

Abstract
The inheritance of the ε4 allele of apolipoprotein E (ApoE4) and cholinergic system dysfunction have long been associated with the pathology of Alzheimer’s disease (AD). Recently, in vitro studies have established a direct link between ApoE and cholinergic function in that synthetic peptides containing segments of the ApoE protein (ApoE133–149 and ApoE141–148) interact with α7 nicotinic acetylcholine receptors (nAChRs) in the hippocampus. This raises the possibility that ApoE peptides may contribute to cognitive impairment in AD in that the hippocampus plays a key role in cognitive functioning. To test this, we acutely infused ApoE peptides into the ventral hippocampus of female Sprague–Dawley rats and assessed the resultant effects on radial-arm maze choice accuracy over a period of weeks after the infusion. Local ventral hippocampal infusion of ApoE peptides caused significant cognitive impairment in radial-arm maze learning that persisted several weeks after the acute infusion. This persisting deficit may be an important model for understanding the relationship between ApoE protein-induced neurotoxicity and cognitive impairment as well as serve as a platform for the development of new therapies to avoid neurotoxicity and cognitive decline.
Keywords: Alzheimer’s disease, Learning, Memory, Radial-arm maze, Hippocampus, ApoE4
1. Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by the progressive loss of cognitive functioning. Apolipoprotein E4 (ApoE4) is of particular interest to understanding AD because it has been identified as a major risk factor for developing AD. People who are homozygous for ApoE4 have a 4.5 times greater risk of acquiring AD and average an earlier age of onset by 16 years [1,20]. While genetic links between AD and ApoE protein have been shown [7], it is not known whether the protein itself directly contributes to the cognitive impairment associated with AD or if the relationship between ApoE proteins and cognitive impairment is less direct.
The hippocampus is of critical importance with regard to the cognitive impairment of AD. A great many studies show the importance of this brain region for integrating learning and memory processes [17]. Additionally, AD is characterized by hippocampal dysfunction, in particular a decrease in the number of hippocampal cholinergic neurons and decreased hippocampal expression of nicotinic acetylcholine receptors (nAChRs) [2]. Thus the progressive loss of cholinergic signaling within the hippocampus may be an important mechanism underlying the cognitive defects associated with AD. Cholinergic systems are also linked to several protein factors associated with AD including β-amyloid (Aβ) [9], the β-amyloid precursor protein (APP) [22], and apolipoprotein E (ApoE) [11,19]. It is our hypothesis that ApoE4 in the hippocampus may provide a critical link between cholinergic systems and the functional impairment associated with AD.
Our hypothesis is based on two separate, but important findings regarding the effects of endogenous ApoE cleavage products and synthetically derived ApoE peptides on hippocampal neurons/receptors. First, an endogenous ApoE cleavage product (tApoE) and ApoE peptides (ApoE277–299) have been found to produce AD-like neurodegeneration in the cortex and hippocampus [16,21]. Additionally, tApoE has been found in the brains and cerebral spinal fluid of AD patients and interact with phosphorylated tau proteins and neurofibrils [8]. The fact that tApoE and small ApoE peptide fragments are cytotoxic and interact with AD-related proteins raises the possibility that ApoE4 contributes to AD and AD-related cognitive impairment by inducing hippocampal dysfunction.
Secondly, ApoE peptides derived from the LDL-receptor binding domain, ApoE133–149 and ApoE141–148, block ACh-evoked maximal current responses of homomeric α7 nAChRs expressed in Xenopus oocytes [4] and native α7 nAChRs in hippocampal slices [10]. The blockade of α7 nAChRs by these ApoE-derived peptides is dose-dependent, voltage-independent, and shows receptor specificity where blockade of α4β2 and α2β2 nAChRs is significantly smaller than that of α7 nAChRs. This is important in that hippocampal α7 nAChRs are critically important to proper spatial cognitive function (for review see [12]). Thus, it is plausible that ApoE peptides through either the neurodegenerative processes or by the blockade of α7 nAChRs may lead to cognitive defects.
The current study was conducted to determine whether ApoE133–149 and ApoE141–148 peptides infused into the rat ventral hippocampus would impair learning and memory as measured in the 8-arm radial maze task. This study focused on the effects of a single bolus dosing of ApoE peptides in an attempt to simulate a pathological state in which AD-related events have existed for a number of years.
2. Methods
2.1. Subjects
Adult female Sprague–Dawley rats (Taconic Labs, Germantown, NY, N = 45) were singly housed in plastic cages with corn-cob shavings. The rats lived in a vivarium (AAALAC-approved facility) immediately adjacent to the behavioral test facility, and maintained on a reverse 12:12 light–dark cycle with testing during the behaviorally active, dark phase. All rats had ad libitum access to water and with one daily meal being administered after behavioral testing. These studies were conducted under approved procedures of the Animal Care and Use Committee of Duke University.
2.2. ApoE peptide infusion
ApoE-derived peptides were synthesized by Sigma–Genosys (The Woodlands, TX, USA) at a purity of 95% and reconstituted in sterile, deionized water yielding stock concentrations of 15–20 mM. The peptides used in this study were acetylated at the amino terminus and amide-capped at the carboxyl terminus. Stock solutions were stored at −20 °C and diluted to desired concentrations in sterile filtered artificial cerebral spinal fluid (150 mM NaCl, 3 mM KCl, 1.4 mM CaCl2, 0.8 mM MgCl2, 1 mM NaH2PO4, pH 7.4) on the day of the experiment.
A guide cannula (Plastics One, Roanoke, VA) was stereotaxically implanted into the ventral hippocampus according to Pellegrino et al. [18] while rats were anesthetized with ketamine (0.6 mg/kg) and domitor (0.15 mg/kg). One week after cannula implantation ApoE peptides (1 or 5 μg per side) was infused into the ventral hippocampus at a flow rate of 0.126 μL/min (a total volume of 0.378 μL was infused over 3 min). Control animals were infused with equal volumes of artificial cerebral spinal fluid. Fig. 1 shows representative bilateral cannula placements in the ventral hippocampus.
Fig. 1.

Representative localization of cannula placements within the ventral hippocampus. Infusion cannula were stereotaxically implanted bilaterally into the ventral hippocampus according to coordinates of Pellegrino et al. [18]. The coordinates were −3.2 anterior/posterior, ±5.0 medial/lateral, and −7.0 dorsal/ventral.
2.3. 8-Arm radial maze
Behavioral testing began 3 days after ApoE peptide infusion. The behavioral task consisted of 18 sessions on the 8-arm radial maze. The rats were tested an average of two times per week. The 8-arm radial maze was constructed of wood and consisted of a central platform 50 cm in diameter, elevated 30 cm from the floor, with eight arms (10 cm × 60 cm) extending radially. Each arm was baited with a treat, and the rats were placed in a plastic cylinder (30 cm in diameter and 20 cm high) on the central platform. To begin the session, the cylinder was lifted allowing the rat to move freely about the maze. Arm choices were recorded when the rat placed all of its paws into the arm. Since the arms were not rebaited during the session, only the first entry into an arm was rewarded. Subsequent entries into an arm previously entered were counted as an error. The session was continued until either the rat entered all baited arms or 5 min elapsed. The choice accuracy measurement of working memory function is entries to repeat (ETR) defined as the number of correct arm entries in a session before the first error was made. Higher ETR scores indicate better memory of entering more correct arms as the session progressed and the task became more difficult. The response latency was measured by determining the seconds taken per arm entry, calculated by dividing the total session length by the number of arm entries. Higher scores indicated slower responding.
2.4. Cannula placement verification
After completion of the behavioral task the brains were removed and cannula placements were verified. Briefly, the rats were anesthetized with sodium pentobarbital. The rats were then perfused with a 0.9% phosphate-buffered saline solution followed by 4% paraformaldehyde solution. The brains were removed and preserved in 4% formaldehyde. Before being sliced on a cryostat to make histological slides, the brains were frozen on dry ice. Histological slides were then made and studied under a microscope for placement verification.
2.5. Statistical analysis
The data from the behavioral studies were assessed for significance by a mixed design analysis of variance for repeated measures and between subjects factors. Significant interactions were followed-up by tests of the simple main effects. Post hoc Dunnett’s tests were used to test the hypothesis that ApoE treatment impaired performance. An alpha level of p < 0.05 was used as a cutoff for statistical significance.
3. Results
To determine the causative effects of hippocampal ApoE peptide infusion on spatial learning, peptide fragments ApoE133–149 (ApoE17) and ApoE141–149 (ApoE8) were infused bilaterally into the ventral hippocampus of adult female Sprague–Dawley rats and the resultant effects on learning were assessed in the 8-arm radial maze task. Cannula placements were verified at the completion of behavioral testing. Fig. 1 illustrates a representative sampling of cannula placements of control and ApoE-infused rats (Fig. 1).
Fig. 2 shows the effect that ApoE peptide infusion had on cognitive functioning averaged over the course of the 18 sessions of training. There was a significant main effect of ApoE infusion (F(4,40) = 3.88, p < 0.01). Dunnett’s tests showed that each of the four ApoE-treated groups had significantly (p < 0.05) lower accuracy scores than the vehicle-infused controls. Fig. 2A shows that infusion of ApoE8 at doses of 1 and 5 μg/side caused significant defects in choice accuracy during the 18 sessions of training on the radial-arm maze. As with the shorter ApoE peptide fragment, infusion of ApoE17 also induced learning defects at 1 and 5 μg/side during the 18 sessions of training on the radial-arm maze.
Fig. 2.

(A) Mean choice accuracy (entries to repeat, ETR) over 18 sessions of training in the radial-arm maze (mean ± S.E.M.). ApoE8 (left panel) and ApoE17 (right panel) peptide fragments induced significant cognitive impairment over the total 18 sessions on the 8-arm radial maze. Each of the ApoE treated groups had significantly (p < 0.05) lower accuracy than controls. (B) Choice accuracy (entries to repeat, ETR) during each six-session phase of acquisition on the radial-arm maze (mean ± S.E.M.). There was a significant (p < 0.0001) main effect of training session block on accuracy but no significant interaction of training session block with ApoE treatment (N = 8–10/group).
The overall analysis of variance included not only the between subjects factor of ApoE fragment infusion, but also the repeated measure of training sessions on the radial-arm maze. To determine the effect of peptide infusion during early, middle, and latter portions of the task, the data were pooled into six-session blocks for statistical analysis. There was a significant (F(2,80) = 62.07, p < 0.0001) main effect of training session block on accuracy showing significant learning by the rats. However there was no significant interaction of training session block with ApoE treatment on rate of learning. The ApoE fragment-induced impaired choice accuracy did not significantly differ with continued training. However, at the very beginning of training, on session 1, there was not a significant ApoE fragment-induced choice accuracy impairment. The controls showed learning rates similar to rats in previous research [13].
To address the possibility that ApoE peptide infusion and cannula implantation could cause motor defects, which could account for differences in cognitive performance, an internal control was included where we compared 8-arm radial maze latency responses. The response latency is a measurement of seconds per arm entry, which was calculated by dividing the total session length by the number of arm entries. Higher scores indicate slower responding. Fig. 3 shows mean latency scores of control, ApoE8, and ApoE17 treated groups. We observed no significant differences in mean latency time between the groups. This indicates that cognitive defects caused by ApoE peptide infusion are not attributed to slower response times on the maze due to motor defects.
Fig. 3.
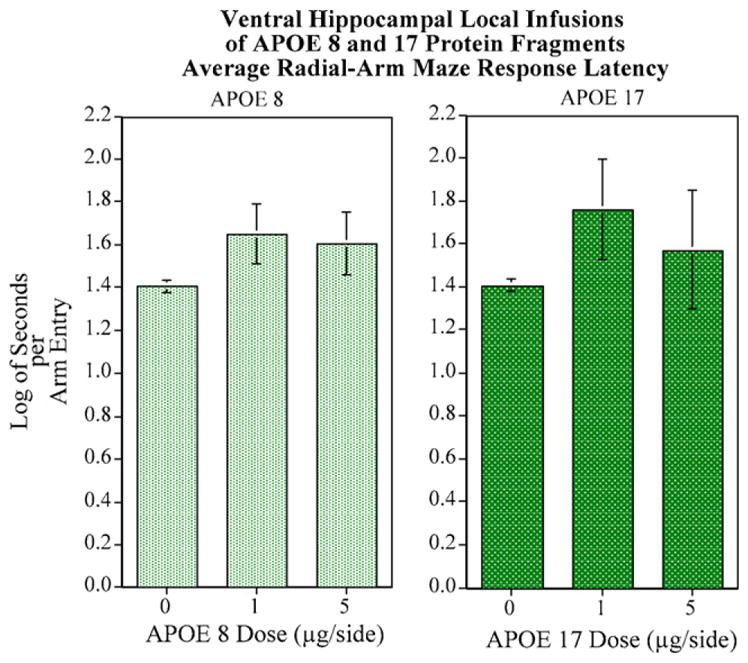
No significant effects on response latency were seen with either ApoE8 and ApoE17 peptide fragments: (N = 8–10/group).
4. Discussion
The current study shows for the first time that ApoE-derived peptides have an effect on cognitive performance. Rats receiving hippocampal infusions of ApoE peptides derived from the LDL-receptor binding domain have lower choice accuracy in the 8-arm radial maze relative to vehicle-infused controls. Moreover, acute peptide infusion causes cognitive impairment which persists for several weeks after the single local infusion. The rats in the peptide-exposed groups began to learn, but still lagged behind performance in the control group. Within the range of doses tested, the cognitive impairment associated with ApoE peptide infusion was not dose-dependent as the 1 μg/side ApoE peptide dosing caused as much impairment as the 5 μg/side dosing. It is also important to note that the longer peptide (ApoE133–149), which has higher potency with respect to blocking α7 nAChRs [10], was not found to be any more effective at inducing cognitive defects than the shorter peptide. This may have been due to a maximal cognitive deficit reached for these mechanisms of impairment. Lastly, the cognitive defects are not attributed to decreased locomotor activity since ApoE peptides do not cause significant change in mean latency scores.
This study demonstrates that a single local hippocampal infusion of ApoE peptides at high levels causes significant cognitive impairment, which persists for several weeks after the acute treatment. This supports our hypothesis that ApoE proteins in the hippocampus may be important to the cognitive impairment associated with AD. We based our hypothesis on two separate data sets; ApoE peptides induce hippocampal cell loss [16,21] and ApoE peptides block hippocampal α7 nAChRs [4,10]. However, the current data demonstrate lasting cognitive impairment results after a single acute dosing. These data argue against cognitive impairment resulting exclusively from pharmacological blockade of hippocampal α7 nAChRs and suggest that cognitive impairment from ApoE peptide infusion involves complex mechanisms beyond simple one-to-one receptor-antagonist pharmacology. It is more likely that cognitive impairment from hippocampal infusion of ApoE peptides results via necrotic/apoptotic and/or inflammatory events within the hippocampus.
Endogenous ApoE cleavage products and synthetically derived ApoE peptides have been found in several studies to induce hippocampal cell death [16,21]. In the cases of endogenous cleavage products, this neurotoxicity appears to result from ApoE interactions with thrombin. Studies have reported that thrombin concentrations are relatively high in Aβ plaques of AD patients [3]. Thrombin cleaves ApoE proteins to produce a bioactive 22 kDa cleavage product (truncated ApoE:tApoE) [16]. tApoE has been identified in the brains and cerebral spinal fluid of AD patients and interacts with phosphorylated tau proteins and neurofibrils [8]. In vitro studies have shown that tApoE is cytotoxic to primary neurons with tApoE4 being more toxic than tApoE3 [16]. ApoE proteins are also cleaved by chymotrypsin serine proteases to produce small ApoE cleavage peptides (residues 277–299) [6]. Like tApoE, these small peptide fragments interact with phosphorylated tau proteins and Aβ neurofibrils and are neurotoxic [8]. Additionally, these peptides produce AD-like neurodegeneration in the cortex and hippocampus [16]. Because tApoE and small ApoE fragments are cytotoxic and have been shown to be associated with AD-linked proteins, it is possible that tApoE and ApoE peptide fragments might contribute to AD and AD-related cognitive impairment. Further studies are needed to determine the effects of the peptides used in the current study (ApoE133–149 and ApoE133–149) on hippocampal cell survival.
The synthetically derived ApoE peptides (ApoE133–149) used in this study are currently being investigated for anti-inflammatory properties. Intravenous injection of these peptides suppresses systemic and brain inflammatory responses in mice by altering levels of TNF α and IL-6 [15]. In addition to reducing inflammatory responses, ApoE-derived peptides also significantly improve behavioral outcomes in a mouse model of closed head injury [14]. There are important differences between these data and the current study which may explain the cognitive impairment reported here. First, previous studies examined the effects of ApoE peptides in a pathological state (closed head injury) while our study administered the peptides during homeostatic conditions. Secondly, we directly infused the peptides into the hippocampus and at much higher doses than the previous studies. While purely speculative, either of these could have significant effects on the properties of the peptides as it relates to inflammation.
ApoE peptides could directly modulate inflammation via the activation of microglia or alternatively by blocking anti-inflammatory responses. In support of direct microglial activation, Guo et al. [5] showed that ApoE proteins have dual roles with respect to neuroinflammatory responses. In anti-inflammatory responses, ApoE3 and ApoE4 suppress Aβ-induced nitric oxide and cyclo-oxygenase-2 release from rat glial cells. ApoE3 and ApoE4 also suppress Aβ-induced ApoE release from rat glial cells. ApoE release is believed to be a feedback mechanism in response to inflammation. In pro-inflammatory responses, ApoE3 and ApoE4 induce the release of the inflammatory cytokine interleukin-1β in the absence of Aβ. ApoE4 is much more effective at inducing the release of IL-1β than ApoE3, further supporting the involvement of ApoE4 in AD. ApoE peptides have anti-inflammatory properties in the presence of a pathological insult, but can induce inflammation when present in normal homeostatic situations. It is also possible that the increased release of ApoE proteins from activated glial cells could convert protective anti-inflammatory responses into detrimental pro-inflammatory responses.
This study lays the groundwork for future mechanistic and therapeutic research concerning the ApoE-related cognitive deficits of AD. This behavioral study is the first step in what we hope is a series of investigations to unravel the bases by which the ApoE peptides have their effects on cognition. The epidemiological literature has shown conclusively the association of ApoE4 genotype and AD [1]. In vitro work has shown the disruption of cholinergic function induced by these ApoE peptide fragments [4,10]. The current study showed in a cause and effect manner that local application of the same ApoE fragments caused a persistent cognitive impairment. This proof of principle that there is a long-lasting cognitive impairment induced by hippocampal infusion of ApoE peptides is an important step in the process of determining how ApoE peptides cause neurobehavioral impairment.
Future studies should assess the neurochemical interactions of the ApoE peptides with cholinergic neurochemistry. The effects of these peptides on inflammatory processes should be evaluated. The generality of the effect to the specific peptide length and make-up needs to be characterized to determine what the critical constituents of the peptide are for inducing the cognitive impairment. The persistent cognitive impairment from local infusion of ApoE peptides into the ventral hippocampus opens the way for investigation of whether necrotic/apoptotic, inflammatory pathways or other mechanisms underlie ApoE-related cognitive impairment. This model can also serve to test new treatments to provide symptomatic relief or even better prevent the neurodegeneration observed in AD.
Acknowledgments
Research support was provided in part by the Merck & Co. Inc./United Negro College Fund (UNCF/Merck) Science Initiative and NIEHS. We thank the Intramural Research Program of the NIEHS, NIH.
References
- 1.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 2.Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219:1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 3.Festoff BW, Rao JS, Chen M. Protease nexin I, thrombin- and urokinase-inhibiting serpin, concentrated in normal human cerebrospinal fluid. Neurology. 1992;42:1361–1366. doi: 10.1212/wnl.42.7.1361. [DOI] [PubMed] [Google Scholar]
- 4.Gay E, Klein RC, Yakel JL. Apolipoprotein E-derived peptides block alpha7 neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Pharmacol Exp Therap. 2006;316:835–842. doi: 10.1124/jpet.105.095505. [DOI] [PubMed] [Google Scholar]
- 5.Guo L, LaDu VELJ. A dual role for apolipoprotein E in neuroinflammation: anti- and pro-inflammatory activity. J Mol Neurosci. 2004;23:205–212. doi: 10.1385/JMN:23:3:205. [DOI] [PubMed] [Google Scholar]
- 6.Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, et al. Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer’s disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA. 2003;100:10966–10971. doi: 10.1073/pnas.1434398100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang W, Qiu C, von Strauss E, Winblad B, Fratiglioni L. APOE genotype, family history of dementia, and Alzheimer disease risk: a 6-year follow-up study. Arch Neurol. 2004;61:1930–1934. doi: 10.1001/archneur.61.12.1930. [DOI] [PubMed] [Google Scholar]
- 8.Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW. Apolipoprotein E fragments present in Alzheimer’s disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA. 2001;98:8838–8843. doi: 10.1073/pnas.151254698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kar S, Slowikowski SP, Westaway D, Mount HT. Interactions between beta-amyloid and central cholinergic neurons: implications for Alzheimer’s disease. J Psychiatry Neurosci. 2004;29:427–441. [PMC free article] [PubMed] [Google Scholar]
- 10.Klein RC, Yakel JL. Inhibition of nicotinic acetylcholine receptors by apolipoprotein E-derived peptides in rat hippocampal slices. Neuroscience. 2004;127:563–567. doi: 10.1016/j.neuroscience.2004.05.045. [DOI] [PubMed] [Google Scholar]
- 11.Lai MK, Tsang SW, Garcia-Alloza M, Minger SL, Nicoll JA, Esiri MM, et al. Selective effects of the APOE epsilon4 allele on presynaptic cholinergic markers in the neocortex of Alzheimer’s disease. Neurobiol Dis. 2006;22:555–561. doi: 10.1016/j.nbd.2005.12.016. [DOI] [PubMed] [Google Scholar]
- 12.Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification and anatomic localization. Psychopharmacology. 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- 13.Levin ED, Wilkerson A, Jones JP, Christopher NC, Briggs SJ. Prenatal nicotine effects on memory in rats: pharmacological and behavioral challenges. Dev Brain Res. 1996;97:207–215. doi: 10.1016/s0165-3806(96)00144-7. [DOI] [PubMed] [Google Scholar]
- 14.Lynch J, Wang RH, Mace B, Leinenweber S, Warner DS, Bennett ER, et al. A novel therapeutic derived from apolipoprotein E reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192:109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 15.Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, et al. APOE genotype and an ApoE-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- 16.Marques MA, Tolar M, Harmony JA, Crutcher KA. A thrombin cleavage fragment of apolipoprotein E exhibits isoform-specific neurotoxicity. Neuroreport. 1996;7:2529–2532. doi: 10.1097/00001756-199611040-00025. [DOI] [PubMed] [Google Scholar]
- 17.O’Keefe J. A computational theory of the hippocampal cognitive map. Prog Brain Res. 1990;83:301–312. doi: 10.1016/s0079-6123(08)61258-3. [DOI] [PubMed] [Google Scholar]
- 18.Pellegrino LJ, Pellegrino AS, Cushman AJ. A stereotaxic atlas of the rat brain. Plenum Press; New York: 1979. [Google Scholar]
- 19.Poirier J, Delisle MC, Quirion R, Aubert I, Farlow M, Lahiri D, et al. Apolipoprotein e4 allele as a predictor of cholinergic deficits and treatment outcome in Alzheimer disease. Proc Natl Acad Sci USA. 1995;92:12260–12264. doi: 10.1073/pnas.92.26.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saunders AM, Schmader K, Breitner JC, Benson MD, Brown WT, Goldfarb L, et al. Apolipoprotein E epsilon 4 allele distributions in late-onset Alzheimer’s disease and in other amyloid-forming diseases. Lancet. 1993;342:710–711. doi: 10.1016/0140-6736(93)91709-u. [DOI] [PubMed] [Google Scholar]
- 21.Tolar M, Marques MA, Harmony JA, Crutcher KA. Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci. 1997;17:5678–5686. doi: 10.1523/JNEUROSCI.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmermann M, Borroni B, Cattabeni F, Padovani A, Di Luca M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol Dis. 2005;19:237–242. doi: 10.1016/j.nbd.2005.01.002. [DOI] [PubMed] [Google Scholar]