Ala10 α‐helix to 1685 cm−1 for the Ala25 α‐helix; for the same reason, the amide II band position shifts from 1528 cm−1 to 1547 cm−1. In Figure 3 c, IR spectra for extended C5 conformations are shown. Their amide I and II signatures do not depend significantly on size and show a split amide I band at 1721 cm−1 and 1698 cm−1 as well as an amide II band at 1466 cm−1. Interestingly, and in contrast to the spectra calculated for the α‐helices, the amide II band in the spectra of the C5‐type structures is much stronger than the amide I band. This effect originates from the directions of the transition dipoles of the N–H and C=O oscillators (see insets in Figures 3 b and 3 c). For the C=O stretching vibration, the transition dipole is oriented in parallel to the C=O bond and in helical structures, individual transition dipoles can add up constructively. On the other hand, the transition dipole of the amide II N–H bending vibration is perpendicular to the N−H bond and parallel with the peptide chain. For a helical structure, this results in transition dipoles that do not add up constructively, thus leading to a reduced overall macro transition dipole. For an extended C5 structure, on the other hand, the individual N–H transition dipoles are again parallel to each other.
The experimental results show a red shift of about 70 cm−1 for the amide IIa with respect to the amide IIb band. In the calculations, the red shift due to the transition from a helix to an extended structure is about 80 cm−1. It also should be noted that the calculations are performed for an idealized structure with no charges, containing only alanine. Taking this into account, the agreement between experiment and theory is remarkable and the calculated IR spectra thus support an unzipping as an explanation for the observed change in line positions. Moreover, in the experiment, the intensity of the amide IIa band is observed to strongly increase with the number of charges until it dominates the spectra of proteins with very high charge states as predicted by theory.
In conclusion, the experimentally observed charge‐state dependence of the IR spectra can be interpreted by the unzipping of a helix to form a stable secondary structure that is stabilized by C5 hydrogen bonds. Support comes from a simple electrostatic model as well as from IR spectral simulations, which qualitatively predict the observed changes in peak position and relative intensity. The unzipping is observed for two different proteins, ubiquitin and cytochrome c. In ion‐mobility studies, results also indicate such a transition, however rather as a gradual increase in cross‐section with charge state, not quite approaching the calculated limit for a completely linear structure.1b, 2, 3e In this work, it is also not clear whether the overall structure is fully linear, and the presence of kinks and bends caused by interactions of charged side‐chain groups with the protein backbone cannot be fully excluded. However, the spectroscopic data presented here shows that for high charge states, the structure is at least locally extended and linear with the absence of helical structure. In such an extended conformation, the structure is dominated by Coulomb repulsion and only weak C5‐type hydrogen bonds are present. While such single‐strand β‐sheet structures are not expected to be relevant in the condensed phase, it is likely a universal form of secondary structure in highly charged gas‐phase protein ions. As such, the presented results are of fundamental importance for mass spectrometry experiments in which highly charged peptide or protein ions are investigated, for example for protein sequencing.
A. I. González Flórez, E. Mucha, D.-S. Ahn, S. Gewinner, W. Schöllkopf, K. Pagel, G. von Helden, Angew. Chem. Int. Ed. 2016, 55, 3295.
References
- 1.
- 1a. Clemmer D. E., Hudgins R. R., Jarrold M. F., J. Am. Chem. Soc. 1995, 117, 10141–10142; [Google Scholar]
- 1b. Shelimov K. B., Clemmer D. E., Hudgins R. R., Jarrold M. F., J. Am. Chem. Soc. 1997, 119, 2240–2248. [Google Scholar]
- 2.
- 2a. Valentine S. J., Clemmer D. E., J. Am. Soc. Mass Spectrom. 2002, 13, 506–517; [DOI] [PubMed] [Google Scholar]
- 2b. Going C. C., Williams E. R., Anal. Chem. 2015, 87, 3973–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Valentine S. J., Counterman A. E., Clemmer D. E., J. Am. Soc. Mass Spectrom. 1997, 8, 954–961; [DOI] [PubMed] [Google Scholar]
- 3b. Koeniger S. L., Merenbloom S. I., Clemmer D. E., J. Phys. Chem. B 2006, 110, 7017–7021; [DOI] [PubMed] [Google Scholar]
- 3c. Koeniger S. L., Merenbloom S. I., Sevugarajan S., Clemmer D. E., J. Am. Chem. Soc. 2006, 128, 11713–11719; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Koeniger S. L., Clemmer D. E., J. Am. Soc. Mass Spectrom. 2007, 18, 322–331; [DOI] [PubMed] [Google Scholar]
- 3e. Wyttenbach T., Bowers M. T., J. Phys. Chem. B 2011, 115, 12266–12275. [DOI] [PubMed] [Google Scholar]
- 4. Barth A., Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1073–1101. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Bierau F., Kupser P., Meijer G., von Helden G., Phys. Rev. Lett. 2010, 105, 133402; [DOI] [PubMed] [Google Scholar]
- 5b. Filsinger F., Ahn D. S., Meijer G., von Helden G., Phys. Chem. Chem. Phys. 2012, 14, 13370–13377. [DOI] [PubMed] [Google Scholar]
- 6. Schöllkopf W., Gewinner S., Junkes H., Paarmann A., von Helden G., Bluem H., Todd A. M. M., in Advances in X-Ray Free-Electron Lasers Instrumentation III, Proceedings of SPIE, Vol. 9512 (Ed.: S. G. Biedron), 2015, p. 95121L. [Google Scholar]
- 7.
- 7a. Smolarek S., Brauer N. B., Buma W. J., Drabbels M., J. Am. Chem. Soc. 2010, 132, 14086–14091; [DOI] [PubMed] [Google Scholar]
- 7b. González Flórez A. I., Ahn D. S., Gewinner S., Schöllkopf W., von Helden G., Phys. Chem. Chem. Phys. 2015, 17, 21902–21911. [DOI] [PubMed] [Google Scholar]
- 8. Zhang X., Brauer N. B., Berden G., Rijs A. M., Drabbels M., J. Chem. Phys. 2012, 136, 044305. [DOI] [PubMed] [Google Scholar]
- 9. Oomens J., Sartakov B. G., Meijer G., von Helden G., Int. J. Mass Spectrom. 2006, 254, 1–19. [Google Scholar]
- 10.
- 10a. Tkatchenko A., Rossi M., Blum V., Ireta J., Scheffler M., Phys. Rev. Lett. 2011, 106, 118102; [DOI] [PubMed] [Google Scholar]
- 10b. Hua S., Xu L., Li W., Li S., J. Phys. Chem. B 2011, 115, 11462–11469. [DOI] [PubMed] [Google Scholar]
- 11. Oomens J., Polfer N., Moore D. T., van der Meer L., Marshall A. G., Eyler J. R., Meijer G., von Helden G., Phys. Chem. Chem. Phys. 2005, 7, 1345. [DOI] [PubMed] [Google Scholar]
- 12. Schnier P. D., Gross D. S., Williams E. R., J. Am. Soc. Mass Spectrom. 1995, 6, 1086–1097. [DOI] [PubMed] [Google Scholar]
- 13. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 14.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian, Inc., Wallingford, CT, USA, 2009.