

Abstract
The development of methods for expression and purification of seven-transmembrane receptors has led to an increase in structural and biophysical data and greatly improved the understanding of receptor structure and function. For chemokine receptors, this has been highlighted by the determination of crystal structures of CXCR4 and CCR5 in complex with small molecule antagonists, followed recently by two receptor/chemokine complexes; CXCR4 in complex with vMIP-II and US28 in complex with the CX3CL1. However, these studies cover only a few of the many chemokines and chemokine receptors and production of stable receptor/chemokine complexes remains a challenging task. Here, we present a method for producing purified complexes between chemokine receptors and chemokines by co-expression in Sf9 cells. Using the complex between atypical chemokine receptor 3 (ACKR3) and its native chemokine CXCL12 as an example, we describe the virus production, protein expression and purification process as well as reconstitution into different membrane mimics. This method provides an efficient way of producing pure receptor/chemokine complexes and has been used to successfully produce receptor/chemokine complexes for CXC as well as CC receptors.
Keywords: chemokine, chemokine receptor, ACKR3, CXCL12, Sf9, membrane protein, co-expression, protein complex
1. Introduction
Chemokines are ∼10 kDa signaling proteins that control cell migration in the context of development, immune surveillance and inflammation by binding to chemokine receptors at the cell membrane (Allen, Crown, Handel, 2007). Most chemokine receptors belong to the large family of G protein-coupled receptors (GPCRs) that bind extracellular ligands and transmit signals by coupling to G proteins in the cytoplasm (Fredriksson, Lagerstrom, Lundin, Schioth, 2003, Pierce, Premont, Lefkowitz, 2002). GPCRs are ubiquitous in human cells and comprise a large fraction of the current drug targets (Salon, Lodowski, Palczewski, 2011). All members of the GPCR family share a common fold with 7 transmembrane helices (TM) and a C-terminal amphipathic helix 8 and are therefore also commonly referred to as 7TM receptors (Katritch, Cherezov, Stevens, 2013, Venkatakrishnan, Deupi, Lebon, Tate, Schertler, Babu, 2013). Except for rhodopsin, that can be purified in large amounts from natural source (Palczewski, Kumasaka, Hori, Behnke, Motoshima, Fox, et al., 2000), structural and biophysical studies of GPCRs have long been hampered by the difficulties of producing stable, functional samples in reconstituted systems. In the last few years the development of new methodology for expression and purification of GPCRs has facilitated the production of purified receptors and enabled studies of GPCRs using a range of structural, biophysical and biochemical methods that were previously not feasible (Chun, Thompson, Liu, Roth, Griffith, Katritch, et al., 2012, Tate, Schertler, 2009, Venkatakrishnan, et al., 2013). Together with advances in data acquisition and sample preparation methods (Caffrey, Cherezov, 2009, Cherezov, Liu, Griffith, Hanson, Stevens, 2008, Liu, Wacker, Gati, Han, James, Wang, et al., 2013, Rosenbaum, Cherezov, Hanson, Rasmussen, Thian, Kobilka, et al., 2007, Steyaert, Kobilka, 2011) this has greatly improved the understanding of receptor structure and function and lead to a large increase in the number of high resolution GPCR structures available (Katritch, et al., 2013, Venkatakrishnan, et al., 2013). The structure of CXCR4, solved both in complex with a small molecule antagonist and a cyclic antagonist peptide using lipid cubic phase (LCP) crystallization (Caffrey, et al., 2009), was the first structure of a chemokine receptor (Wu, Chien, Mol, Fenalti, Liu, Katritch, et al., 2010). This structure was followed by an NMR structure of CXCR1(Park, Das, Casagrande, Tian, Nothnagel, Chu, et al., 2012) and a crystal structure of CCR5 solved in complex with the FDA approved small molecule antagonist maraviroc (Tan, Zhu, Li, Chen, Han, Kufareva, et al., 2013), and recently, structures of CXCR4 in complex with the viral chemokine vMIP-II (Qin, Kufareva, Holden, Wang, Zheng, Zhao, et al., 2015) and the viral chemokine receptor US28 in complex with CX3CL1 (Burg, Ingram, Venkatakrishnan, Jude, Dukkipati, Feinberg, et al., 2015). In addition, there have been a number of studies where molecular details of recombinantly expressed and purified chemokine receptors and chemokines have been studied using other methods (Kofuku, Yoshiura, Ueda, Terasawa, Hirai, Tominaga, et al., 2009, Kufareva, Stephens, Holden, Qin, Zhao, Kawamura, et al., 2014). Still, these studies cover only a few of the more than 20 chemokine receptors and 50 chemokines (Allen, et al., 2007) and the production of chemokine receptors, especially in complex with chemokines remains a challenging task. Consequently, there is a great need for development of methods for the production of receptor/chemokine complexes.
Chemokines are small, soluble proteins and can generally be produced using expression in E. coli or by chemical synthesis (Allen, Hamel, Handel, 2011, Veldkamp, Peterson, Hayes, Mattmiller, Haugner, de la Cruz, et al., 2007). However, chemokine receptors, like other 7TM receptors, generally require eukaryotic expression systems for proper folding and membrane insertion (Allen, Ribeiro, Horuk, Handel, 2009, Burg, et al., 2015, Wu, et al., 2010). In addition, several chemokine receptors require post-translational modifications such as tyrosine sulfation and/or glycosylation for efficient ligand binding (Bannert, Craig, Farzan, Sogah, Santo, Choe, et al., 2001, Fong, Alam, Imai, Haribabu, Patel, 2002, Veldkamp, Seibert, Peterson, De la Cruz, Haugner, Basnet, et al., 2008). Production of stable receptor/chemokine complexes poses additional challenges. For example, receptors and chemokines may prefer different solvent conditions (pH, salt etc) for optimal stability in vitro. Also, the Kd of chemokines for their receptors varies from the pM to high nM range and can be dependent on multiple factors including membrane lipids (Nguyen, Taub, 2002, Nguyen, Taub, 2003) and the presence of G proteins bound to the intracellular side of the receptor (Nijmeijer, Leurs, Smit, Vischer, 2010). Furthermore, purification of 7TM receptors generally requires small molecule ligands to efficiently extract receptor from membranes and keep them stable during purification (Grisshammer, 2009). This means large amounts of high affinity small molecules, which may be not be commercially available or prohibitively expensive will likely be required. Furthermore, to prepare receptor/chemokine complexes, ligand exchange steps may be needed to replace the small molecule for purified chemokine. The alternative is to produce large quantities of purified chemokine and use them as ligands throughout the extraction and purification process, but this is not practical.
Here, we will give detailed instructions for each step involved in production of complexes between chemokine receptors and chemokines through co-expression in Sf9 cells for structural and biophysical studies without the need for small molecule ligands or separately purified chemokines. Using the production of a complex between atypical chemokine receptor 3, ACKR3 (CXCR7) with CXCL12 (SDF-1) as an example, we show important quality controls and point out potential pitfalls associated with the various steps of the expression and purification process. Fig 1 outlines the major steps involved in the process; and each step is described in detail in the following section.
Figure 1.
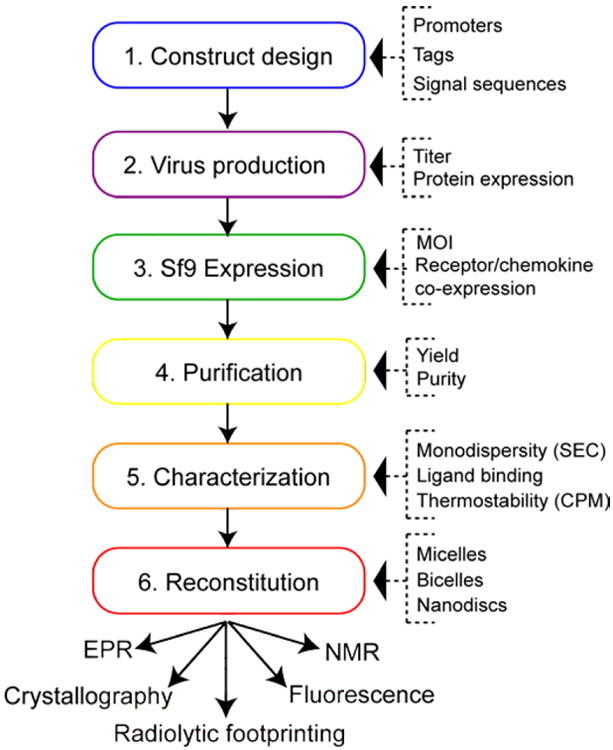
Flow chart for production of complexes.
2. Methods
2.1 Design of constructs
2.1.1 Receptor Constructs
Expression constructs were designed with the receptor sequence cloned into a pFastBac vector. A schematic of the receptor construct in the vector is shown in Fig 2A. This construct utilizes a GP64 or polH promoter to drive receptor expression, an HA signal sequence (HAss) to enhance localization in the Sf9 cell membrane and a C-terminal Prescission protease (PP) cleavage site followed by FLAG and His tags which permit detection and purification, respectively. Restriction sites before and after the receptor sequence facilitates transfer of receptor sequences between plasmids with different promoters, signal sequences and tags. In our experience the choice of promoter and the location and identity of the tags can have a major effect on the yield of receptor. For example, Fig 2B shows the yield of purified ACKR3 with four different combinations of tags and promoters. In this case a C-terminal FLAG tag gives a significantly higher yield than a construct in which the same tag is placed at the N-terminus of the receptor. Furthermore, a GP64 promoter gives a better yield than a polH promoter. The presence and position of the tag can also sometimes inhibit proteolysis of the receptor termini (data not shown).
Figure 2.
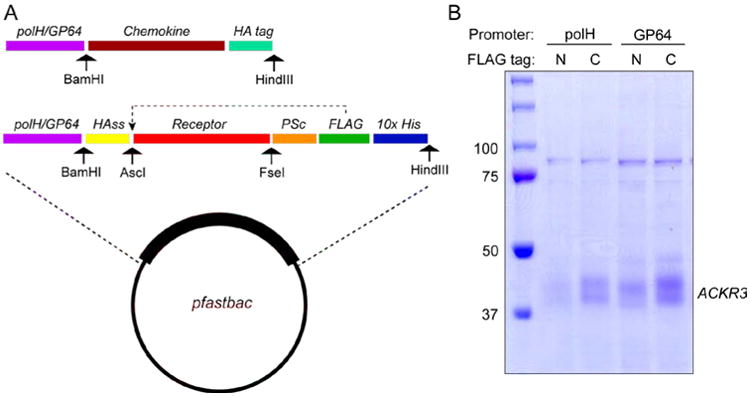
Construct design of receptor and chemokine samples. A.) Schematic overview of chemokine and receptor constructs. B.) SDS-PAGE of purified ACKR3 showing the effect of promoter and tag placement on the final yield of ACKR3 protein. The doubling and fuzziness of the ACKR3 band is due to glycosylation.
2.1.2 Chemokine Constructs
Chemokine constructs with native signal sequences are cloned into pFastBac vectors with a polH promoter and a C-terminal HA tag. A schematic is shown in Fig 2A. In our experience, the endogenous chemokine signal sequence is sufficient to enable export of the chemokine out of the Sf9 cell but utilizing other signal sequences (HA, other chemokines etc) give identical results.
2.2 Baculovirus production
2.2.1 Equipment
Temperature controlled shaker
Heat block
Centrifuge
Vi-CELL XR Cell Viability Analyzer (Beckman Coulter)
Guava EasyCyte 8HT Benchtop Flow Cytometer (Millipore)
Well Plate 24 10.4mL PP Individually Wrapped (Thomson Instrument Company, 931565-G-1X)
Breathe-Easy sealing membrane (USA Scientific, 9123-6100)
Costar Clear Polystyrene 96-Well Plates (Corning, 3788)
125mL polycarbonate Erlenmeyer flask with vent cap (Corning, 431143)
2.2ml Deep Well Plate, 96 Square Well, PP, 50/Cs, Round Bottom (Phenix Research Products, M-0661)
2.2.2 Reagents
pFastBac vectors with receptor and chemokine of interest
MAX Efficiency DH10Bac Chemically Competent Cells (Thermo Fisher)
SOC medium
LB Agar plates with Gentamicin, Kanamycin, Tetracycline, Bluo gal, IPTG (Teknova, L1919)
Kanamycin
Gentamicin
Tetracycline
Buffer P1 (Qiagen)
Buffer P2 (Qiagen)
Buffer P3 (Qiagen)
X-tremeGENE HP DNA Transfection Reagent (Roche, 6366244001)
Transfection medium (Expression Systems, 95-020-20)
ESF 921 Insect Cell Culture Medium, Protein-Free (Expression systems, 96-001-01)
Sf9 cells (ATCC® CRL-1711™)
gp64-PE Antibody (Expression Systems, 97-201)
Monoclonal ANTI-FLAG® M2-FITC antibody produced in mouse (Sigma-Aldrich, F4049)
Monoclonal Anti-HA–FITC antibody produced in mouse (Sigma-Aldrich, H7411)
7-AAD Viability Staining Solution (eBioscience, 00-6993-50)
Triton X-100
Fungizone® Antimycotic (Thermo Fisher, 15290-018)
2.2.3 Protocol
Baculovirus for expression of chemokines and receptors is produced using the Invitrogen Bac-to-Bac system. For a detailed explanation of virus production using this system see the manual (https://tools.thermofisher.com/content/sfs/manuals/bactobac_man.pdf). With some exceptions the steps described below follow the manual.
- Transformation of pFastBac vectors into DH10 bac cells
- Add the pfastbac vector to DH10 bac cells (Thermo Fisher) and incubate on ice for 15 min. The amount of DNA needed is highly dependent on the transformation efficiency of the DH10 bac cells. In our case, 50 ng of DNA is typically added to 20 μl of DH10 bac cells.
- Heat shock at 42°C for 45 s and transfer back to ice for 2 min.
- Add 170 μl of SOC media and incubate for 4 h at 37°C. Simultaneously, incubate LB agar plates with 50 μg/mL kanamycin, 7 μg/mL gentamicin, 10 μg/mL tetracycline, 100 μg/mL Bluo-gal, and 40 μg/mL IPTG (Teknova) at 37°C.
- After 4 h plate cells onto the LB agar plates. Again, the amount of cells to plate is highly cell-dependent and needs to be optimized. In our hands 15-25 μl is generally sufficient.
- Cover the plates from light and incubate at 37°C for ∼40h or until the color of the colonies (blue/white) can be clearly distinguished. Store plates at 4°C.
- Bacmid purification
- Prepare overnight cultures by adding antibiotics (50 μg/mL kanamycin, 7 μg/mL gentamicin, and 10 μg/mL tetracycline) to 5 mL of LB media
- Inoculate a single white colony from the LB agar plates in the media and grow over night at 37°C.
- Centrifuge the culture (10 min, 1800 × g) and discard the supernatant.
- Resuspend the pellet in 300 μl of cold buffer P1 (Qiagen) and transfer to a 2 mL microcentrifuge tube
- Add 300 μL of buffer P2 (Qiagen), mix by inverting the tube and incubate for 5 min at room temperature.
- Add 300 μl of cold buffer P3 (Qiagen), incubate on ice for 5 min and centrifuge for 10 min at 15,000 × g.
- Transfer 900 μl of supernatant to a new 2 mL microcentrifuge tube.
- Add 700 μl of isopropanol, invert gently to mix and incubate for 15 min on ice.
- Centrifuge for 15 min at 15,000 × g.
- Remove the supernatant and add 500 μl of 70% ethanol to wash the pellet
- Centrifuge for 5 min 15,000 × g.
- Carefully remove the supernatant and leave the tube open for ∼5 min to allow the remaining ethanol to evaporate. Add 40 μL of TE buffer (10 mM TRIS pH 8, 1 mM EDTA), flick the tube gently to solubilize the bacmid and store at 4°C.
- P0 virus production
- Make a transfection mixture by combining 100 μl of transfection medium (Expression Systems), 3 μl of X-tremeGENE HP DNA Transfection Reagent (Roche) and 5 μl of bacmid (from step 2l) in low-bind DNA microcentrifuge tubes and incubate for 30 min.
- Stocks of Sf9 cells are cultured in ESF 921 Insect Cell Culture Medium (Expression systems) at 27°C shaking at 140 rpm and passaged every 72h. Transfer 2.5 mL of Sf9 cells at a concentration of 1.2-1.4 cells/mL to a 24-well block (Thomson Instrument Company). Use one well per virus sample and one additional well to use as a control sample of untransfected cells.
- Add the transfection mixture (108 μl total) to the cells, cover the block with a sealing membrane (USA Scientific) and incubate at 27°C for 96 h shaking at 300 rpm.
- Transfer 10 μl of cells and 10 μl of anti-GP64 PE conjugated antibody (Expression Systems) to a 96-well plate (Corning) and incubate for 20 min.
- While the 96-well plate is incubating, cover the block and centrifuge at 2000 × g for 15 min.
- Transfer the supernatant to microcentrifuge tubes and save at 4°C covered from light until use. This is the P0 virus.
- Add 180 μl of TBS buffer (50 mM TRIS pH 7.5, 150 mM NaCl) to the 96-well plate and read samples using a flow cytometer. Quantify the percent of cells infected by virus by using the untransfected cells as a control. At this point >80% of the cells should be infected by virus (Fig 3A).
- P1 virus production
- Prepare 125 mL growth flasks (Corning) with 40 mL of Sf9 cells in early log phase (cell density of 2-3 × 106 cells/mL). Add 400 μl of P0 virus and include a control flask with untransfected cells. Incubate at 27°C shaking at 140 rpm.
- After 20-24 h of incubation count the cells and measure viability using trypan blue. At this stage cell growth should be arrested (total cell count ∼3 × 106 cells/mL) and the cell diameter increased by ∼20-25% compared to control cells, but the viability should remain above 90%. Measure virus infection using a GP64 assay (see steps 3d and g above). The percent of cells infected at this stage is typically above 90% for successful virus production (Fig 3B). Transfer flasks back to the shaker and incubate for 24 more hours.
- After 48 h total incubation count the cells and measure the viability. At this point the viability will be lowered (usually 60-80% for chemokines and 50-70% for receptors) and the total cell count is typically ∼3 × 106 cells/mL.
- Add 10 μl of cells to a 96-well plate (Corning), two wells per virus sample. To the first well add 10 μl of anti-FLAG-FITC antibody (Sigma-Aldrich) for receptor samples or anti-HA–FITC antibody (Sigma-Aldrich) for chemokine samples and 2 μl of 7-AAD staining solution (eBioscience). To the second well add 10 μl of anti-FLAG antibody (receptor) or anti-HA antibody (chemokine), 2 μl of 7-AAD and 0.0075%(v/v) μl of triton X-100. Incubate for 20 min
- Add 178 μl of TBS buffer to the 96-well plate. Read samples using a flow cytometer. Separate live and dead populations using the 7-AAD dye (red fluorescence) and quantify protein expression using FITC (green fluorescence). Fig 3C shows a summary of the expected FITC results for chemokine samples and for receptor samples with N- and C-terminal tags. In our experience, even if chemokines are secreted from the cell, detection of intracellular depots of chemokines at this stage is important since it confirms that the chemokine is produced by the Sf9 cells. Fig 3D shows experimental data for ACKR3 and CXCL12 samples. Triton X-100 was added to the samples in the bottom row to permeabilize the cells, and allow for detection of C-terminally tagged receptors and intracellular chemokines. Note that for chemokines and C-terminally tagged receptors, cell permeabilization is necessary to quantify expression.
- To harvest the virus, transfer the cells to 50 mL conical tubes and centrifuge for 15 min at 2000 × g. Transfer the supernatant to dark conical tubes, add 400 μl of Fungizone (Thermo Fisher) to prevent contamination, and store at 4°C. This is the P1 virus stock.
- Titering
- Prepare 1:10 dilutions of viruses in microcentrifuge tubes by mixing 10 μl of P1 virus with 90 μl ESF 921 insect cell culture medium (Expression Systems)
- Add 96 μl of medium and 4 μl of the 1:10 virus dilution in column 1 of a 96 well-plate (one well per P1 virus sample). Make serial dilutions by mixing 50 μl of column 1 with 50 μl of medium in column 2 and repeat for columns 3 and 4.
- Transfer 100 μl of Sf9 cells at log phase density (2-3 × 106 cells/mL) to a 96-well deep well block (Phenix Research Products). Copy the configuration of the dilution plate so that all wells that have virus in the dilution plate have cells in the corresponding well of the deep-well block. In addition add cells to one additional well to use a control.
- Add 20 μl of the virus dilutions to the cells, seal with a Breathe-Easy sealing membrane (USA Scientific) and incubate at 27°C, 300 rpm.
-
After 18-24 h measure the percent of infected cells using a GP64 PE antibody as described in steps 3d and 3g above.To calculate the virus titer (measured as infectious units (IU) per mL) insert the virus dilution factors (250, 500, 1000 or 2000), number of cells in the well at the time of transfection, percent infected cells from the GP64 assay (%GP64) and the volume of inoculum (0.02 mL) into the equations below. For samples with <30% of the cells infected use:For samples with >30% of the cells infected, use:This titering method and calculations are described in more detail by Gueret et al. (Gueret, Negrete-Virgen, Lyddiatt, Al-Rubeai, 2002)and Li et al.(Li, Ling, Liu, Laus, Delcayre, 2010). Using this method we will typically obtain titers of 1*109-4*109 IU/mL with chemokine viruses usually having slightly higher titer than receptor viruses.
Figure 3.
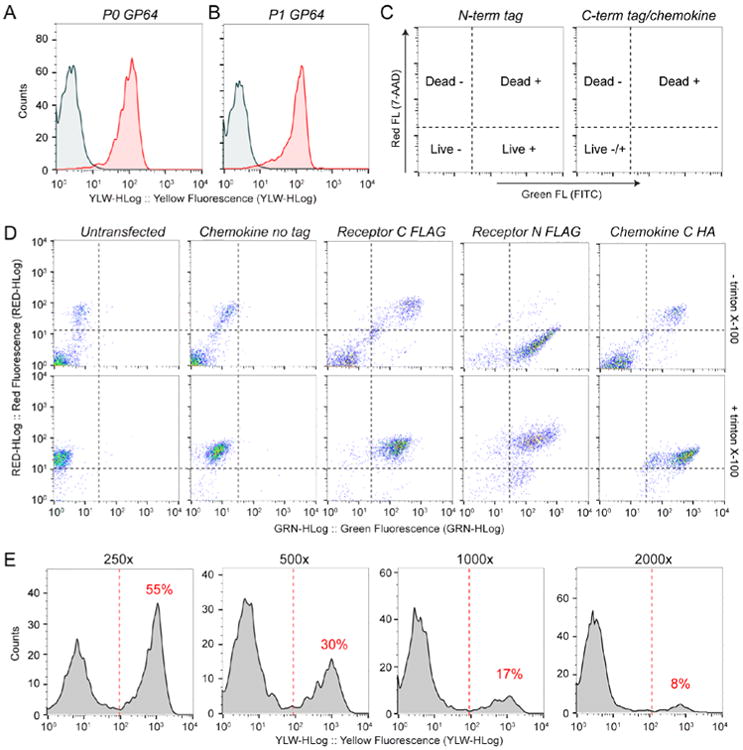
Baculovirus production detected by flow cytometry. A-B.) P0 and P1 virus expression detected by PE conjugated anti-GP64 antibody. C.) Interpretation of FITC assay for expression of receptor and chemokine. D.) FITC assay results for receptor and chemokine samples. Control experiments (columns 1 and 2) were acquired with an anti-FLAG FITC conjugated antibody, identical results were obtained with anti-HA FITC antibody (data not shown). Note that due to their larger size, cells expressing untagged chemokine have higher non-specific antibody binding than untransfected cells. Receptor and chemokine experiments (columns 3-5) were acquired with FITC conjugated FLAG or HA antibodies as indicated in the figure. Samples in the bottom row contained 0.0075% triton X-100 to permeabilize the cells. E.) Titer of P1 virus using the GP64 assay and serial virus dilutions as indicated above each plot. The % infected cells of each dilution is shown in red above the infected population in each plot.
2.3 Expression
2.3.1 Equipment
Temperature controlled shaker
125mL polycarbonate Erlenmeyer flask with vent cap (Corning, 431143)
1L Polycarbonate Erlenmeyer Flask with Vent Cap (Corning, CLS431147)
Optimum Growth 5L Flasks (Thomson, 931116)
Vi-CELL XR Cell Viability Analyzer (Beckman Coulter)
Costar Clear Polystyrene 96-Well Plates (Corning, 3788)
Centrifuge
Vi-CELL XR Cell Viability Analyzer (Beckman Coulter)
Guava EasyCyte 8HT Benchtop Flow Cytometer (Millipore)
2.3.2 Reagents and solutions
ESF 921 Insect Cell Culture Medium, Protein-Free (Expression systems)
Monoclonal ANTI-FLAG® M2-FITC antibody produced in mouse (Sigma-Aldrich, F4049)
Monoclonal Anti-HA–FITC antibody produced in mouse (Sigma-Aldrich, H7411)
7-AAD Viability Staining Solution (eBioscience, 00-6993-50)
Triton X-100
2.3.3 Protocol
-
Transfect Sf9 cells when the cells are in log phase (density of ∼2.5 ×106 mL-1) by directly adding virus from the receptor and chemokine P1 stocks to the same growth flask. Growth flasks should be chosen based on the volume of expression. We utilize Optimum Growth 5L Flasks (Thomson) for larger scale expressions (1-2 L), 1 L Polycarbonate Erlenmeyer Flask with Vent Cap (Corning) for intermediate scale expressions (150-350 mL), and 125mL Polycarbonate Erlenmeyer Flasks with Vent Cap (Corning) for smaller scale expressions (20-40 mL). Calculate the volume of virus to add using:
The virus multiplicity of infection (MOI) is typically 5-10 but needs to be optimized for each receptor/chemokine pair separately. This can be done through a titration of receptor and chemokine virus MOI in a co-expression experiment (for an example see Kufareva et al, chapter 18 of this issue). In the case of ACKR3/CXCL12 the optimal MOI for production of the receptor/chemokine complex was determined to be 6 for the chemokine when an MOI of 6 is used for the receptor and higher chemokine MOI suppressed receptor expression. In addition to the co-expressions, transfect control samples, one with receptor virus and one with chemokine virus using the same MOI as for the receptor/chemokine co-expression. The controls are used to confirm receptor and chemokine expression in step 2.
48 hours after transfection, count the cells and measure the cell viability by staining with trypan blue. At this point cell growth should have been arrested, the cell diameter should be ∼20-25% larger than for untransfected cells and the cell viability dropped to <80% due to virus infection and receptor/chemokine co-expression. To test for expression, transfer 10 μl of each of the co-expression and control cell cultures into a 96-well assay plate (two wells per sample) for flow cytometry. Harvest the rest of the cells by centrifugation at 2000 × g for 15 min and store the pellets at -80°C. Add anti-HA FITC-conjugated antibody (Sigma-Aldrich) to the first well of the 96-well assay plate to detect chemokine, anti-FLAG FITC-conjugated antibody (Sigma-Aldrich) to the second well to detect receptor and the dye 7AAD (eBioscience) to both wells to separate live and dead cells. In addition, for cases where the receptor has a C-terminal FLAG tag add Triton X-100 to a final concentration of 0.0075%(v/v) to the second well (calculated based on a final volume of 200 μl) to enable the antibody to access the epitope on the receptor, which is on the cytoplasmic side of the cell membrane. Incubate the plate for 20 minutes in the dark at 4 °C, dilute to 200 μl with TBS buffer and read the FITC fluorescence using a flow cytometer. Compare the receptor and chemokine histograms of the co-expressed cells to those of control cells to quantify the expression of the proteins. Detection of the FLAG tag confirms expression of the receptor and detection of the HA-tagged chemokine on the cell surface confirms that the chemokine is expressed and interacting with the receptor. Fig 4 shows a representative example of flow cytometry data for a co-expression of C-terminally tagged ACKR3-FLAG co-expressed with CXCL12-HA and appropriate controls.
Figure 4.
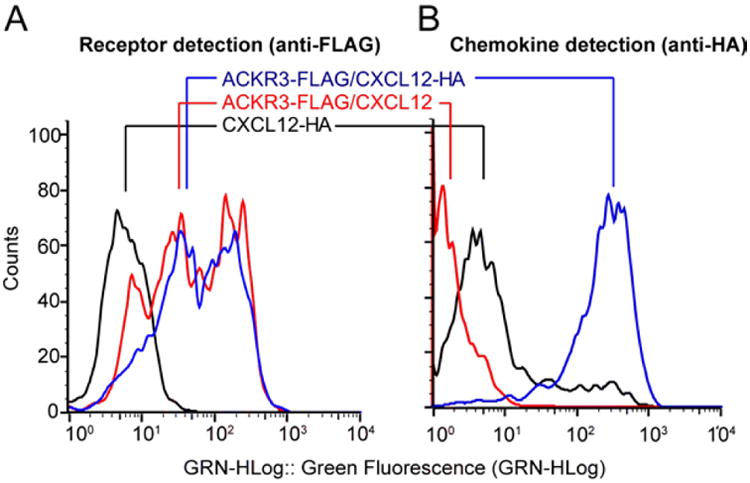
Expression of ACKR3/CXCL12 complex. A.) Detection of total receptor expression in co-expressed samples using anti-FLAG FITC antibody in the presence of triton X-100 to a final concentration of 0.0075% (v/v). B.) Detection of chemokine binding to receptor at the cell surface using anti-HA FITC antibody. Co-expression with the receptor gives a large signal increase for CXCL12-HA, indicating specific binding of the chemokine to the receptor.
2.4 Purification
2.4.1 Equipment
Centrifuge
Kimble-Chase Kontes™ Dounce Tissue Grinders 100, 15, 1 mL (Fisher Scientific, 8853000100, 8853000040, 8853000001)
Poly-Prep Chromatography Columns (Bio-rad, 7311550)
Micro Bio-Spin Columns (Bio-rad, 7326204)
PD-10 desalting column (GE Healthcare, 17-0851-01)
2.4.2 Reagents
cOmplete EDTA-free protease inhibitor tablets (Roche, 5056489001)
Iodoacetamide (GE Healthcare, RPN6302)
n-Dodecyl-β-D-Maltopyranoside (DDM) (Anatrace, D310)
Cholesteryl hemisuccinate (CHS) (Sigma-Aldrich, C6512)
TALON Superflow Metal Affinity Resin (Clontech, 635507)
Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-100 membrane (EMD Millipore, UFC510096)
Amicon Ultra-4 Centrifugal Filter Unit with Ultracel-100 membrane (EMD Millipore, UFC810096)
HRV 3C Protease (Human Rhinovirus 3C Protease, PreScission Site) (Sino Biological, S3CP01)
PNGaseF (New England Biolabs, P0704S)
2.4.3 Buffers
Hypotonic buffer
10 mM HEPES pH 7.5
10 mM MgCl2
20 mM KCl
cOmplete EDTA-free protease inhibitor tablets (Roche, 5056489001)
Wash buffer 1
25 mM HEPES pH 7.5
400 mM NaCl
10% glycerol
10 mM imidazole
0.1/0.02% DDM/CHS
Wash buffer 2
25 mM HEPES pH 7.5
400 mM NaCl
10% glycerol
10 mM imidazole
0.025/0.005% DDM/CHS
Elution buffer
Wash buffer 2
250 mM imidazole
Buffer exchange buffer
25 mM HEPES pH 7.5
400 mM NaCl
10% glycerol
0.025/0.005% DDM/CHS
2.4.4 Protocol
This protocol describes the purification of a receptor/chemokine complex from a 1 L culture of Sf9 cells. Typically, 40 mL of cells is a good starting point for a small scale expression of a chemokine receptor construct. In the case of a small scale purification volumes of buffers and resin can to be scaled down by 25-fold. Throughout the purification all steps should be performed on ice or at 4°C.
Thaw the cell pellets, resuspend in ∼120 mL hypotonic buffer and homogenize with 40 strokes in a dounce homogenizer. Transfer the suspension to 40 ml centrifuge tubes, centrifuge at 50,000 × g for 30 min, discard the supernatant, and repeat the process once with hypotonic buffer and three times using high salt buffer (hypotonic buffer + 1 M NaCl). For small scale purifications one round of douncing in hypotonic buffer and two rounds in high salt buffer are usually sufficient. After the last centrifugation, resuspend in hypotonic buffer with 30% glycerol to a final volume of 25 mL, dounce with 40 strokes, transfer to conical tubes and flash freeze in liquid nitrogen and store the membranes at -80°C until the next step of the purification.
-
Thaw the membranes, add protease inhibitors and dilute with 25 mL of hypotonic buffer. For receptors with exposed cysteine residues, 2 mg/mL iodoacetamide can be added to avoid aggregation during the purification. Incubate for 30 min on a rotisserie.
Alternative strategy: If large amounts of purified chemokine is available, receptor can be expressed without chemokine in Sf9 cells and chemokine can be added in step 2, at the same time as the protease inhibitors. In this case the concentration of chemokine used for extraction needs to be optimized. An example of ACKR3 extracted with 3 μM of a CXCL12 mutant (KPV at the N-terminus replaced by LRHQ, LRHQ-CXCL12) (Hanes, Salanga, Chowdry, Comerford, McColl, Kufareva, et al., 2015) or with 100 μM of a small molecule compound is shown in Fig 5.
Prepare 50 mL of 2× solubilization buffer (100 mM HEPES pH 7.5, 800 mM NaCl, 1.5/0.3% n-Dodecyl-β-D-Maltopyranoside (DDM) (Anatrace)/Cholesteryl hemisuccinate (CHS) (Sigma-Aldrich). To facilitate the solubilization of DDM and CHS in the buffer, 10/2% (w/v) stocks of DDM/CHS should prepared in advance by weighing out DDM and CHS, dissolving in 200 mM TRIS pH 8 and filtering the solution to remove insoluble particles.
Split the sample into two tubes, add 25 mL of 2× solubilization buffer to each tube and incubate for another three hours. Centrifuge the solubilized samples (50,000 × g, 30 min), transfer the supernatant (containing the solubilized receptor/chemokine complex) to two 50 mL conical tubes and add 1 mL of 50% Talon resin and 10 mM of imidazole to each tube. Incubate at 4°C at a rotisserie over night.
Centrifuge the samples for 5 min at 350 × g, discard the supernatant and transfer the resin to Poly-Prep Chromatography Columns (Bio-rad). Wash the resin with 20mL of wash buffer 1 and 10 mL of wash buffer 2 by adding 2 mL buffer at a time and use gravity flow to run the buffer through the column. After the last wash elute with 3 mL of elution buffer in 1 mL fractions.
Concentrate the eluted sample to 500 μL using Amicon Ultra-4 100 kDa molecular weight cutoff spin concentrators (EMD Millipore).
-
Equilibrate a PD-10 desalting column (GE Healthcare) with 3 × 2 mL of buffer exchange buffer. Apply 500 μL of sample to the column and discard the flow-through. Add 1 mL of buffer and collect the flow-through. At this stage the sample should be pure enough (as assessed by SDS-PAGE, Fig 5) to be used directly for basic characterization experiments and for certain sample preparations where the presence of tags is required (i.e nanodiscs reconstitution, see section 2.6.5). However, for other applications it is necessary to cleave off the C-terminal His and FLAG tags on the receptor. This will also serve as an additional purification step.
Alternative strategy: For small scale purifications steps 4 and 5 can be replaced by buffer exchanging samples using Amicon Ultra-0.5 100 kDa molecular weight cutoff spin concentrators (EMD Millipore). In this case, spin down to ∼50 μL and bring the volume back to 500 μL three times. After a fourth and final spin transfer the sample to a new tube and bring the volume to 100 μL.
To cleave off the His and FLAG tags, incubate the sample (1 mL) with Prescission Protease (Sino Biological, PP) over night at 4°C. For glycosylated receptors and chemokines, Pngase F (New England Biolabs) can be added to remove N-linked glycosylation. The amounts of PP and Pngase F needed for cleavage and deglycosylation will vary depending on the receptor and the activity of the enzyme. After over-night incubation, add 500 μL of Talon resin slurry and incubate for 90 min at 4°C. Transfer the sample to a Micro Bio-Spin column (Bio-rad) and collect the flow through. Add 500 μl of buffer exchange buffer and again collect the flow through for a total sample volume of ∼1.8 mL. Tag cleavage and deglycosylation can be followed from band shifts using SDS-PAGE (Fig 5, see next section for experimental details).
After step 8 the complex can be concentrated to the desired concentration and characterized for purity, stability and monodispersity (see next section). In the case of ACKR3/CXCL12, samples can be stored for weeks in detergent or kept frozen at -80°C for longer periods. However, the sample stability can be highly receptor/chemokine dependent and needs to be tested for each complex individually.
Figure 5.
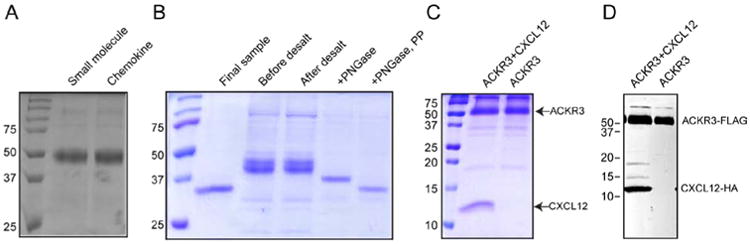
Purification of ACKR3/CXCL12 complex. A.) 10% SDS-PAGE of an bril-ACKR3 fusion protein extracted with the small molecule CCX777 or with a mutant of CXCL12 where KPV at the N-terminus was replaced with LRHQ (LRHQ-CXCL12) (Hanes, et al., 2015). B.) Different stages of purification characterized by SDS-PAGE (10%). C.) 18% SDS-PAGE of a co-expressed and co-purified ACKR3/CXCL12 sample (left lane). ACKR3 expressed alone and purified in complex with a small molecule compound is shown in the right lane. D.) Western blot detecting FLAG-tagged ACKR3 and HA-tagged CXCL12 in samples from C.
2.5 Characterization of Receptor/Chemokine Complex Purity, Homogeneity and Stability
2.5.1 Equipment
RotorGene Q 6-plex RT-PCR machine (Qiagen)
HPLC system
Equipment for SDS-PAGE setup (gel chambers, gels, power source)
2.5.2 Reagents and solutions
Standard solutions for SDS-PAGE (loading buffer, staining/destaining solutions, running buffers)
Monoclonal ANTI-FLAG M2 antibody produced in mouse (Sigma Aldrich, F3165)
Anti-HA High Affinity from rat IgG1 (Roche, 11867423001)
IRDye 800CW goat anti-rat IgG (LI-COR Biosciences, 925-32219)
IRDye 680RT donkey anti-mouse IgG (LI-COR Biosciences, 925-68072)
Sepax SRT-C 300 column, 5um, 300 A 4.6 × 250 mm (Sepax Technologies, 235300-4625)
N-[4-(7-diethylamino-4-methyl-3-coumarinyl)phenyl]maleimide (CPM) dye (Thermo Fisher, D-10251)
2.5.3 Buffers
SEC Buffer
25 mM HEPES pH 7.5
400 mM NaCl
2.5% glycerol
10 mM imidazole
0.025/0.005% DDM/CHS
2.5.4 SDS-PAGE/western blot
SDS-PAGE is used to analyze the purity and quantity of the purified complexes while western blots can confirm the presence of chemokine and receptor in the sample. To get sufficient resolution of both receptor and chemokine it is useful to run a 10% polyacrylamide gel to detect the receptor and an 18% gel to detect the chemokine. Load ∼5-10 μg of protein for detection with coomassie stain and 0.5-1 μg for western blot detection with antibody. Fig 5 shows typical coomassie stained gels and western blots for the purified ACKR3/CXCL12 complex. For a pure sample the receptor and chemokine bands should be easily detectable by SDS-PAGE and be the most prominent bands on the gel. However, if the sample yield is low and/or the sample is not pure, western blots can be used to confirm the presence of receptor and chemokine. For western blots, receptor is detected with a mouse anti-Flag M2 primary antibody (Sigma Aldrich) and an IRDye 680 conjugated donkey anti-mouse IgG (LI-COR Biosciences); the presence of chemokine in the complex is confirmed using a rat anti-HA 3F10 primary antibody (Roche) and an IRDye 800 conjugated goat anti-rat IgG (LI-COR Biosciences). The procedure for detecting receptor/chemokine complexes by western blotting is described in more detail in chapter 18 of this issue (see Kufareva et al).
2.5.5 Size exclusion chromatography (SEC)
The quality of an SEC trace is indicative of the monodispersity of the receptor/chemokine complex since aggregated samples will give broad, asymmetric peaks that elute earlier than well-folded non-aggregated complexes (Fig 6A). For SEC experiments, equilibrate a Sepax SRT-C 300 column with SEC buffer and inject 5-10 μg of protein onto the column. Measure the absorbance at 280nm to detect elution of the complex from the column. Fig 6B shows an SEC trace for ACKR3:CXCL12 in DDM/CHS micelles. The elution volume and the sharp symmetric peak indicate that the complex is not aggregated in solution. The elution volume of the complex will be column-dependent.
Figure 6.
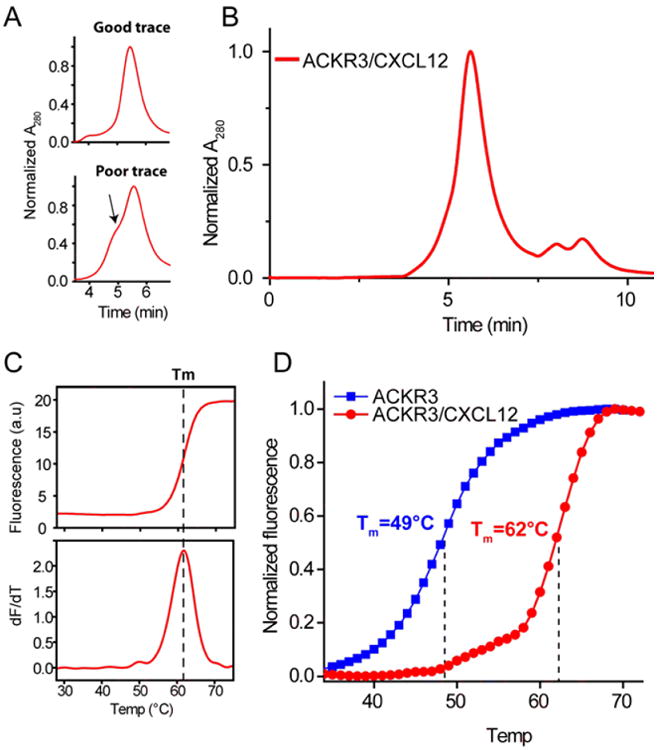
Characterization of receptor/chemokine complexes. A.) Analytical SEC traces of varying quality. The top trace has a sharp, symmetric peak while the peak in the bottom trace has a shoulder (indicated by arrow), which is a sign that the sample is partially aggregated. B.) Analytical SEC trace of the ACKR3/CXCL12 complex. C.) CPM measurements are used to determine the midpoint of thermal unfolding (Tm) of the receptor. Data can be plotted either as CPM fluorescence as a function of temperature (top) or as the derivative of the fluorescence (bottom) D.) CPM experiments with apo ACKR3 and the ACKR3/CXCL12 complex.
2.5.6 Thermal unfolding
The midpoint of unfolding or Tm is a useful metric for stability of the protein complex. Unfolding assays utilizing the cysteine-reactive CPM dye is a well-established method to measure the Tm and thereby assess the relative stability of 7TM receptors (Alexandrov, Mileni, Chien, Hanson, Stevens, 2008). The CPM assay has been applied to a large number of receptors, especially in the context of selecting stable constructs and complexes for crystallization. Since chemokines lack free cysteine residues the assay will report on the stability of the receptor, which is highly dependent on the binding of its chemokine ligand. A high Tm indicates a stable complex that can be used for further studies. In our experience stable complexes between receptors and chemokine have Tm values above ∼60°C and may reach as high as 75°C in some cases. However, this will be highly dependent on the receptor and chemokine. For CPM experiments 0.2-0.4 μg of complex is incubated for 20 minutes with 20 μl of buffer exchange buffer containing 1 μg/mL of CPM dye. Thermal unfolding is measured using a RotorGene Q 6-plex RT-PCR machine (Qiagen) and the Tm is determined from non-linear fitting of the unfolding curves (Fig 6C) (Alexandrov, et al., 2008). Representative examples of unfolding curves for ACKR3, free and in complex with CXCL12, are shown in Fig 6D.
2.6 Reconstitution
2.6.1 Equipment
Bath sonicator
Desiccator
FPLC system
Superdex 200 10/300 GL column (GE Healthcare, 17-5175-01)
2.6.2 Reagents and solutions
1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC, 14:0 PC) (Avanti)
Amberlite® XAD®-2 beads (Sigma Aldrich, 10357)
1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC, 6:0 PC) (Avanti)
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, 16:0-18:1 PC) (Avanti, 850457)
Sodium cholate (Sigma Aldrich, C6445)
MSP (∼150-200 μM in 25mM HEPES pH 7.5, 150 mM NaCl)
2.6.3 Buffers
Bicelle reconstitution buffer/nanodisc buffer
25mM HEPES pH 7.5
150 mM NaCl
Nanodisc reconstitution buffer
25mM HEPES pH 7.5
150 mM NaCl
200mM cholate
Nanodisc elution buffer
25mM HEPES pH 7.5
150 mM NaCl
250 mM imidazole
Different biophysical and biochemical methods require reconstitution of the receptor/chemokine complex into different membrane mimics. Many measurements can be done directly in detergent micelles. However, micelles are usually not the ideal environment for receptor stability and are inferior mimics of a native membrane compared to other systems like nanodiscs or bicelles. Reconstitution of receptor/chemokine complexes into these systems may require extensive optimization of reconstitution methods, lipids and buffers. Below are protocols for the reconstitution of ACKR3/CXCL12 into bicelles and nanodiscs.
2.6.4 Bicelles
Bicelles are composed of a mixture of long chain and short chain lipids where the long chain lipid form bilayers with short chain lipids coating the edges (Whiles, Deems, Vold, Dennis, 2002). Membrane proteins insert into the bilayer portion and thus bicelles provide a better mimic of a true membrane bilayer than micelles. Bicelles have been used in a number of membrane protein studies, most notably in solid state NMR, where bicelles with a high q (molar ratio of long to short chain lipid, >3) can be used to align membrane proteins in the magnetic field (Durr, Gildenberg, Ramamoorthy, 2012), and solution NMR where low q (∼0.33-1), isotropic bicelles are utilized (Poget, Girvin, 2007). Several conditions need to be fulfilled for bicelles to form. The long-chain lipids must have saturated chains and a (phosphocholine (PC) head group but a portion (up to ∼25%) can be exchanged for a lipid with a different head group and chain saturation. This is especially relevant for ACKR3 that is stabilized by the presence of negatively charged head groups. The choice of short chain lipid is less stringent and several detergents can be utilized including 1,2-dihexanoyl-glycero-3-phosphocholine (DHPC), Triton X-100 and 3-[(3-Cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesulfonate (CHAPSO) (Park, Opella, 2010, Whiles, et al., 2002). In addition, the concentration of the short chain lipid needs to be above its CMC and the q value has to be 0.05-1 for the bicelle to have an isotropic behavior in solution as opposed to an aligned bicelle that is formed at q values of ∼3 or higher (Whiles, et al., 2002). This protocol describes the formation of a 200 μl bicelle sample with q = 0.6 using DMPC as the long chain and DHPC as the short chain lipid. The concentration of DHPC is 20 mM, which is above the CMC (15mM) and the total lipid concentration is 1.7% (w/v).
Weigh out 1.6 mg of DMPC in a glass tube, solubilize in chloroform, and dry away the chloroform under a stream of nitrogen gas until the lipid forms a film at the bottom of the tube. Desiccate over night to remove traces of chloroform.
Add 132 μl of reconstitution buffer to the lipids and use vortexing and sonicating (in a bath sonicator) to resuspend the lipid. At this point the sample should become cloudy and there should be no lipid stuck to the bottom of the tube.
Add 40 μl of 10 % (w/v) DDM and alternate vortexing (carefully to avoid foam) and sonicating until the solution is clear and the lipid is completely solubilized.
Add 10 μl of protein and mix by vortexing (if a different volume of protein is used the volume of reconstitution buffer in step 2 should be adjusted accordingly to get a total sample volume of 182 μl).
Add ∼70 mg of Amberlite XAD-2 beads (Sigma-Aldrich) and a micro stir bar.
Incubate at room temperature on a stir plate for 3-4 h.
Remove the sample from biobeads using a gel loading tip. At this time the solution should be cloudy since the DDM has been absorbed by the biobeads and the sample consists of the receptor/chemokine complex in multi-lamellar DMPC vesicles.
Add 18 μl of D-6-PC from a 10 % stock to form the final sample. At this point the sample should turn completely clear.
2.6.5 Nanodiscs
Nanodiscs (Denisov, Grinkova, Lazarides, Sligar, 2004) have emerged in the last few years as a membrane mimic that can be used for a number of applications including NMR and fluorescence experiments (Bayburt, Vishnivetskiy, McLean, Morizumi, Huang, Tesmer, et al., 2013, Raschle, Hiller, Yu, Rice, Walz, Wagner, 2009, Whorton, Jastrzebska, Park, Fotiadis, Engel, Palczewski, et al., 2008). Nanodiscs provide a more native-like membrane mimic than a micelle. Compared to bicelles, nanodiscs avoid the need for detergent in the samples and allows for significantly more flexibility in the choice of lipids. This protocol describes the production of MSP1E3D1 (Bayburt, Sligar, 2010) nanodiscs containing ACKR3 in complex with CXCL12. For simplicity a single lipid (POPC) is used but similar results are obtained with other lipid mixtures (see Fig 7A). The protocol is based on methods published by the Sligar lab; see Richie et al (Ritchie, Grinkova, Bayburt, Denisov, Zolnerciks, Atkins, et al., 2009), for a protocol describing expression and purification of MSP and a detailed explanation of the reconstitution step.
Weigh out 7.6 mg of POPC in a glass tube and add chloroform to dissolve the lipids. Use nitrogen gas to evaporate the chloroform and form a lipid film at the bottom of the tube. Desiccate over night to remove traces of chloroform.
Add 120 μl of nanodisc reconstitution buffer. Vortex and sonicate in a bath sonicator (∼30s each) until the solution is clear and the lipid is completely solubilized.
-
Make a reconstitution mixture by combining the following components to get a ACKR3/CXCL12:MSP:POPC molar ratio of 1:10:1250.
- 4.5 mg lipid (70 μl of lipid/detergent mixture from step 2)
- 1.4 mg of MSP1E3D1 (260 μl from a 175 μM solution in 25 mM HEPES, 150 mM NaCl)
- 200 μg ACKR3/CXCL12 (120 μl from a 36 μM solution in DDM/CHS)
The volumes and concentrations of MSP and receptor/chemokine stock solutions can be varied. However, if the cholate concentration in the final mixture does not exceed the CMC, additional cholate needs to be added. In this example the cholate concentration is ∼30 mM, which is safely above the CMC of ∼15 mM.
Incubate for 90 min on ice
Add Amberlite XAD-2 beads (Sigma-Aldrich) and incubate over night at 4°C stirring.
Remove the beads using a gel loading tip
Spin down for 10 min at 25,000 × g and transfer the supernatant to a new tube
Inject the supernatant onto a Superdex 200 10/300 GL column that has been equilibrated with nanodisc buffer and use a fraction collector to collect the elution from the column. In our experience nanodiscs will elute at an elution volume of ∼9-14 mL but this should be confirmed by SDS-PAGE/western blot of the fractions.
Pool the fractions containing nanodiscs and add 400 μl of 50% Talon slurry and 10 mM Imidazole. Incubate over night at 4°C in rotisserie. Only receptor-containing nanodiscs should bind to the resin.
Centrifuge (350 × g, 5 min) and transfer the resin to a Micro Bio-Spin Columns (Bio-rad). Wash with 5 × 1 mL of nanodisc buffer using gravity flow.
Elute the nanodiscs containing ACKR3/chemokine with 3 × 0.4 mL of nanodisc elution buffer.
Buffer exchange the sample using 100 kDa cut-off spin concentrators by concentrating to 50 μl, bringing the volume to 500 μl and repeating the process three times to get the final sample.
Run 10% SDS-PAGE to confirm the nanodisc composition (Fig 7B). The final sample should consist of only protein-containing nanodiscs and thus have a 2:1 receptor:MSP molar ratio since each nanodisc is composed of two chains of MSP.
Figure 7.
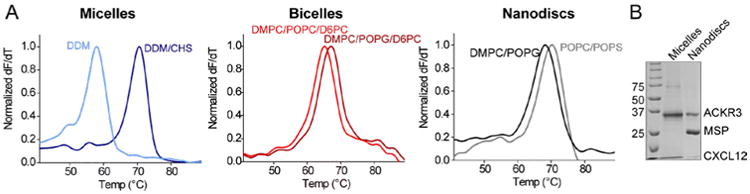
The ACKR3/CXCL12 complex reconstituted into different membrane mimics. A.) The ACKR3/CXCL12 complex is stable in all three membrane mimics as indicated by the sharp peak (derivative of the CPM unfolding curves, see Fig 6C) and the Tm above 60°C. B.) SDS-PAGE of ACKR3/CXCL12 sample in nanodiscs and DDM/CHS micelles.
Fig 7A shows unfolding curves of the ACKR3CXCL12 complex in micelles, bicelles and nanodiscs acquired using the CPM assay (see section 2.5.6). In this case, midpoints of unfolding are similar in all three membrane mimics showing that the complex is stable in all three systems. The large increase in Tm after addition of CHS illustrates the importance of the lipid/micelle environment to receptor stability.
3. Summary and conclusions
The methods described in this chapter provide an efficient way of producing receptor/chemokine and has been used in our lab to successfully produce large amounts of receptor/chemokine complexes for CXC as well as CC receptors. Still, it is important to point out that complex formation through co-expression is not a universal method and will not be suitable for all combinations of chemokines and receptors. In the case of ACKR3, co-expression works well for complex formation with CXCL12, which has an affinity of ∼0.2 nM in cell membranes(Burns, Summers, Wang, Melikian, Berahovich, Miao, et al., 2006). However, complexes between ACKR3 and CXCL11 (ITAC) cannot be produced using the same method due to a ∼10-fold lower affinity(Burns, et al., 2006) and thus faster off-rate of the chemokine. Another example is the CXCR4/CXCL12, which requires G protein for optimal affinity. In these cases the protocol above will provide an excellent starting point for modified protocols that implement ligand exchange after purification or co-expression with G proteins that may be needed for successful reconstitution of a receptor/chemokine complex.
Acknowledgments
This work was supported by grants to TMH (NIAID R01 AI37113, NIGMS U01GM094612 and NIAID R01AI118985) and a Robertson Foundation/Cancer Research Institute Irvington post-doctoral fellowship to MG.
References
- Alexandrov AI, Mileni M, Chien EY, Hanson MA, Stevens RC. Microscale fluorescent thermal stability assay for membrane proteins. Structure. 2008;16:351–9. doi: 10.1016/j.str.2008.02.004. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Crown SE, Handel TM. Chemokine: receptor structure, interactions, and antagonism. Annu Rev Immunol. 2007;25:787–820. doi: 10.1146/annurev.immunol.24.021605.090529. [DOI] [PubMed] [Google Scholar]
- Allen SJ, Hamel DJ, Handel TM. A rapid and efficient way to obtain modified chemokines for functional and biophysical studies. Cytokine. 2011;55:168–73. doi: 10.1016/j.cyto.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen SJ, Ribeiro S, Horuk R, Handel TM. Expression, purification and in vitro functional reconstitution of the chemokine receptor CCR1. Protein Expr Purif. 2009;66:73–81. doi: 10.1016/j.pep.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannert N, Craig S, Farzan M, Sogah D, Santo NV, Choe H, et al. Sialylated O-glycans and sulfated tyrosines in the NH2-terminal domain of CC chemokine receptor 5 contribute to high affinity binding of chemokines. J Exp Med. 2001;194:1661–73. doi: 10.1084/jem.194.11.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS Lett. 2010;584:1721–7. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, et al. Monomeric rhodopsin is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem. 2013;286:1420–8. doi: 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg JS, Ingram JR, Venkatakrishnan AJ, Jude KM, Dukkipati A, Feinberg EN, et al. Structural biology. Structural basis for chemokine recognition and activation of a viral G protein-coupled receptor. Science. 2015;347:1113–7. doi: 10.1126/science.aaa5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns JM, Summers BC, Wang Y, Melikian A, Berahovich R, Miao Z, et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J Exp Med. 2006;203:2201–13. doi: 10.1084/jem.20052144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey M, Cherezov V. Crystallizing membrane proteins using lipidic mesophases. Nat Protoc. 2009;4:706–31. doi: 10.1038/nprot.2009.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherezov V, Liu J, Griffith M, Hanson MA, Stevens RC. LCP-FRAP Assay for Pre-Screening Membrane Proteins for in Meso Crystallization. Cryst Growth Des. 2008;8:4307–15. doi: 10.1021/cg800778j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun E, Thompson AA, Liu W, Roth CB, Griffith MT, Katritch V, et al. Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure. 2012;20:967–76. doi: 10.1016/j.str.2012.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. Directed self-assembly of monodisperse phospholipid bilayer Nanodiscs with controlled size. J Am Chem Soc. 2004;126:3477–87. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- Durr UH, Gildenberg M, Ramamoorthy A. The magic of bicelles lights up membrane protein structure. Chem Rev. 2012;112:6054–74. doi: 10.1021/cr300061w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong AM, Alam SM, Imai T, Haribabu B, Patel DD. CX3CR1 tyrosine sulfation enhances fractalkine-induced cell adhesion. J Biol Chem. 2002;277:19418–23. doi: 10.1074/jbc.M201396200. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–72. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Grisshammer R. Purification of recombinant G-protein-coupled receptors. Methods Enzymol. 2009;463:631–45. doi: 10.1016/S0076-6879(09)63036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueret V, Negrete-Virgen JA, Lyddiatt A, Al-Rubeai M. Rapid titration of adenoviral infectivity by flow cytometry in batch culture of infected HEK293 cells. Cytotechnology. 2002;38:87–97. doi: 10.1023/A:1021106116887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanes MS, Salanga CL, Chowdry AB, Comerford I, McColl SR, Kufareva I, et al. Dual Targeting of the Chemokine Receptors CXCR4 and ACKR3 with Novel Engineered Chemokines. J Biol Chem. 2015;290:22385–97. doi: 10.1074/jbc.M115.675108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–56. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuku Y, Yoshiura C, Ueda T, Terasawa H, Hirai T, Tominaga S, et al. Structural basis of the interaction between chemokine stromal cell-derived factor-1/CXCL12 and its G-protein-coupled receptor CXCR4. J Biol Chem. 2009;284:35240–50. doi: 10.1074/jbc.M109.024851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kufareva I, Stephens BS, Holden LG, Qin L, Zhao C, Kawamura T, et al. Stoichiometry and geometry of the CXC chemokine receptor 4 complex with CXC ligand 12: molecular modeling and experimental validation. Proc Natl Acad Sci U S A. 2014;111:E5363–72. doi: 10.1073/pnas.1417037111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Ling L, Liu X, Laus R, Delcayre A. A flow cytometry-based immuno-titration assay for rapid and accurate titer determination of modified vaccinia Ankara virus vectors. J Virol Methods. 2010;169:87–94. doi: 10.1016/j.jviromet.2010.07.003. [DOI] [PubMed] [Google Scholar]
- Liu W, Wacker D, Gati C, Han GW, James D, Wang D, et al. Serial femtosecond crystallography of G protein-coupled receptors. Science. 2013;342:1521–4. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DH, Taub D. Cholesterol is essential for macrophage inflammatory protein 1 beta binding and conformational integrity of CC chemokine receptor 5. Blood. 2002;99:4298–306. doi: 10.1182/blood-2001-11-0087. [DOI] [PubMed] [Google Scholar]
- Nguyen DH, Taub DD. Inhibition of chemokine receptor function by membrane cholesterol oxidation. Exp Cell Res. 2003;291:36–45. doi: 10.1016/s0014-4827(03)00345-8. [DOI] [PubMed] [Google Scholar]
- Nijmeijer S, Leurs R, Smit MJ, Vischer HF. The Epstein-Barr virus-encoded G protein-coupled receptor BILF1 hetero-oligomerizes with human CXCR4, scavenges Galphai proteins, and constitutively impairs CXCR4 functioning. J Biol Chem. 2010;285:29632–41. doi: 10.1074/jbc.M110.115618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–45. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Park SH, Das BB, Casagrande F, Tian Y, Nothnagel HJ, Chu M, et al. Structure of the chemokine receptor CXCR1 in phospholipid bilayers. Nature. 2012;491:779–83. doi: 10.1038/nature11580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Opella SJ. Triton X-100 as the "short-chain lipid" improves the magnetic alignment and stability of membrane proteins in phosphatidylcholine bilayers for oriented-sample solid-state NMR spectroscopy. J Am Chem Soc. 2010;132:12552–3. doi: 10.1021/ja1055565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–50. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- Poget SF, Girvin ME. Solution NMR of membrane proteins in bilayer mimics: small is beautiful, but sometimes bigger is better. Biochim Biophys Acta. 2007;1768:3098–106. doi: 10.1016/j.bbamem.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Kufareva I, Holden LG, Wang C, Zheng Y, Zhao C, et al. Structural biology. Crystal structure of the chemokine receptor CXCR4 in complex with a viral chemokine. Science. 2015;347:1117–22. doi: 10.1126/science.1261064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raschle T, Hiller S, Yu TY, Rice AJ, Walz T, Wagner G. Structural and functional characterization of the integral membrane protein VDAC-1 in lipid bilayer nanodiscs. J Am Chem Soc. 2009;131:17777–9. doi: 10.1021/ja907918r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie TK, Grinkova YV, Bayburt TH, Denisov IG, Zolnerciks JK, Atkins WM, et al. Chapter 11 - Reconstitution of membrane proteins in phospholipid bilayer nanodiscs. Methods Enzymol. 2009;464:211–31. doi: 10.1016/S0076-6879(09)64011-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–73. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- Salon JA, Lodowski DT, Palczewski K. The significance of G protein-coupled receptor crystallography for drug discovery. Pharmacol Rev. 2011;63:901–37. doi: 10.1124/pr.110.003350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyaert J, Kobilka BK. Nanobody stabilization of G protein-coupled receptor conformational states. Curr Opin Struct Biol. 2011;21:567–72. doi: 10.1016/j.sbi.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341:1387–90. doi: 10.1126/science.1241475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tate CG, Schertler GF. Engineering G protein-coupled receptors to facilitate their structure determination. Curr Opin Struct Biol. 2009;19:386–95. doi: 10.1016/j.sbi.2009.07.004. [DOI] [PubMed] [Google Scholar]
- Veldkamp CT, Peterson FC, Hayes PL, Mattmiller JE, Haugner JC, 3rd, de la Cruz N, et al. On-column refolding of recombinant chemokines for NMR studies and biological assays. Protein Expr Purif. 2007;52:202–9. doi: 10.1016/j.pep.2006.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, et al. Structural basis of CXCR4 sulfotyrosine recognition by the chemokine SDF-1/CXCL12. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatakrishnan AJ, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–94. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- Whiles JA, Deems R, Vold RR, Dennis EA. Bicelles in structure-function studies of membrane-associated proteins. Bioorg Chem. 2002;30:431–42. doi: 10.1016/s0045-2068(02)00527-8. [DOI] [PubMed] [Google Scholar]
- Whorton MR, Jastrzebska B, Park PS, Fotiadis D, Engel A, Palczewski K, et al. Efficient coupling of transducin to monomeric rhodopsin in a phospholipid bilayer. J Biol Chem. 2008;283:4387–94. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–71. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]