

Abstract
A copper complex has proven to be a potent catalyst for forming a C–B bond via diborylation of arynes and alkynes, affording vic‐diborylarenes and vic‐diborylalkenes with high efficiency. A boryl‐substituted organocopper species, which is intermediately generated in the diborylation, has been found to be captured by a tin or a carbon electrophile, leading to three‐component borylstannylation or carboboration, in which C–B and C–Sn (or C) bonds are constructed simultaneously. Furthermore, reducing the Lewis acidity of the boron center with 1,8‐diaminonaphthalene decisively alters the regiochemical behavior of the borylcopper species, enabling the installation of a boryl moiety to occur at an internal carbon of terminal alkynes in borylstannylation and protoboration. Copper catalysis for C–Sn and C–Si bond‐forming processes via distannylation, hydrostannylation and silylstannylation, as well as silver catalysis for a C–B bond‐forming reaction, is also described.
Keywords: boron, copper, metalation, silicon, tin
1. Introduction
Main‐group organometallics containing boron,1 tin2 or silicon3 are highly important carbon nucleophiles utilized for introduction of functional groups and carbon–carbon bond‐forming processes, especially via cross‐coupling reactions4 (B: Suzuki–Miyaura, Sn: Migita–Kosugi–Stille, Si: Hiyama), because of their bench‐stable and easy‐to‐handle properties arising from their inertness toward oxygen and moisture, moderate reactivities suitable for chemoselective transformations, high functional group compatibility, etc. Whilst B‐, Sn‐ or Si‐containing organometallics with multifarious structures are accessible by such established methods as metal‐exchange reactions between highly nucleophilic organometallics (organolithiums, Grignard reagents) and metal electrophiles, hydrometalation of unsaturated C–C bonds and catalytic metalation of C–X bonds (X: hydrogen, halogen/pseudohalogen), the search for new synthetic methods that open up facile approaches to B‐, Sn‐ or Si‐containing organometallics of structural complexity with defined stereochemistry and/or high functionalities continues to be a central subject in chemical synthesis.
We have directed our attention to aryne‐based organic synthesis,5, 6 because this offers potent and unique methods for constructing aromatic frameworks that would not be achievable with stable compounds. In addition to successful insertion reactions into Nu–El σ bonds7 and multicomponent couplings8 based on the highly electrophilic character of arynes, their salient reactivities were found to be controllable under transition‐metal catalysis: arynes could be directly transformed into diboryl‐,9 distannyl‐10 and disilylarenes11 by dimetalation reactions catalyzed by group 10 transition‐metal complexes (Pd, Pt), demonstrating that arynes are promising building blocks in forming B‐, Sn‐ or Si‐containing organometallics. Furthermore, we have also exploited unique copper catalysis for converting arynes into biaryl derivatives via hydroalkynylation12 or bromoalkynylation.13 During the course of these studies, we became especially interested in the ambiphilic behavior of a copper species, which has both transition‐metal and main‐group‐metal character, and in its utilization as a catalyst for the transformation of arynes into B‐, Sn‐ or Si‐containing organometallics. This paper presents an overview of our recent study on the copper‐catalyzed borylation reaction of arynes and its application to other unsaturated C–C bonds. Related stannylation and silylation reactions are also discussed.
2. Borylation
2.1. Diborylation
At first our attention was riveted on the use of nucleophilic borylcopper species, arising from σ‐bond metathesis between Cu(I) complexes and diborons,14, 15, 16 as key intermediates, because they were expected to have high affinity for arynes of soft electrophilic character, leading to the occurrence of facile C–B bond‐forming processes.
We carried out the reaction of in situ generated benzyne (from 2‐(trimethylsilyl)phenyl triflate and KF/18‐crown‐6) with bis(pinacolato)diboron (1, (pin)B–B(pin)) in the presence of (PPh3)3CuOAc, and observed that the B–B bond was smoothly added across the C–C triple bond, affording a diborylation product (2a) in 68% yield (Scheme 1).17, 18 A variety of monosubstituted and disubstituted arynes reacted smoothly with the diboron to afford the respective vic‐diborylarenes (2b–2e) in good yields. It should be noted that the trimethylsilyl moiety of 4‐(trimethylsilyl)benzyne was totally tolerated regardless of the presence of the fluoride ion, leading to the selective formation of 2f.
Scheme 1.
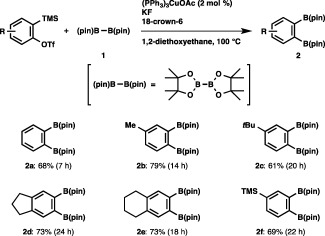
Diborylation of arynes.
As illustrated in Scheme 2, a key intermediate of the diborylation would be a borylcopper species (3) generated from copper acetate with 1. Subsequent insertion of the C–C triple bond of an aryne into the Cu–B bond to give a boryl‐substituted arylcopper species (4) followed by σ‐bond metathesis with 1 provides a diborylation product with regeneration of 3. The validity of the second step of the catalytic cycle was confirmed through the stoichiometric reaction of mesitylcopper(I) with 1, in which the C(sp2)–Cu bond was indeed transformed into the C(sp2)–B bond (Scheme 3).
Scheme 2.
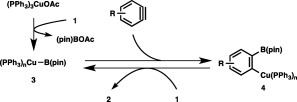
A catalytic cycle for the diborylation of arynes.
Scheme 3.
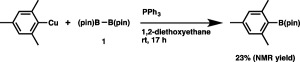
The stoichiometric reaction of mesitylcopper with (pin)B–B(pin).
The results imply that alkynes may also undergo the diborylation under similar conditions, since alkynes have been known to insert into the Cu–B bond of 3, giving boryl‐substituted alkenylcopper species.16 As depicted in Scheme 4, cis‐vic‐diborylalkene (5a) was produced in 82% yield when 4‐octyne was treated with 1 in the presence of PCy3 and Cu(OAc)2. Other aliphatic alkynes including 2‐octyne and 4‐methyl‐2‐pentyne could also participate in the diborylation to afford high yields of the respective vic‐diborylalkenes (5b and 5c), and treatment of a homopropargyl ether resulted in the formation of 5d, whose C–OMe bond remained intact throughout the reaction. The broad substrate scope for alkynes was demonstrated by extension to diarylalkynes and aryl(alkyl)alkynes to successfully produce 5e–5h.
Scheme 4.
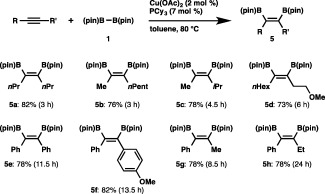
Diborylation of alkynes.
The most striking feature of the copper catalysis was observed in the reaction of a propargyl ether (Scheme 5). Thus, with the aid of the copper catalyst, 1,4‐dimethoxy‐2‐butyne was found to be solely convertible into a tetraborylation product, 1,2,3,4‐tetraboryl‐2‐butene (5i), in which four C–B bonds had been formed all in one pot by replacing the MeO groups with the boryl groups. This result was totally different from that using the established platinum catalyst (Pt(PPh3)4),19 which furnished the usual diborylation product (5′i).
Scheme 5.
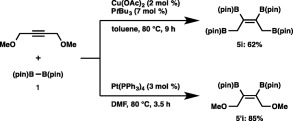
Cu‐ or Pt‐catalyzed borylation of 1,4‐dimethoxy‐2‐butyne.
We supposed that formation of borylallene 7 via β‐oxygen elimination from alkenylcopper species 6 should trigger the tetraborylation (Scheme 6).20 The resulting borylallene then inserted into the Cu–B bond of 3 to furnish π‐allyl copper species 8, which was transformed into 2,3‐diborylbutadiene (10) by a second β‐oxygen elimination from σ‐allyl copper species 9. Finally, 1,4‐diborylation of 10 occurred under the copper catalysis to provide 5i.
Scheme 6.
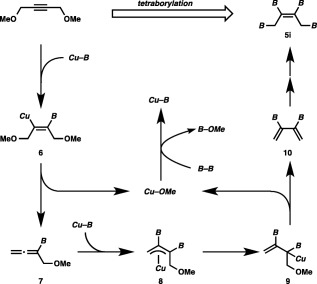
A catalytic cycle for the tetraborylation.
2.2. Borylstannylation
In view of the nucleophilicity of organocopper species, we envisaged that a boryl‐substituted alkenylcopper species (6), being an intermediate in the diborylation of alkynes, could serve as a key intermediate in a new type of borylation reaction by capturing suitable electrophiles, and found that the use of a tin alkoxide as a third component enabled the first catalytic three‐component borylstannylation of unsaturated carbon linkages to occur, giving diverse vic‐borylstannylalkenes in a regio‐ and stereodefined manner.21
We first carried out the reaction of diphenylacetylene with 1 and tributyltin methoxide (11a) in the presence of Cu(OAc)2–PCy3, and observed that three‐component borylstannylation took place in a cis fashion to afford 12a in 87% yield (Scheme 7).22 Variously substituted diarylalkynes smoothly participated in the borylstannylation, in which almost equal amounts of regioisomers (12c and 12′c) were generated from an unsymmetrical alkyne. In marked contrast, the reaction of a diarylalkyne bearing an electron‐donating (MeO) and electron‐withdrawing (CF3) groups proceeded with perfect regioselectivity, and the stannyl moiety was attached to the geminal position relative to the 4‐methoxyphenyl moiety. The exclusive formation of a single regioisomer (12e–12g), whose stannyl moiety and the aryl moiety were invariably geminal, was also observed in the reaction of alkyl(aryl)alkynes, and moreover a trimethylstannyl moiety could be introduced into the product (12g) with the same regioselectivity by using trimethyltin tert‐butoxide 11b. Aliphatic alkynes were also found to be convertible into the products (12h and 12i), and it should be noted that the propargylic C–O bonds in 1,4‐dimethoxy‐2‐butyne remained intact throughout the borylstannylation, in contrast with the result obtained in the above tetraborylation. In addition, the reaction was applicable to terminal alkynes to afford the products bearing the stannyl moiety at the more substituted site. The functional group compatibility of the borylstannylation was appreciably high, and an aryl–Br bond, an alkyl–Cl bond and a cyano group were totally tolerated under the reaction conditions.
Scheme 7.
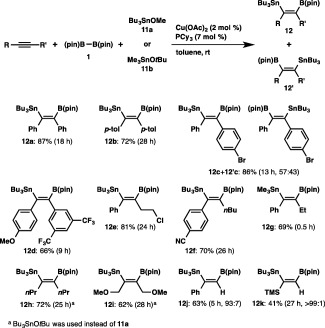
Borylstannylation of alkynes.
A key intermediate of the borylstannylation is expected to be a boryl‐substituted alkenylcopper species (6), which is generated by addition of borylcopper species 3 to an alkyne (step A, Scheme 8). Subsequent capture of 6 with a tin alkoxide (11) provides a borylstannylated product and a copper alkoxide (step B), which is transformed into 3 upon reaction with 1 (step C). Because steps A and C are well‐known processes in the copper‐catalyzed borylation reactions of alkynes, we conducted a reaction of 11a with an alkenylcopper species (13), which can be formed in situ by transmetalation between borylstilbene and Cu–X (X: OAc or OMe).23 As depicted in Scheme 9, the reaction led to the smooth formation of stannylstilbene, which confirms the validity of step B and the proposed catalytic cycle.
Scheme 8.
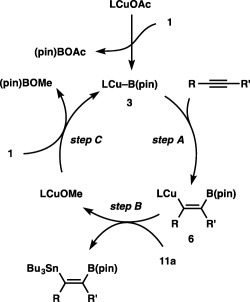
A catalytic cycle for the borylstannylation of alkynes.
Scheme 9.
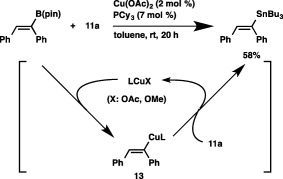
The reaction of an alkenylcopper with a tin alkoxide.
The borylstannylation has turned out to be applicable to alkenes: the reaction of methyl acrylate regioselectively afforded an 84% yield of vic‐borylstannylalkane 14a having a boryl moiety at the β‐position of the ester group (Scheme 10).24 Diastereoselectivity in the reaction using β‐substituted α,β‐unsaturated esters was sufficiently high, and thus (E)‐methyl crotonate selectively gave anti‐14b (anti:syn = 93:7) in 74% yield. Furthermore, (E)‐ or (Z)‐methyl cinnamate underwent the diastereoselective borylstannylation to afford anti‐14c as the major product irrespective of the stereochemistry of the starting alkenes, implying intermediary formation of a copper enolate species in the reaction (vide infra). It is worth noting that the borylstannylation of methyl 3‐methoxyacrylate was accompanied by replacement of the methoxy moiety with a boryl moiety, similar to that observed with 1,4‐dimethoxy‐2‐butyne, furnishing the 3,3‐diborylated product (14d) in 79% yield.
Scheme 10.
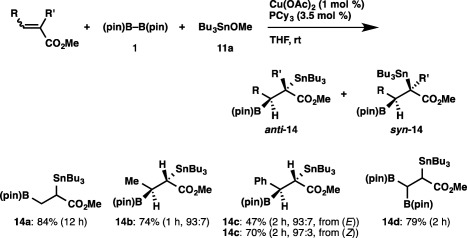
Borylstannylation of α,β‐unsaturated esters.
An N‐heterocyclic carbene (NHC)‐coordinated copper complex ((SIMes)CuCl) was an effective catalyst for the borylstannylation of simple alkenes (Scheme 11). Treatment of a vinylborane with 1 and 11a in the presence of (SIMes)CuCl led to the formation of 15a in 88% yield with perfect regioselectivity, where the stannyl moiety was installed into the geminal position of the parent boryl moiety. Similar regioselectivity was observed with vinylsilanes, and trimetalated alkanes (15b and 15c) were formed as the sole products. Furthermore, norbornene was solely transformed into the exo‐borylstannylation product (15d). The substrate scope was further extended to allenes: the reaction of 1‐phenylallene or 1‐octylallene with the aid of the catalyst (IMes)CuCl proceeded regio‐ and stereoselectively, leading to the exclusive formation of 16a and 16b having allylstannane and alkenylborane units (Scheme 12). The regioselective borylstannylation also occurred with 1,1‐diphenylallene to give 16c in 76% yield.
Scheme 11.
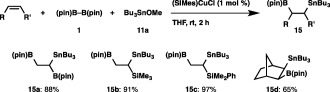
Borylstannylation of alkenes.
Scheme 12.
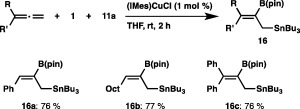
Borylstannylation of allenes.
The perfect regioselectivity observed in the reaction of α,β‐unsaturated esters and boryl‐ or silyl‐substituted alkenes can be explained by selective installation of the copper into the gem‐position of the ester, boryl and silyl groups in the borylcupration step, induced by the electronic directing effect of these substituents, and the selective production of the same diastereomer (anti‐14c) regardless of the (E) or (Z) stereochemistry of methyl cinnamate should suggest the intermediary formation of copper enolate species (anti‐17, syn‐17 and 17′) and fast interconversion between them (Scheme 13). As was the case with the copper‐catalyzed proto(deuterio)boration of alkenes,25 the capture of 17 with 11a may proceed with retention of the stereochemistry, and thus fast isomerization to anti‐17 should be responsible for the preferential formation of anti‐14.
Scheme 13.
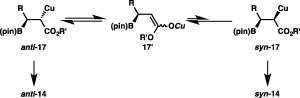
Intermediacy of a copper enolate species.
In a similar manner to the above tetraborylation, β‐alkoxy elimination from β‐borylalkylcopper species 18 should lead to the formation of methyl 3‐borylacylate, which then undergoes borylstannylation to provide diborylstannylated product 14d (Scheme 14).
Scheme 14.
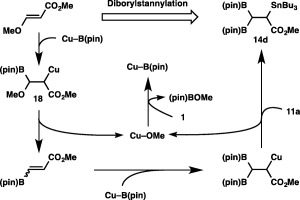
A catalytic cycle for the diborylstannylation.
2.3. Carboboration
Having succeeded in the three‐component borylstannylation with a tin electrophile, our attention was next turned toward the use of a carbon electrophile, which should lead to three‐component carboboration.26 As shown in Scheme 15, benzyl chloride was found to be adequate for this purpose: treatment of diphenylacetylene with 1 and benzyl chloride in DMF in the presence of Cu(OAc)2–PCy3 and KOtBu resulted in the simultaneous formation of B–C and C–C bonds to afford the cis‐carboboration product (19a) in 67% yield.27, 28 The carboboration was also applicable to a diarylalkyne and a dialkylalkyne to give the respective products (19b and 19c) in moderate to good yields, and exclusive formation of a single regioisomer (19d and 19e), whose benzyl moiety was attached to the geminal position relative to the phenyl moiety, was observed in the reaction of alkyl(aryl)alkynes. The reaction of phenylacetylene proceeded with perfect regioselectivity to afford 19f bearing the benzyl moiety at the more substituted site, whereas 1‐octyne furnished a mixture of regioisomers.
Scheme 15.
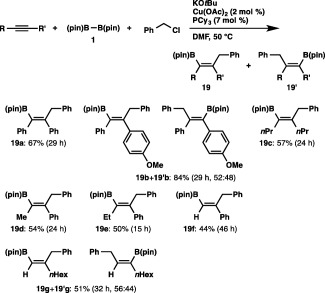
Carboboration of alkynes.
The broad substrate scope of carbon electrophiles was exemplified by the use of substituted benzyl chlorides and alkyl (pseudo)halides, and the formation of the cyclopropylmethylated product (19l) in the reaction using cyclopropylmethyl bromide suggests that a radical pathway is not operative in the carboboration (Scheme 16).
Scheme 16.
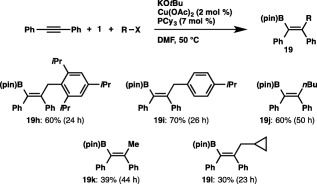
Carboboration of diphenylacetylene with various carbon electrophiles.
We next investigated the carboboration of alkenes, and found through catalyst screening that (SIMes)CuCl was the optimum choice.29 As depicted in Scheme 17, the carboboration of styrene derivatives,30 2‐vinylpyridine, vinylborane and vinylsilane with benzyl chloride took place smoothly to give borylalkanes (20a–20e) in good to high yield, where the benzyl moiety was preferentially attached to the internal carbon of the alkenes. In marked contrast, such aliphatic alkenes as 1‐octene or vinylcyclohexane underwent the reaction with the inverse regioselectivity to furnish 20′f and 20′g as the major products having the benzyl moiety at the terminal carbon of the alkenes.
Scheme 17.
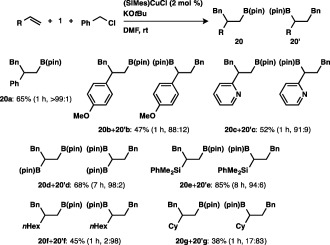
Carboboration of monosubstituted alkenes.
Disubstituted alkenes could also participate in the carboboration: perfect regioselectivity was observed in the reaction of methyl methacrylate, and the boryl and benzyl moieties were introduced into 1,2‐dihydronaphthalene regio‐ and stereoselectively, affording syn‐adduct 20i in 75% yield (Scheme 18).
Scheme 18.
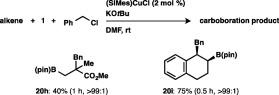
Carboboration of disubstituted alkenes.
The reaction of substituted benzyl chlorides with a vinylsilane and 1 furnished high yields of the respective products, irrespective of the electronic character of the substituents, and 1‐naphthylmethyl chloride could also act as a carbon electrophile (Scheme 19). The sole formation of the cyclopropylmethylated product was observed in the reaction with cyclopropylmethyl bromide, and moreover a chemoselective reaction with 1,5‐dibromopentane or ethyl 4‐bromobutyrate occurred, leaving the reactive functional groups (C–Br and ester moieties) intact.
Scheme 19.
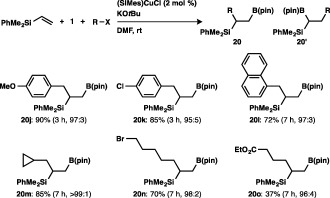
Carboboration of a vinylsilane with various carbon electrophiles.
As was the case with the diborylation and borylstannylation, a boryl‐substituted organocopper intermediate should form from 3 and an unsaturated C–C bond (Scheme 20). Subsequent interaction with KOtBu produces cuprate 21, which is then captured by a carbon electrophile to provide a carboboration product and CuOtBu.
Scheme 20.
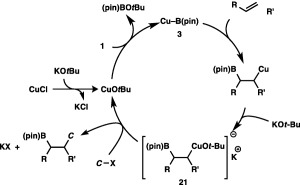
A catalytic cycle for the carboboration.
2.4. Protoboration with a Masked Diboron
The borylcopper species Cu–B(pin) has been proven to operate effectively as a key intermediate in synthesizing diverse organoboron compounds through diborylation, borylstannylation and carboboration of unsaturated C–C bonds, and we then became especially interested in the regiochemical outcomes of these reactions. These are definitively governed by the mode of addition of Cu–B(pin) across the unsaturated C–C bonds, and β selectivities are usually observed with terminal alkynes (and alkenes), as depicted in Scheme 21. Similarly to the conventional uncatalyzed hydroboration, one of the chief factors affecting the orientation of the approaching borylcopper species should be the Lewis acidic character of the B(pin) moiety, which favors addition to the terminal carbons to generate the more stable carbocationic transition state. We thus hypothesized that reducing the Lewis acidity of a boron center with appropriate substituents may alter the regiochemical behavior of a borylcopper species in the borylcupration step, leading to α selectivity. In this regard, an unsymmetrical diboron (22, (pin)B–B(dan)),31 containing a boryl moiety masked with 1,8‐diaminonaphthalene (dan),32 caught our attention, because the masked diboron was expected to produce a borylcopper species of diminished Lewis acidity, Cu–B(dan), via selective interaction between the Lewis acidic B(pin) moiety and the alkoxy moiety of Cu–OR in the σ‐bond metathesis step.
Scheme 21.
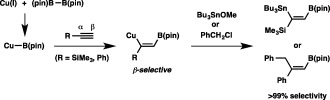
Borylstannylation and carboboration of terminal alkynes with β selectivity.
We first carried out the reaction of 22 with phenylacetylene in THF in the presence of an NHC‐coordinated copper complex ((SIPr)CuCl, 2 mol %), KOtBu and MeOH, and observed that the B(dan) moiety was introduced with exclusive α selectivity to afford a branched borylstyrene (23a) in 81% yield (Scheme 22).33 The ortho‐ and para‐methoxy‐substituted arylalkynes were also smoothly converted into the respective α‐borylstyrenes (23b and 23c) in excellent yields, which is in marked contrast to Hoveyda's results where a mixture of α‐ and β‐borylalkenes was produced (41:59 for ortho‐methoxyphenylacetylene; 62:38 for para‐methoxyphenylacetylene).34 It should be noted that the protoboration (formal hydroboration) proceeded with a high degree of α selectivity, irrespective of the steric and electronic properties of the terminal alkynes employed, giving the boryl‐substituted aliphatic alkenes (23d and 23e), silylalkene (23f) and allyl ether (23g) in high yields. Furthermore, the high functional group compatibility of the reaction was demonstrated by the use of 4‐bromo‐1‐butyne, where the C–Br bond remained intact (23h), and the reaction was also applicable to 1,7‐octadiyne, both of whose triple bonds underwent the α‐selective protoboration to give diborylation product 23i.
Scheme 22.
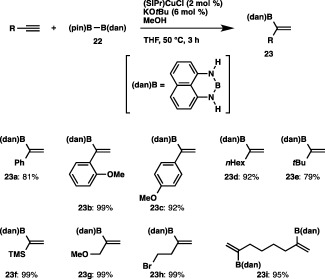
The α‐selective protoboration of terminal alkynes with a masked diboron.
The protoboration would be triggered by exclusive formation of a masked borylcopper species, Cu–B(dan) (24). Subsequent insertion of an alkyne into the Cu–B(dan) bond, which generates a β‐borylalkenylcopper species (25), followed by protonation with MeOH provides the protoboration product with regeneration of Cu–OMe (Scheme 23). Owing to the diminished Lewis acidity of the masked B(dan) moiety in 24, the interaction between an incoming terminal alkyne and the boron center, which may be a contributory factor of β‐selective addition through the more stable carbocationic transition state (vide supra), would be negligible in the borylcupration step. Therefore, the orientation of 24 should simply be controlled by steric repulsion between the alkyne substituent and the bulkier copper moiety, which results in the sole introduction of the B(dan) moiety into the internal carbon of terminal alkynes.
Scheme 23.
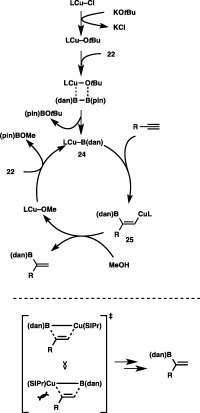
A catalytic cycle for the α‐selective protoboration of terminal alkynes.
The direct construction of a C–B(dan) bond by the Cu‐catalyzed protoboration using 22 was also achievable with internal alkynes (Scheme 24).35 Thus, treatment of 1‐phenyl‐1‐propyne with 22 and MeOH in the presence of (Ph3P)3CuCl (2 mol %) furnished cis‐protoboration product 23j with perfect regioselectivity, where the B(dan) moiety was attached to the geminal position of the methyl group. Functionalized alkyl(aryl)alkynes having ketone, ester, nitrile, bromo or hydroxy groups were likewise converted into the respective products (23k–23o) regioselectively, leaving these functional groups intact. As depicted in Scheme 25, the copper catalysis was also effective in installing the B(dan) moiety into the C–C double bonds of methyl acrylate and styrene to afford excellent yields of alkylborons (26a and 26b) with perfect regioselectivity.
Scheme 24.
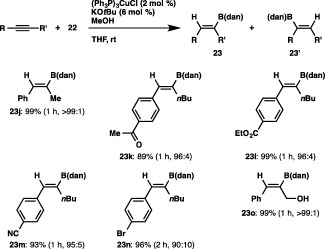
Protoboration of internal alkynes with a masked diboron.
Scheme 25.
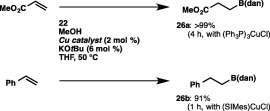
Protoboration of alkenes with a masked diboron.
2.5. Borylstannylation with a Masked Diboron
The exclusive generation of 25 by the use of a masked diboron directed our attention to its utilization for the borylstannylation (cf. Section 2.2), because terminal alkynes always accepted regioselective addition of a boryl group at a terminal carbon and a stannyl group at an internal carbon in the previous borylstannylation, irrespective of the catalytic systems and the reaction modes.21, 22
We conducted the reaction of 1‐octyne with 22 and tributyltin methoxide (11a) in THF at room temperature in the presence of (SIPr)CuCl, and found that the cis‐borylstannylation took place with regioselectivity inverse to that of the previous borylstannylation (86% yield, 27a:27′a = 96:4), leading to the introduction of the boryl group at the internal carbon and the stannyl group at the terminal carbon (Scheme 26).36 The reaction could also be applied to other simple aliphatic alkynes and functionalized alkynes, whose functional groups (C–Br bond and cyano group) remained intact. Similar regioselectivity was also observed using an enyne and phenylacetylene, and furthermore the reactions of a THP‐protected propargyl alcohol, propargyl amine and trimethylsilylacetylene, resulting in the exclusive formation of 27b–27j. The versatility of the borylstannylation was further expanded by application to allenes: the regio‐ and stereoselective reaction proceeded to provide (Z)‐1‐stannyl‐2‐boryl‐2‐alkenes (28a and 28b) as the sole product (Scheme 27).
Scheme 26.
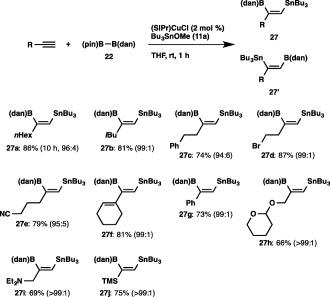
Borylstannylation of terminal alkynes with a masked diboron.
Scheme 27.
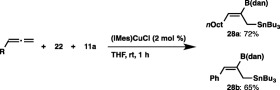
Borylstannylation of allenes with a masked diboron.
2.6. Ag‐Catalyzed Protoboration
In marked contrast to the high potency of copper catalysis in the borylation reactions, the catalytic use of silver, another group 11 element, for synthesizing organoboron compounds remains almost totally unexplored, except for the diborylation of alkenes with bis(catecholato)diboron.37 In view of the differences between copper and silver in fundamental properties such as atomic radius, electronegativity, softness as a Lewis acid, etc., we envisaged that borylation reactions of different modes and/or selectivities should be feasible under silver catalysis.
When the reaction of 1‐octyne with (pin)B–B(pin) (1) in the presence of MeOH using (IMes)AgCl (2 mol %) and KOtBu (6 mol %) was carried out, a protoboration product (29a) formed in 89% yield with high β selectivity (Scheme 28).38 The intriguing feature of the silver catalysis in the protoboration is higher regioselectivity compared to that obtained with a copper catalyst:14 c, 34 aliphatic terminal alkynes having i‐amyl or phenethyl groups were readily convertible into the respective linear borylalkenes (29b and 29c). The reaction could also be applied to cyclopentyl‐ and tert‐butylacetylene to provide high yields of the products (29d and 29e), irrespective of the steric congestion around the triple bonds. Only the C–C triple bond of an enyne underwent the protoboration to afford boryldiene 29f, and the high chemoselectivity of the present system was further demonstrated by the reactions of functionalized alkynes bearing cyano, halogen or silyl ether moieties, in which these reactive groups remained intact. In contrast, the reaction of propargyl ether or N‐propargylphthalimide gave α‐adducts to some extent. Internal alkynes such as diphenylacetylene and 1‐phenyl‐1‐propyne also underwent the regio‐ and stereoselective protoboration.
Scheme 28.
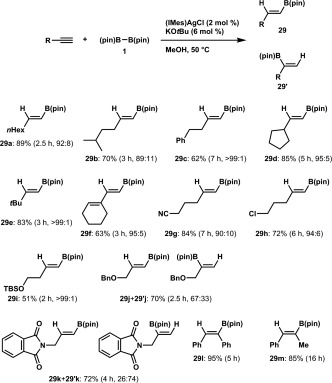
Ag‐catalyzed protoboration of alkynes.
The protoboration may be triggered by formation of a borylsilver(I) species (30) arising from σ‐bond metathesis between a silver(I) alkoxide and 1, as in the copper(I)‐catalyzed borylation reactions (step A, Scheme 29). Subsequent addition of 30 across the C–C triple bond (step B, borylargentation), which produces a boryl‐substituted alkenylsilver species (31), followed by protonation with MeOH gives a protoboration product with regeneration of the silver alkoxide (step C).
Scheme 29.
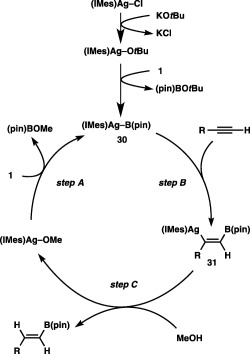
A catalytic cycle for the Ag‐catalyzed protoboration of alkynes.
3. Stannylation
3.1. Distannylation
Having demonstrated that a copper catalyst is extremely versatile for the C–B bond‐forming reactions, which produce diverse organoboron compounds, our attention was naturally riveted on developing C–Sn bond‐forming reactions under copper catalysis; however, the lack of practical approaches to the catalytic generation of Cu–Sn species discouraged the widespread utilization of this potential methodology.39 Through thorough screening of reaction conditions, we developed the unprecedented activation of a distannane with a copper(I) complex, which leads to the efficacious generation of a Cu–Sn species, and its synthetic application to the first copper‐catalyzed distannylation of alkynes.40, 41
The distannylation was found to take place smoothly to afford cis‐vic‐distannyloctene 33a, when we conducted the reaction of hexamethyldistannane (32) with 1‐octyne in DMF in the presence of (PPh3)3CuOAc (2 mol %) and Cs2CO3 (20 mol %) (Scheme 30). Other aliphatic terminal alkynes bearing a primary or secondary alkyl group were converted into the respective distannylalkenes (33b–33d) in high yields. The prominent copper catalysis toward the electronically non‐biased aliphatic terminal alkynes is worth noting, because the reaction using a conventional palladium catalyst was sluggish and led to a low product yield (Scheme 31).41 a,b The distannylation was also applicable to 5‐chloro‐1‐pentyne and 5‐hexynenitrile to afford the products (33e and 33f) without damaging these functional groups. The course of the distannylation was little affected by coordinating groups in the propargylic amine and ether, although the yields became moderate. In addition, phenylacetylene and THP‐protected propargylic alcohol could easily be coupled with 32 by the use of a strong base (KOtBu or LiOtBu) instead of Cs2CO3.
Scheme 30.
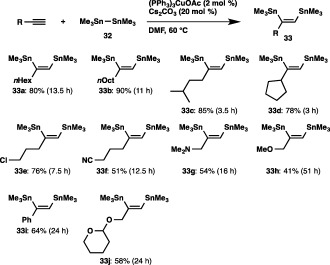
Distannylation of alkynes.
Scheme 31.
Pd‐catalyzed distannylation of 1‐octyne.
A key intermediate of the distannylation should be a Cu–Sn species (34) derived from the CuOR′ complex and a distannane (32) (Scheme 32). Subsequent addition of 34 to the C–C triple bond of an alkyne affords stannyl‐substituted alkenylcopper species 35, which is then captured by Me3SnOR′ to give the distannylation product with regeneration of the CuOR′ complex.
Scheme 32.
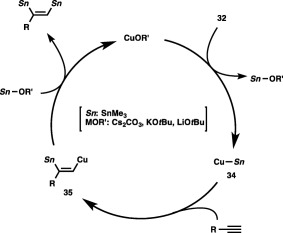
A catalytic cycle for the distannylation of alkynes.
3.2. Hydrostannylation
The intermediary formation of a stannyl‐substituted alkenylcopper species in the distannylation prompted us to investigate its capture with another electrophile, and the use of water as a protic reagent was found to be quite efficient for synthesizing alkenylstannanes with excellent α selectivity.42, 43
The α‐selective hydrostannylation has proven to be a feasible method, providing branched (36a) and linear (36′a) alkenylstannanes in 75% yield (36a:36′a = 98:2), when we treated 1‐tetradecyne with hexamethyldistannane (32) and water in toluene at 110°C in the presence of Cu(OAc)2–PtBu3 catalyst (Scheme 33). The reaction was also applicable to 1‐dodecyne and an imide‐substituted alkyne, giving 36b and 36c in excellent yield with high degrees of α selectivity, and functionalized aliphatic alkynes bearing acetal, silyl ether, hydroxy or amine groups smoothly underwent the α‐selective hydrostannylation, leaving these reactive moieties intact. The regioselective installation of a stannyl group into the internal carbon of aromatic terminal alkynes was achieved under the present conditions as well, and thus pyridyl, naphthyl and phenyl acetylenes were efficiently transformed into the respective branched alkenylstannanes (36h–36j). An internal alkyne, diphenylacetylene, also participated in the reaction to furnish (E)‐trimethylstannylstilbene (36k) as the sole product, showing that the hydrostannylation completely proceeds in a cis fashion.
Scheme 33.
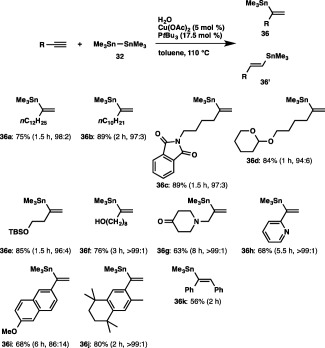
The α‐selective hydrostannylation of terminal alkynes with a distannane.
With the successful synthesis of diverse branched alkenylstannanes having a trimethylstannyl moiety, we next investigated the α‐selective installation of a tributylstannyl moiety, and found that a silylstannane, tributyl(trimethylsilyl)stannane (37), turned out to serve as a more effective and reactive stannylating reagent compared with hexabutyldistannane, affording a 73% yield of 38a and 38′a (38a:38′a = 86:14) (Scheme 34). It should be noted that the silyl moiety was not incorporated into the alkyne at all in the reaction with the silylstannane, which is in marked contrast to the copper‐catalyzed selective silyl‐incorporation reactions into unsaturated hydrocarbons with silylboranes.44 The hydrostannylation using a silylstannane also took place smoothly with various aliphatic terminal alkynes to provide 38b and 38c with high α selectivity, and functionalized terminal alkynes bearing cyano, chloro or benzyloxy moieties were transformed into the respective branched alkenylstannanes as well.
Scheme 34.
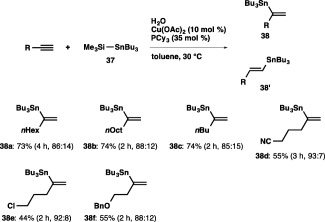
The α‐selective hydrostannylation of terminal alkynes with a silylstannane.
Formation of a stannylcopper species (34) via σ‐bond metathesis between a distannane (or silylstannane) and Cu–OH would initiate the hydrostannylation (Scheme 35). This stannylcopper species (34) then adds across the C–C triple bond of the terminal alkyne (stannylcupration) to produce a stannyl‐substituted alkenylcopper species (35), which is finally transformed into the hydrostannylation product through protonation with water. The formation of branched alkenylstannanes with high α selectivity should be attributable to the regioselective generation of 35, possessing the stannyl moiety at the internal carbon, in the stannylcupration step, which has already been well documented to be kinetically favored in the stoichiometric reaction of a stannylcopper species with a terminal alkyne.45
Scheme 35.
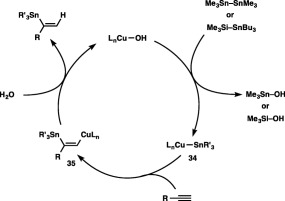
A catalytic cycle for the α‐selective hydrostannylation of terminal alkynes.
4. Silylation
4.1. Silylstannylation
Through the above studies on the stannylation reactions, where a boryl‐ or stannyl‐substituted organocopper species was facilely trapped by a tin alkoxide, we envisaged that silylstannylation of unsaturated C–C bonds would also be feasible under copper catalysis by the use of a suitable silylating reagent44, 46 to allow the smooth generation of silylcopper and silyl‐substituted organocopper species.
The three‐component silylstannylation was found to readily occur to afford 40a and 40′a in 86% yield with regioselectivity inverse to that of the previous Pd‐catalyzed silylstannylation (40a:40′a = 93:7),47 when the reaction of 1‐octyne, a silylborane (PhMe2Si–B(pin), 39) and tributyltin tert‐butoxide (11c) was carried out in acetonitrile at room temperature in the presence of CuCl–PtBu3 catalyst (Scheme 36).48 Linear and branched aliphatic terminal alkynes could undergo the regioselective silylstannylation, where the stannyl moieties were predominantly attached to the terminal carbon of the alkynes. This unique regioselectivity was also achievable with functionalized alkynes bearing cyano, bromo or hydroxy groups, and the result that these reactive moieties remained intact demonstrates the high functional group compatibility of the silylstannylation. In contrast, the stannyl moiety was selectively introduced into the internal carbon of THP‐protected propargyl alcohol and propargyl ether to provide 40′g and 40′h as the major products.
Scheme 36.
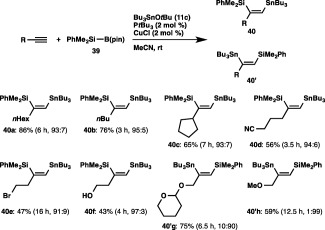
Silylstannylation of alkynes.
The Cu‐catalyzed silylstannylation of allenes was also found to proceed smoothly with regioselectivity inverse to that of the previous silylstannylation under palladium catalysis (Scheme 37).49 Thus, treatment of pentadeca‐1,2‐diene with 39 and tributyltin methoxide (11a) in the presence of ClIMesCuCl catalyst afforded an 87% yield of (E)‐ and (Z)‐41a (ratio = 78:22), where the stannyl moiety had been exclusively installed into the central carbon of the allene. The regioselective formation of silylstannylated products (41b and 41c) bearing allylsilane and alkenylstannane units was observed with 5‐phenylpenta‐1,2‐diene and cyclohexylallene, and functionalized allenes possessing a silyl ether or a theobromine moiety also underwent the silylstannylation with a similar regioselectivity to provide the respective products (41d and 41e) without damaging these functional groups.
Scheme 37.
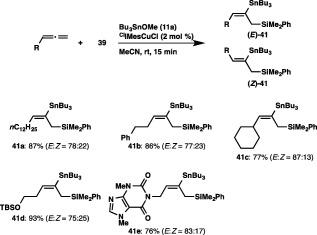
Silylstannylation of allenes.
Generation of a silylcopper species, Cu–SiMe2Ph 42, via σ‐bond metathesis between a copper alkoxide and a silylborane would trigger the silylstannylation (Scheme 38, step A). Then, the alkyne or allene inserts into the Cu–Si bond to give a silyl‐substituted alkenylcopper species (43 or 44, step B), which is subsequently trapped by a tin alkoxide to furnish the silylstannylation product with regeneration of the copper alkoxide (step C). The regiochemical outcome of the reaction with an alkyne or an allene should be ascribable to the regioselective formation of 43 50 or 44, the latter of which has been demonstrated to be kinetically favored in the stoichiometric reaction using a silylcopper species.51 On the other hand, the electronic directing effect of a propargylic functional group, which induces the addition of the copper moiety to the internal carbon of the alkyne in step B, should become dominant to provide 40′ as the major product.
Scheme 38.
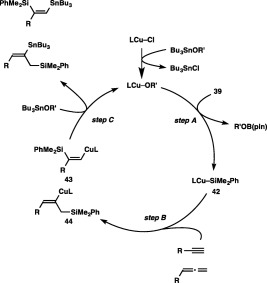
A catalytic cycle for the silylstannylation.
5. Conclusion
We have developed copper‐catalyzed dimetalation, carbometalation and hydrometalation reactions of unsaturated C–C bonds, which allow direct access to various main‐group organometallics containing B, Sn and Si of high synthetic significance. The reaction system, which utilizes the ambiphilic character of a copper species between a transition metal and a main‐group metal, can basically assemble three components, namely, a main‐group metal, an unsaturated C–C bond and an electrophile, and hence main‐group organometallics can be synthesized and diversified more readily compared with conventional two‐component reactions. Moreover, we have demonstrated that the Lewis acidity of the boron center in a borylcopper species governs its regiochemical behavior in the borylcupration, which leads to a regioselectivity reversal in the reaction of terminal alkynes, depending upon the diboron employed. Further studies on catalytic metalation reactions for synthesizing main‐group organometallics are in progress.
Acknowledgements
I would like to express my sincerest gratitude to Prof. K. Takaki for his helpful advice, and to my co‐workers, whose names are cited in the references, for their invaluable contributions to the chemistry described herein. This work was financially supported by Research for Promoting Technological Seeds from the Japan Science and Technology Agency (JST), the Electric Technology Research Foundation of Chugoku, The Mazda Foundation, the Furukawa Technology Promotion Foundation, and the SEI Group CSR Foundation.
Biography
Hiroto Yoshida was born in Fukuoka, Japan, in 1973. He graduated from Kyoto University in 1996 and received his Ph.D. from Kyoto University under the supervision of Professors Tamejiro Hiyama and Eiji Shirakawa in 2001. He then became an Assistant Professor at Hiroshima University in 2001 and was promoted to an Associate Professor in 2006. He received the BCSJ Award (2001), the Japan Combinatorial Chemistry Focus Group Award in Synthetic Organic Chemistry, Japan (2005), the Tetrahedron Letters Most Cited Paper 2003–2006 Award (2006), The Chemical Society of Japan Award for Young Chemists (2007), The Society of Silicon Chemistry Award for Young Chemists (2008), and The Young Scientists’ Prize, The Commendation for Science and Technology by the Minister of Education, Culture, Sports, Science and Technology (2009). His research interests include (1) catalytic reactions for the synthesis of main‐group organometallics containing boron, tin or silicon, (2) aryne‐based organic synthesis, and (3) their application to organic electronics materials.

REFERENCES
- 1. Boronic Acids (Ed.: Hall D. G.), Wiley‐VCH, Weinheim, 2011. [Google Scholar]
- 2.a) Orita A., Otera J., in Main Group Metals in Organic Synthesis (Eds.: Yamamoto H., Oshima K.), Wiley‐VCH, Weinheim, 2004, pp. 621–720; [Google Scholar]; b) Davies A. G., Organotin Chemistry, Wiley‐VCH, Weinheim, 2004. [Google Scholar]
- 3.a) Miura K., Hosomi A., in Main Group Metals in Organic Synthesis (Eds.: Yamamoto H., Oshima K.), Wiley‐VCH, Weinheim, 2004, pp. 409–592; [Google Scholar]; b) Hiyama T., in Organometallics in Synthesis, Third Manual (Ed.: Schlosser M.), Wiley, Hoboken, 2013, Chapter 3. [Google Scholar]
- 4. Metal‐Catalyzed Cross‐Coupling Reactions and More (Eds.: de Meijere A., Brase S., Oestreich M.), Wiley‐VCH, Weinheim, 2014. [Google Scholar]
- 5.a) Yoshida H., in Transition‐Metal‐Mediated Aromatic Ring Construction (Ed.: Tanaka K.), John Wiley & Sons, Hoboken, 2013, pp. 773–796; [Google Scholar]; b) Yoshida H., in Comprehensive Organic Synthesis (Eds.: Knochel P., Molander G. A.), 2nd edition, Elsevier, Amsterdam, 2014, Vol. 4, pp. 517–579; [Google Scholar]; c) Yoshida H., in Multicomponent Reactions in Organic Synthesis (Eds.: Zhu J., Wang Q., Wang M.‐X.), Wiley‐VCH, Weinheim, 2015, pp. 39–71. [Google Scholar]
- 6.a) Yoshida H., Ohshita J., Kunai A., Bull. Chem. Soc. Jpn. 2010, 83, 199; [Google Scholar]; b) Yoshida H., Takaki K., Heterocycles 2012, 85, 1333; [Google Scholar]; c) Yoshida H., Takaki K., Synlett 2012, 23, 1725. [Google Scholar]
- 7.Representative examples: ; a) Yoshida H., Terayama T., Ohshita J., Kunai A., Chem. Commun. 2004, 1980; [DOI] [PubMed] [Google Scholar]; b) Yoshida H., Minabe T., Ohshita J., Kunai A., Chem. Commun. 2005, 3454; [DOI] [PubMed] [Google Scholar]; c) Yoshida H., Mimura Y., Ohshita J., Kunai A., Chem. Commun. 2007, 2405; [DOI] [PubMed] [Google Scholar]; d) Yoshida H., Kishida T., Watanabe M., Ohshita J., Chem. Commun. 2008, 5963; [DOI] [PubMed] [Google Scholar]; e) Yoshida H., Yoshida R., Takaki K., Angew. Chem. Int. Ed. 2013, 52, 8629. [DOI] [PubMed] [Google Scholar]
- 8.Representative examples: ; a) Yoshida H., Fukushima H., Ohshita J., Kunai A., Tetrahedron Lett. 2004, 45, 8659; [Google Scholar]; b) Yoshida H., Fukushima H., Morishita T., Ohshita J., Kunai A., Tetrahedron 2007, 63, 4793; [Google Scholar]; c) Yoshida H., Morishita T., Fukushima H., Ohshita J., Kunai A., Org. Lett. 2007, 9, 3367; [DOI] [PubMed] [Google Scholar]; d) Morishita T., Fukushima H., Yoshida H., Ohshita J., Kunai A., J. Org. Chem. 2008, 73, 5452; [DOI] [PubMed] [Google Scholar]; e) Yoshida H., Ito Y., Ohshita J., Chem. Commun. 2011, 47, 8512. [DOI] [PubMed] [Google Scholar]
- 9. Yoshida H., Okada K., Kawashima S., Tanino K., Ohshita J., Chem. Commun. 2010, 46, 1763. [DOI] [PubMed] [Google Scholar]
- 10.a) Yoshida H., Tanino K., Ohshita J., Kunai A., Angew. Chem. Int. Ed. 2004, 43, 5052; [DOI] [PubMed] [Google Scholar]; b) Yoshida H., Tanino K., Ohshita J., Kunai A., Chem. Commun. 2005, 5678. [DOI] [PubMed] [Google Scholar]
- 11.a) Yoshida H., Ikadai J., Shudo M., Ohshita J., Kunai A., J. Am. Chem. Soc. 2003, 125, 6638; [DOI] [PubMed] [Google Scholar]; b) Yoshida H., Ikadai J., Shudo M., Ohshita J., Kunai A., Organometallics 2005, 24, 156; [Google Scholar]; c) Ikadai J., Yoshida H., Ohshita J., Kunai A., Chem. Lett. 2005, 34, 56. [Google Scholar]
- 12. Yoshida H., Morishita T., Nakata H., Ohshita J., Org. Lett. 2009, 11, 373. [DOI] [PubMed] [Google Scholar]
- 13. Morishita T., Yoshida H., Ohshita J., Chem. Commun. 2010, 46, 640. [DOI] [PubMed] [Google Scholar]
- 14.Pioneering works: ; a) Ito H., Yamanaka H., Tateiwa J., Hosomi A., Tetrahedron Lett. 2000, 41, 6821; [Google Scholar]; b) Takahashi K., Ishiyama T., Miyaura N., Chem. Lett. 2000, 29, 982; [Google Scholar]; c) Takahashi K., Ishiyama T., Miyaura N., J. Organomet. Chem. 2001, 625, 47. [Google Scholar]
- 15.A study on a borylcopper species: Laitar D. S., Müller P., Sadighi J. P., J. Am. Chem. Soc. 2005, 127, 17196. [DOI] [PubMed] [Google Scholar]
- 16.Reviews on Cu‐catalyzed borylation reactions: ; a) Yun J., Asian J. Org. Chem. 2013, 2, 1016; [Google Scholar]; b) Fujihara T., Semba K., Terao J., Tsuji Y., Catal. Sci. Technol. 2014, 4, 1699; [Google Scholar]; c) Barbeyron R., Benedetti E., Cossy J., Vasseur J.‐J., Arseniyadis S., Smietana M., Tetrahedron 2014, 70, 8431; [Google Scholar]; d) Semba K., Fujihara T., Terao J., Tsuji Y., Tetrahedron 2015, 71, 2183. [Google Scholar]
- 17. Yoshida H., Kawashima S., Takemoto Y., Okada K., Ohshita J., Takaki K., Angew. Chem. Int. Ed. 2012, 51, 235. [DOI] [PubMed] [Google Scholar]
- 18.Cu‐catalyzed diborylation of styrene, phenylacetylene and diphenylacetylene with bis(catecholato)diboron was reported: Lillo V., Fructos M. R., Ramírez J., Braga A. A. C., Maseras F., Díaz‐Requejo M. M., Pérez P. J., Fernández E., Chem. Eur. J. 2007, 13, 2614. [DOI] [PubMed] [Google Scholar]
- 19.a) Ishiyama T., Matsuda N., Miyaura N., Suzuki A., J. Am. Chem. Soc. 1993, 115, 11018; [Google Scholar]; b) Ishiyama T., Matsuda N., Murata M., Ozawa F., Suzuki A., Miyaura N., Organometallics 1996, 15, 713; [Google Scholar]; c) Lesley G., Nguyen P., Taylor N. J., Marder T. B., Scott A. J., Clegg W., Norman N. C., Organometallics 1996, 15, 5137; [Google Scholar]; d) Thomas R. L., Souza F. E. S., Marder T. B., J. Chem. Soc., Dalton Trans. 2001, 1650; [Google Scholar]; e) Burks H. E., Kliman L. T., Morken J. P., J. Am. Chem. Soc. 2009, 131, 9134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Ito H., Ito S., Sasaki Y., Matsuura K., Sawamura M., J. Am. Chem. Soc. 2007, 129, 14856; [DOI] [PubMed] [Google Scholar]; b) Ito H., Sasaki Y., Sawamura M., J. Am. Chem. Soc. 2008, 130, 15774. [DOI] [PubMed] [Google Scholar]
- 21.Previous reports on the borylstannylation: ; a) Onozawa S.‐Y., Hatanaka Y., Sakakura T., Shimada S., Tanaka M., Organometallics 1996, 15, 5450; [Google Scholar]; b) Weber L., Wartig H. B., Stammler H. G., Stammler A., Neumann B., Organometallics 2000, 19, 2891; [Google Scholar]; c) Singidi R. R., RajanBabu T. V., Org. Lett. 2010, 12, 2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takemoto Y., Yoshida H., Takaki K., Chem. Eur. J. 2012, 18, 14841. [DOI] [PubMed] [Google Scholar]
- 23.a) Whittaker A. M., Rucker R. P., Lalic G., Org. Lett. 2010, 12, 3216; [DOI] [PubMed] [Google Scholar]; b) Shintani R., Takatsu K., Hayashi T., Chem. Commun. 2010, 46, 6822. [DOI] [PubMed] [Google Scholar]
- 24. Takemoto Y., Yoshida H., Takaki K., Synthesis 2014, 46, 3024. [Google Scholar]
- 25. Lee Y., Hoveyda A. H., J. Am. Chem. Soc. 2009, 131, 3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Previous reports on the carboboration: ; a) Suginome M., Yamamoto A., Murakami M., J. Am. Chem. Soc. 2003, 125, 6358; [DOI] [PubMed] [Google Scholar]; b) Suginome M., Yamamoto A., Murakami M., Angew. Chem. Int. Ed. 2005, 44, 2380; [DOI] [PubMed] [Google Scholar]; c) Suginome M., Yamamoto A., Murakami M., J. Organomet. Chem. 2005, 690, 5300; [Google Scholar]; d) Yamamoto A., Suginome M., J. Am. Chem. Soc. 2005, 127, 15706; [DOI] [PubMed] [Google Scholar]; e) Suginome M., Shirakura M., Yamamoto A., J. Am. Chem. Soc. 2006, 128, 14438; [DOI] [PubMed] [Google Scholar]; f) Suginome M., Yamamoto A., Sasaki T., Murakami M., Organometallics 2006, 25, 2911; [Google Scholar]; g) Daini M., Yamamoto A., Suginome M., J. Am. Chem. Soc. 2008, 130, 2918; [DOI] [PubMed] [Google Scholar]; h) Daini M., Suginome M., Chem. Commun. 2008, 5224. [DOI] [PubMed] [Google Scholar]
- 27. Yoshida H., Kageyuki I., Takaki K., Org. Lett. 2013, 15, 952. [DOI] [PubMed] [Google Scholar]
- 28.Cu‐catalyzed carboboration of similar type: ; a) Alfaro A., Parra A., Alemán J., Ruano J. L. G., Tortosa M., J. Am. Chem. Soc. 2012, 134, 15165; [DOI] [PubMed] [Google Scholar]; b) Zhang L., Cheng J., Carry B., Hou Z., J. Am. Chem. Soc. 2012, 134, 14314; [DOI] [PubMed] [Google Scholar]; c) Zhou Y., You W., Smith K. B., Brown M. K., Angew. Chem. Int. Ed. 2014, 53, 3475; [DOI] [PubMed] [Google Scholar]; d) Semba K., Nakao Y., J. Am. Chem. Soc. 2014, 136, 7567; [DOI] [PubMed] [Google Scholar]; e) Smith K. B., Logan K. M., You W., Brown M. K., Chem. Eur. J. 2014, 20, 12032; [DOI] [PubMed] [Google Scholar]; f) Semba K., Bessho N., Fujihara T., Terao J., Tsuji Y., Angew. Chem. Int. Ed. 2014, 53, 9007. [DOI] [PubMed] [Google Scholar]
- 29. Kageyuki I., Yoshida H., Takaki K., Synthesis 2014, 46, 1924. [Google Scholar]
- 30.Cu‐catalyzed borylation of styrene derivatives: ; a) Ito H., Toyoda T., Sawamura M., J. Am. Chem. Soc. 2010, 132, 5990; [DOI] [PubMed] [Google Scholar]; b) Zhong C., Kunii S., Kosaka Y., Sawamura M., Ito H., J. Am. Chem. Soc. 2010, 132, 11440 See also reference [25]. [DOI] [PubMed] [Google Scholar]
- 31. Iwadate N., Suginome M., J. Am. Chem. Soc. 2010, 132, 2548. [DOI] [PubMed] [Google Scholar]
- 32.a) Noguchi H., Hojo K., Suginome M., J. Am. Chem. Soc. 2007, 129, 758; [DOI] [PubMed] [Google Scholar]; b) Noguchi H., Shioda T., Chou C.‐M., Suginome M., Org. Lett. 2008, 10, 377; [DOI] [PubMed] [Google Scholar]; c) Iwadate N., Suginome M., Chem. Lett. 2010, 39, 558. [Google Scholar]
- 33. Yoshida H., Takemoto Y., Takaki K., Chem. Commun. 2014, 50, 8299. [DOI] [PubMed] [Google Scholar]
- 34. Jang H., Zhugralin A. R., Lee Y., Hoveyda A. H., J. Am. Chem. Soc. 2011, 133, 7859. [DOI] [PubMed] [Google Scholar]
- 35. Yoshida H., Takemoto Y., Takaki K., Asian J. Org. Chem. 2014, 3, 1204. [Google Scholar]
- 36. Yoshida H., Takemoto Y., Takaki K., Chem. Commun. 2015, 51, 6297. [DOI] [PubMed] [Google Scholar]
- 37. Ramírez J., Corberán R., Sanaú M., Peris E., Fernández E., Chem. Commun. 2005, 3056. [DOI] [PubMed] [Google Scholar]
- 38. Yoshida H., Kageyuki I., Takaki K., Org. Lett. 2014, 16, 3512. [DOI] [PubMed] [Google Scholar]
- 39.Reviews on the formation of C–Sn bonds using stoichiometric Cu–Sn species: ; a) Barbero A., Pulido F. J., Chem. Soc. Rev. 2005, 34, 913; [DOI] [PubMed] [Google Scholar]; b) Pulido F. J., Barbero A., in The Chemistry of Organocopper Compounds (Eds.: Rappoport Z., Marek I.), John Wiley & Sons, Chichester, 2009, pp. 775–856. [Google Scholar]
- 40. Yoshida H., Shinke A., Takaki K., Chem. Commun. 2013, 49, 11671. [DOI] [PubMed] [Google Scholar]
- 41.Previous reports on the distannylation: ; a) Mitchell T. N., Killing A. A. H., Rutschow D., J. Organomet. Chem. 1983, 241, C45; [Google Scholar]; b) Mitchell T. N., Killing A. A. H., Rutschow D., J. Organomet. Chem. 1986, 304, 257; [Google Scholar]; c) Piers E., Skerlj R. T., J. Chem. Soc., Chem. Commun. 1986, 626; [Google Scholar]; d) Piers E., Skerlj R. T., Can. J. Chem. 1994, 72, 2468; [Google Scholar]; e) Mancuso J., Lautens M., Org. Lett. 2003, 5, 1653; [DOI] [PubMed] [Google Scholar]; f) Braune S., Kazmaier U., Angew. Chem. Int. Ed. 2003, 42, 306. [DOI] [PubMed] [Google Scholar]
- 42. Yoshida H., Shinke A., Kawano Y., Takaki K., Chem. Commun. 2015, 51, 10616. [DOI] [PubMed] [Google Scholar]
- 43.Previous reports on the α‐selective hydrostannylation: ; a) Hibino J., Matsubara S., Morizawa Y., Oshima K., Nozaki H., Tetrahedron Lett. 1984, 25, 2151; [Google Scholar]; b) Kazmaier U., Schauss D., Pohlman M., Org. Lett. 1999, 1, 1017; [Google Scholar]; c) Shibata I., Suwa T., Ryu K., Baba A., J. Am. Chem. Soc. 2001, 123, 4101; [DOI] [PubMed] [Google Scholar]; d) Hamze A., Veau D., Provot O., Brion J.‐D., Alami M., J. Org. Chem. 2009, 74, 1337. [DOI] [PubMed] [Google Scholar]
- 44.Representative examples: ; a) Vyas D. J., Oestreich M., Angew. Chem. Int. Ed. 2010, 49, 8513; [DOI] [PubMed] [Google Scholar]; b) Wang P., Yeo X.‐L., Loh T.‐P., J. Am. Chem. Soc. 2011, 133, 1254; [DOI] [PubMed] [Google Scholar]; c) Fujihara T., Tani Y., Semba K., Terao J., Tsuji Y., Angew. Chem. Int. Ed. 2012, 51, 11487. [DOI] [PubMed] [Google Scholar]
- 45. Singer R. D., Hutzzinger M. W., Oehlschlager A. C., J. Org. Chem. 1991, 56, 4933. [Google Scholar]
- 46.Cu‐catalyzed silylation with a disilane: ; a) Ito H., Ishizuka T., Tateiwa J., Sonoda M., Hosomi A., J. Am. Chem. Soc. 1998, 120, 11196; [Google Scholar]; b) Clark C. T., Lake J. F., Scheidt K. A., J. Am. Chem. Soc. 2004, 126, 84; [DOI] [PubMed] [Google Scholar]; c) Ito H., Horita Y., Sawamura M., Adv. Synth. Catal. 2012, 354, 813; [Google Scholar]; d) Iannazzo L., Molander G. A., Eur. J. Org. Chem. 2012, 4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.a) Chenard B. L., Laganis E. D., Davidson F., RajanBabu T. V., J. Org. Chem. 1985, 50, 3666; [Google Scholar]; b) Mitchell T. N., Killing H., Dicke R., Wickenkamp R., J. Chem. Soc., Chem. Commun. 1985, 354; [Google Scholar]; c) Mitchell T. N., Wickenkamp R., Amamria A., Dicke R., Schneider U., J. Org. Chem. 1987, 52, 4868; [Google Scholar]; d) Hemeon I., Singer R. D., Chem. Commun. 2002, 1884; [DOI] [PubMed] [Google Scholar]; e) Murakami M., Matsuda T., Itami K., Ashida S., Terayama M., Synthesis 2004, 1522. [Google Scholar]
- 48. Yoshida H., Hayashi Y., Ito Y., Takaki K., Chem. Commun. 2015, 51, 9440. [DOI] [PubMed] [Google Scholar]
- 49.a) Mitchell T. N., Schneider U., J. Organomet. Chem. 1991, 407, 319; [Google Scholar]; b) Barrett A. G. M., Wan P. W. H., J. Org. Chem. 1996, 61, 8667; [Google Scholar]; c) Shin S., RajanBabu T. V., J. Am. Chem. Soc. 2001, 123, 8416; [DOI] [PubMed] [Google Scholar]; d) Jeganmohan M., Shanmugasundaram M., Chang K.‐J., Cheng C.‐H., Chem. Commun. 2002, 2552. [Google Scholar]
- 50.Regioselective formation of this alkenylcopper species was also observed in the copper‐catalyzed hydrosilylation of terminal alkynes. See reference [43b].
- 51. Barbero A., Pulido F. J., Acc. Chem. Res. 2004, 37, 817. [DOI] [PubMed] [Google Scholar]