

Abstract
Probucol (PB), an antioxidant drug, is commonly used as a lipid concentration lowering drug to reduce blood plasma cholesterol levels in the clinic. However, the therapeutic effects of this drug are negatively impacted by its poor water solubility and low oral absorption efficiency. In this study, a PEGylated G5 PAMAM dendrimer (G5-PEG) modified nanoliposome was employed to increase water solubility, transepithelial transport, and oral absorption of PB. The uptake mechanism was explored in vitro in Caco-2 cells with the results suggesting that the absorption improvement of G5-PEG modified PB-liposome (PB-liposome/G5-PEG) was related to P-glycoprotein (P-gp) efflux pump, but was independent of caveolae endocytosis pathways. Additionally, plasma lipid concentration lowering effects of PB-liposome/G5-PEG were evaluated in vivo in a LDLR−/− hyperlipidemia mouse model. Compared with saline treated group, treatment with PB-liposome/G5-PEG significantly inhibited the increase of plasma total cholesterol (TC) and triglyceride (TG) of mice induced by a high fat diet. Moreover, its lipid concentration lowering effects and plasma drug concentration were greater than PB alone or commercial PB tablets. Our results demonstrated that PB-liposome/G5-PEG significantly increased the oral absorption of PB and therefore, significantly improved its pharmacodynamic effects.
Keywords: probucol, G5-PEG PAMAM dendrimer, nanoliposome, transepithelial absorption and mechanism, in vivo pharmacodynamic effects
1. Introduction
In recent research, probucol (PB), originally an antioxidant drug, has been found to decrease the concentrations of plasma total cholesterol and very low density lipoprotein cholesterol (VLDL-c), while also reducing atherosclerosis (AS) plaques and improving coronary restenosis by enhancing reverse cholesterol transport (RCT)1–3. Animal studies have also demonstrated the therapeutic effects of PB on cardiovascular diseases1, 4, 5. In addition to a lipid concentration lowering effect, PB also remarkably reduced lipid oxidation, delaying the progression of plaque formation. These effects make it a more promising drug than statins for prevention and cure of hyperlipidemia and AS in clinic.
Unfortunately, the therapeutic effects of this drug are significantly reduced by its poor water solubility (only 5 ng/mL in water) and low oral absorption efficiency (bioavailability only 2–8%)6. To achieve satisfactory lipid concentration lowering effects, patients have to increase PB dose at the expense of aggravating side effects including lowering high density lipoprotein cholesterol (HDL-c) and increasing the electrocardiogram (ECG) Q-T interval7. Although combining PB with statins can reduce the dose and negative side effects of PB, this strategy does not ameliorate the decreased concentration of HDL-c8–10. Therefore, improved strategies to increase the oral absorption efficiency, and thereby reduce side effects from high doses, are greatly needed for the clinic.
Preparation of nano-drug delivery systems is a general strategy to overcome problems of solubility and absorption of hydrophobic drugs, thereby improving their oral bioavailability. There are several kinds of PB nanoparticles reported in literature based on PB/polyvinylpyrrolidone/sodium dodecyl sulfate11, 12 or PB/sodium dodecyl sulfate/methacrylic acid co-grinding systems13, 14, self-microemulsion15–17 and nano-suspension18. Chitosan was also used as a carrier and trimeric sodium phosphate as a cross-linking agent to prepare PB containing nanoparticles19. Although all of these PB nano-delivery systems made improvements to some extent, for example improving the solubility of PB, there are still many issues remaining unresolved with these strategies including residues of organic solvent from preparation, surfactant related toxicity, and storage stability of liquid pharmaceuticals. Perhaps the largest challenge stems from the fact that the therapeutic dose of PB is about 500 mg per day for an adult with a normal body weight, but drug loading percentages of the present PB nano-delivery systems are usually low (5 to 10 wt.%), making it difficult to achieve the dose demands in clinic10–17. Therefore, further pharmaceutical studies on nano-delivery systems for PB are in great demand.
Liposomes are vesicular structures formed by lecithin bilayers with cholesterol insertion to increase membrane rigidity20. Hydrophobic drugs can be easily encapsulated in liposome bilayers with a high loading percentage21. Encapsulation of hydrophobic drugs in liposomes not only significantly increases solubility of the drug, but also releases the drug at a sustained and controlled rate22. In addition, components of liposome bilayer are very similar to that of the cell membrane, and therefore drugs encapsulated in liposomes can be easily delivered into the cells through confluence effects of the liposome with the cell membranes23. Although liposomes possess specific advantages in encapsulation and delivery of hydrophobic drugs, to the best of our knowledge there is no research related to PB liposomes to date.
Poly (amidoamine) (PAMAM) dendrimers, a class of highly branched polymers, have been demonstrated potential as drug delivery carriers due to their low polydispersity and nanoscopic size. Hydrophobic drugs can be attached on surface of dendrimer molecules electrostatically or be encapsulated in their non-polar interiors, resulting in increased water solubility and a sustained release of the encapsulated drugs24. In addition, PAMAM dendrimer can improve transepithelial permeation of the encapsulated drugs in the intestinal tract, although the mechanism remains unclear24–28. Despite these potential advantages, simple mixing of PB and dendrimers still results in a limited drug loading percentage and low storage stability29.
A combined drug delivery system composed of dendrimer and liposome was designed to overcome drawbacks of traditional single drug delivery system and offer advantages to the system. This combined drug delivery system can increase encapsulation efficacy30, 31 and modify the release rates of the loaded drugs32, 33, improve bioactivities of the therapeutics34, and promote the stability of the liposome membrane35.
Our previous studies demonstrate that modification with 8% polyethylene glycol (PEG, MW 5000) not only reduced the cytotoxicity and hemolysis toxicity of generation 5 (G5) PAMAM dendrimer, but also significantly increased gene delivery efficiency of this material36, 37. In this study, PEG 5000 conjugated G5 PAMAM dendrimer (G5-PEG) was utilized to modify liposome as a combined formulation for the improvement of solubility, encapsulation, release, transepithelial transport and therapeutic effects of PB. Studies probing the mechanism of permeability enhancement of liposomal formulation of G5-PEG modified PB-liposome (PB-liposome/G5-PEG) were explored in Caco-2 cell monolayers (an intestinal absorption model) and plasma lipid lowering effects of PB-liposome/G5-PEG were studied in vivo in low density lipoprotein receptor deficient (LDLR−/−) mice.
2. Materials and methods
2.1. Materials
PAMAM dendrimers of generation 5 with –NH2, –OH, and –COOH termination were purchased from Dendritech Inc (Midland, MI, USA). Cyclosporin A (CsA) and all other reagents at analytical grade were purchased from Sigma-Aldrich Chemical Company (Saint Louis, MO, USA). Probucol was purchased from Japanese Otsuka Corporation (Chiyodakum, Tokyo, Japan). Caco-2 cell line was bought from American Type Culture Collection (ATCC, Manassas, VA, USA). FITC tagged cholera toxin B subunit (CTB-FITC) was from Invitrogen (Carlsbad, CA, USA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS) and other cell culture related reagents were all purchased from GIBCO (New York, NY, USA). Rabbit-anti-caveolin-1 polyclonal antibody and goat-anti-rabbit IgG-HRP were from Santa Cruz Biotechnology (Dallas, TX, USA). Ad-CAV-1 and vector Ad-Null were from Vector Biolabs (Philadelphia, PA, USA). Polystyrene 24-well Transwell® with filters of 3.0 μm mean pore size was from COSTAR (Acton, MA, USA).
2.2. PEGylated G5 PAMAM dendrimer
G5 PAMAM dendrimers were purified by dialyzing (cut off: 10 KDa) against distilled water (8 media changes), then centrifuged by Millipore tube (Amicon Ultra, cut off: 5 KDa). Purified G5 was collected by lyophilization.
8% of the terminal amine groups of G5-NH2 PAMAM dendrimer were PEGylated by PEG 5000 to generate PEGylated G5 PAMAM dendrimer (G5-PEG) according to a previously published procedure36.
2.3. Water solubility of PB in the presence of G5
To investigate the influence of different charges and terminal groups of dendrimers on solubility of PB, saturation concentration of PB in water was measured in the presence of different G5 PAMAM with different end-group terminations (G5-PEG, G5-NH2, G5-COOH, and G5-OH). Briefly, excess PB (around 5 mg) was added to 2 mL water, which ensured a saturated condition. G5 dendrimers were added into the PB suspensions with equal molar concentrations of 174 nM dendrimer. Each suspension was mechanically shaken at 37 °C for 48 h and then centrifuged at 10000 rpm. PB concentrations in the supernatant were measured by HPLC (λ = 242 nm). Three repeats were conducted for each experiment.
2.4. Preparation and characterization of PB-liposome and G5-PEG modified PB-liposome
PB-liposomes were prepared by using a thin film dispersion method38. To prepare the lipid films, soybean lecithin (225 mg), cholesterol (25 mg) and PB (25 mg) were mixed in a 9:1:1 ratio and dissolved in 3 mL chloroform. Solvent was slowly removed by evaporation at 37 °C for 30 min in a rotary evaporator. The lipid films were then dried under vacuum overnight at room temperature. Multilamellar vesicles (MLVs) were prepared by hydrating the lipid film with 6 mL phosphate buffered saline (PBS, pH 7.5) and the resultant suspensions were sonicated for 30 min using a probe sonicator (at 25 HZôUP 50H, Hiescher, Germany) to achieve small unilamellar vesicles (SUVs) of PB-liposome.
To prepare G5-PEG modified PB liposome (PB-liposome/G5-PEG), the lipid film with PB was formed by the same method described above; however, 174 nM or 696 nM (4 × 174 nM) of G5-PEG in PBS was used to hydrate the lipid film to prepare PB-liposome/G5-PEG or PB-liposome/4(G5-PEG), respectively.
An Amicon® Ultra-15 3K ultrafiltration centrifugal tube (Millipore, USA) was used to remove unloaded free PB from the liposomes. Briefly, 6 mL PB-liposome or G5-PEG modified PB-liposome was added into the internal vial of the tube. Then 6 mL PBS (pH 7.5) was added in the liposomes followed by centrifugation (Eppendorf 5810R, USA) at 1672 g for 15 min. Free PB filtered out in PBS was discarded, and the PBS washing step was repeated for 5 to 6 times to remove all free PB from the liposomes.
The size and ζ-potential of PB-liposome and G5-PEG modified PB-liposome were measured using a Malvern Zetasizer Nano-ZS (Malvern Instruments Ltd, Worcestershire, UK), and the encapsulated percentage of PB in the liposomes was measured by HPLC.
2.5. Stability of the formulations in artificially gastrointestinal juice
To evaluate the stability of the PB/4(G5-PEG), PB-liposome and PB-liposome/4(G5-PEG) in the gastrointestinal environment of human body, artificial gastric juice (pH 1.89) and intestinal juice (pH 6.8) were prepared according to the China Pharmacopoeia, and in vitro stability of the three formulations in the above two juices were investigated. Briefly, 1 mL of each formulation was added into 9 mL of the artificial gastric or intestinal juice, and the mixture was stirred at 37 °C using a magnetic stirrer (MS-H280-Pro, China). At the time points of 2 h in the artificial gastric juice or 24 h in the artificial intestinal juice, 200 μL of samples were taken out and then demulsified with 800 μL methanol. The suspension was centrifuged (Eppendorf 5810R, USA) at 10450 g for 25 min, and the concentrations of PB in the supernatant were analyzed by HPLC. The stability of the three formulations were evaluated using a ratio of M/M0 (M was the mass of PB at 2 h in the artificial gastric juice or 24 h in the artificial intestinal juice, and M0 was the initial mass of PB in the three formulations before the experiment.
2.6. In vitro PB release
Three formulations of PB/4(G5-PEG), PB-liposome and PB-liposome/4(G5-PEG) were adjusted into an equal PB concentration of 1 mg/mL, and were aliquot into three dialysis bags (10 KD) respectively. The dialysis bags with 1 mL of the formulations were placed in glass vials containing 10 mL of artificial intestinal juice (pH 6.8 PBS) and maintained at 37°C in a shaker bath. At predetermined time intervals, 1 mL samples were taken out from the release medium and 1 mL fresh PBS (pH 6.8) were replaced in the vials. The concentrations of PB in the samples were determined by HPLC, and the accumulative release percentages of PB from the formulations were calculated and plotted.
2.7. Caco-2 Cell Culture
Caco-2 cells were grown in Dulbecco’s Modified Eagle’s Medium (DMEM) at 37 °C with an atmosphere of 95% oxygen and 5% CO2. To make a complete DMEM, 10% fetal bovine serum (FBS), 1% nonessential amino acids, 1% glutamine, 10,000 units/mL penicillin and 10,000 μg/mL streptomycin were supplemented in the cell culture medium. The cells were passaged at 80–90% confluence by using a 0.25% trypsin/ethylenediamine tetraacetic acid (EDTA) solution. Cell passages from 35 to 45 were used for the following experiments.
2.8. Cytotoxicity Assay
Cytotoxicity of PB, PB/G5-PEG, PB/4(G5-PEG), PB-liposome and PB-liposome/4(G5-PEG) was evaluated by MTT assay. Briefly, Caco-2 cells were seeded on a 96-well plate with a cell density of 2×l04 per well and grew to 80–90% confluence in the complete DMEM. The cell culture medium was changed to Hank’s balanced salt solution (HBSS). PB (dissolved in dimethyl sulfoxide (DMSO)), PB/G5-PEG, PB/4(G5-PEG), PB-liposome and PB-liposome/4(G5-PEG) were added respectively in HBSS with a PB concentration of 20 mM and 174 nM of G5-PEG or 696 nM of 4(G5-PEG). After a 72 h incubation, the incubation medium was removed and 10 μL of MTT stock solution (5 mg/mL) in 200 μL fresh HBSS was added in each well, followed by a four-hour-incubation. Then, supernatant in each well was removed and 200 μL of DMSO was added into the well. The plate was shaken for 10 min at 37 °C to dissolve MTT crystals. Absorbance at 590 nm was measured by utilizing a SpectraMax M2 microplate reader (Bio-rad550, USA). Cell viability with each treatment was expressed as a percentage compared to cells grown in HBSS only, which was considered as 100% cell viability. Six repeats were conducted for each sample.
2.9. Transepithelial Transport
Caco-2 cell monolayers were used as an in vitro intestinal absorption model to study the transepithelial permeability of PB, PB/G5-PEG, PB-liposome, and PB-liposome/G5-PEG. Caco-2 cells were seeded onto a 24-well Transwell® with a density of 5×105 cells/mL. The monolayer integrity, or ability of the cells to provide their barrier function, was monitored by transepithelial electrical resistance (TEER). After the cells had been cultured for 21 days, monolayers with a TEER of 350–450 Ω•cm2 were used for the transport studies.
Prior to transport experiments, the complete DMEM was changed to 500 and 1,500 μL HBSS buffer (with Mg2+ and Ca2+) in the apical (AP) and basolateral (BL) compartments of the Transwell, respectively. Samples to be tested were added to the AP side of compartment and 200 μL samples were collected from the BL side of compartment at 0, 30, 60, 90, 120, 180 and 240 min, followed by supplying 200 μL of fresh HBSS. PB concentrations in the collected samples were determined by HPLC. The apparent permeability coefficients (Papp) were calculated according to following formula:
dQ/dt : amount of drug transported across the monolayers per unit of time (mol • s−1); A: surface area of polycarbonate film of the Transwell, in this model equivalent to an area of support film of 1.13 cm2; C0: the initial drug concentration in the AP or BL side of the monolayers.
2.10. Mechanism of G5-PEG and liposome in improving the transport of PB across epithelium
The mechanism of G5-PEG and liposomal formulation to enhance the permeability of PB was explored using Caco-2 cells with a focus on the role of the P-glycoprotein (P-gp) efflux pump and caveolae endocytosis pathway.
2.10.1. P-glycoprotein (P-gp) efflux pump
Cyclosporin A (CsA), a P-gp inhibitor, was used to investigate the role of the P-gp efflux pump on transepithelial transport of PB and the liposome formulation across the Caco-2 cells.
Caco-2 cells were seeded onto a 24-well Transwell® and grew to be monolayers according to the procedure described above. Prior to experiments, 20 μM CsA was added in both the AP and BL side of the Transwell for 0.5 h. The medium was then replaced with fresh HBSS. Samples to be tested were added in the AP or BL side of the Transwells in order to study their transepithelial transport from AP to BL side (AP→BL) or BL to AP side (BP→AL), respectively. Samples (100 μL) from the opposite side of the addition compartment of the Transwell were collected at 60, 120, 180 and 240 min, followed by supplying 100 μL fresh HBSS in the well. PB concentration in the collected samples was determined by HPLC. Efflux ratio (ER, P ratio) was calculated using the following formula: P ratio = Papp (BL→AP) / Papp (AP→BL).
2.10.2. Caveolae
To study the possibility of transepithelial transport of PB and PB-liposome/G5-PEG by the caveolae endocytosis pathway, Caveolin-1 (Cav-1), the most crucial protein to maintain the structure and function of caveolae, was upregulated by adenovirus (Ad-Cav-1) infection of Caco-2 cells.
Briefly, Caco-2 cells were seeded on 6-well plates at a cell density of 5×104 per well. After 24h cell culture, the cells were infected by Ad-CAV-1 (titer: 1×1010 pfu/mL) at multiplicity of infection (MOI) of 10 or 30. The infected cells were then incubated for 48 h to express Cav-1 protein. To evaluate the influence of adenovirus vector alone on the cells, parallel experiments were conducted by using an empty adenovirus vector (Ad-null, titer: 1×1010 pfu/mL) to infect the cells at an equal dose of MOI 30.
After 48 h infection, the cells were collected and western blot (WB) was employed to detect expression of Cav-1 in cell lysates by using a primary antibody of rabbit-anti-caveolin-1 polyclonal antibody (1:1500 dilution) and a second antibody of goat-anti-rabbit IgG-HRP (1:2000 dilution).
CTB, a marker of the caveolae endocytosis pathway, was used to confirm normal endocytosis functions of caveolae regulated by Ad-Cav-1 on the Caco-2 cells. The effects of upregulation of caveolae pathway on uptake of PB or PB-liposome/G5-PEG were measured by flow cytometry.
Transwell experiments were done under the same conditions as described above to analyze the role of Cav-1 and the caveolae pathway in the process of transepithelial transport of PB/G5-PEG and PB-liposome/G5-PEG across the Caco-2 cell monolayers.
2.11. In vivo studies
Male LDLR−/− mice with an age of 7 to 8 weeks were obtained from the animal department of Peking University Health Science Center (Beijing, China). The Laboratory Animal Care Principles (NIH publication no. 85–23, revised 1996) were followed, and the experimental protocol was approved by Animal Care Committee, Peking University Health Science Center. All mice were raised under a 12-hour light/dark cycle with free access to food and water.
LDLR−/− mice were randomly divided into 4 groups with 6 mice in each group. To develop a hyperlipidemia disease model, all of mice were fed for 4 weeks with a western-type diet (TD 96125, Harlan-Teklad; 42% of calories from fat, 43% from carbohydrates, 15% from protein). From day 0, the 4 groups of mice were intragastric administrated twice a day with saline (NS), PB, commercial PB tablets, or PB-liposome/4(G5-PEG), respectively. All groups of mice receiving PB were treated at an equal PB dose of 133 mg/kg body weight per day. At the end of 4 weeks, blood samples of mice from each group were collected and plasma total cholesterol (TC) and triglyceride (TG) were determined. Plasma PB concentrations in the blood samples were measured by HPLC.
2.12. Statistical analysis
All data are presented as mean values ± standard error of mean (SEM) and analyzed by Student’s t-tests. One-way analysis of variance (ANOVA) with Bonferroni correction was used for comparison among multiple groups. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Solubilization of PB by G5 dendrimers
Characterization data for G5-PEG are shown in Fig. S1. A molecular structure of PB (Fig. 1A) and a cartoon of a PB/G5 dendrimer complex (Fig. 1B) are shown in Fig. 1. After forming complexes with G5 dendrimers, the water solubility of PB was improved to different extents depending upon the surface functionalities. Fig. S2 shows the effects of surface groups and charges of the G5 PAMAM dendrimers on the aqueous solubility of PB. G5-PEG and G5-NH2 showed a five to six folds improvement of aqueous solubility of PB as compared to G5-OH and G4.5-COOH. Fig. 1C illustrates that solubilization of PB by G5-PEG was highly concentration dependent. When the molar concentrations of G5-PEG increased to four times (696 nM) of the initial one (174 nM), the aqueous solubility of PB had a significant increase (3.7 fold), and reached to 1.5 μg/mL (a 300 fold increase compared to PB alone).
FIG. 1. Chemical structure of PB (A), cartoon of PB/G5 complex (B) and concentration-dependent solubilization of G5-PEG to PB (C).
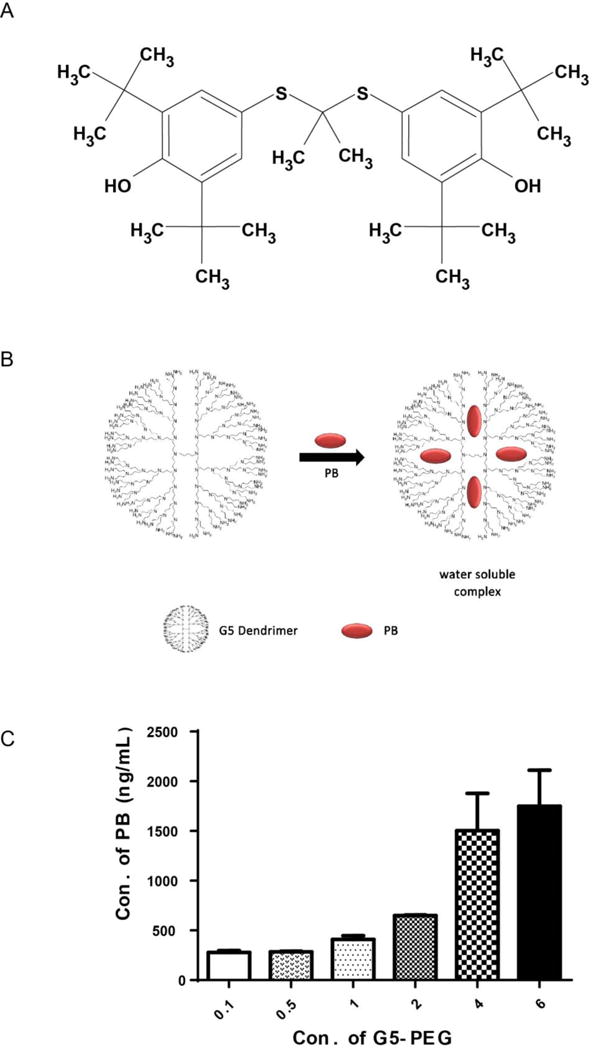
In cartoon B, red dots represent PB, blue scaffold is schematic dendrimer molecule. In Fig (C), the horizontal axis is the fold of molar concentration of G5-PEG. For example: 1 fold equals to 174 nM, 4 folds equal to 696 nM, and so on.
3.2. PB-liposome with or without G5-PEG modification
The cartoon in Fig. 2A illustrates the preparation process of G5-PEG modified PB-liposome. The average size of the PB-liposome and G5-PEG modified PB-liposome was determined as 115 ± 4.2 nm and 119 ± 3.7 nm, respectively. PB encapsulation efficiency in the PB-liposome and G5-PEG modified PB-liposome was 85 ± 1.45% and 84.3 ± 1.53%, respectively.
FIG. 2. In vitro PB release profiles.
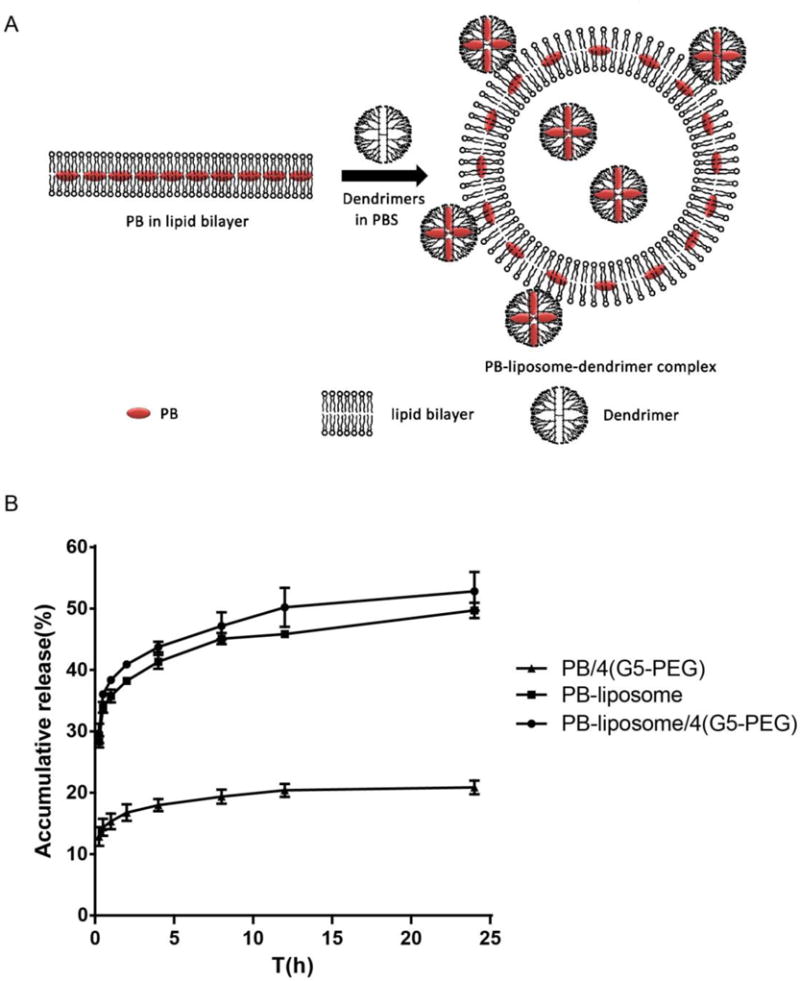
(A) Cartoon of preparation process of G5-PEG modified PB-liposome. Red dots represent PB. (B). In vitro PB release profiles of PB/4(G5-PEG), PB-liposome, and PB-liposome/4(G5-PEG) in an artificial intestine juice (pH 6.8 PBS). ***p<0.005 PB-liposome/4(G5-PEG) or PB-liposome vs. PB/4(G5-PEG) at all the investigated time points. n=3.
3.3. Stability of the formulations in artificially gastrointestinal juice
As shown in Table 1, the M/M0 ratios of the three formulations are all above 85%, which indicates they are relatively stable in the gastrointestinal fluid. After 2 h stirring in the artificial gastric juice (pH 1.89), PB-liposome had relatively lower M/M0 ratio (87.5±0.04% vs 94.7±0.01%, P=0.0545) than G5-PEG modified PB-liposome. In the artificial intestinal juice for 24 h, the M/M0 ratios remained above 90% for all the three formulations, indicating their great stability in the intestinal tract.
Table 1.
The stability of the three formulations in the artificially digestive tract fluid. (n=3, mean±SD)
| Formulations | M/M0 ratios of the formulations (%)
|
|
|---|---|---|
| Artificial gastric juice (pH1.89, 2 h) |
Artificial intestinal juice (pH6.8, 24 h) |
|
| PB/4(G5-PEG) | 91.9±0.14 | 91.1±0.002 |
| PB-liposome | 87.5±0.04 | 95.4±0.07 |
| PB-liposome/4(G5-PEG) | 94.7±0.01 | 93.4±0.10 |
3.3. In vitro PB release
Fig. 2B demonstrates that the accumulative PB release rate from PB/4(G5-PEG) in 24 h was 20.9 ± 1.1%, while PB-liposome and G5-PEG modified PB-liposome significantly increased PB release rate, and reached to 49.7 ± 1.2% (P<0.005) and 52.8 ± 3.2% (P<0.005), respectively. There was no difference between PB-liposome and PB-liposome/4(G5-PEG) in PB release rate.
3.4. Effects of G5 and liposomal formulation on transepithelial transport of PB
An MTT assay (Fig. 3A) shows that viabilities of Caco-2 cells were all above 85% after incubating the cells with 20 mM PB or the PB formulations (PB concentration in the formulations was 20 mM). PB forming complexes with 174 nM or 696 nM G5-PEG (described as PB/G5-PEG or PB/4(G5-PEG), respectively) did not obviously affect the activity of the Caco-2 cells. Modification of the PB-liposome with 696 nM of G5-PEG (described as PB-liposome/4(G5-PEG)) did not result in acute toxicity to the Caco-2 cells. The results demonstrate that the formulations containing high concentration (696 nM) of G5-PEG, such as PB/4(G5-PEG) and PB-liposome/4(G5-PEG), employed in the cellular experiments were all safe to Caco-2 cells.
FIG. 3. Effects of G5-PEG and liposomal formulation on the transepithelial transport of PB across Caco-2 cell monolayers.
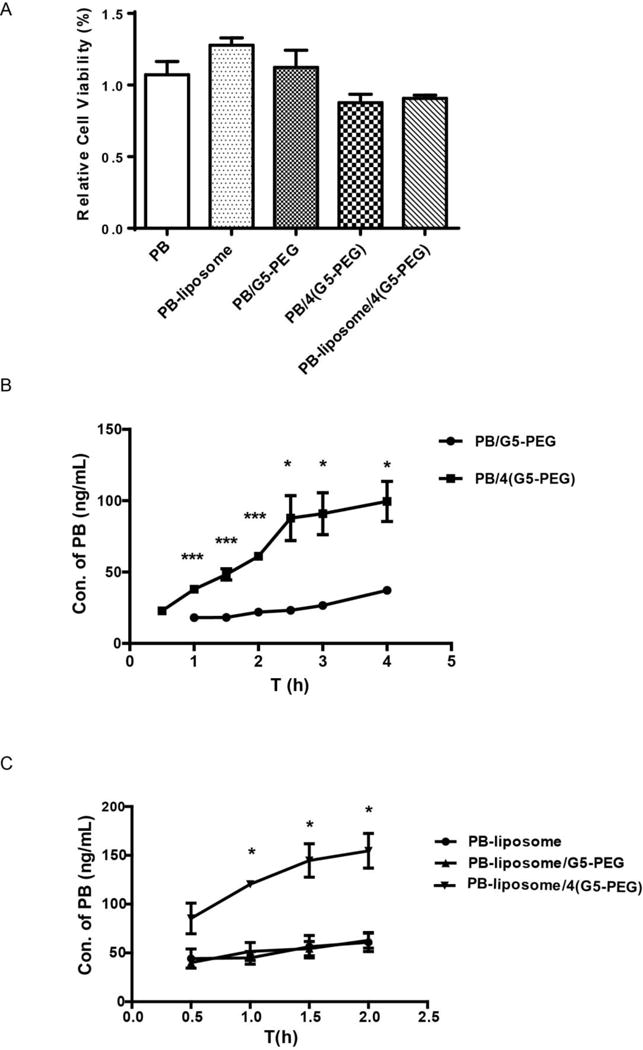
(A) Caco-2 cell viabilities in the presence of PB, PB-liposome, PB/G5-PEG, PB/4(G5-PEG) and PB-liposome/4(G5-PEG). n=6. (B) Effect of G5-PEG concentrations on the transport of PB across Caco-2 cell monolayers over a period of 4 h. *p<0.05, ***p<0.001 vs. PB/G5-PEG. n=3. (C) Effects of G5-PEG concentrations on transport of PB-liposome across Caco-2 cell monolayers over a period of 2 h. *p<0.05 vs. PB-liposome. n=3.
Fig. 3B illustrates that the transepithelial transport of PB across Caco-2 cell monolayers was significantly increased when 4(G5-PEG) (696 nM) was applied to form complexes with PB. Although it significantly increased water solubility of PB (Fig. S2), G5-NH2 (174 nM) failed to improve transepithelial transport of PB, even when higher molar concentration of 4(G5-NH2) (696 nM) was applied (Fig. S3). Comparing Fig. 3C with Fig. 3B, transepithelial transport of PB was increased about 2 folds by liposomal formulation, and modification with 4(G5-PEG) (696 nM) more significantly enhanced the transepithelial transport of PB-liposome (Fig. 3C). These enhancement effects not only depended on the concentrations of G5-PEG, but were also related to the incubation time. With the presence of 4(G5-PEG), the transepithelial transport of PB (Fig. 3B) and its liposome (Fig. 3C) was greater increased within the first than the second 2h of incubation (data of PB-liposome are only shown the first 2h of incubation).
3.5. Mechanism of G5-PEG and liposomal formulation in improving the transport of PB across the epithelium
3.5.1. P-gp efflux pump
Fig. 4A shows that 20 μM CsA significantly improved Papp (AP→BL) of PB-liposome and PB-liposome/4(G5-PEG). In addition, Efflux P ratios of PB/4(G5-PEG) (Fig. 4B) and PB-liposome (Fig. 4C) were significantly reduced by addition of 20 μM CsA, which suggested increased PB transport from AP to BL side, and/or a decreased PB transport from BL to AP side of Caco-2 monolayers. The sum of these results demonstrates that the transepithelial transport of PB/4(G5-PEG), PB-liposome, and PB-liposome/4(G5-PEG) were all related to the P-gp efflux pump on Caco-2 cells.
FIG. 4. Effects of CsA on transepithelial transport of PB/4(G5-PEG), PB-liposome and PB-liposome/4(G5-PEG) in Caco-2 cell monolayers.
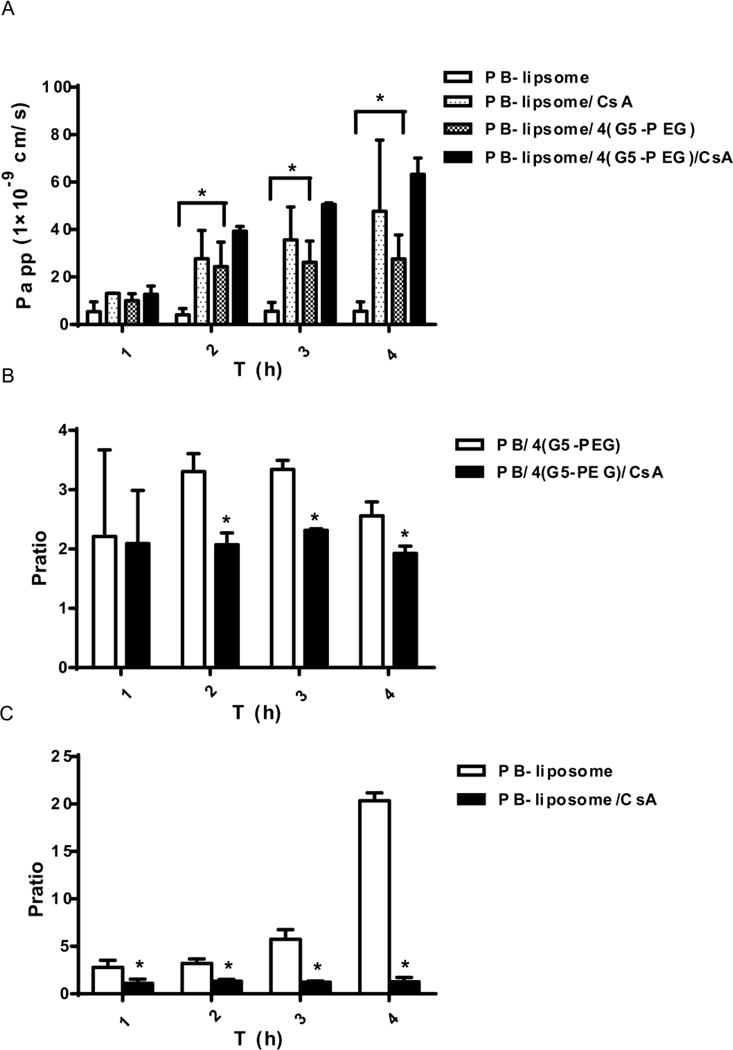
(A) Papps of PB-liposome and PB-liposome/4(G5-PEG) before and after 20 μM CsA being applied. (B & C) Efflux P ratios of PB/4(G5-PEG) (B) and PB-liposome (C) before and after 20 μM CsA being applied. P ratio=Papp (BL-AP)/Papp (AP-BL). *p<0.05 vs. data without CsA. n=3.
3.5.2. Cav-1 protein and caveolae endocytosis pathway
WB results in Fig. 5A indicate that Cav-1 expression was successfully upregulated by Ad-Cav-1 at a dose of MOI 30, while adenovirus vector itself (Ad-null) almost had no influence on the expression of Cav-1 at an equal dose. Endocytosis functions of caveolae were confirmed through a significant increase of CTB-FITC uptake in Caco-2 cells facilitated by the adenovirus expressed Cav-1 (Fig. 5B). By way of comparison, Transwell experiments indicated that transepithelial transport of PB/4(G5-PEG) (Fig. 6A), and PB-liposome/4(G5-PEG) (Fig. 6C) were not influenced by upregulation of Cav-1 expression on Caco-2 cells. Even the transport of PB-liposome had a decreasing trend after Cav-1 expression was upregulated, there was no significant difference (Fig. 6B). The results demonstrate that the permeability enhancement of G5-PEG and liposomal formulation to PB was not related to Cav-1 and the caveolae endocytosis pathway in Caco-2 cells.
FIG. 5. Effects of upregulation of Cav-1 on uptakes of CTB in Caco-2 cells.

(A) Cav-1 expression detected by western blot before and after the cells being infected by Ad-Cav-1 and with GAPDH as an internal control. (B) Uptakes of CTB-FITC before and after Cav-1 being upregulated in Caco-2 cells.
FIG. 6. Effects of upregulation of Cav-1 on transepithelial transport of PB/4(G5-PEG) (A), PB-liposome (B), and PB-liposome/4(G5-PEG) (C) in Caco-2 cell monolayers.
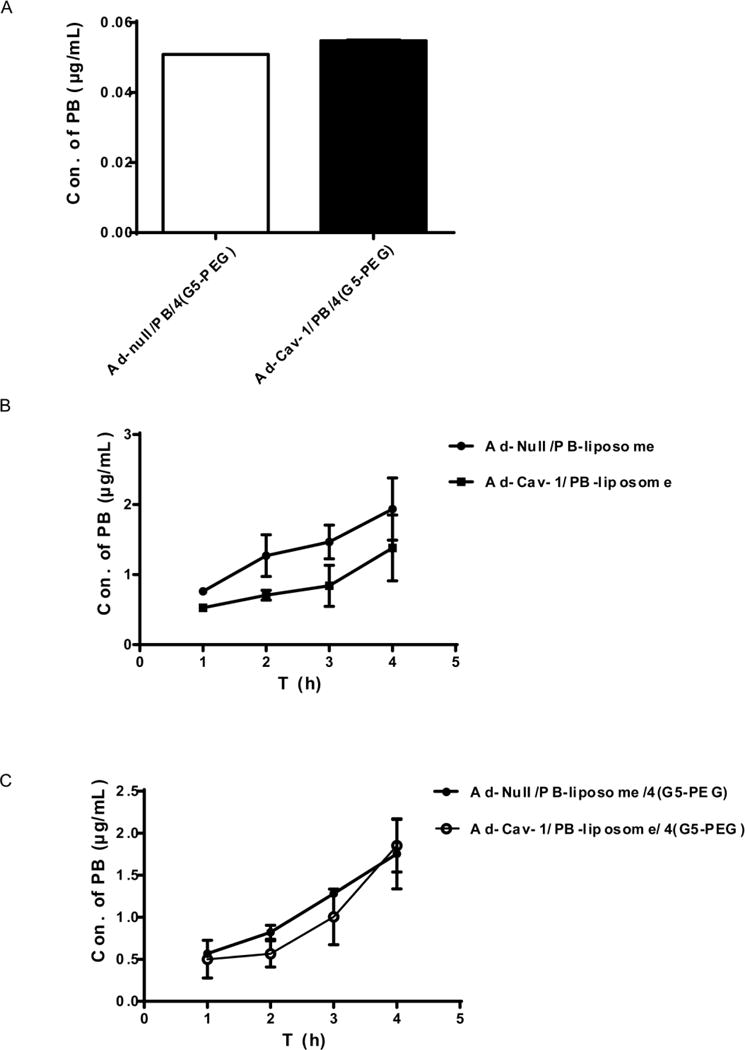
Fig (A) was a sum of 4h transport.
3.6. In vivo lipid concentration lowering effects of G5-PEG modified PB-liposome
Fig. 7B&C demonstrate that plasma TC (B) and TG (C) levels of LDLR−/− mice were significantly decreased in the PB-liposome/4(G5-PEG) treated group compared with that of the saline treated group, but both TC and TG levels did not change in the groups treated with either PB or commercial PB tablets. Plasma PB concentrations from the PB-liposome/4(G5-PEG) treated group were 5 (P<0.005) and 2 (P<0.01) times higher than those from the PB and the commercial PB tablets treated groups, respectively (Fig. 7A). The results demonstrate that G5-PEG modified liposome significantly increased the oral absorption of PB, and therefore, significantly improved its lipid concentration lowering effects.
FIG. 7. Plasma PB concentrations and lipid concentration lowering effects of G5-PEG modified PB-liposome in LDLR.

−/− mice. (A) Plasma PB concentrations of mice from each group determined by HPLC. *p<0.05, ***p<0.001 vs. PB. (B) Plasma total cholesterol level of mice from each group. *p<0.05 vs. NS. (C) Plasma TG level of mice from each group. **p<0.01 vs. NS. NS means saline-treated group. n=6. All data were measured after the mice having been treated with the formulations in company with high fat diet for 4 weeks.
4. Discussion
In this research, we investigated and reported the effects of G5-PEG modified PB-liposome by in vitro transepithelial transport and in vivo oral absorption of PB. First, we found that all four surface functionalities of G5 dendrimers investigated improved the solubility of PB in water. In all cases, the hydrophobic drug could be encapsulated in the internal hydrophobic cavities of the G5 dendrimer molecules. Positively charged G5-NH2 and G5-PEG improved the water solubility of PB substantially more than negatively charged G4.5-COOH or neutral G5-OH did. This agrees with our previous results, which indicate that positively charged dendrimers expand in water whereas the neutral dendrimers collapse on themselves39, 40, that might make the internal spaces of positively charged dendrimers more available than neutral dendrimers. Therefore, PB is much more soluble in G5-NH2 or G5-PEG than in G4.5-COOH or G5-OH.
Although it was less effective than G5-NH2, PEG 5000 could also increase PB solubility in water because of its hydrophilic effects (Fig. S2). Therefore, modification of G5-NH2 with PEG 5000 resulted in the material with the best PB solubilization capability (Fig. S2).
The solubilization of PB by G5-PEG was concentration dependent, since more dendrimer molecules would be available to encapsulate more drugs at higher concentrations. It was seen that when molar concentrations of G5-PEG were elevated to four times higher than the initial one, its solubilization effect increased rapidly (Fig. 1C).
Although G5-NH2 and G5-PEG solubilized PB to a similar degree (Fig S2), G5-NH2 failed to improve transepithelial transport of PB on Caco-2 cell monolayers, even when a high concentration of 696 nM G5-NH2 was employed to form PB/4(G5-NH2) complex (Fig S3). By comparison, G5-PEG not only significantly improved solubility of PB, but it also significantly increased transepithelial transport of PB (Fig 3B). The results suggest that absorption enhancement of G5-PEG to PB also related to mechanisms other than solubilization. For example, inhibition of P-gp on the Caco-2 cell membranes, which was investigated in this work.
Liposomes are an effective approach solving the problems of poor water solubility and low oral absorption for hydrophobic drugs20. Our results demonstrate that liposomal formulation significantly increased encapsulation, release rate (Fig. 2B) and transepithelial transport (Fig. 3C) of PB. G5-PEG was chosen to modify PB-liposome, since it gave the best improvement to the solubility (Fig. S2) and permeability (Fig. S3) of PB as compared to the other three investigated G5 dendrimers. Although modification of PB-liposome with G5-PEG did not significantly change encapsulation and release rate (Fig. 2B) of PB, it protected PB-liposome from being degraded in the gastric fluid (Table 1). Besides, the hydrophilic characteristic of G5-PEG might enable it to attach to the external aqueous phase and also enter the internal water phase of the liposome (Fig. 2A), which stabilized the liposomal formulation36. On the other hand, PAMAM dendrimer has good permeability across cell membranes, which might help to promote transmembrane transport of liposomes41, 42. Our results show modification of PB-liposome with 696 nM G5-PEG significantly increased the transepithelial transport of PB (Fig. 3C).
Strategies for PAMAM dendrimer platforms promoting drug absorption have been mainly focused on two mechanisms: 1) intracellular endocytosis into cells via proteins or receptors on the cell membranes43–45, and 2) intercellular transport by disturbing and opening tight junctions of the cells46. Liposomes provide a third mechanism, namely amalgamation or fusion of the liposome bilayers with the cell membrane. However, specific mechanisms of how G5-PEG modified liposome promote drug transmembrane transport has remained unknown. The roles of the P-gp efflux pump and caveolae pathway in endocytosis were investigated in this paper.
P-gp is a 170 kD transmembrane glycoprotein belonging to the ATP binding cassette (ABC) transporter carrier protein family. It inhibits absorption of drugs by an energy dependent discharge of drug from the intracellular side to extracellular side of cells47. Correlation of drug transport with P-gp can be determined by testing Papp and P ratio of the drug. If the Papp value remains constant over the entire concentration range, passive diffusion is considered to be the main transport mechanism. In this study, the Papp value of PB increased with concentration, suggesting that the permeability enhancement of PB/G5-PEG did not follow a simple passive diffusion mechanism. Moreover, with 20 μM CsA, Papp (AP→BL) significantly increased for both PB-liposome and PB-liposome/4(G5-PEG). Besides, P ratios of PB/4(G5-PEG) and PB-liposome decreased. The results demonstrate that absorption of PB promoted by G5-PEG, liposome, or G5-PEG modified liposome was all modulated by P-gp efflux pump. Inhibition of P-gp by CsA contributed to increase the transepthelial transport of PB/G5-PEG, PB-liposome and PB-liposome/G5-PEG.
Caveolae is a concave structure of the cytoplasm membrane with a variety of forms and can be found in many types of cells48. Caveolin-1 (Cav-1) is the most important protein to maintain the structure and functions of caveolae. Therefore, in order to study the relationship of the transmembrane transport process of G5-PEG modified PB-liposome and the caveolae endocytic pathway, Cav-1 expression was upregulated in Caco-2 cells by adenovirus infection. The uptake of CTB, a caveolae endocytosis pathway marker, significantly increased after Ad-CAV-1 infection in Caco-2 cells (Fig. 5B), which indicates that upregulation of Cav-1 by the adenovirus can facilitate the normal endocytosis function of caveolae. By way of comparison, transwell studies (Fig. 6) indicate that the transport of PB-liposome/4(G5-PEG) was not proceeded by the caveloae pathway. Additional mechanistic studies are needed to further clarify the other possible mechanism for transepithelial transport of G5-PEG modified PB-liposome.
As PB is commonly used as a lipid-lowering drug to reduce plasma cholesterol and triglyceride concentrations in clinic, we studied oral pharmacodynamics of PB-liposome/4(G5-PEG) in vivo in LDLR−/− mice with PB and commercial PB tablets as two controls. LDLR−/− mice can be induced to hyperlipidemia after being fed with a high fat diet for one month. The plasma TC level from the saline treated mice group increased to 600–900 mg/dL after four weeks, which meant severe hypercholesterolemia was successfully developed in LDLR−/− mice. By way of comparison, treatment with PB-liposome/4(G5-PEG) significantly inhibited the increase of plasma TC (Fig. 7B) and TG (Fig. 7C) levels of mice, while treatment with either PB or commercial tablets resulted in no significant difference in plasma TC and TG levels (Fig. 7). The results demonstrate that PB-liposome/4(G5-PEG) had better lipid concentration lowering effects than PB or commercial PB tablets. Additionally, mice body weights from each group showed no difference (Fig. S4), which meant food intake in each group was equal. Therefore, the greater lipid concentration lowering effects of PB-liposome/4(G5-PEG) than PB or commercial PB tablets were not from weight loss of the mice, but from the greater solubilization, encapsulation, stability, PB release, and absorption of PB improved by G5-PEG modified liposome. Plasma drug concentration also confirmed this point, which showed that plasma PB concentrations from PB-liposome/4(G5-PEG) treated mice group were nearly five times higher than those from the PB treated group (Fig. 7A).
5. Conclusions
G5-PEG modified liposome formulation significantly increased the water solubility and transepithelial transport of PB. Enhancement mechanisms were related to P-gp efflux pump, but were independent of caveolae endocytosis pathways. Compared with PB alone and commercial PB tablets, PB-liposome/4(G5-PEG) significantly improved plasma concentration of PB in LDLR−/− mice after oral administration, which resulted in a dramatic inhibition of the elevation of plasma TC and TG levels induced by high fat diet.
Supplementary Material
Acknowledgments
This study was supported by grants from the National Basic Research Program of China (No. 2009CB930300), National Natural Science Foundation of China (No. 81270368ô81360054), and federal funds from the National Institute of Biomedical Imaging and Bioengineering (RO1-EB005028).
Footnotes
Supporting Information
Supporting information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Yamamoto S, Tanigawa H, Li X, Komaru Y, Billheimer JT, Rader DJ. Pharmacologic suppression of hepatic ATP-binding cassette transporter 1 activity in mice reduces high-density lipoprotein cholesterol levels but promotes reverse cholesterol transport. Circulation. 2011;124:1382–1390. doi: 10.1161/CIRCULATIONAHA.110.009704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sawayama Y, Shimizu C, Maeda N, Tatsukawa M, Kinukawa N, Koyanagi S, Kashiwagi S, Hayashi J. Effects of probucol and pravastatin on common carotid atherosclerosis in patients with asymptomatic hypercholesterolemia. Fukuoka Atherosclerosis Trial (FAST) J Am Coll Cardiol. 2002;39:610–616. doi: 10.1016/s0735-1097(01)01783-1. [DOI] [PubMed] [Google Scholar]
- 3.Wakeyama T, Ogawa H, Iida H, Takaki A, Iwami T, Mochizuki M, Tanaka T. Effects of candesartan and probucol on restenosis after coronary stenting: results of insight of stent intimal hyperplasia inhibition by new angiotensin II receptor antagonist (ISHIN) trial. Circ J. 2003;67:519–524. doi: 10.1253/circj.67.519. [DOI] [PubMed] [Google Scholar]
- 4.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor SR-BI/apolipoprotein E double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu BJ, Di Girolamo N, Beck K, Hanratty CG, Choy K, Hou JY, Ward MR, Stocker R. Probucol [4,4′-[(1-methylethylidene)bis(thio)]bis-[2,6-bis(1,1-dimethylethyl)phenol]] inhibits compensatory remodeling and promotes lumen loss associated with atherosclerosis in apolipoprotein E-deficient mice. J Pharmacol Exp Ther. 2007;321:477–484. doi: 10.1124/jpet.106.118612. [DOI] [PubMed] [Google Scholar]
- 6.Betge S, Lutz K, Roskos M, Figulla HR. Oral treatment with probucol in a pharmacological dose has no beneficial effects on mortality in chronic ischemic heart failure after large myocardial infarction in rats. Eur J Pharmacol. 2007;558:119–127. doi: 10.1016/j.ejphar.2006.11.049. [DOI] [PubMed] [Google Scholar]
- 7.Dujovne CA, Harris WS. The pharmacological treatment of dyslipidemia. Annu Rev Pharmacol Toxicol. 1989;29:265–288. doi: 10.1146/annurev.pa.29.040189.001405. [DOI] [PubMed] [Google Scholar]
- 8.Yoshikawa T, Mitani K, Kotosai K, Nozako M, Miyakoda G, Yabuuchi Y. Antiatherogenic effects of cilostazol and probucol alone, and in combination in low density lipoprotein receptor-deficient mice fed with a high fat diet. Horm Metab Res. 2008;40:473–478. doi: 10.1055/s-2008-1065348. [DOI] [PubMed] [Google Scholar]
- 9.Ko YG, Kim BK, Lee BK, Kang WC, Choi SH, Kim SW, Lee JH, Lee M, Honda Y, Fitzerald PJ, Shim WH, Investigators, S Study design and rationale of “Synergistic effect of combination therapy with cilostazol and ProbUcol on plaque stabilization and lesion REgression (SECURE)” study: a double-blind randomised controlled multicenter clinical trial. Trials. 2011;12:10. doi: 10.1186/1745-6215-12-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim JH, Park SH, Bae SS, Hong KW, Kim YD, Park KP, Choi BT, Shin HK. Combinatorial effect of probucol and cilostazol in focal ischemic mice with hypercholesterolemia. J Pharmacol Exp Ther. 2011;338:451–457. doi: 10.1124/jpet.111.181180. [DOI] [PubMed] [Google Scholar]
- 11.Zhang J, Higashi K, Limwikrant W, Moribe K, Yamamoto K. Molecular-level characterization of probucol nanocrystal in water by in situ solid-state NMR spectroscopy. Int J Pharm. 2012;423:571–576. doi: 10.1016/j.ijpharm.2011.11.028. [DOI] [PubMed] [Google Scholar]
- 12.Wanawongthai C, Pongpeerapat A, Higashi K, Tozuka Y, Moribe K, Yamamoto K. Nanoparticle formation from probucol/PVP/sodium alkyl sulfate co-ground mixture. Int J Pharm. 2009;376:169–175. doi: 10.1016/j.ijpharm.2009.04.034. [DOI] [PubMed] [Google Scholar]
- 13.Io T, Fukami T, Yamamoto K, Suzuki T, Xu J, Tomono K, Ramamoorthy A. Homogeneous nanoparticles to enhance the efficiency of a hydrophobic drug, antihyperlipidemic probucol, characterized by solid-state NMR. Mol Pharm. 2010;7:299–305. doi: 10.1021/mp900254y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fukami T, Ishii T, Io T, Suzuki N, Suzuki T, Yamamoto K, Xu J, Ramamoorthy A, Tomono K. Nanoparticle processing in the solid state dramatically increases the cell membrane permeation of a cholesterol-lowering drug, probucol. Mol Pharm. 2009;6:1029–1035. doi: 10.1021/mp9000487. [DOI] [PubMed] [Google Scholar]
- 15.Nielsen FS, Petersen KB, Mullertz A. Bioavailability of probucol from lipid and surfactant based formulations in minipigs: influence of droplet size and dietary state. Eur J Pharm Biopharm. 2008;69:553–562. doi: 10.1016/j.ejpb.2007.12.020. [DOI] [PubMed] [Google Scholar]
- 16.Fatouros DG, Nielsen FS, Douroumis D, Hadjileontiadis LJ, Mullertz A. In vitro-in vivo correlations of self-emulsifying drug delivery systems combining the dynamic lipolysis model and neuro-fuzzy networks. Eur J Pharm Biopharm. 2008;69:887–898. doi: 10.1016/j.ejpb.2008.01.022. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen FS, Gibault E, Ljusberg-Wahren H, Arleth L, Pedersen JS, Mullertz A. Characterization of prototype self-nanoemulsifying formulations of lipophilic compounds. J Pharm Sci. 2007;96:876–892. doi: 10.1002/jps.20673. [DOI] [PubMed] [Google Scholar]
- 18.Shou-lei LV, W-x J, Cheng-gang WANG. Preparation and in vivo evaluation of probucol nanosuspensions. China Pharmaceutical Journal. 2010;45:3405–3409. [Google Scholar]
- 19.Li GAO, A-j W. Preparation of probucol loaded chitosan nanoparticles andin vitrorelease study. Chinese Journal of New Drugs. 2009;6:1892–1896. [Google Scholar]
- 20.Ramishetti S, Huang L. Intelligent design of multifunctional lipid-coated nanoparticle platforms for cancer therapy. Ther Deliv. 2012;3:1429–1445. doi: 10.4155/tde.12.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weiner AL. Liposomes for protein delivery: selecting manufacture and development processes. Immunomethods. 1994;4:201–209. doi: 10.1006/immu.1994.1021. [DOI] [PubMed] [Google Scholar]
- 22.Honda M, Asai T, Oku N, Araki Y, Tanaka M, Ebihara N. Liposomes and nanotechnology in drug development: focus on ocular targets. Int J Nanomedicine. 2013;8:495–503. doi: 10.2147/IJN.S30725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hougeir FG, Kircik L. A review of delivery systems in cosmetics. Dermatol Ther. 2012;25:234–237. doi: 10.1111/j.1529-8019.2012.01501.x. [DOI] [PubMed] [Google Scholar]
- 24.Kitchens KM, El-Sayed ME, Ghandehari H. Transepithelial and endothelial transport of poly (amidoamine) dendrimers. Adv Drug Deliv Rev. 2005;57:2163–2176. doi: 10.1016/j.addr.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 25.Sweet DM, Kolhatkar RB, Ray A, Swaan P, Ghandehari H. Transepithelial transport of PEGylated anionic poly(amidoamine) dendrimers: implications for oral drug delivery. J Control Release. 2009;138:78–85. doi: 10.1016/j.jconrel.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kitchens KM, Kolhatkar RB, Swaan PW, Eddington ND, Ghandehari H. Transport of poly(amidoamine) dendrimers across Caco-2 cell monolayers: Influence of size, charge and fluorescent labeling. Pharm Res. 2006;23:2818–2826. doi: 10.1007/s11095-006-9122-2. [DOI] [PubMed] [Google Scholar]
- 27.Morgan MT, Nakanishi Y, Kroll DJ, Griset AP, Carnahan MA, Wathier M, Oberlies NH, Manikumar G, Wani MC, Grinstaff MW. Dendrimer-encapsulated camptothecins: increased solubility, cellular uptake, and cellular retention affords enhanced anticancer activity in vitro. Cancer Res. 2006;66:11913–11921. doi: 10.1158/0008-5472.CAN-06-2066. [DOI] [PubMed] [Google Scholar]
- 28.Ke W, Zhao Y, Huang R, Jiang C, Pei Y. Enhanced oral bioavailability of doxorubicin in a dendrimer drug delivery system. J Pharm Sci. 2008;97:2208–2216. doi: 10.1002/jps.21155. [DOI] [PubMed] [Google Scholar]
- 29.Holden CA, Tyagi P, Thakur A, Kadam R, Jadhav G, Kompella UB, Yang H. Polyamidoamine dendrimer hydrogel for enhanced delivery of antiglaucoma drugs. Nanomedicine. 2012;8:776–783. doi: 10.1016/j.nano.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 30.Khopade AJ, Caruso F, Tripathi P, Nagaich S, Jain NK. Effect of dendrimer on entrapment and release of bioactive from liposomes. Int J Pharm. 2002;232:157–162. doi: 10.1016/s0378-5173(01)00901-2. [DOI] [PubMed] [Google Scholar]
- 31.Kontogiannopoulos KN, Assimopoulou AN, Hatziantoniou S, Karatasos K, Demetzos C, Papageorgiou VP. Chimeric advanced drug delivery nano systems (chi-aDDnSs) for shikonin combining dendritic and liposomal technology. Int J Pharm. 2012;422:381–389. doi: 10.1016/j.ijpharm.2011.09.031. [DOI] [PubMed] [Google Scholar]
- 32.Papagiannaros A, Dimas K, Papaioannou GT, Demetzos C. Doxorubicin-PAMAM dendrimer complex attached to liposomes: cytotoxic studies against human cancer cell lines. Int J Pharm. 2005;302:29–38. doi: 10.1016/j.ijpharm.2005.05.039. [DOI] [PubMed] [Google Scholar]
- 33.Gardikis K, Tsimplouli C, Dimas K, Micha-Screttas M, Demetzos C. New chimeric advanced Drug Delivery nano Systems (chi-aDDnSs) as doxorubicin carriers. Int J Pharm. 2010;402:231–237. doi: 10.1016/j.ijpharm.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Gardikis K, Hatziantoniou S, Bucos M, Fessas D, Signorelli M, Felekis T, Zervou M, Screttas CG, Steele BR, Ionov M, Micha-Screttas M, Klajnert B, Bryszewska M, Demetzos C. New drug delivery nanosystem combining liposomal and dendrimeric technology (liposomal locked-in dendrimers) for cancer therapy. J Pharm Sci. 2010;99:3561–3571. doi: 10.1002/jps.22121. [DOI] [PubMed] [Google Scholar]
- 35.Gardikis K, Hatziantoniou S, Signorelli M, Pusceddu M, Micha-Screttas M, Schiraldi A, Demetzos C, Fessas D. Thermodynamic and structural characterization of Liposomal-Locked in-Dendrimers as drug carriers. Colloids Surf B Biointerfaces. 2010;81:11–19. doi: 10.1016/j.colsurfb.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 36.Qi R, Gao Y, Tang Y, He RR, Liu TL, He Y, Sun S, Li BY, Li YB, Liu G. PEG-conjugated PAMAM dendrimers mediate efficient intramuscular gene expression. AAPS J. 2009;11:395–405. doi: 10.1208/s12248-009-9116-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tang Y, Li YB, Wang B, Lin RY, van Dongen M, Zurcher DM, Gu XY, Banaszak Holl MM, Liu G, Qi R. Efficient in vitro siRNA delivery and intramuscular gene silencing using PEG-modified PAMAM dendrimers. Mol Pharm. 2012;9:1812–1821. doi: 10.1021/mp3001364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Elhissi AM, O’Neill MA, Roberts SA, Taylor KM. A calorimetric study of dimyristoylphosphatidylcholine phase transitions and steroid-liposome interactions for liposomes prepared by thin film and proliposome methods. Int J Pharm. 2006;320:124–130. doi: 10.1016/j.ijpharm.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 39.Kelly CV, Leroueil PR, Orr BG, Banaszak Holl MM, Andricioaei I. Poly(amidoamine) dendrimers on lipid bilayers II: Effects of bilayer phase and dendrimer termination. J Phys Chem B. 2008;112:9346–9353. doi: 10.1021/jp8013783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly CV, Leroueil PR, Nett EK, Wereszczynski JM, Baker JR, Jr, Orr BG, Banaszak Holl MM, Andricioaei I. Poly(amidoamine) dendrimers on lipid bilayers I: Free energy and conformation of binding. J Phys Chem B. 2008;112:9337–9345. doi: 10.1021/jp801377a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kolhatkar RB, Swaan P, Ghandehari H. Potential oral delivery of 7-ethyl-10-hydroxy-camptothecin (SN-38) using poly(amidoamine) dendrimers. Pharm Res. 2008;25:1723–1729. doi: 10.1007/s11095-008-9572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ebrahimnejad P, Dinarvand R, Sajadi A, Jaafari MR, Nomani AR, Azizi E, Rad-Malekshahi M, Atyabi F. Preparation and in vitro evaluation of actively targetable nanoparticles for SN-38 delivery against HT-29 cell lines. Nanomedicine. 2010;6:478–485. doi: 10.1016/j.nano.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 43.El-Sayed M, Ginski M, Rhodes C, Ghandehari H. Transepithelial transport of poly(amidoamine) dendrimers across Caco-2 cell monolayers. J Control Release. 2002;81:355–365. doi: 10.1016/s0168-3659(02)00087-1. [DOI] [PubMed] [Google Scholar]
- 44.Kitchens KM, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis inhibitors prevent poly(amidoamine) dendrimer internalization and permeability across Caco-2 cells. Mol Pharm. 2008;5:364–369. doi: 10.1021/mp700089s. [DOI] [PubMed] [Google Scholar]
- 45.Kitchens KM, Foraker AB, Kolhatkar RB, Swaan PW, Ghandehari H. Endocytosis and interaction of poly (amidoamine) dendrimers with Caco-2 cells. Pharm Res. 2007;24:2138–2145. doi: 10.1007/s11095-007-9415-0. [DOI] [PubMed] [Google Scholar]
- 46.Goldberg DS, Ghandehari H, Swaan PW. Cellular entry of G3.5 poly (amido amine) dendrimers by clathrin- and dynamin-dependent endocytosis promotes tight junctional opening in intestinal epithelia. Pharm Res. 2010;27:1547–1557. doi: 10.1007/s11095-010-0153-3. [DOI] [PubMed] [Google Scholar]
- 47.Barrand MA, Bagrij T, Neo SY. Multidrug resistance-associated protein: a protein distinct from P-glycoprotein involved in cytotoxic drug expulsion. Gen Pharmacol. 1997;28:639–645. doi: 10.1016/s0306-3623(96)00284-4. [DOI] [PubMed] [Google Scholar]
- 48.Parton RG. Caveolae and caveolins. Curr Opin Cell Biol. 1996;8:542–548. doi: 10.1016/s0955-0674(96)80033-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.