

Alzheimer’s disease (AD) pathology, characterized by Aβ deposition in the brain as insoluble extracellular plaques and intracellular tau aggregation in paired helical filaments, begins to develop ~10–15 years before the onset of memory impairment (1). By the time cognitive symptoms and signs are present, substantial synaptic and neuronal injury has already occurred. In recent years, one focus of AD research has been to define the preclinical phase of AD, when Aβ with or without neocortical tau deposition has begun to occur, but before clear cognitive decline. The ultimate goal is therapeutic intervention at this stage to prevent progression to symptomatic AD (2). AD pathology has long been associated with memory impairment in older adults, but recent evidence has also shown Aβ deposition to be associated with disruption in sleep quality even in the absence of cognitive impairment (3). Furthermore, recent evidence supports a role for sleep in the development of AD, at least in part by influencing Aβ. Aβ fluctuates diurnally: soluble Aβ levels are higher during wakefulness and lower during sleep (4,5). Sleep deprivation accelerates Aβ deposition in APP transgenic mice (4), whereas orexin deficiency, which increases sleep, decreases it (6). In addition, amyloid deposition disrupts sleep in APP transgenic mice (7). The relationship between sleep and Aβ deposition has thus been proposed to be bidirectional: sleep disruption leads to protein deposits and protein deposits result in sleep disturbance (8). The relationship among Aβ deposition, other aspects of AD pathology, sleep and memory impairment are not well defined in humans.
In this issue of Nature Neuroscience, Mander et al. (9) report a link between brain Aβ deposition, sleep and memory dysfunction. They hypothesize that Aβ accumulation in the medial prefrontal cortex (mPFC) is associated with diminished slow-wave activity (SWA) during non-REM (NREM) sleep that further correlates with the extent of impaired overnight hippocampus-dependent memory consolidation in older adults. Previous work from this group has shown that mPFC atrophy is associated with reduced NREM SWA and that this association correlates with overnight memory retention (10). To further investigate whether amyloid deposition rather than brain atrophy has a similar effect, the authors recruited 26 cognitively normal older adults who underwent positron emission tomography imaging with Pittsburgh compound B to determine the amount of fibrillar Aβ deposited in the brain. To assess memory function, all participants trained on a set of word pairs in the evening. Then sleep was monitored overnight with polysomnography to assess different sleep stages, such as NREM sleep, and to obtain electroencephalography (EEG) for power analysis. Participants took the word-pair test again in the morning during a functional MRI scanning session.
The authors found that higher amyloid burden in the mPFC correlated with decreased NREM SWA in this brain region, but not with higher frequencies of EEG activity or with decreased NREM SWA in other regions. This decrease in mPFC NREM SWA further correlated with worse overnight memory retention, even after controlling for age and sex. The authors then sought to determine the interaction of these factors using path analysis. Three models were constructed to determine the nature of the interactions between the factors. The standardized metrics used to determine the interactions were root-mean-square residual, goodness of fit and Bayesian information criterion. Using parameters for mPFC amyloid, SWA, hippocampal activation and memory retention, the model with the best statistical fit to emerge was a sleep-dependent model in which the influence of mPFC fibrillar Aβ deposition on impaired memory retention was not direct, but was mediated through sleep, implying a causative relationship (Fig. 1a).
Figure 1.
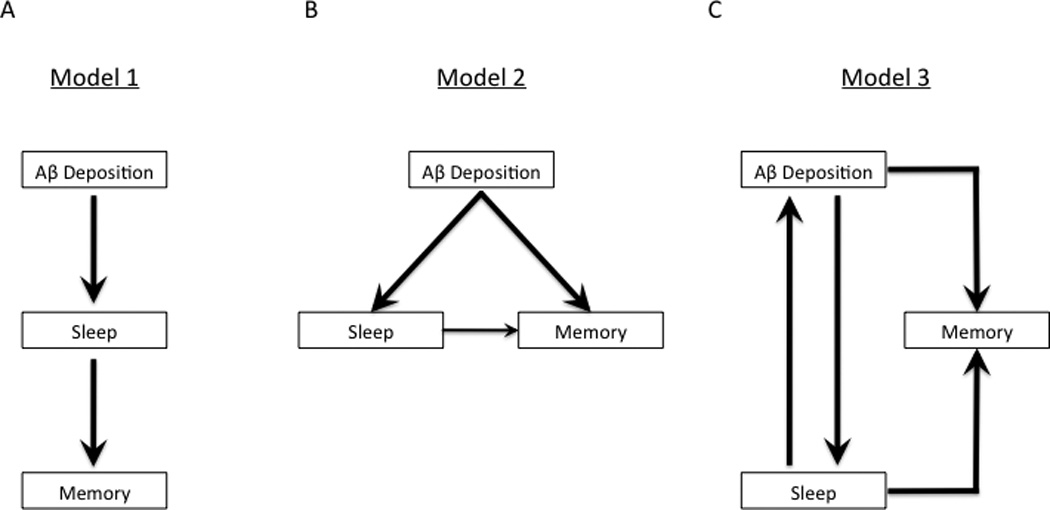
Models for the relationship among amyloid, sleep and memory. (a) Model proposed by Mander et al. (9). Aβ deposition leads to changes in sleep, such as decreased NREM SWA. This then affects memory function that is dependent on sleep. This model suggests a linear relationship from amyloid to sleep to memory. (b) Alternative model. Aβ deposition independently affects both sleep and memory function. Sleep has additional adverse effects on memory. This model suggests that there are parallel relationships between amyloid and sleep, and amyloid and memory. (c) Alternative model. Aβ deposition and sleep have a bidirectional interaction resulting in a feedback loop, and both affect memory function.
Sleep and memory are both network processes in the brain and are not the properties of one specific brain region. For example, the encoding and recognition phases of working memory and long-term memory involve the prefrontal cortex and many other brain regions, including cingulate, parietal and temporal lobe regions (11). Similarly, functional neuroimaging has shown sleep stage–dependent changes in regional brain connectivity (12). Furthermore, there are changes in other brain networks during sleep. The transition from wakefulness to progressively deeper stages of sleep, for example, has been associated with decreased connectivity of the mPFC to the default mode network (13).
Cognitive decline in AD progression involves memory dysfunction coincident with disruption of entorhinal-hippocampal connectivity, but disruption of brain networks such as the default mode network has also been shown, even during the preclinical phase (14). Recent work has also shown that sleep quality is worse in cognitively normal older adults with amyloid deposition than in those without (3). These findings support the hypothesis that Aβ deposition disrupts sleep as a network process. Furthermore, the interaction between sleep and memory is likely more complicated than the authors’ model suggests. For instance, their research group previously reported that age-related mPFC atrophy was associated with decreased NREM SWA and sleep-dependent memory retention (10), suggesting that processes related to normal aging are involved.
Mander et al. (9) provide associative evidence of a possible sleep-mediated mechanism that leads linearly from amyloid deposition to slow-wave sleep disruption to abnormal hippocampal activation to memory impairment in AD (Fig. 1a). As the authors acknowledge, this study is cross-sectional and correlational, limiting its interpretation in terms of establishing causation. The proposed sleep-dependent model fits the study data statistically, but there are plausible alternative hypotheses. For example, amyloid deposition may affect memory and sleep simultaneously by disrupting their respective networks (Fig. 1b). AD risk is related to increasing age, genetics and environmental factors (such as exercise and diet) that influence AD pathology. Age is also associated with changes in sleep such as decreased NREM slow-wave sleep and modification of sleep has been associated with changes in Aβ in both mice and humans (4,15). Sleep disturbances related to aging may impair memory while also precipitating and/or amplifying Aβ deposition, leading to a feedback loop of amyloid plaque formation causing further sleep disruption in addition to memory loss (Fig. 1c). Tau deposition may contribute to the observations here and will be especially important to follow, as it initially deposits and accumulates in the medial temporal lobe, begins to spread to the neocortex as amyloid deposition progresses and is strongly correlated with cognitive decline in AD. The question may best ultimately be answered with a longitudinal approach following Aβ, other AD pathologies including tau, sleep, memory and brain network function in older adults over several years.
In addition to decreasing sleep quality, amyloid effects on sleep may also hinder adaptability to new environments. Mander et al. (9) assessed sleep in the home setting with sleep logs and then monitored it overnight in a sleep laboratory with polysomnography. Although participants were permitted to sleep at times similar to their home sleep periods, the authors did not compare participant sleep quality during the sleep study compared with self-report nor assess the sleep-wake cycle by a constant measure across the entire study period, as would be possible with methods such as actigraphy. Without these data, a possible explanation for the study findings is that Aβ deposition results in poor adaptability to new sleep environments (that is, the sleep laboratory) and that decreased sleep quality from baseline led to the observed changes in NREM SWA and memory. As the authors note, EEG-based sleep stage monitoring in the home with memory testing would be more representative of the participants’ sleep quality.
Despite these issues, the study by Mander et al. (9) provides important new insights into the changes in sleep and memory in preclinical AD, as well as indicating potential new avenues for investigations. Future longitudinal studies will be helpful to define the cascade of changes in brain functions, such as memory and sleep, caused by AD pathologies, including amyloid and tau deposition as well as neuroinflammation. Hypothetically, sleep could be used as a diagnostic marker for Aβ deposition and/or a disease-modifying treatment. Further understanding the relationships between AD pathology, sleep and memory may lead to important diagnostic and therapeutic interventions for patients at risk for AD.
Acknowledgments
Competing Financial Interests:
B.P.L. was supported by the Washington University Institute of Clinical and Translational Sciences grant UL1TR000448, sub-award KL2TR000450, from the National Center for Advancing Translational Sciences (NCATS) of the National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official view of the NIH.
D.M.H. was supported by 1P01NS074969. D.M.H. is also a co-founder of C2N Diagnostics LLC and is on the scientific advisory boards of, or consults for, AstraZeneca, Genentech, Eli Lilly, Neurophage and C2N Diagnostics. Washington University receives research grants to the laboratory of D.M.H. from Cure Alzheimer's Fund, the JPB Foundation, Tau Consortium, Eli Lilly and C2N Diagnostics.
References
- 1.Perrin RJ, Fagan AM, Holtzman DM. Nature. 2009;461:916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sperling RA, et al. Alzheimers Dement. 2011;7:280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ju YE, et al. JAMA Neurol. 2013;70:587–593. doi: 10.1001/jamaneurol.2013.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang JE, et al. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang Y, et al. Arch. Neurol. 2012;69:51–58. doi: 10.1001/archneurol.2011.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roh JH, et al. J. Exp. Med. 2014;211:2487–2496. doi: 10.1084/jem.20141788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roh JH, et al. Sci. Transl. Med. 2012;4:150ra22. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ju Y-E, Lucey BP, Holtzman DM. Nat. Rev. Neurol. 2014;10:115–119. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mander BA, et al. Nat. Neurosci. 2015;18:1051–1057. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mander BA, et al. Nat. Neurosci. 2013;16:357–364. doi: 10.1038/nn.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranganath C, Johnson MK, D’Esposito M. Neuropsychologia. 2003;41:378–389. doi: 10.1016/s0028-3932(02)00169-0. [DOI] [PubMed] [Google Scholar]
- 12.Dang-Vu TT, et al. Sleep. 2010;33:1589–1603. doi: 10.1093/sleep/33.12.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sämann PG, et al. Cereb. Cortex. 2011;21:2082–2093. doi: 10.1093/cercor/bhq295. [DOI] [PubMed] [Google Scholar]
- 14.Sperling RA, et al. Neuron. 2009;63:178–188. doi: 10.1016/j.neuron.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ooms S, et al. JAMA Neurol. 2014;71:971–977. doi: 10.1001/jamaneurol.2014.1173. [DOI] [PubMed] [Google Scholar]