

Abstract
Amyloid-like fibrils formed by huntingtin exon-1 (httex1) are a hallmark of Huntington's Disease (HD). The structure of these fibrils is unknown and determining their structure is an important step towards understanding the misfolding processes that cause HD. In HD a polyglutamine (polyQ) domain in httex1 is expanded to a degree that it gains the ability to form aggregates comprising the core of the resulting fibrils. Despite the simplicity of this polyQ sequence the structure of httex1 fibrils has been difficult to determine. The current study provides a detailed structural investigation of fibrils formed by httex1 using solid-state NMR spectroscopy. We show that the polyQ domain of httex1 forms the static amyloid core similar to polyQ model peptides. The Gln residues of this domain exist in two distinct conformations that are found in separate domains or monomers but are relatively close in space. The rest of httex1 is relatively dynamic on an NMR time scale, especially the proline-rich C-terminus, which we found to be in a polyproline II helical and random coil conformation. We observed a similar dynamic C-terminus in a soluble form of (httex1 indicating that the conformation of this part of httex1 is not changed when aggregating into an amyloid fibril. From these data we propose a bottlebrush model for the fibrils formed by httex1. In this model, the polyQ domains form the center and the proline-rich domains the bristles of the bottlebrush.
Introduction
Huntington's disease (HD) is a heritable, fatal neurodegenerative disease with symptoms of motor dysfunctions, cognitive impairments, and psychiatric disorders.1 HD is the most common of a class of diseases in which a polyglutamine (polyQ) domain is pathologically extended above a certain threshold (36 repeats in the case of HD).2 Besides changing the flexibility of the monomeric state,3 pathologically expanded polyQ domains have the tendency to form fibrillar, amyloid-like aggregates in vivo and in vitro. Fibril forming kinetics and the onset of HD are faster the longer the polyQ domain.4,5 In HD, the polyQ domain is part of the protein huntingtin (htt) and is located on the htt exon-1 (httex1).6 Furthermore, httex1 has been shown to be significant for HD since it is prominently found in the amyloid deposits of postmortem brains7 and can be produced by an aberrant splice variant.8 httex1 has an N-terminal amphiphilic domain often termed N17, followed by the polyQ domain, whose aggregation is aided by the presence of the N17 domain.9–11 The C-terminus of httex1 has two pure polyproline stretches interrupted by a proline-rich sequence (see Figure 1). Such polyproline flanking sequences were shown to have an inhibitory effect on polyQ aggregation.12 How the polyQ expansion results in HD is unknown. The mechanism of htt toxicity is an active field of research and there are non-toxic and toxic fibril species. Furthermore, there are toxic protofibrils and oligomeric forms of htt.13–15 In order to understand the molecular origins of toxicity and protein misfolding in HD it is important to know the molecular structure and the dynamic properties of the fibrils that are the end product of this misfolding process.
Figure 1.

Sequence of httex1 Q46. The N17, polyQ, and C-terminal domain are highlighted in orange, blue, and green, respectively.
Until recently, structural studies on htt fibrils have focused on simple polyQ model peptides and httex1 mimics with polyQ domains shorter than those found in HD.16–21 A recent EPR study done on fibrils formed by httex1 Q46 showed that the N17 and the polyQ domain are relatively static, whereas the Pro rich domain becomes increasingly dynamic towards the C-terminus. Interestingly, EPR also showed that, contrary to many other amyloid fibrils, the polyQ domain is not in an in-register β-sheet conformation.22 However, the precise structural organization of httex1 fibrils remains unknown.
To provide detailed structural information, the present study uses solid-state NMR data on httex1 fibrils grown at 4°C, the same types fibrils employed in the previous EPR study. Temperature was shown to modulate the mechanism of misfolding, the saturation concentrations, and fibril forming kinetics of htt.11 Moreover, fibrils grown at 4°C were previously shown to be more toxic and less rigid than fibrils grown at 37°C.14 Our data on the polyQ domain of httex1 allow the comparison with the polyQ domain of htt model peptides and we show that the proline-rich domain of httex1 is dynamically and structurally more complex than previously thought.
Materials and Methods
Protein Expression, Purification, and Fibril Formation
Uniformly 13C, 15N labeled wild-type httex1 Q46 fibrils were expressed, and purified as described by Fodale et al.23 with modification following a protocol by Marley et al.24 that allows the efficient isotope labeling. Overnight cultures of BL21(DE3) transformed with the pET32a-HDx46Q plasmid were diluted 50-fold into LB medium and grown at 37°C to 0.6 A600. Pellets were collected by centrifugation at 3500 g, resuspended in M9 wash buffer, pelleted again, and resuspended in a quarter of the original volume using M9 medium containing 4 g/l U13C glucose and 0.5 g/l 15N ammonium chloride. After 1 h of incubation at 30°C, 200 rpm, protein expression was initiated by adding 1 mM Isopropyl 1-thio-D-galactopyranoside. Cell pellets were collected by centrifugation after 4 hours of incubation and resuspended in 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, and 10 mM imidazole containing 1 × CelLytic B Cell Lysis reagent (Sigma-Aldrich), and incubated for 20 min at room temperature on a shaker. Lysates were clarified by centrifugation at 21,000 g for 10 min and incubated with nickel-nitrilotriacetic acid-agarose beads (Qiagen) for 1 h at 4°C on a shaker. Beads were decanted into an Econo-Pac chromatography column (Bio-Rad) and washed with several column volumes of 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole. The pure protein was eluted with 20 mM Tris-HCl, pH 8.0, 300 mM NaCl, 250 mM imidazole.
Following concentration of the proteins via Amicon Ultra-15 10,000 MWCO centrifugal filters (Millipore), proteins were re-diluted in 20 mM Tris, pH 7.4, then purified on a HiTrap Q XL column (GE Healthcare) with an AKTA FPLC system (Amersham Pharmacia Biotech) using a NaCl gradient. The protein eluted at about 150 mM NaCl and was consequently diluted to 20μM (560 μg/ml). To cleave the N-terminal thioredoxin tag and initiate fibril formation, EKMax (Invitrogen) was added to 1 unit/280 μg of protein. The reaction was incubated without agitation at 4 °C for 3 days. Finally, fibril samples were washed with deionized water, sedimented by ultracentrifugation (150,000 g, 20 minutes) and packed into 1.6 mm magic angle spinning (MAS) rotors.
Uniformly 13C-15N labeled httex1 Q7 was expressed and purified as described above but without cleaving the thioredoxin tag. The protein was concentrated with an Amicon Ultra-15 10,000 MWCO centrifugal filter and concentration was determined via absorbance at 280 nm. 25% glycerol was added to the final buffer of 20 mM Tris-HCl, pH 7.4 150 mM NaCl and the protein solution was pipetted in 4 mm NMR rotors.
Solid-state NMR Spectroscopy
All spectra were recorded on an Agilent DD2 600 MHz solid-state NMR spectrometer. Fibril spectra were recorded using a T3 1.6 mm probe operating at 25 kHz MAS. 200 kHz and 100 kHz hard pulses were applied on 1H and 13C, respectively. The recycle delay was 3 s for all spectra. 1H-13C cross polarizations (CPs) were done using a Hartman-Hahn match of 60 kHz on 13C and 85 kHz on 1H with a 10% amplitude ramp. 120 kHz two pulse phase modulation (TPPM) 1H decoupling was applied during direct and indirect detection. 13C-13C spectra were recorded using a 25 kHz 1H recoupling field during the 50 ms mixing time (DARR25) and no recoupling field for the spectrum with 500 ms mixing time (PDSD). The 13C spectral width was 50 kHz in both dimensions and 400 complex t1 increments and 16 and 32 acquisitions were co-added per increment for the spectra with 50 ms and 500 ms mixing time, respectively. 15N-13C correlation spectra were recorded using a SPECIFIC double CP (DCP) pulse sequence26 with rf-fields of 40 kHz on 15N and 65 kHz on 1H for the first and 40 kHz on 15N and 15 kHz on 13C for the second CP. The NCA spectrum was recorded with a transmitter at 52 ppm, the NCOCX spectrum with a 13C transmitter at 191 ppm and an additional 13C-13C DARR mixing of 50 ms. Both 15N-13C correlation spectra were recorded with a 50 kHz spectral width in the direct 13C dimension and 10 kHz spectral width in the indirect 15N dimension and 32 and 64 acquisitions were co-added for each of the 96 complex t1 increments for the NCA and the NCOCX spectra, respectively. Direct polarization (DP) Constant-time uniform-sign cross- peak (CTUC) correlation spectroscopy (COSY) spectra were recorded using hard pulses for the CA-CO correlations and using 420 μs and 180 μs r-SNOB pulses on carbonyl and aliphatic regions, respectively, for the aliphatic correlations.27,28 The spectral width of these DP CTUC COSY spectra was 50 kHz in both dimensions and 32 acquisitions were co-added for each of the 340 and 400 complex t1 increments for the aliphatic and CA-CO spectra, respectively. The 1H-13C heteronuclear correlation (HETCOR) spectrum of the fibrils was recorded using a refocused INEPT transfer. The indirect 1H dimension had an 180° 13C refocusing pulse and a spectral width of 10 kHz; the direct 13C dimension had a spectral width of 50 kHz and 16 acquisitions were co-added for each of the 128 complex t1 increments.
The refocused INEPT HETCOR spectrum of soluble httex1 Q7 with N-terminal thioredoxin tag was recorded in a 4 mm T3 probe at a MAS frequency of 3 kHz. 75 kHz and 50 kHz hard pulses were applied on 1H and 13C respectively. Twenty kHz TPPM decoupling was applied in the direct 13C dimension. The spectral width was 50 kHz for the direct 13C dimension and 10 kHz for the indirect 1H dimension and 80 acquisitions were co-added for each of the 128 complex t1 increments. All chemical shifts were referenced externally to DSS using adamantane.29
Results
For this NMR study, we expressed uniformly 13C-15N labeled httex1 with a 46 amino acid long polyQ domain in E.coli. The N-terminal thioredoxin tag was cleaved off to induce fibril formation and fibrils were formed at 4°C. As can be seen from the electron microscopy (EM) image in Figure S1 of the Supporting Information, these conditions lead to relatively homogenous fibril preparations with occasional bundling. Frequently fibrils appeared to be hindered from crossing over in the EM images and were spaced approximately 28 to 33 nm (center-to-center) apart. Mature fibrils were packed in a 1.6 mm MAS rotor. To study the homogeneity and structure of the httex1 fibril core, we recorded dipolar CP based solid-state NMR spectra that are sensitive to the static domains of the httex1 fibrils.30 As can be seen from the 2D DARR spectrum shown in Figure 2, the resolution of the spectrum was very good. For example the Pro Cγ linewidth highlighted in Figure 2 is 0.7 ppm and one of the Gln Cα lines has a width of 1.3 ppm, which is comparable to linewidths observed for other homogenous amyloid preparations31 even though both of these lines arise from several different sites. These narrow lines indicate a high structural homogeneity of the static parts of our httex1 fibrils. The amino acid type assignment of this spectrum identifies three different forms of Gln, two different forms of Pro, and a Glu. Two forms of Gln, termed Gln A and Gln B, have roughly the same intensity and match the previously observed chemical shifts assignments17,18 where Gln B is compatible with an extended β-strand sheet conformation, and Gln A has a chemical shifts different from average Cα, Cβ, C' chemical shifts reported for Gln in an α-helical, β-sheet, or random coil conformation.32 From Gln C only the Cγ-Cδ can be identified in the 2D DARR spectrum. The other peaks of Gln C partially overlap with Gln B but are the dominant form of Gln and better visible in the spectra focusing on the dynamic domains of the httex1 fibrils discussed below. The two forms of Pro observed in this spectrum have very different intensities, where Pro A gives intense cross peaks and Pro B is barely detectable. The chemical shift of Pro A, which has been previously reported on spectra of N17Q30P9K2,19 is in good agreement with a polyproline II helix bound to an SH3 domain33 but fits less well to the Pro shift measured in polyproline powder samples.34,35 Pro B, which has not been reported previously, has a chemical shift compatible with a random coil conformation.32
Figure 2.
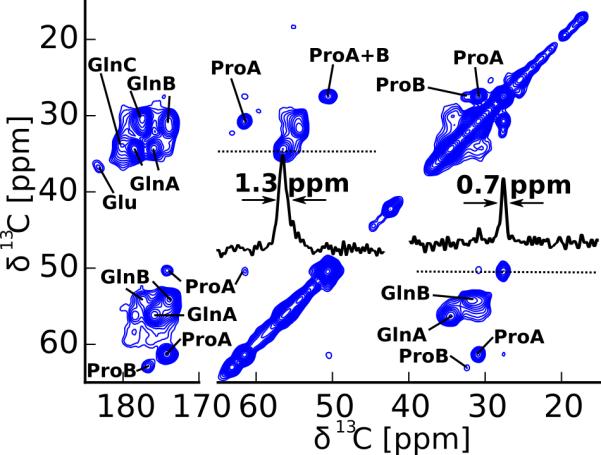
The fibril core of httex1 Q46 is very homogenous. 2D DARR spectrum of httex1 fibrils recorded at 14.1 T, 25 kHz MAS, 0°C, and a mixing time of 50 ms. The amino acid assignments of the two types of Gln and Pro are indicated and the linewidth of Gln A Cα and Pro A Cγ is illustrated via 1D slices in the direct dimension. The thus determined linewidth of 0.8 ppm for Pro A Cγ and 1.3 ppm for Gln A Cα are remarkably narrow considering that these peaks result from the superposition of several resonances.
An important question regarding the two forms of Gln that dominate the 2D DARR of httex1 fibrils is whether they are either located in separate domains or fibrils, or alternate in sequence (i.e. type A is followed by type B, which is followed by type A etc.), or are randomly distributed in sequences (see Figure 3A). Distinguishing these three possibilities will allow us to exclude and confirm different structural models. For example an alternate arrangement of the two Gln types could point towards an α-sheet structure that has been proposed as a possible conformation of polyQ fibrils.36 In α-sheets the backbone dihedral angles alternate between two different regions of the Ramachandran plot, which is an interesting model since it would explain that we observe two different types of Gln at a 1:1 ratio. To distinguish between the three arrangements of Gln A and B, we recorded the NCA and NCOCX spectra. The NCA spectrum shows cross peaks between the backbone nitrogen (N) and the Cα and other aliphatic carbons of the same residue. The NCOCX spectrum shows cross peaks between N and the C' and aliphatic carbons of the preceding residue in sequence (see Figure 3B). If the two Gln forms alternate in sequence, the NCOCX spectrum would show cross peaks of the N of Gln A with the aliphatic carbons of Gln B and of the N of Gln B with the aliphatic carbons of Gln A. In the case of random distribution, we would expect cross peaks from N of Gln A and Gln B with the aliphatic carbons of both Gln A and Gln B. If Gln A and Gln B are separated in sequence, we expect to see only cross peaks from Gln A to Gln A and from Gln B to Gln B. As can be seen from Figure 3C, the latter is correct since the NCA and the NCOCX spectra overlap very well and no backbone Gln A to Gln B cross peaks were observed.
Figure 3.

Gln A and Gln B are sequentially clustered. (A) Illustration of three possible arrangements of Gln A and B in the primary sequence of httex1. (B) Schematic showing the magnetization pathway of the NCA (red) and NCOCX (blue) experiment. (C) 2D NCA (red) spectra correlate the backbone nitrogen to the Cα of the same residue, NCOCA spectra (blue) correlate the backbone nitrogen with the Cα of the preceding residue. The almost perfect overlap of the N-Cα peaks in these spectra and also the overlap of the weaker N-Cβ/γ peaks indicate that Gln A has only Gln A as sequence neighbor and Gln B only Gln B. Cross peaks that would be expected in different arrangements are indicated with dashed circles. Despite the clear separation of Gln A and B in sequence, weak side chain cross peaks between Gln A and Gln B were detected in the NCOCX spectrum. Spectra were recorded under conditions similar to the spectrum in Figure 2 using a SPECIFIC DCP pulse sequence for the NCA spectrum and a SPECIFIC DCP followed by a 50 ms 13C-13C DARR mixing period for the NCOCX experiment.
The above experiments show that Gln A and Gln B are separated in sequence, or even found in different monomers. However, are the Gln A and Gln B also separated in space e.g. found in separate fibrils? To answer this question, we recorded 2D 13C-13C DARR spectra with a long mixing time of 500 ms. DARR spectra of this kind can have cross peaks between amino acid residues that are up to 7 Å apart in space. However, nucleus-specific distance information is often obscured by relayed magnetization transfer that occurs during the long mixing time.37 As can be seen from Figure 4, we observed clear cross peaks between the Gln A and Gln B carbons, for example the strong Cα-Cα cross peak close to the diagonal. If Gln A and Gln B were found in completely separate fibrils, no such cross peaks would be detectable. In addition to these Cα-Cα cross peaks, our NCOCX spectrum in Figure 3C shows weak Nε2-carbon cross peaks compatible with Gln A-Gln B side chain-side chain contacts besides the stronger Nε2-Cγ, Nε2-Cβ cross peaks within the same amino acid type. Together these data show that although the two forms of Gln are in separate domains or even monomers, they are relatively close in space.
Figure 4.
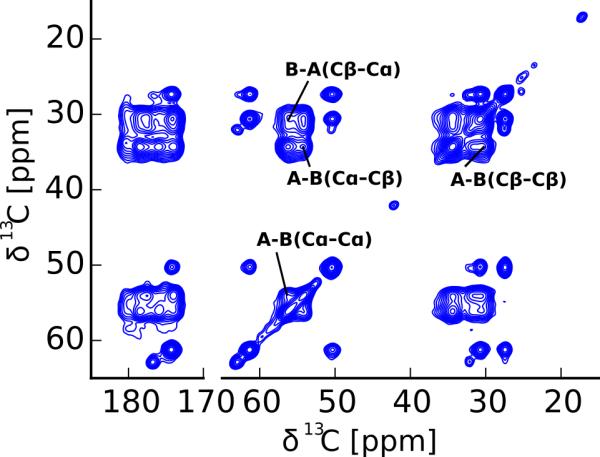
Although not neighboring in sequence, Gln A and Gln B are close in space. 13C-13C DARR spectrum similar to the one shown in Figure 2 but with a mixing time of 500 ms. For clarity, only additional cross peaks belonging to Gln A-Gln B correlations are indicated. These cross peaks show that Gln A and Gln B are within 7 Å in space. Interestingly, no additional cross peaks between the two types of Pro nor between Gln and Pro are detected in these spectra.
Besides the static domains of httex1, we investigated the dynamic domains of httex1 using J-coupling based spectroscopy that works best in the case of narrow, dynamically averaged 1H and 13C lines.30 As can be seen from the direct excitation (DE) 13C-13C CTUC-COSY spectrum27,28 shown in Figure 5, we were able to detect an additional Cβ-Cγ cross peak for Glu and the full spin system of Gln C, as well as Pro A and Pro B, which were also detected using CP based spectroscopy. In addition, we were able to detect cross peaks that we assigned to His, Ala, Leu, Val, and Lys that could not be observed in the 2D DARR spectrum. From these residues, His (from the His-tag), and the only Val (V105) are exclusive to the C terminus, Glu, Ala, Leu can be found in both the C-terminus and N17 domain, and Lys is exclusive to the N17 domain. Interestingly, Pro A and Pro B are observed with comparable intensity in these spectra indicating that Pro A might be found in the static as well as dynamic domains of httex1 whereas Pro B is mostly located in dynamic domains (see Supplementary Fig. S2).
Figure 5.
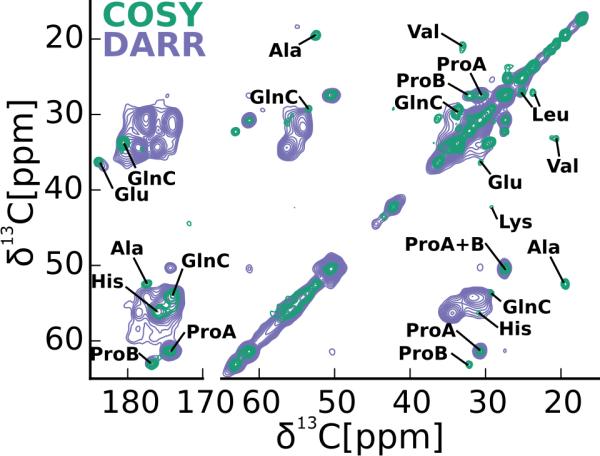
The dynamic parts of httex1 include both types of Pro, the third form of Gln (Gln C), and a Glu residue as well as resonances compatible with Leu, His, Ala, Val, and Lys that were not previously detected using dipolar-based spectroscopy. Overlay of 13C-13C DP-CTUC-COSY (green) and 13C-13C DARR (purple) spectra of httex1 fibrils recorded at 25 kHz MAS. The residues identified in the 13C-13C DP-CTUC-COSY, which is sensitive to the more dynamic parts of the fibril, are indicated.
These dynamic residues are also observed in the 1H-13C INEPT-HETCOR spectrum shown in Figure 6. In addition, the INEPT-HETCOR also has resonances we assigned to Arg and Met. Met and Lys are the only dynamic residues that are exclusive to the N-terminus of httex1 and only signals from atoms located towards the ends of their side chains (i.e. Lys Cε-Hε and Met Cε-Hε) could be detected. The rest of the dynamic residues are either found throughout httex1 or are C-terminus specific where in this case also backbone resonances were observable (i.e. His Hα-Cα, Pro A+B Hα-Cα). Of the amino acid types that could not be identified in any of our spectra, three are exclusive to the N-terminus (Thr, Ser, Phe) and only one is found in the C-terminus (Gly). Taken together these data indicate that most of the dynamic residues are located at the C-terminus of httex1 and only a few side chains of the N17 peptide are dynamic enough or static enough to be detected by J-coupling based or dipolar-coupling based spectroscopy, respectively.
Figure 6.
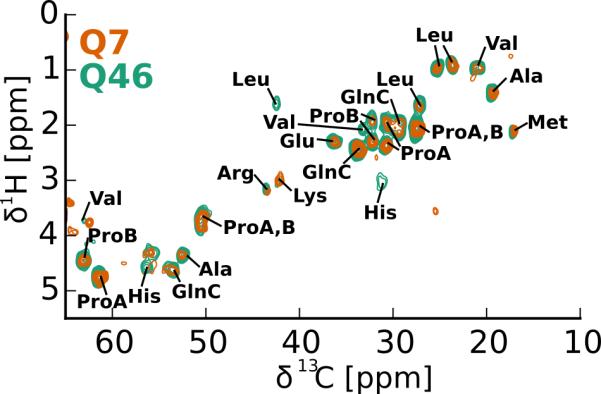
The dynamic parts of httex1 fibrils are compatible with the C-terminal proline-rich domain. Overlay of 1H-13C INEPT-HETCOR spectra of httex1 Q46 fibrils (green) and soluble httex1 Q7 with N-terminal thioredoxin tag (orange). The Q46 fibril spectrum was recorded at 14.1 T, 25°C, 25 kHz MAS in a 1.6 mm rotor. The soluble httex1 Q7 (300 μM protein) was recorded at 14.1 T, 5°C, in a 4 mm rotor spinning at 3 kHz MAS. The tentative amino acid type assignments of the httex1 fibril spectrum are indicated. Except for Met and Lys side-chain resonances, all residues identified are found in the proline-rich C-terminus of httex1. The good overlap with the spectrum of soluble httex1 Q7 indicates that in the most dynamic domains the fibrils and the soluble form are the same.
The chemical shifts of all the dynamic residues detected using J-based spectroscopy are compatible with a random coil conformation except for Gln C which has a particular set of chemical shifts different from Gln A and Gln B also not matching any of the shift patterns observed for α-helical, β-sheet, or random coil conformation.32 (See Tables S2 and S3 of the Supporting Information for all chemical shifts reported in this paper.)
Is this dynamic C-terminus a particular feature of the fibril formed by httex1 or similar in the soluble state of httex1?10,13 To answer this question, we recorded a 1H-13C INEPT-HETCOR spectrum on soluble httex1. We used httex1 that still had its N-terminal thioredoxin tag, contained a polyQ domain of only 7 residues in length, and was dissolved in 25% (v/v) glycerol to prevent aggregation. This spectrum is shown in orange in Figure 6 and overlaps well with the equivalent spectrum recorded on httex1 Q46 fibrils. No signals specific to the N-terminal thioredoxin tag (e.g. Asp or Asn) could be detected in these spectra. The notable differences between these two spectra are limited to a few but not all of the Leu, His, and Val resonances as well as the glycerol peaks that come from the solvent of httex1 Q7. The striking overlap of the two spectra indicates that the C-termini of the soluble httex1 Q7 and the httex1 Q46 fibril are conformationally related.
Discussion
Our experiments on the static domains of 4°C httex1 fibrils show that these are dominated by two different conformations of Gln (Gln A and Gln B) in a 1:1 ratio and a Pro that is compatible with a polyproline II conformation. Furthermore, signals of a third Gln (Gln C), a second Pro (Pro B) and a Glu could be detected in these spectra. Except for Gln A and Gln B all resonances detected in the static domains of our httex1 fibrils were also detected in the dynamic domains (i.e. in the CTUC-COSY and INEPT-HETCOR spectra) with the same chemical shifts. This suggests that the residues flanking the polyQ domain of h httex1 experience some dynamical heterogeneity that was observed with EPR especially at the interface of the polyQ and Pro rich domain.22
Gln A and B were also observed in fibrils formed by selectively labeled N17Q30P9K2 and on polyQ peptides of different lengths.17–19 Latter fibrils also showed a third Gln conformation. However, the Gln C observed in our spectra had different backbone chemical shifts and was the dominant Gln observed in the dynamic domains of the fibril and less dominant in the 2D DARR spectra. NMR data on selectively labeled N17Q30P9K2 samples showed that all Gln of the polyQ domain expect for the last show the splitting into Gln A and Gln B.18,19 These data, however, did not give any information about whether this splitting occurs randomly, alternatingly, or in separate domains. Our NCA and NCOCX spectra of httex1 fibrils refine this assessment by showing no evidence for Gln A-Gln B contacts in these sequential backbone walks, which indicates that the two types of Gln are either in large separated domains of one monomer or found in different monomers. These data already exclude the possibility that httex1 is forming an α-sheet structure which was suggested as a possible structure of polyQ fibril structures.36 Nevertheless by using 13C-13C 2D DARR spectra, we observed through-space couplings between Gln A and Gln B in our httex1 fibrils. These data show that, although separated in primary structure, both forms of Gln must be located within about 7 Å. The weak side chain cross peaks between Gln A and Gln B we detected in the NCOCX spectrum indicate that these contacts come from side chain rather than backbone interactions. Combined with the previous observation that all Gln in the polyQ domain can adopt both conformations, our data show that Gln A and Gln B are either located in separate monomers or inside large separate domains of the same monomer that have no fixed location in the primary structure (see Figure 7A). In the latter case, the one possible sequential Gln A and Gln B contact would give signals that would be too weak to be detected in our spectra. One possibility is that Gln A and Gln B are clustered in different β-sheets and the through-space couplings observed between Gln A and Gln B are inter-sheet contacts. Such sheet to sheet interactions could occur (1) within a monomer, (2) due to lateral association of different filaments that constitute a fibril, or (3) lateral association of fibrils in a fibril bundle. However, option (3) is unlikely since most of the fibrils shown in Figure S1 of the Supporting Information do not bundle and the 1:1 ratio observed for Gln A and B would require that there are independently formed type A and B fibrils that have exactly the same conformational free energy.
Figure 7.

(A) Model illustrating the dynamic and structural heterogeneity of the httex1 monomers found in 4°C fibrils. The N17 fragment, which was shown to be in an α-helical conformation, was difficult to detect in our spectra probably due to intermediate dynamics. The polyQ domain is static and features two types of Gln, A and B, which are separate in sequence but relatively close in space. The C-terminus has Pro residues in a polyproline II conformation and Pro and other non-Pro residues in a random coil conformation. The C-terminus is the most dynamic domain in httex1 fibrils albeit with considerable dynamic heterogeneity due to relatively static Pro residues as detected in our spectra. (B) Bottlebrush model of httex1 4°C fibrils. The N17 (orange) and polyQ (blue) domains form the center of the fibril. The Proline-rich C-termini (green) are flexible, partially disordered and pointing away from the center forming the bristles in this bottlebrush model.
But what is the origin of the different chemical shifts observed for Gln A and Gln B? One possibility could be a difference in side-chain dihedral angles as suggested by Schneider et al.17 Another possibility could be that the chemical shift difference is the result of a difference in β-sheet twist e.g. found in different filaments or on different sides of a β-sandwich.
It is interesting to note that Gln alone has a relatively low propensity to form amyloid especially when arranged in an in-register parallel β-sheet conformation.38,39 The different conformations of Gln A and Gln B might improve the complementarity of Gln in a β-sheet environment thereby compensating for its low aggregation propensity. However, further experiments are needed to determine the origins of the two Gln forms.
Gln C gave weak signals in the CP based spectra but was the dominant signal in the J-coupling based spectroscopy whereas Gln A and Gln B were not detected in the latter spectra. The chemical shift pattern of Gln C is, similarly to Gln A, not well compatible with the average chemical shift patterns observed for either α-helical, β-sheet, or random coil conformation. Due to their dynamics, we think that these Gln residues are found either at the edge of the polyQ domain or amid the 7 Gln residues that are not part of the polyQ domain but found in the proline-rich C-terminal domain between the first and second polyproline stretch and briefly after the second polyproline stretch. The origin of this particular chemical shift pattern is not clear. One possibility could be that these Gln residues are part of the C-terminal polyproline II helical structure. Another possibility could be that these Gln residues are transiently forming part of the structure formed by the polyQ domain. Both possibilities would explain the partial immobilization of these Gln residues.
The other amino acid types we detected in the dynamic domains of our httex1 fibrils using J-coupling based spectroscopy match the C-terminus more than the N17 domain of httex1. Due to the absence of Thr, Ser, and Phe resonance in any of our spectra, we conclude that the dynamic residues we detect in our J-based spectra are located at the C-terminus of httex1. Except for two side-chain resonances that are often relatively flexible, the N17 domain is not visible in our spectra, possibly due to their intermediate dynamics described by Hoop et al. that might make their detection with both our CP and J-coupling based experiments difficult.19
Our CP and J-coupling based experiments indicate that Pro is found in both the static and dynamic parts of our httex1 fibrils. While these residues are certainly not present in fibrils formed by polyQ peptides, Hoop et al. detected relatively immobile Pro in fibrils formed by N17Q30P9K2 peptides and assigned it to a polyproline II helical structure, correlating with previous circular dichroism (CD) spectroscopy data on httex1.19,22,33 Our NMR spectra, in contrast, show two different confirmations of Pro, Pro A and Pro B. Pro A, which gives intense signals in both CP and J-coupling based spectra, matches the Pro described by Hoop et al. Pro B, which has not been described previously, has a chemical shift compatible with a random coil conformation. Pro B gives only faint cross peaks in the CP-based spectra that highlight the static domains of the fibril, but gives strong cross peaks comparable in intensity to Pro B in the J-coupling based spectra, which are dominated by the dynamic domains of the fibril. This can also be seen from 1D spectra (see Figure S2 of the Supporting Information), where the Pro B:Pro A ratio is 1:2.2 using dipolar CP and 1:1.1 in the refocused INEPT spectrum. In the direct pulsed spectrum, which excites all spins independent of dynamics, the Pro B:Pro A ratio is 1:1.7 which is comparable to the ratio of Pro that has non-Pro residues as neighbors (12) to Pro that is surrounded by other Pro (18). Taken together with previous EPR and NMR observations19,22 our data suggest that the Pro A is found closer to the polyQ domain whereas Pro B is found in the more dynamic parts towards the C-terminus. Alternatively, Pro B is the preferred conformation of Pro that has non-Pro residues as neighbors. Both of these models would also explain why previous studies on N17Q30P9K2 only observed Pro A since this construct only contains the first 9 Pro residues following the polyQ domain.
Interestingly, the highly dynamic C-terminal residues are found not only in fibrils of httex1 but also in our non-fibrillar soluble preparations of httex1 as shown by the almost identical INEPT-HETCOR spectra we recorded on fibrillar httex1 Q46 and soluble httex1 Q7, which served as a reference state of non-fibrillized httex1. These data show that the dynamic, mixed polyproline II helical and random coil C-terminus is not specific to the fibrillar form of httex1 and that the most dynamic domains of httex1 are structurally not affected by fibril formation under these conditions.
We consequently propose a bottlebrush model for the structure of httex1 fibrils that is shown in Figure 7B. In this model, the static, β-sheet rich polyQ domains form the center of the brush. The N17 peptides with intermediate dynamics are appended to the polyQ domain, similar to a previous model from Williamson and co-workers.40 The proline-rich C-terminal domains form the bristles of the bottlebrush pointing to the outside of the fibril. This model is supported not only by the solid-state NMR data presented in this paper but also by previous EPR data22 as described in the following: (1) The EPR linewidths of labeled sites in the C-terminus were much narrower than those observed in the N17 and polyQ domain of httex1 indicating that the C-terminus is the most dynamic domain of the fibril. (2) Our solid-state NMR confirm that the proline-rich domain much more dynamic than the relatively static polyQ domain. (3) The EPR linewidth of the proline-rich domain decrease towards the C-terminus until they are below 2 Gauss and comparable to EPR linewidths found in soluble protein domains lacking tertiary or quaternary contacts.41,42 (4) The chemical shifts measured with solid-state NMR show that the proline-rich domain is in a polyproline II helical and random coil conformation. (5) Previous CD data22 confirm the existence of a polyproline II helix in httex1. (6) Polyproline II helices are known to be extended and act as bristles.43 For that reason, they are often used as molecular rulers. (7) These bristles could principally be facing into the fibril or they could be on the outside. Were they to face inward, the interior of the fibrils would have to have a large aqueous cavity long enough to accommodate bristles of more than 12 nm in length assuming fully extended polyproline II helices (~40 amino acids at 3.1 Å per amino acid in polyproline II helix). Our EM data show no evidence of such a cavity, whose diameter would exceed that size of most fibrils (about 10–13 nm, Figure S1). Moreover, the crowding in such a cavity would also likely lead to tertiary or quaternary contacts, which would be inconsistent with the EPR and NMR data. Thus, the polyproline region is likely to face outward. Disordered protein domains outside the fibril core are usually not detected using negatively stained EM and, thus, we cannot directly detect the presence of polyproline bristles via EM.44 We did note, however, that fibrils are often appear to have a regular spacing between them. This center-to-center spacing is about 28–33 nm, a distance range that is consistent with fibrils being held apart by polyproline bristles.
Polyproline or proline-rich domains have been previously suggested to act as entropic spacers or bristles and a recent molecular dynamics simulation of polyproline showed that these polymers can form heterogeneous conformational ensembles that feature relatively rigid extended segments that are interrupted by flexible kinks.45 This heterogeneity could explain our findings that the C-terminal Pro residues of httex1 can be found in both the static and dynamic parts of the fibril.
In summary, we described the structural and dynamic properties of fibrils formed by httex1. The polyQ domain is dominated by two different forms of Gln that are sequentially separate, but still relatively close in space. The proline-rich domain has Pro in two different conformations one being compatible with a polyproline II helix, the other with a random coil conformation. Parts of the polyproline II Pro A are relatively static, while other parts of the polyproline II Pro A are found in the dynamic domains of httex1 together with Pro B, which is compatible with a random coil conformation. Most of the other amino acids identified in the dynamic domains are compatible with the C-terminus confirming that the proline-rich domain of httex1 fibrils becomes increasingly dynamic towards the C-terminus.
Supplementary Material
Acknowledgements
We would like to thank Franziska Meier for initial work on the expression of labeled httex1, Natalie C. Kegulian for carefully proofreading the manuscript, Alexander Falk for fruitful discussions, and Erin N. Johnson for making the bottlebrush model in Figure 7.
Funding Information This work was supported by the Hereditary Disease Foundation (R.L.), startup funds by the University of Southern California (A.B.S), and by the National Institute Of Neurological Disorders and Stroke of the National Institutes of Health under Award Number R01NS084345.
Abbreviations
- htt
huntingtin
- httex1
huntingtin exon 1
- HD
Huntington's Disease
- polyQ
polyglutamine
- NMR
nuclear magnetic resonance
- EPR
electron paramagnetic resonance
- rpm
rotations per minute
- MAS
magic angle spinning
- MWCO
molecular weight cut-off
- TPPM
two pulse phase modulation
- CP
cross polarization
- DARR
dipolar assisted rotational resonance
- PDSD
proton driven spin diffusion
- DCP
double cross polarization
- HETCOR
heteronuclear correlation
- DP
direct polarization
- CTUC
constant-time uniform-sign cross-peak
- COSY
correlation spectroscopy
- INEPT
insensitive nuclei enhanced by polarization transfer
- EM
electron microscopy
- CD
circular dichroism;
Footnotes
Figure S1, S2 and Tables S3 and S4. This material is available free of charge via the Internet at http://pubs.acs.org
References
- (1).Vonsattel JP, DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- (2).Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- (3).Caron NS, Desmond CR, Xia J, Truant R. Polyglutamine domain flexibility mediates the proximity between flanking sequences in huntingtin. Proc. Natl. Acad. Sci. U. S. A. 2013;110:14610–14615. doi: 10.1073/pnas.1301342110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am. J. Hum. Genet. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- (5).Chen S, Ferrone FA, Wetzel R. Huntington's disease age-of-onset linked to polyglutamine aggregation nucleation. Proc. Natl. Acad. Sci. U. S. A. 2002;99:11884–11889. doi: 10.1073/pnas.182276099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- (7).DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- (8).Sathasivam K, Neueder A, Gipson T. a., Landles C, Benjamin AC, Bondulich MK, Smith DL, Faull RLM, Roos R. a. C., Howland D, Detloff PJ, Housman DE, Bates GP. Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. U. S. A. 2013;110:2366–2370. doi: 10.1073/pnas.1221891110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kokona B, Rosenthal ZP, Fairman R. Role of the coiled-coil structural motif in polyglutamine aggregation. Biochemistry. 2014;53:6738–6746. doi: 10.1021/bi500449a. [DOI] [PubMed] [Google Scholar]
- (10).Jayaraman M, Kodali R, Sahoo B, Thakur AK, Mayasundari A, Mishra R, Peterson CB, Wetzel R. Slow Amyloid Nucleation via α-Helix-Rich Oligomeric Intermediates in Short Polyglutamine-Containing Huntingtin Fragments. J. Mol. Biol. 2011;415:881–899. doi: 10.1016/j.jmb.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Crick SL, Ruff KM, Garai K, Frieden C, Pappu RV. Unmasking the roles of N- and C-terminal flanking sequences from exon 1 of huntingtin as modulators of polyglutamine aggregation. Proc. Natl. Acad. Sci. U. S. A. 2013;110:20075–20080. doi: 10.1073/pnas.1320626110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Darnell GD, Derryberry J, Kurutz JW, Meredith SC. Mechanism of cis-inhibition of polyQ fibrillation by polyP: PPII oligomers and the hydrophobic effect. Biophys. J. 2009;97:2295–2305. doi: 10.1016/j.bpj.2009.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Hoffner G, Djian P. Monomeric, Oligomeric and Polymeric Proteins in Huntington Disease and Other Diseases of Polyglutamine Expansion. Brain Sci. 2014;4:91–122. doi: 10.3390/brainsci4010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nekooki-Machida Y, Kurosawa M, Nukina N, Ito K, Oda T, Tanaka M. Distinct conformations of in vitro and in vivo amyloids of huntingtin-exon1 show different cytotoxicity. Proc. Natl. Acad. Sci. U. S. A. 2009;106:9679–9684. doi: 10.1073/pnas.0812083106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Pieri L, Madiona K, Bousset L, Melki R. Fibrillar α-Synuclein and Huntingtin Exon 1 Assemblies Are Toxic to the Cells. Biophys. J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wetzel R. Physical chemistry of polyglutamine: Intriguing tales of a monotonous sequence. J. Mol. Biol. 2012 Aug 24;:466–490. doi: 10.1016/j.jmb.2012.01.030. Elsevier Ltd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schneider R, Schumacher MC, Mueller H, Nand D, Klaukien V, Heise H, Riedel D, Wolf G, Behrmann E, Raunser S, Seidel R, Engelhard M, Baldus M. Structural characterization of polyglutamine fibrils by solid-state NMR spectroscopy. J. Mol. Biol. 2011;412:121–136. doi: 10.1016/j.jmb.2011.06.045. [DOI] [PubMed] [Google Scholar]
- (18).Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, Van Der Wel PCA. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J. Am. Chem. Soc. 2011;133:4558–4566. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hoop C, Lin H-K, Kar K, Hao Z, Poirier M, Wetzel R, Wel P. C. A. Van Der. Polyglutamine Amyloid Core Boundaries and Flanking Domain Dynamics in Huntingtin Fragment Fibrils Determined by Solid-State Nuclear Magnetic Resonance. Biochemistry. 2014;53:6653–6666. doi: 10.1021/bi501010q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Michalek M, Salnikov ES, Werten S, Bechinger B. Membrane interactions of the amphipathic amino terminus of huntingtin. Biochemistry. 2013;52:847–858. doi: 10.1021/bi301325q. [DOI] [PubMed] [Google Scholar]
- (21).Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–1212. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Bugg CW, Isas JM, Fischer T, Patterson PH, Langen R. Structural features and domain organization of huntingtin fibrils. J. Biol. Chem. 2012;287:31739–31746. doi: 10.1074/jbc.M112.353839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fodale V, Kegulian NC, Verani M, Cariulo C, Azzollini L, Petricca L, Daldin M, Boggio R, Padova A, Kuhn R, Pacifici R, Macdonald D, Schoenfeld RC, Park H, Isas JM, Langen R, Weiss A, Caricasole A. Polyglutamine- and temperature-dependent conformational rigidity in mutant huntingtin revealed by immunoassays and circular dichroism spectroscopy. PLoS One. 2014;9:e112262. doi: 10.1371/journal.pone.0112262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Marley J, Lu M, Bracken C. A method for efficient isotopic labeling of recombinant proteins. J Biomol NMR. 2001;20:71–75. doi: 10.1023/a:1011254402785. [DOI] [PubMed] [Google Scholar]
- (25).Takegoshi K, Nakamura S, Terao T. 13C-1H dipolar-assisted rotational resonance in magic-angle spinning NMR. Chem. Phys. Lett. 2001;344:631–637. [Google Scholar]
- (26).Baldus M, Petkova AT, Herzfeld J, Griffin RG. Cross polarization in the tilted frame: Assignment and spectral simplification in heteronuclear spin systems. Mol. Phys. 1998;95:1197–1207. [Google Scholar]
- (27).Chen L, Olsen RA, Elliott DW, Boettcher JM, Zhou DH, Rienstra CM, Mueller LJ. Constant-time through-bond 13C correlation spectroscopy for assigning protein resonances with solid-state NMR spectroscopy. J Am Chem Soc. 2006;128:9992–9993. doi: 10.1021/ja062347t. [DOI] [PubMed] [Google Scholar]
- (28).Comellas G, Lemkau LR, Zhou DH, George JM, Rienstra CM. Structural intermediates during α-synuclein fibrillogenesis on phospholipid vesicles. J. Am. Chem. Soc. 2012;134:5090–5099. doi: 10.1021/ja209019s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Harris RK, Becker ED, Menezes S. M. C. De, Granger P, Hoffman RE, Zilm KW, publication by, P. Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008): international Union of Pure and Applied Chemistry Physical and Biophysical Chemistry Division. Magn Reson Chem. 2008;46:582–598. doi: 10.1002/mrc.2225. [DOI] [PubMed] [Google Scholar]
- (30).Siemer AB, Arnold AA, Ritter C, Westfeld T, Ernst M, Riek R, Meier BH. Observation of highly flexible residues in amyloid fibrils of the HET-s prion. J. Am. Chem. Soc. 2006;128:13224–13228. doi: 10.1021/ja063639x. [DOI] [PubMed] [Google Scholar]
- (31).Lu J-X, Qiang W, Yau W-M, Schwieters CD, Meredith SC, Tycko R. Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell. 2013;154:1257–1268. doi: 10.1016/j.cell.2013.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Candel AM, Conejero-Lara F, Martinez JC, van Nuland N. a J., Bruix M. The high-resolution NMR structure of a single-chain chimeric protein mimicking a SH3-peptide complex. FEBS Lett. 2007;581:687–692. doi: 10.1016/j.febslet.2007.01.032. [DOI] [PubMed] [Google Scholar]
- (34).Sone M, Yoshimizu H, Kurosu H, Ando I. Side-chain conformation of poly(l-proline) form II in the crystalline state as studied by high-resolution solid-state 13C NMR spectroscopy. J. Mol. Struct. 1994;317:111–118. [Google Scholar]
- (35).Graf R, Spiess HW, Floudas G, Butt H-J, Gkikas M, Iatrou H. Conformational Transitions of Poly(l -proline) in Copolypeptides with Poly(γ-benzyl- l -glutamate) Induced by Packing. Macromolecules. 2012;45:9326–9332. [Google Scholar]
- (36).Babin V, Roland C, Sagui C. The α-sheet: A missing-in-action secondary structure? Proteins Struct. Funct. Bioinforma. 2011;79:937–946. doi: 10.1002/prot.22935. [DOI] [PubMed] [Google Scholar]
- (37).Gehman JD, Paulson EK, Zilm KW. The influence of internuclear spatial distribution and instrument noise on the precision of distances determined by solid state NMR of isotopically enriched proteins. J. Biomol. NMR. 2003;27:235–259. doi: 10.1023/a:1025439606001. [DOI] [PubMed] [Google Scholar]
- (38).Pawar AP, DuBay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM. Prediction of “aggregation-prone” and “aggregation- susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005;350:379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- (39).Margittai M, Langen R. Side chain-dependent stacking modulates tau filament structure. J. Biol. Chem. 2006;281:37820–37827. doi: 10.1074/jbc.M605336200. [DOI] [PubMed] [Google Scholar]
- (40).Williamson TE, Vitalis A, Crick SL, Pappu RV. Modulation of Polyglutamine Conformations and Dimer Formation by the N-Terminus of Huntingtin. J. Mol. Biol. 2010;396:1295–1309. doi: 10.1016/j.jmb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Mchaourab HS, Lietzow M. a, Hideg K, Hubbell WL. Motion of spin-labeled side chains in T4 lysozyme. Correlation with protein structure and dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- (42).Isas JM, Langen R, Haigler HT, Hubbell WL. Structure and dynamics of a helical hairpin and loop region in annexin 12: A site-directed spin labeling study. Biochemistry. 2002;41:1464–1473. doi: 10.1021/bi011856z. [DOI] [PubMed] [Google Scholar]
- (43).Ruff KM, Khan SJ, Pappu RV. A Coarse-Grained Model for Polyglutamine Aggregation Modulated by Amphipathic Flanking Sequences. Biophys. J. 2014;107:1226–1235. doi: 10.1016/j.bpj.2014.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wegmann S, Medalsy ID, Mandelkow E, Müller DJ. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. U. S. A. 2013;110:E313–21. doi: 10.1073/pnas.1212100110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Radhakrishnan A, Vitalis A, Mao AH, Steffen AT, Pappu RV. Improved atomistic Monte Carlo simulations demonstrate that poly-l-proline adopts heterogeneous ensembles of conformations of semi-rigid segments interrupted by kinks. J. Phys. Chem. B. 2012;116:6862–6871. doi: 10.1021/jp212637r. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.