

Abstract
Impaired blood-brain barrier function represents an important component of hypoxic-ischemic brain injury in the perinatal period. Proinflammatory cytokines could contribute to ischemia-related blood-brain barrier dysfunction. IL-6 increases vascular endothelial cell monolayer permeability in vitro. However, contributions of IL-6 to blood-brain barrier abnormalities have not been examined in the immature brain in vivo. We generated pharmacologic quantities of ovine-specific neutralizing anti-IL-6 mAbs and systemically infused mAbs into fetal sheep at 126 days of gestation after exposure to brain ischemia. Anti–IL-6 mAbs were measured by ELISA in fetal plasma, cerebral cortex, and cerebrospinal fluid, blood-brain barrier permeability was quantified using the blood-to-brain transfer constant in brain regions, and IL-6, tight junction proteins, and plasmalemma vesicle protein (PLVAP) were detected by Western immunoblot. Anti–IL-6 mAb infusions resulted in increases in mAb (P < 0.05) in plasma, brain parenchyma, and cerebrospinal fluid and decreases in brain IL-6 protein. Twenty-four hours after ischemia, anti–IL-6 mAb infusions attenuated ischemia-related increases in blood-brain barrier permeability and modulated tight junction and PLVAP protein expression in fetal brain. We conclude that inhibiting the effects of IL-6 protein with systemic infusions of neutralizing antibodies attenuates ischemia-related increases in blood-brain barrier permeability by inhibiting IL-6 and modulates tight junction proteins after ischemia.—Zhang, J., Sadowska, G. B., Chen, X., Park, S. Y., Kim, J.-E., Bodge, C. A., Cummings, E., Lim, Y.-P., Makeyev, O., Besio, W. G., Gaitanis, J., Banks, W. A., Stonestreet, B. S. Anti–IL-6 neutralizing antibody modulates blood-brain barrier function in the ovine fetus.
Keywords: development, ischemia, monoclonal, permeability, tight junction proteins
Hypoxic-ischemic encephalopathy is a critical neuropathophysiologic disorder originating during the prenatal and perinatal periods that can lead to serious long-term neurodevelopmental impairment or death in affected infants (1). Injury to the fetal and/or neonatal CNS can disrupt normal developmental processes in the brain and can be associated with such disorders as cerebral palsy (CP), mental retardation, and seizures, which are among the most common severe lifelong disabilities originating in childhood (2). Reduced cerebral blood flow and oxygen delivery predisposes to hypoxia-ischemia and, consequently, to hypoxic-ischemic encephalopathy, which can originate before birth (3).
The blood-brain barrier (BBB) is a selective diffusion barrier composed of endothelial cells, basement membranes, pericytes, and astrocyte end-feet processes, which separates the brain parenchyma from the systemic circulation, limits the entry of substances from the systemic circulation into brain, and maintains a critical ionic environment for normal brain function. The permeability of the BBB is regulated by 2 major routes: the paracellular pathway controlled by the tight junction complex (4–6) and transcellular pathways modulated by transporters and channels such as caveolin-1 (7, 8), plasma lemma vesicle protein (PLVAP) (9), etc. The tight junction complex is composed of tight junction proteins including the transmembrane proteins of the claudin family and occludin and the cytoplasmic plaque proteins of the zonula occludens (ZO) family (4–6).
We have recently shown that impaired BBB function represents an important component of hypoxic-ischemic brain injury in the fetus and that changes in the tight junction molecular composition are associated with increases in BBB permeability after ischemia (10). In adult subjects, the BBB is also impaired during the initial phase of hypoxic-ischemic insults, and increases in BBB permeability can predispose to the formation of brain edema (11). During the evolution of hypoxic-ischemic injury in adults, functional changes in the BBB are influenced by numerous biologic modulating factors including proinflammatory cytokines, VEGF, and nitric oxide in response to brain injury (12–15). The effects of proinflammatory cytokines such as IL-1, TNF-α, and IL-6 have received particular attention because they are often detected in high levels after severe hypoxic-ischemic brain injury and can be accompanied by increases in BBB permeability in adults (12, 13). In addition, we have recently shown that infusions of anti–IL-1β mAb attenuate ischemia-reperfusion–related increases in BBB permeability in sheep fetuses (16). However, the role of IL-6 after injury in the immature brain has been studied much less extensively than those of IL-1β and TNF-α.
IL-6 was originally identified as a B cell-stimulating factor (17). It is one of the major cytokines in the CNS secreted by microglia, astrocytes, or infiltrated leukocytes (18, 19) and plays an essential role both in brain development and injury (20, 21). IL-6 affects the CNS by stimulating the activity of the hypothalamus-adrenocortical axis, inducing fever, and promoting neuronal differentiation (22, 23). IL-6 also induces phospholipase A2 expression that in turn stimulates the production of prostanoids, leukotrienes, and platelet activating factor, all of which potentially contribute to the development of brain injury (24). Increased expression of IL-6 in the brain has been reported in neonatal rats after exposure to experimental hypoxic-ischemic injury (25, 26). IL-6 accentuates inflammatory responses and, potentially, hypoxic-ischemic brain injury in preterm and full-term infants (27, 28). Elevated IL-6 levels in cord blood, cerebrospinal fluid (CSF), or amniotic fluid consistently predicts brain injury in both full-term and preterm neonates with hypoxia-ischemia and/or inflammatory conditions, including diffuse white matter injury determined by MRI, periventricular leukomalacia, CP, and intellectual impairment (29–32). Furthermore, elevated levels of IL-6 have been identified in a variety of systemic inflammatory states that are associated with dysfunction of the endothelium through its effects on tight junction proteins (33, 34). However, the functional role of IL-6 and its influence on BBB function have not been previously studied in vivo in the immature brain.
We recently generated pharmacologic quantities of a highly selective, ovine-specific anti–IL-6 mAb and anti–IL-1β mAb. The neutralizing abilities of these mAbs have previously been confirmed in vitro in ovine splenic mononuclear cell cultures (35). Moreover, we recently demonstrated that infusions of an anti–IL-1β mAb result in the uptake of the anti–IL-1β mAb into the brain and attenuate ischemia-reperfusion–related increases in BBB permeability in ovine fetal brain using the preclinical translational fetal sheep model (16). The fetal sheep brain has many similarities to the human fetal and premature brain with regard to completion of neurogenesis, white matter development, onset of cerebral sulcation, and onset of cortical-evoked potentials (36–41). Procedures such as administration of antibodies via a clinically relevant intravenous route and quantitative kinetic studies of BBB function are not feasible in fetuses and neonates of small animals such as rodents. In addition, the development of the ovine brain at 126 days of gestation is approximately similar to that of the near-term human infant (36, 38). Therefore, we tested the hypotheses that systemic infusions of neutralizing IL-6 mAb attenuate ischemia-related abnormalities in BBB function of the ovine fetus in part by modulating tight junction protein expression at the BBB. To study this, we used a well-established model of brain ischemia in the ovine fetus (36).
MATERIALS AND METHODS
This study was conducted after approval by the Institutional Animal Care and Use Committees of Brown University and Women & Infants Hospital of Rhode Island and according to the U. S. National Institutes of Health Guidelines for use of experimental animals.
Surgical procedures and study design
Surgery was performed on 24 mixed-breed ewes from 118 to 124 days of gestation as previously described in detail (10, 16, 42–44). Ewes were administered isoflurane (1–2%) and oxygen for anesthesia. The uterus was exposed, and the fetus was operated through a hysterectomy (44). Polyvinyl catheters were placed into a fetal brachial artery for blood sampling, heart rate, and blood pressure monitoring and into a fetal brachial vein for antibody (mAb) or placebo administration. An amniotic fluid catheter was placed for the measurement of amniotic fluid pressure as a referent for the fetal arterial blood pressure. The carotid arteries were exposed and the vertebral-occipital anastomoses and lingual arteries ligated to restrict blood flow from the vertebral and noncerebral blood vessels, respectively (36). Two inflatable 4-mm vascular occluders (In Vivo Metric, Healdsburg, CA, USA) were placed around each carotid artery along with a perivascular flow probe (Transonic System, Inc., Ithaca, NY, USA) caudal to the occluders (45, 46). Two pairs of stainless steel screws (Small Parts Inc., Miami Lakes, FL, USA) were implanted onto the dura with a reference electrode sewn to the scalp to detect the electrocorticograms (ECoG) and connected to a recorder (AD Instruments, Colorado Springs, CO, USA) via insulated wires (Alpha Wire Co., Elizabeth, NJ, USA) to ensure the adequacy of the brain ischemia (36, 45, 46).
The ewes were permitted 5–8 days to recover from surgery. At the time of study, the fetal sheep were at 85% of gestation (range, 125–130 d of gestation). Full-term gestation in this breed of sheep is 145–147 days of gestation. The fetal sheep were assigned to 3 groups: instrumented sham-operated (Sham, n = 5), placebo-treated ischemic (Isch-PL, n = 9), and mouse anti-ovine IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups. After baseline determinations, brain ischemia was induced by inflating the carotid artery occluders with sterile water for 30 minutes. Anti–IL-6 mAb (9.7 ± 1.0 mg/kg) or equivalent volumes of placebo (0.154 M NaCl, 15 ml) were infused intravenously into the fetuses 15 minutes and 4 hours after brain ischemia. Two anti–IL-6 mAb infusions were given to achieve sustained elevations in mAb plasma concentrations before the onset of the BBB permeability studies occurring 24 hours after ischemia. At the termination of the BBB permeability studies, a hysterectomy was performed under intravenous pentobarbital anesthesia, the fetus was withdrawn from the uterus, and the brain was quickly removed. The fetus and ewe were then killed with pentobarbital (100–200 mg/kg). Before removal of the brain, 0.5–1 ml of CSF was obtained via a direct puncture of the allantoic membrane into the cisterna magnum of the fetus with a 23-gauge needle. CSF samples that had visible blood contamination were not saved for analysis. The fetal brain was dissected into different regions to measure regional BBB permeability as described below (43), and the remainder of the brain was dissected and frozen to measure cerebral cortical concentrations of anti–IL-6 mAb and the expressions of IL-6, tight junction proteins, and PLVAP.
Physiologic measurements
Fetal heart rate, mean arterial blood pressure, and amniotic fluid pressures were measured with pressure transducers and recorded on a PowerLab Data Acquisition System (AD Instruments) at baseline and sequentially after the end of ischemia. Carotid arterial blood flow was measured with the flow probes (Small Animal Blood Flow Meter: T206; Transonic Systems Inc.) as previously described (10). Arterial pH and blood gases were measured with a blood gas analyzer (model 248; Siemens, Washington, DC, USA) corrected to 39.5°C. Studies were performed on the fetus if the fetal arterial blood gases were within the normal range (pH > 7.30, PaO2 > 17 mmHg, and PaCO2 < 60 mmHg). Hematocrit was measured by the microhematocrit method.
The ECoG was recorded on a PowerLab Data Acquisition System (AD Instruments). The ECoG files were visually inspected by a board-certified epileptologist (J.G.) who was not aware of the group designation (10). The recorded ECoG signals were digitized (16 bits, 1000 Hz) and stored. Signal processing was performed with MATLAB (Mathworks, Natick, MA, USA) on data segments from each study period. The duration of each ECoG segment in each group averaged 15 minutes. The length of the analyzed ECoG segments did not differ among the groups. The grand average power spectral densities (PSDs) were used to determine the frequency ranges yielding the largest differences among the study periods. Preliminary examination indicated that a discriminatory frequency band of 10–100 Hz was ideal for analysis (10). Segments in this band were subdivided into 10-second nonoverlapping windows to calculate the power of the extracted records. A 10-second window was selected based on the requirements for the window to be long enough for accurate power analysis estimates and short enough to eliminate potential artifacts. A fifth-order Butterworth band-pass filter of 10–100 Hz and a 60 Hz notch filter were applied to each window. The window mean was subtracted, and the variance of the de-meaned windows was used as the window power because the power of a sample with a zero mean is equal to the sample variance (10). The power of each segment was calculated as an average power for the windows within the segment that did not contain discontinuities caused by artifacts. The power of the segments was normalized to a zero mean and unit standard deviation for each fetus to account for offset power differences among the fetuses. The baseline power for each group was subtracted from the power for the rest of the segments before analysis.
IL-6 protein and anti–IL-6 mAb production and purification
Recombinant ovine IL-6 protein encoded by the pQE30 vector was generated and purified as previously described (47) and further purified on diethylaminoethyl-convective interaction media anion-exchange monolithic resins (BIASeparations, Villach, Austria). The contaminants were bound tightly to the DEAE column, and the IL-6 proteins were collected in the unbound fraction. An additional separation of high-molecular-weight contaminants was performed on a TSKgel G3000SW Size Exclusion chromatographic column (Tosoh Bioscience, King of Prussia, PA, USA). Pure IL-6 was obtained after these 2 chromatographic procedures (35).
The anti–IL-6 mAb was produced using mouse hybridoma cells and purified from the cell culture supernatants (48, 49). Additional purification procedures included affinity chromatography on Protein G Sepharose (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). Bound antibodies were eluted using 0.1 M glycine-HCl (pH 2.3) and neutralized to pH 7.4 after elution by adding 1 M Tris buffer. The eluted antibodies were passed through an anion-exchange column (CIMmultus QA; BIASeparations, Wilmington, DE, USA) and eluted from the column by a buffer containing 200 mM NaCl to remove potential endotoxin contamination. Endotoxin concentrations were tested and confirmed to be <0.5 EU/ml. Finally, the antibody solution was concentrated on ultrafiltration devices with 30 kDa cutoff membranes and passed through a 0.2 μm syringe sterile filter (Millipore Corp., Chicago, IL, USA). The pQE30 vector and mouse hybridoma cells were generously provided by Commonwealth Scientific and Industrial Research Organization (Livestock Industries, Victoria, Australia).
Direct ELISA for IL-6 protein
A direct sandwich ELISA was developed to detect IL-6 protein concentration in fetal plasma. A mouse anti–IL-6 mAb (Millopore, Billerica, MA, USA) was used as the capture antibody, diluted 1:400 in coating buffer (0.1 M carbonate/bicarbonate, pH 9.6), and added to a 96-well ELISA plate. After overnight incubation at 4°C and washing with washing buffer [0.1% bovine albumin serum (BSA); Sigma-Aldrich, Inc., St. Louis, MO, USA], the coated wells were blocked with 2% BSA and 1% normal goat serum in PBS for 1 hour at room temperature. After washing, the purified ovine IL-6 protein was used to create a standard curve with a range from 10 to 20,000 pg/ml across the plate. Thereafter, 100 μl of diluted plasma samples (1:2 dilutions) in blocking solution was added to the remaining wells in duplicate. The plate was then incubated for 2 hours at room temperature. The binding of the antibody was detected using a signal enhancing ABC kit for rabbit IgG (Thermal Fisher Scientific) following the manufacturer’s protocol. Plates were then washed and developed using a BD OptEIA 3,3′5,5′-tetramethylbenzidine substrate solution (BD Biosciences, Franklin Lakes, NJ, USA) and read on a microplate reader (Bio-Rad Laboratories, Hercules, CA, USA) at 450 nm. The lower limit of detection was 10 pg/ml. A standard curve was plotted using a polynomial equation. The optical densities of samples were read against the standard curve to determine sample IL-6 concentration. Experiments were repeated at least 3 times and the data were accepted when the correlation coefficient (r2) of standard curve was greater than 0.99. The inter- and intra-assay coefficients of variation were 17.3% and 7.3%, respectively.
Indirect ELISA for anti–IL-6 mAb
An indirect ELISA was developed to detect the anti–IL-6 mAb in plasma, CSF, and the parietal cortex. Purified ovine IL-6 protein was diluted to 2 μg/ml in coating buffer (0.1 M carbonate/bicarbonate, pH 9.6), and coated onto a 96-well ELISA plate. After overnight incubation at 4°C and washing with a buffer (0.1% BSA in PBS with 0.05% Tween-20), the coated wells were blocked with 2% BSA in PBS for 2 hours at room temperature. A standard curve of the mouse anti–IL-6 mAb, which was the same antibody as used for the intravenous infusions into the sheep fetuses, was produced with a range from 1 to 1600 ng/ml by serial dilutions and placed in duplicate across the plate. Thereafter, 100 μl of diluted plasma (1:100 or 1:400), CSF (without dilution), or extract of the cerebral cortical samples (1:10) in blocking buffer was added to the remaining wells in duplicate. The plate was then incubated for 1.5 hours at room temperature. Thereafter, the binding of the antibody was detected using a signal enhancing ABC kit (Thermal Fisher Scientific) following the manufacturer’s protocol. Plates were then washed and developed using 3,3′5,5′-tetramethylbenzidine substrate solution (Invitrogen, Frederick, MD, USA). The lower limit of detection was 1 ng/ml.
Plates were read on a microplate reader (Model 680; Bio-Rad Laboratories) at 450 nm. A standard curve was plotted using 3-parameter logistic regression analysis (SigmaPlot; Systat Software, Inc., San Jose, CA, USA). A class-matched normal mouse IgG1 against sheep (AbD Serotec, Raleigh, NC, USA) was used as a negative control for incubating with precoated IL-6 protein in the indirect ELISA. Experiments were repeated ≥3 times, and the data were accepted when the correlation coefficient (r2) of the standard curve was >0.99. The inter- and intra-assay coefficients of variation were <10% and 15%, respectively.
Calculation of anti–IL-6 mAb concentrations in fetal brain parenchyma
Frozen brain tissue was homogenized in a lysis buffer containing 50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 0.1 mM benzethonium chloride (Sigma-Aldrich), 0.1% SDS (Bio-Rad Laboratories), 1% Igepal (Sigma-Aldrich), and 0.5% sodium deoxycholate (Sigma-Aldrich). The homogenate was incubated on ice for 30 minutes and centrifuged at 16,000 g for 30 minutes. The supernatant was aliquoted and frozen at −80°C until analysis. The quantities of anti–IL-6 mAb in the brain parenchyma were determined using previously published methods (50, 51). The brain vascular space measured by technetium-99m-labeled (Cardinal Health, East Providence, RI, USA) red blood cells (RBCs), and the hematocrit was obtained as described above and in our earlier report (10). These measurements facilitated corrections for the anti–IL-6 mAb remaining within the brain vascular space and were calculated according to the following equations (16):
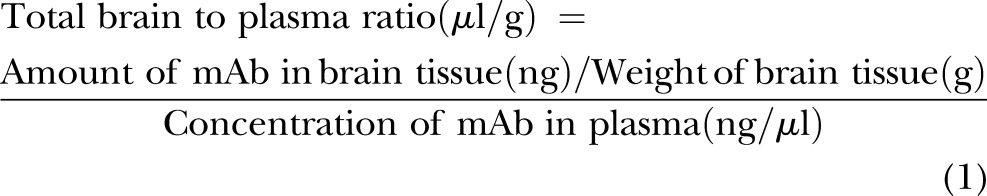 |
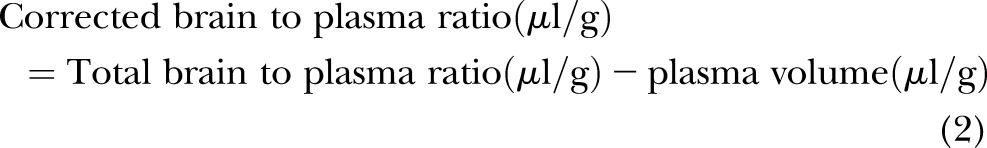 |
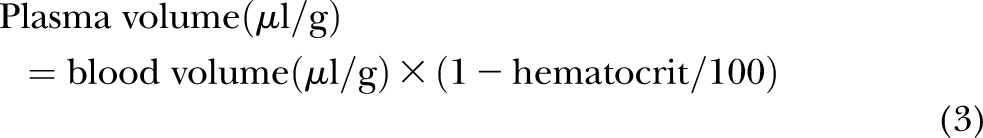 |
 |
Western immunoblot
The protein extraction procedures for the IL-6 and tight junction proteins from brain tissue have been described in detail (10, 52, 53). Briefly, brain tissue was extracted in buffer F (10 mM Tris-HCl pH 7.05, 50 mM NaCl, 30 mM sodium pyrophosphate, 50 mM NaF, 5 µM ZnCl2, 0.1 mM NaVO4, 1% Triton X-100) with 1% complete protease inhibitor cocktail (Roche, Nutley, NJ, USA) for the IL-6 protein analysis. Cell membrane fractions of the brain samples for occludin, claudin-1, claudin-5, and PLVAP protein analyses were extracted in Triton/deoxycholate/SDS (100 mM NaCl, 1% Triton X-100, 0.5 g sodium deoxycholate, 0.2% SDS, 2 mM EDTA, and 1 mM benzamidine) with 1% complete protease inhibitor cocktail (Roche). The cytosolic cell fraction for ZO-1 was extracted in urea buffer (6 M urea, 150 mM NaCl, 5 mM MgCl2, 5 mM EGTA, 10 mM Tris, pH 8.0, and 1% Triton X-100) with 1% complete protease inhibitor cocktail. Protein concentrations were determined with a bicinchoninic acid protein assay (Thermal Fisher Scientific).
The extracted proteins were fractionated by SDS-PAGE and transferred onto polyvinylidene diflouride membranes (Bio-Rad Laboratories) using a semidry technique. Membranes were probed with primary antibodies: rabbit polyclonal IL-6 antibody (Millipore Corp., Chicago, IL, USA) at a dilution of 1:1000, mouse monoclonal occludin antibody (Zymed, San Francisco, CA, USA) at a dilution of 1:2500; rabbit polyclonal claudin-1 antibody (Zymed) at a dilution of 1:6000; mouse monoclonal claudin-5 and ZO-1 antibodies (Zymed) at a dilution of 1:5000, mouse polyclonal PLVAP (Abcam, Cambridge, MA, USA) at a dilution of 1:10,000, and mouse anti-vinculin antibody (Thermo Fisher Scientific) at a dilution of 1:10,000. Peroxidase-labeled secondary antibodies were used: goat anti-mouse secondary antibodies (Zymed) at a dilution of 1:5000 for occludin, 1:10,000 for claudin-5, ZO-1, PLVAP, and vinculin, and goat anti-rabbit secondary antibody (Alpha Diagnostic, San Antonio, TX, USA) for IL-6 and claudin-1 at a dilution of 1:5000.
All experimental samples were normalized to a protein extract obtained from a homogenate pool from the cerebral cortex of 1 adult sheep. As we have previously described, these samples served as internal control protein standards (54, 55). The use of the internal control standard is unique to our laboratory, allows for comparisons among large groups of study subjects examined on multiple immunoblots, and facilitates accurate quantification of the Western immunoblots (10, 52, 54, 56). Vinculin expression was also used as a loading control to ensure that equal amounts of protein were applied to each lane. The optical densities of experimental IL-6, tight junction, and PLVAP proteins were expressed as a ratio to the internal control values, thus facilitating normalized comparisons among the different experimental groups and immunoblots. Band intensities were analyzed with a Gel-Pro Analyzer (Media Cybernetics, Silver Spring, MD, USA). All experimental samples were normalized to the average of 3 internal control samples on each immunoblot. The final values represented the ratio of the experimental to average internal control sample densitometry values obtained from ≥3 different immunoblots.
BBB permeability measurements
BBB function was measured with α-[14C]-aminoisobutyric acid (AIB, 103 Da; American Radiolabeled Chemicals, Inc., St. Louis, MO, USA) as previously described in detail (10, 16, 43, 57). Briefly, 24 hours after ischemia, α-[14C]-AIB (27.6 ± 5.7 µCi/kg, mean ± sd) was rapidly injected intravenously, and arterial plasma α-[14C]-AIB concentrations were obtained at fixed intervals over a 1-hour study period. Brain vascular volume was determined by administering technetium-99m-([99mTc]) radiolabeled RBCs (10).
Fetal brains were dissected into different brain regions (43). Plasma and tissue samples were prepared and radioactivity quantified for α-[14C]-AIB (43, 58). Knowledge of the plasma concentration profile and tracer concentrations in the parenchyma allows calculation of the blood-to-brain transfer constant (43, 59) (Ki; µl/g brain per minute) given by
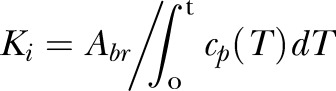 |
where Abr is the amount of tracer that crossed the BBB from blood to brain during the tracer study (dpm/g), and cp is the tracer concentration in plasma (dpm/µl) at time t (minutes). Abr is obtained by correcting the total amount of isotope measured in the tissue Am (dpm/g) for the residual part remaining in the brain vasculature space, which is measured by [99mTc]-labeled RBCs. Thus, Abr = Am − Vpcp, where Vp is the blood volume of brain tissue (µl/g) and cp is the concentration of tracer in the terminal plasma sample (dpm/g). Vp = A†m/c†p, where A†m and c†p have the same definitions as Am and cp except that they apply to the [99mTc]-labeled RBCs (10, 43, 60).
Statistical analysis
Results are expressed as means ± sd, with the exception of the ECoG, which is expressed as median ± sd. The median was used for the ECoG because the ECoG values in the placebo- and anti–IL-6 mAb-treated ischemic groups were not normally distributed. The Mann-Whitney U test was used to compare ECoG recordings from the ischemia and reperfusion study periods that were normalized to the baseline ECoG values between the groups. Two-factor ANOVA for repeated measures was used to compare Ki values for regional BBB permeability, serial measurements of physiologic variables, and the IL-6 mAb concentrations in the plasma over time within and between the groups. One-way ANOVA was used to detect differences between the 3 groups for the IL-6 mAb, IL-6 protein, tight junction, and PLVAP proteins in the brain parenchyma, and IL-6 mAb in CSF. If a significant difference was found by ANOVA, the Fisher least significant difference test was used to detect specific differences between the study groups and brain regions. A value of P < 0.05 was considered statistically significant.
RESULTS
Physiologic measures
Significant differences in gestational ages, brain weight, or body weight were not observed among the study groups (Table 1). Likewise, fetal arterial pH, blood gases, heart rate, arterial blood pressure, hematocrit, and lactate values also did not differ between the study groups at baseline or after ischemia. However, the base excess was lower at some study measurement periods after ischemia in the placebo- and anti–IL-6 mAb-treated ischemic groups compared with the sham control group (ANOVA main effects for groups, F = 3.75, P < 0.05; Table 2).
TABLE 1.
Days of gestation, body, and brain weights of the ovine fetuses
| Group | Sham | Isch-PL | Isch-IL-6 mAb |
|---|---|---|---|
| Gestation (d) | 127 ± 2 | 126 ± 1 | 126 ± 2 |
| Brain weight (g) | 42.7 ± 9.6 | 38.0 ± 8.4 | 38.4 ± 6.1 |
| Body weight (kg) | 3.28 ± 0.80 | 2.94 ± 0.34 | 3.29 ± 0.56 |
Values are mean ± sd; n = number of animals in the sham (n = 5), placebo-treated ishemic (Isch-PL, n = 9), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups. The gestational ages, brain weights, and body weights did not differ among the study groups.
TABLE 2.
Fetal arterial pH, blood gases, base excess, heart rate, mean arterial blood pressure, hematocrit, and lactate values by the study group
| Variable | Group | Time after ischemia |
|||||
|---|---|---|---|---|---|---|---|
| Baseline | 15 min | 1 h | 2 h | 4 h | 24 h | ||
| pH | Sham | 7.36 ± 0.06 | 7.35 ± 0.03 | 7.36 ± 0.02 | 7.34 ± 0.04 | 7.36 ± 0.02 | 7.37 ± 0.03 |
| Isch-PL | 7.34 ± 0.03 | 7.33 ± 0.03 | 7.33 ± 0.03 | 7.34 ± 0.03 | 7.34 ± 0.03 | 7.36 ± 0.05 | |
| Isch-anti–IL-6 mAb | 7.35 ± 0.03 | 7.34 ± 0.03 | 7.33 ± 0.03 | 7.32 ± 0.04 | 7.31 ± 0.05 | 7.35 ± 0.03 | |
| Arterial PO2 (mmHg) | Sham | 23 ± 2 | 23 ± 3 | 23 ± 2 | 23 ± 4 | 23 ± 2 | 24 ± 1 |
| Isch-PL | 21 ± 3 | 21 ± 3 | 21 ± 3 | 21 ± 4 | 21 ± 4 | 21 ± 2 | |
| Isch-anti–IL-6 mAb | 22 ± 2 | 22 ± 2 | 23 ± 3 | 21 ± 2 | 21 ± 2 | 21 ± 4 | |
| Arterial PCO2 (mmHg) | Sham | 54 ± 3 | 54 ± 3 | 56 ± 2 | 56 ± 2 | 55 ± 4 | 54 ± 2 |
| Isch-PL | 51 ± 4 | 51 ± 5 | 52 ± 6 | 51 ± 6 | 53 ± 4 | 48 ± 6 | |
| Isch-anti–IL-6 mAb | 53 ± 4 | 53 ± 3 | 53 ± 4 | 54 ± 4 | 54 ± 4 | 51 ± 3 | |
| Arterial base excess (mEq/L) | Sham | 3.9 ± 4.1 | 4.2 ± 2.6 | 4.1 ± 2.1 | 2.9 ± 3.4 | 3.7 ± 3.2 | 4.2 ± 2.9 |
| Isch-PL | 0.3 ± 3.7 | −0.2 ± 2.7* | 0.1 ± 3.3* | 0.4 ± 2.1 | 1.4 ± 2.5 | 0.2 ± 3.6* | |
| Isch-anti–IL-6 mAb | 2.1 ± 2.1 | 1.2 ± 2.1* | 1.0 ± 1.4* | 0.5 ± 2.5 | −0.6 ± 3.2* | 1.5 ± 1.7 | |
| Heart rate (beats/min) | Sham | 155 ± 17 | 151 ± 17 | 155 ± 14 | 170 ± 19 | 163 ± 7 | 162 ± 22 |
| Isch-PL | 166 ± 19 | 165 ± 22 | 165 ± 24 | 166 ± 14 | 169 ± 17 | 174 ± 11 | |
| Isch-anti–IL-6 mAb | 166 ± 8 | 164 ± 9 | 166 ± 18 | 162 ± 16 | 178 ± 15 | 187 ± 28 | |
| Mean arterial blood pressure (mmHg) | Sham | 44 ± 3 | 45 ± 6 | 43 ± 3 | 44 ± 5 | 45 ± 5 | 47 ± 7 |
| Isch-PL | 48 ± 8 | 48 ± 8 | 45 ± 9 | 47 ± 9 | 47 ± 8 | 45 ± 6 | |
| Isch-anti–IL-6 mAb | 43 ± 9 | 44 ± 7 | 47 ± 5 | 48 ± 6 | 45 ± 8 | 47 ± 9 | |
| Hematocrit (%) | Sham | 33 ± 4 | ND | ND | ND | ND | 31 ± 3 |
| Isch-PL | 37 ± 4 | ND | ND | ND | ND | 35 ± 4 | |
| Isch-anti–IL-6 mAb | 36 ± 4 | ND | ND | ND | ND | 32 ± 3 | |
| Lactate (mg/dl) | Sham | 15.9 ± 3.3 | ND | ND | ND | ND | 13.9 ± 1.7 |
| Isch-PL | 15.7 ± 3.5 | ND | ND | ND | ND | 14.7 ± 2.0 | |
| Isch-anti–IL-6 mAb | 12.9 ± 2.4 | ND | ND | ND | ND | 15.0 ± 8.7 | |
Values are mean ± sd. Sham control fetuses (Sham, n = 5); placebo-treated fetuses (Isch-PL, n = 9) anti–IL-6 mAb-treated fetuses (Isch-IL-6 mAb, n = 10). Arterial PO2 and PCO2 are oxygen and carbon dioxide pressures, respectively. ND, not determined.
P < 0.05 vs. sham control.
Carotid blood flow and ECoG
Carotid blood flow was measured during the study periods to ensure that the blood flow was reduced during ischemia. The carotid blood flow among the 3 study groups was similar at baseline, with an average value of 116 ± 27 ml/min for the 3 groups. The blood flow values approached zero during ischemia in the placebo- and anti–IL-6 mAb-treated ischemic groups and increased to baseline levels during reperfusion (Fig. 1A). Differences in carotid blood flow were not observed between the placebo- and anti–IL-6 mAb-treated ischemic groups. Ischemia was also associated with attenuation in fetal (PSD)-ECoG signals during ischemia in the placebo- and anti–IL-6 mAb-treated ischemic groups (Fig. 1B). Furthermore, (PSD)-ECoG signals did not differ between the placebo- and anti–IL-6 mAb-treated ischemic groups during or after ischemia (Fig. 1B).
Figure 1.
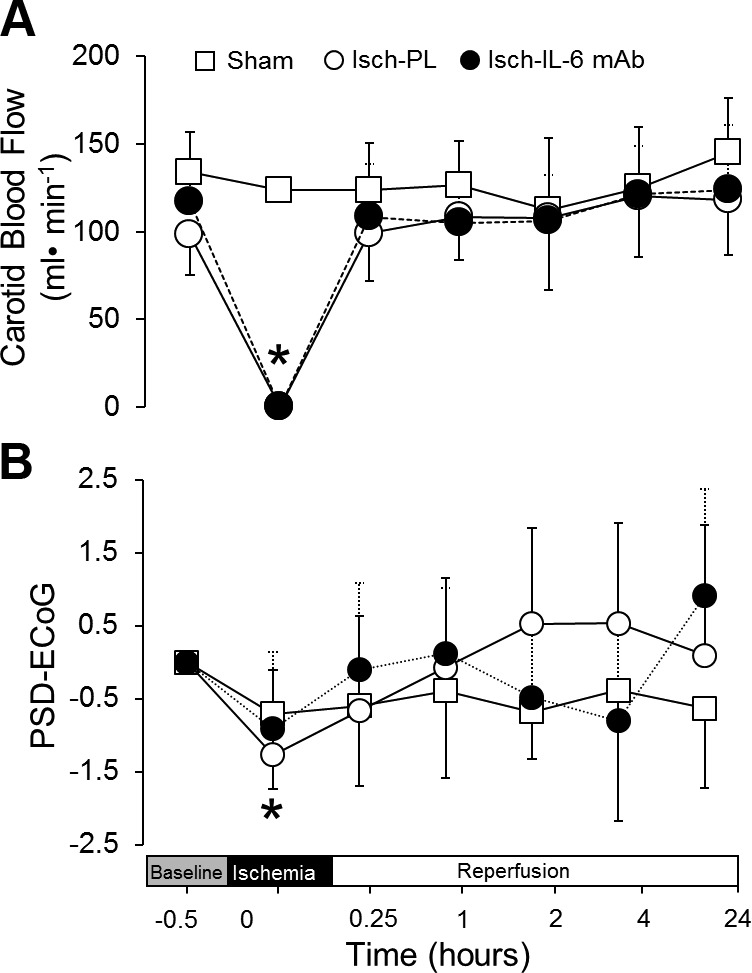
Carotid blood flow and ECoG values before ischemia, during ischemia, and reperfusion. Carotid blood flow (A) in the sham (n = 4), placebo-treated ischemic (Isch-PL, n = 9), and anti-IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups plotted against study time in hours. Power spectral densities, PSD-ECoG (B), for the sham (n = 4), Isch-PL (n = 7), and Isch-IL-6 mAb (n = 9) was plotted as the difference from the individually averaged baseline ECoG values. N = 4 in the sham group because 1 recording was not technically adequate. Values are means ± sd for carotid blood flow, and medians ± sd for ECoG. *P < 0.05 vs. baseline within the ischemic groups (Isch-PL and Isch-IL-6 mAb).
IL-6 protein in the plasma
Sequential levels of IL-6 cytokine were determined in plasma of sham control, placebo-, and anti–IL-6 mAb-treated ischemic groups using a direct sandwich ELISA adapted in our laboratory for sheep plasma. The levels of plasma IL-6 did not differ from baseline values during the study periods in sham control fetuses with an average concentration of 97.8 ± 142.1 pg/ml. Brain ischemia resulted in significant increases in plasma IL-6 concentrations in the placebo-treated ischemic group beginning at 1 h (1850.6 ± 1017.4 pg/ml, mean ± sd) and remained consistently elevated up to 4 hours (1689.8 ± 1901.0 pg/ml, mean ± sd) after the end of ischemia. However, plasma IL-6 concentrations returned to values similar to baseline by 24 h after ischemia. A similar pattern of plasma IL-6 concentrations was also observed in the anti–IL-6 mAb-treated ischemic fetuses. Although major differences in the plasma IL-6 concentrations were not observed between placebo- and mAb-treated-ischemic groups, IL-6 plasma concentrations were higher in the placebo- than the mAb-treated ischemic group 1 hour after the onset of ischemia (Fig. 2).
Figure 2.
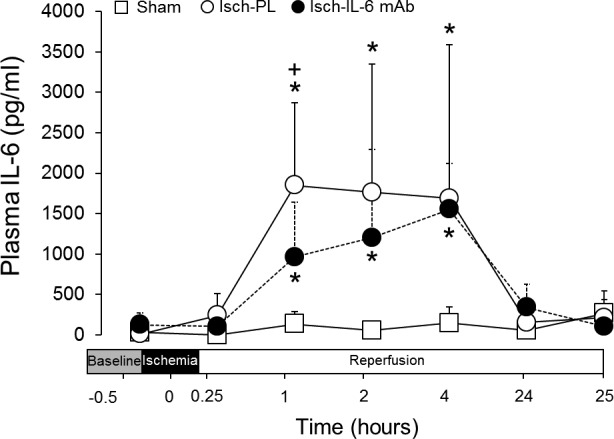
Concentration of IL-6 protein in fetal plasma of sham (n = 5), placebo-treated ischemic (Isch-PL, n = 9), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups plotted against study time in hours. Values are means ± sd. *P < 0.01 vs. the sham group at the same time point; +P < 0.05 vs. Isch-IL-6 mAb group at the same time point.
Anti–IL-6 mAb concentrations in the plasma, brain, and CSF
Concentrations of anti–IL-6 mAb in plasma, brain parenchyma, and CSF were determined by an indirect ELISA established in our laboratory using the recombinant ovine IL-6 as the coating protein, and the identical anti–IL-6 mAb that was infused into the fetal sheep was used to create the standard curve. Plasma anti–IL-6 mAb concentrations increased significantly within 45 minutes after the onset of mAb infusions and increased continuously up to 25 hours after ischemia. The highest concentration (87 ± 48 µg/ml) was detected 25 hours after ischemia (Fig. 3A). Most importantly, intravenous infusions of anti–IL-6 mAb resulted in significant accumulation of the anti–IL-6 mAb within in the brain parenchyma (1.20 ± 0.73 µg/g brain; Fig. 3B) and CSF (0.46 ± 0.10 µg/ml) at the end of the study (Fig. 3C). A representative immunoblot of ovine IL-6 protein specifically detected with the identical anti–IL-6 antibody that was infused into the fetal sheep is contained in Fig. 3D. The single band indicates the specificity of anti–IL-6 antibody produced by mouse hybridoma cells and validates the use of ovine IL-6 protein in this assay and the use of anti–IL-6 antibody for the standard curve.
Figure 3.

Concentrations of anti–IL-6 mAb in fetal plasma (A) in placebo-treated ischemic (Isch-PL, n = 7) and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups. Plasma anti-IL6 mAb plotted against study time in hours at the initiation of ischemia. Arrows indicate the timing of the placebo or anti–IL-6 mAb infusions. Concentrations of anti–IL-6 mAb in brain parenchyma (B) in the sham (n = 5), placebo-treated ischemic (Isch-PL, n = 4), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 6) groups, and concentrations of anti–IL-6 mAb in CSF (C) in the sham (n = 3), placebo-treated ischemic (Isch-PL, n = 4), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 8) groups. Values are means ± sd. A) *P < 0.01 vs. baseline value at zero hours for plasma concentration of the anti–IL-6 mAb. B, C) *P < 0.01. D) Western immunoblot shows the purity of the recombinant ovine IL-6 protein used as the coating protein for the indirect ELISA.
IL-6 protein in the expression in the brain
The expression of IL-6 protein was detected by Western immunoblot in the brain parenchyma. IL-6 protein bands were detected in cerebral cortical extracts from ovine fetuses in the sham, placebo-, and anti–IL-6 mAb-treated ischemic groups. Although ischemia and reperfusion for 24 hours was not associated with significant increases in IL-6 protein expression, treatment with anti–IL-6 mAb significantly decreased the expression of IL-6 protein in the brain parenchyma compared with the placebo-treated ischemic group (Fig. 4).
Figure 4.
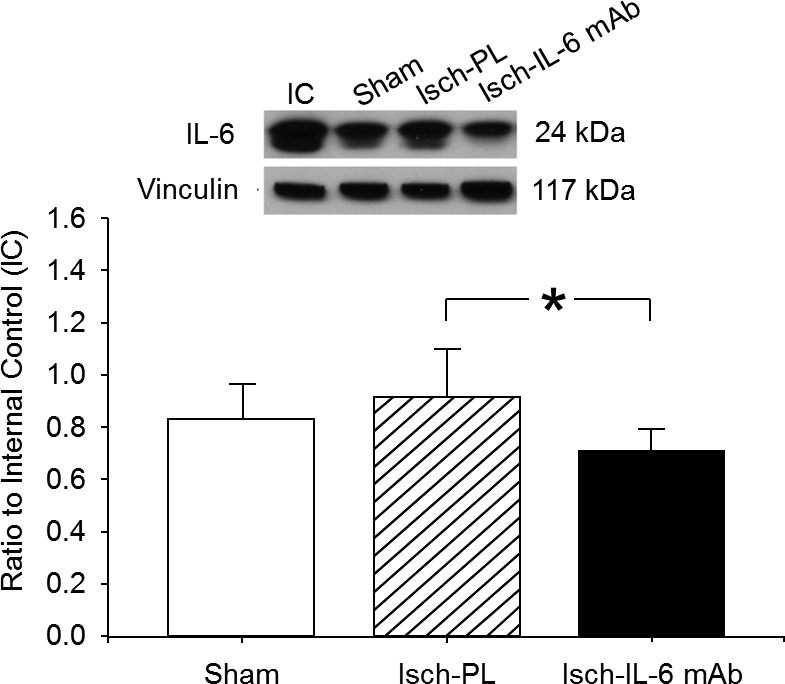
IL-6 expression level in the cerebral cortex of the sham (n = 5), placebo-treated ischemic (Isch-PL, n = 7), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 9) groups. IC indicates the internal control standard protein derived from cerebral cortex from an adult sheep. The immunoblot was selected for illustration that most closely represented the mean value of data illustrated in the bar graphs. Vinculin shown as loading control. Values are the densitometry ratios of the experimental IL-6 to the IC protein. Values are means ± sd, *P < 0.01.
Ki measurements of BBB permeability
The Ki was measured in 11 brain regions with radiolabeled AIB in the sham control, placebo-, and anti–IL-6 antibody-treated fetal sheep (43). Ki values differed across brain regions among the 3 groups (ANOVA: main effects for groups across brain regions, F = 4.51, P < 0.02; Fig. 5). The Ki values were higher across the brain regions of the placebo-treated ischemic than the sham control fetal sheep (ANOVA: main effects for group across brain regions, F = 9.00, P < 0.02) and were higher across the regions in the placebo-treated than in the anti–IL-6 mAb-treated ischemic fetal sheep (ANOVA: main effects for group across brain regions, F = 7.39, P < 0.02), but did not differ between the anti–IL-6 mAb-treated ischemic and sham control fetal sheep (ANOVA: main effects for group across brain regions, F = 0.13, P = 0.72).
Figure 5.

Ki in the sham (n = 5), placebo-treated ischemic (Isch-PL, n = 9), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups plotted for 11 brain regions. Values are means ± sd. *P < 0.05 vs. Sham; +P < 0.05 vs. Isch-PL.
Ki values also differed across cerebral cortical regions (frontal, parietal, and occipital cortices) among the 3 groups (ANOVA: main effects for groups across cerebral cortical regions, F = 3.96, P < 0.05; Fig. 6). Although Ki values were higher across the cerebral cortical regions of the placebo- than the anti–IL-6 mAb-treated ischemic fetal sheep (ANOVA: main effects for group across brain regions, F = 5.19, P < 0.04), differences were not detected across the cortical regions between the sham and placebo-treated ischemic fetal sheep (ANOVA: main effects for group across brain regions, F = 2.56, P = 0.11). Ki values also did not differ across the cortical regions between the anti–IL-6 mAb-treated ischemic and sham fetal sheep (ANOVA: main effects for group across brain regions, F = 0.09, P = 0.77).
Figure 6.

Ki in the sham (n = 5), placebo-treated ischemic (Isch-PL, n = 9), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 10) groups measured in the frontal, parietal, and occipital cerebral cortex. Values are means ± sd. *P < 0.05 vs. Isch-PL; +P < 0.05 vs. Sham.
Tight junction protein expression
To determine the effects of the anti–IL-6 mAb treatment on the tight junction proteins, Western immunoblot was used to detect the expression of 4 key constituent proteins of the endothelial tight junctions in the study groups as shown in Fig. 7. ZO-1 is a cytosolic component of the tight junction complex, and occludin, claudin-1, and claudin-5 are transmembrane components of tight junctions. ZO-1 was significantly lower in the anti–IL-6 mAb-treated group compared with both the sham control and the placebo-treated ischemic groups in the cerebral cortex, cerebellum, and medulla (Fig. 7A). In contrast, claudin-1 expression was significantly higher in the medulla of the anti–IL-6 mAb-treated-ischemic group compared with both the sham and the placebo-treated ischemic groups (Fig. 7B). However, the expression patterns of claudin-5 differed from those of claudin-1. Claudin-5 expression was significantly lower in the caudate nucleus and medulla of the anti–IL-6 mAb-treated ischemic group compared with the sham control and placebo-treated ischemic groups (Fig. 7C). On the other hand, the expression of occludin was significantly higher in the cerebral cortex, caudate nucleus, and medulla of the anti–IL-6 mAb-treated ischemic group than the sham and placebo-treated ischemic groups (Fig. 7D).
Figure 7.

Tight junction protein expression in the cerebral cortex, caudate nucleus, cerebellum, and medulla of sham (n = 5), placebo-treated ischemic (Isch-PL, n = 6), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 7) groups. Representative Western immunoblot shown for each tight junction protein; IC indicates the internal control standard protein derived from the cerebral cortex from an adult sheep. The immunoblot that most closely represented the mean value of the group mean value was selected for illustration above the bar graph. Values indicate the ratio of the densitometry values of IL-6 to the IC protein expression; values are means ± sd. *P < 0.05 vs. Sham, +P < 0.05 vs. Isch-PL.
PLVAP protein expression
To measure the effects of the anti–IL-6 mAb treatment on proteins regulating transcellular BBB function, we measured the protein expression of PLVAP in the cerebral cortex of the fetal sheep using Western immunoblot. PLVAP protein expression was not significantly (P = 0.46) increased after ischemia in the placebo-treated group (Fig. 8). In contrast, PLVAP was significantly (P < 0.01) higher in the anti–IL-6 mAb-treated ischemic group compared with the sham control and placebo-treated ischemic groups.
Figure 8.
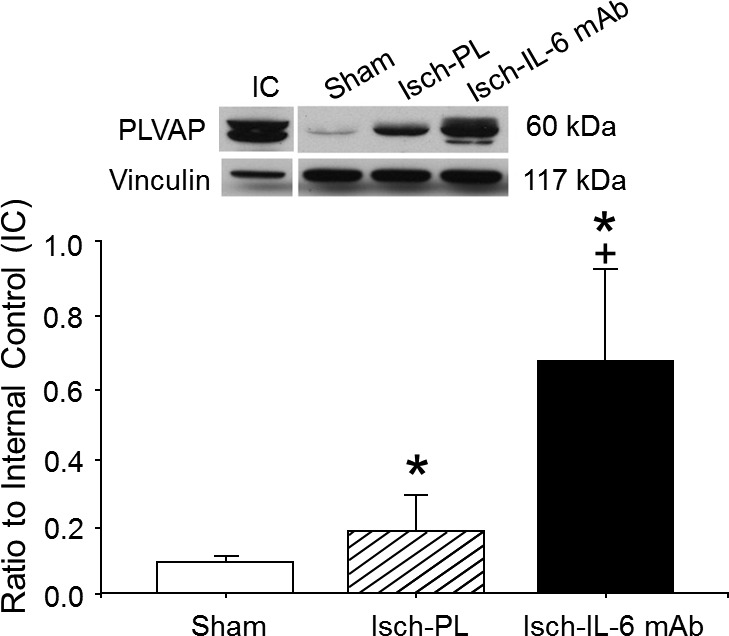
PLVAP expression in the cerebral cortex of the sham (n = 4), placebo-treated ischemic (Isch-PL, n = 5), and anti–IL-6 mAb-treated ischemic (Isch-IL-6 mAb, n = 7) groups. Representative Western immunoblot shown for PLVAP expression; IC indicates the internal control standard protein derived from the cerebral cortex from an adult sheep. The immunoblot that most closely represented the mean value of the group mean value was selected for illustration above the bar graph. Values indicate the ratio of the densitometry values of PLVAP to the IC protein expression; values are means ± sd. *P < 0.01 vs. Sham; +P < 0.01 vs. Isch-PL.
DISCUSSION
The main objective of the present study was to determine the ability of systemic intravenous infusions of neutralizing anti–IL-6 mAb to attenuate ischemia-related increases in BBB permeability in the ovine fetus. There are 3 novel findings in our study. First, systemic anti–IL-6 mAb infusions resulted in considerable mAb accumulation within the fetal brain parenchyma and CSF after ischemia. Second, anti–IL-6 mAb exerted inhibitory effects on IL-6 expression within the fetal brain. Third, the selective anti–IL-6 mAb exerts protective effects on BBB function after ischemia in the fetal brain.
We confirmed similarity of ischemic insults in the placebo- and anti–IL-6 antibody-treated groups by comparable decreases in carotid arterial blood flow (Fig. 1A) and attenuations in the ECoG power (Fig. 1B) (36, 45). In addition, anti–IL-6 mAb infusions did not appear to have major effects on changes in the carotid arterial blood flow, ECoG, or physiologic variables (Fig. 1; Tables 1 and 2), suggesting that the mAb infusions did not affect physiologic, biochemical, metabolic, or hemodynamic homeostasis in the fetuses.
Significant increases in systemic plasma IL-6 concentrations were detected within 1 h after brain ischemia and persisted up to 4 hours after ischemia (Fig. 2). Concentrations of plasma IL-6 returned to values similar to baseline by 24 hours after ischemia, suggesting that the increased plasma IL-6 concentrations were confined to the acute phase of the hypoxic-ischemic brain injury in the fetus. In addition, plasma concentrations of the IL-6 cytokine did not appear to be affected by infusions of anti–IL-6 mAb, even though the ratio of plasma antibody to cytokine concentration reached ∼3000 to 1 (Figs. 2 and 3A). Consistent with our findings, systemic IL-6 concentrations are elevated acutely in adult patients after traumatic brain injury (61–64). In addition, systemic elevations in IL-6 correlate with elevations in matrix metallopeptidase-9 (MMP-9) after traumatic brain injury, suggesting that the elevations in IL-6 could also correlate with damage to vasculature of the neurovascular unit because MMP-9 is an integral component of the BBB basement membrane (5, 64). The increased plasma IL-6 can be produced from the cerebral endothelial cells in injured brain regions or through the activated peripheral immune cells such as neutrophils, monocytes, or T cells. However, our findings of elevations in plasma IL-6 after ischemic brain injury differ from previous findings showing no elevations in IL-6 after umbilical cord occlusion in fetal sheep regardless of the severity of cord occlusion (65, 66). The differences between our findings and the prior work (65, 66) are most likely related to differences in the animal models. Furthermore, our finding showing no major differences between IL-6 plasma concentrations in the placebo-treated and mAb-treated ischemic groups suggests that even though the activity of IL-6 was most likely neutralized by high systemic mAb concentrations, the antigen-antibody complex could still be detected by our ELISA assay.
In the present study, we administered 2 doses of ∼10 mg/kg of anti–IL-6 mAb based on previous work using therapeutic antibodies to treat brain ischemia in adult rats (67) and adjusted the dose for the fetus by accounting for the larger fetal blood volume and potential sequestration of mAb by the placenta (68). The objective of the 2 infusions was to produce rapid and sustained increases in circulating mAb to facilitate continued mAb exposure to the cerebral vasculature. The mAb infusions induced continuous increases in the plasma mAb with a final concentration of 87 µg/ml by 25 hours after ischemia. The total amount of anti–IL-6 mAb remaining in fetal circulation was ∼26 mg. Based on the total plasma volume of the sheep fetus at this age (92 ± 3 ml/kg), 26 mg represents ∼40% of the total mAb administered to the fetus during the 2 infusions (69). These findings suggest this anti–IL-6 mAb could have had a longer half-life >24 hours in the fetal circulation. In addition, although we selected a relatively early treatment time after ischemia (15 minutes) as a proof of principle in our study, infants experiencing hypoxic-ischemic brain injury usually cannot receive immediate treatment (70, 71). Therefore, future work is required to establish the precise half-life of the anti–IL-6 mAb in the sheep fetus and to determine the efficacy of delayed treatment.
Our current findings deserve comparison with our recent report examining the effects of an ovine specific anti–IL-1β mAb on BBB function in the fetus (16). The peak concentration that we achieved in the fetal plasma after infusion of the anti–IL-6 mAb is 30–40 times higher than we had achieved with the anti–IL-1β mAb infusions, although the dose of the anti–IL-6 was only twice that of the anti–IL-1β mAb dose given to the same gestational aged fetal sheep (16). Therefore, the large differences between the achieved plasma concentrations of the ovine-specific anti–IL-6 and anti–IL-β mAbs suggest that there could be differences in the volumes of distributions between the 2 mAbs and/or difference the rate of metabolism of the mAbs. However, direct comparisons of the 2 antibodies await further investigation.
Anti–IL-6 mAb infusions also resulted in significant accumulation of mAb in the fetal brain parenchyma (Fig. 3B) and CSF (Fig. 3C). Likewise, similar to the differences discussed above regarding the plasma concentrations of mAb after the anti–IL-6 and anti–IL-1β mAb infusions (16), the brain uptake of the anti–IL-6 mAb was higher than that of the anti–IL-1β mAb (16). This could have resulted from the higher plasma concentrations of the anti–IL-6 mAb compared with the concentrations of anti–IL-1β mAb and/or better penetration of the anti–IL-6 mAb than anti–IL-1β mAb. Although previous studies have suggested that antibodies do not cross the intact BBB because of their large size and lack of specific transporters, recent evidence suggests that therapeutic antibodies that cross the BBB, even at very low rates, could have efficacy for the treatment of CNS diseases. For example, in adult mice with systemic inflammation, intraperitoneal injections of anti–IFN-α antibodies have been shown to cross the BBB and inhibit IFN-α (72). In addition, a recent study has shown that the anti–small-molecule inhibitor of the β-secretase antibody reduces CNS amyloid-β (Aβ) concentrations in mice and monkeys, consistent with a measurable uptake of antibody across the BBB (73). Our findings are also consistent with work showing that intravenous anti-Aβ protein antibodies can be taken up by the brain in adult rats with experimental Alzheimer’s disease (50) and that even when small quantities of antibodies are taken up by brain via an extracellular route, they can reverse the cognitive impairments of Alzheimer’s disease in mice (51). Hence, consistent with our findings, it appears to be possible to target specific molecules of interest within the CNS via peripheral antibody administration particularly after brain injury.
Our current findings are also consistent with a recent report showing that autism in young children may be associated with high levels of maternal anti–brain-reactive antibodies (74) and that injections of autism-linked maternal IgG antibodies in utero result in abnormal brain growth and behavior in monkeys after birth (75). These findings suggest that maternal antibodies may cross the fetal BBB to influence brain development (74, 75). The mechanisms by which mAbs enter the fetal brain include the possibilities that mAbs may cross the immature developing BBB (43) at low rates, cross the fetal BBB because of ischemic damage (10), and/or enter the brain via extracellular pathways (50). In addition to the above possibilities, receptor-mediated endocytosis or transcytosis could play a role in drug delivery to the brain, particularly for macromolecules such as immunoglobulins. These transport mechanisms could be facilitated by transport via Fc receptors. Although previous work suggests that Fc receptors can mediate the efflux of IgG molecules from the brain side of the vasculature into blood (76, 77), in vitro studies imply that Fc receptor can actually mediate the endocytosis and transcytosis of IgG across the endothelial barrier of blood vessels in both directions (78). Therefore, Fc receptor mediated transport could also potentially facilitate influx of mAb into brain particularly when blood concentrations of IgG are high. However, we cannot comment on the potential role of Fc receptor-mediated mAb transport in our studies because we do not have isolated vascular endothelium from our fetal sheep with which to measure Fc receptors and/or Fc receptor mediated transport. Nevertheless, our study demonstrates substantial anti–IL-6 mAb uptake into brain parenchyma suggesting that this specific mAb enters the brain and therefore can inhibit ischemia related inflammation.
The BBB restricts the passage of proteins and small lipid insoluble molecules between systemic circulation and the brain extracellular fluid space from relatively early in development (43, 79). Previous work has shown that hypoxic insults lead to increased paracellular permeability (80, 81), resulting in vasogenic edema both in adult and neonatal subjects (82, 83). Hypoxic-ischemic BBB dysfunction has been thought to be mediated by several factors including increased nitric oxide production (84, 85), calcium influx (86), and release of inflammatory cytokines either from the brain parenchyma and/or the vasculature (79, 87). Our previous work using the same methodology established significant increases in regional BBB permeability 24 hours after ischemia in the sheep fetus (10). We also have previously reported heterogeneity in regional BBB permeability under a variety of physiologic and pathophysiologic conditions in the fetus (10, 43, 57, 88, 89). Consistent with our previous findings, the regional Ki values were increased 24 hours after brain ischemia compared with sham-operated animals and exhibited regional heterogeneity within the sham control, placebo-treated ischemic, and anti–IL-6 mAb-treated ischemic groups (Fig. 5). In addition, these findings are also consistent with our previous in vitro work showing that IL-6 decreases tight junction protein expression in the cerebral microvasculature of sheep and that neutralization of IL-6 attenuates reductions in claudin-5 expression (34). Furthermore, our findings that systemic treatment with anti–IL-6 mAb protects the fetal BBB from ischemia-induced barrier dysfunction suggest that IL-6 plays a critical role in impaired BBB function after ischemic brain injury in vivo in the fetus.
Ischemia-related brain injury can stimulate neuronal, astrocytic, and microglial IL-6 secretion that can damage the neurovascular unit (90). It has been shown that hypoxia-ischemia increases the expression of IL-6 in the brains of neonatal rats (25) with early peaks of IL-6 expression between 3 and 6 h after the onset of injury (26). Increased CSF levels of IL-6 have been also reported in preterm infants who exhibited MRI-defined cerebral white matter injury (91) and in full-term infants after perinatal asphyxia (14). Increased IL-6 concentrations in amniotic fluid and cord blood have been reported in premature infants who later developed CP and also have been detected within white matter lesions of infants who died with periventricular leukomalacia (30, 31). In human infants with hypoxic-ischemic encephalopathy, elevated IL-6 levels in umbilical cord blood have been consistently associated with severity of hypoxic-ischemic encephalopathy and neurodevelopmental outcomes (92). However, in the current study, we did not detect a significant increase in cerebral cortical IL-6 protein expression 24 hours after ischemia (Fig. 4) or 48 or 72 hours after ischemia in our former work (52). Nonetheless, anti–IL-6 mAb infusions significantly decreased the expression of IL-6 compared with the placebo-treated ischemic group, suggesting that anti–IL-6 mAb penetration into the brain (Fig. 2B) may result in a relative decrease in IL-6 expression, potentially down-regulating the cytokine expression within the brain parenchyma. Therefore, anti–IL-6 mAb infusions could have protected the BBB in part by decreasing the overall amount of IL-6 present within the brain parenchyma and thereby neutralizing the proinflammatory effects of IL-6 (93).
On the other hand, the BBB could be also impaired by circulating or local vascular proinflammatory cytokines, which damage the endothelial cells of the BBB (94). IL-6 has been shown to damage the BBB in several in vitro models. Maruo et al. has shown that treatment with recombinant IL-6 in bovine endothelial vascular cells significantly increased albumin penetration through the endothelial cell layer and that anti–IL-6 antibody completely abolished these effects (93). Desai et al. confirmed that IL-6 increased endothelial permeability in human umbilical vein endothelial cells in a dose- and time-dependent manner via the protein kinase C pathway (33). Consistent with this possibility, we have previously shown in vitro that IL-6 reduces occludin and claudin-5 expression in microvessels from sheep brain (34). Therefore, it remains possible that systemic infusions of anti–IL-6 mAb could have attenuated ischemia-related increases in BBB permeability (Fig. 5) by reducing ischemia-induced increases in endothelial derived cytokines and/or the activity of systemic cytokines (95).
There are 2 major routes for the transport of substrates across the BBB. These routes include paracellular and transcellular pathways. Thus far, most experimental studies including our own studies (10, 16) have focused on pathophysiologic changes as related to the paracellular pathways, which are modulated primarily by tight junctions composed of tight junction proteins located between adjacent endothelial cells in the brain microvasculature (5). As described above, we showed that recombinant ovine IL-6 affected the tight junction expression in isolated microvessels in vitro from sheep and that treatment with anti–IL-6 mAb neutralized these effects (34). In the present study, although anti–IL-6-mAb treatment after brain ischemia modified protein composition of tight junctions, their patterns were not consistent across brain regions or tight junction proteins (Fig. 7). Anti–IL-6 mAb treatment after ischemia was associated with decreases in ZO-1 and claudin-5 expression, but with increases in claudin-1 and occludin in some, but not all brain regions. Therefore, even though treatment with the anti–IL-6 mAb attenuated the ischemia-related increases in the Ki values across the brain regions (Fig. 5), changes in the expression of the tight junction proteins may not entirely account for the observed changes in BBB permeability. The expression levels of the tight junction proteins are not the only determinant of their effectiveness in modulating tight junction function. The localization of the tight junction proteins within the endothelial cells are also important determinants of the efficacy of the tight junction proteins in modulating BBB function (96, 97). For example, ischemia has been associated with translocation of claudin-5 and occludin and with detachment from the tight junction in adult subjects (96, 97). However, we did not have tissue adequately preserved from our studies to measure the immunohistochemical localization of the tight junction proteins. Therefore, we cannot comment on the effects of ischemia and treatment with the anti–IL-6 mAb on the endothelial cellular localization of the tight junction proteins in our fetal sheep.
In addition to paracellular pathways, the transcellular pathways that include passive diffusion, active carrier mediated transport, and transcytosis contribute to the integrity of the BBB. However, it is important to point out that we extensively measured paracellular permeability in this and our previous work (10, 43, 57, 88, 89). Nonetheless, recent work has shown that caveolin-1, a membrane integral protein located at caveolae, can prevent the degradation of tight junction proteins, and protect BBB integrity by inhibiting MMP activity (98, 99). PLVAP is another transmembrane protein that is associated with the caveolae of fenestrated microvascular endothelial cells (9). Although we were not able to determine caveolin-1 expression in the brain tissue from our fetal sheep, PLVAP expression exhibited an unexpected striking increase after anti–IL-6 antibody treatment in the ischemic group (Fig. 8). Although previous work has shown that PLVAP expression can be up-regulated during the later phase after brain ischemia when BBB dysfunction is present (100), these findings cannot explain the relative decrease in Ki that we observed after ischemia in the anti–IL-6 antibody-treated ischemic group (Fig. 5). On the other hand, it has also been suggested that increases in PLVAP may also be associated with revascularization/angiogenesis and wound healing (100–102). We have recently shown that neovascularization occurs after exposure of the fetal brain to ischemia-reperfusion (103). Therefore, combined with our recent work (103), the striking increase in PLVAP that we observed in the anti–IL-6 mAb-treated ischemic group could suggest that anti–IL-6 mAb could enhance revascularization (angiogenesis) by neutralizing the IL-6 activity in the brain vasculature or parenchyma, which may also provide important functional and therapeutic implications for hypoxic-ischemic injury in immature subjects.
Even though elevations in IL-6 have frequently been associated with ischemic brain injury in adult, neonatal, and fetal subjects (20, 25, 52, 104), IL-6 has also been shown to exhibit both pro- and anti-inflammatory properties in the adult brain (105). For example, IL-6 has been shown to exhibit neuroprotective properties by increasing acidic fibroblast growth factor levels and reducing glutamate-induced cytotoxicity in adult subjects (106, 107) and therefore exert a neuroprotective properties against several types of brain injury including cerebral ischemia (108–110). However, IL-6 can also predispose to neurodegeneration, increase the sensitivity to glutamatergic neuronal lethality, and amplify the damaging effects of neurotoxic insults (106). In contrast, IL-6 exerts neurotoxic rather than neuroprotective effects in most instances in the developing brain after hypoxic-ischemic injury (111). The findings of the current study support the contention that IL-6 appears to be one of the factors contributing to the impaired BBB function after ischemia in the fetus.
Multiple lines of evidence support the contention that proinflammatory cytokines contribute to ischemia and inflammatory related brain damage in premature infants (27, 28, 112, 113). Furthermore, inflammatory related processes that begin in utero are often antecedents of brain injury in premature infants because elevations in inflammation-related proteins including cytokines in the first days after preterm birth predict the risk of brain injury in these infants (114). Several cytokines including IL-1β and TNF-α have previously been shown to increase the permeability of the BBB in a variety of adult and newborn animal models (115–117). In addition, we have recently shown that impaired BBB function is an important component of hypoxic-ischemic brain injury in the fetus (10). Taken together with previous work (115–117), our findings suggest that IL-6 and IL-1β (16) are also critical contributors to BBB dysfunction after ischemia in the fetus. Furthermore, the major proinflammatory cytokines including TNF-α, IL-6, and IL-1β most likely contribute to BBB dysfunction associated with both ischemic and inflammatory brain injury in the fetus and neonate (16, 27, 28, 112, 113).
In conclusion, the present study provides important findings showing that systemic infusions of anti–IL-6 mAb results in accumulation of the monoclonal antibody within the fetal brain parenchyma, decreases brain parenchymal IL-6 levels, and attenuates increases in BBB permeability after brain ischemia in the ovine fetus. These findings suggest that the proinflammatory cytokine, IL-6, contributes to the impaired BBB function after ischemic injury in fetus and that treatment with an anti–IL-6 antibody may protect the developing fetal brain and could provide an effective prevention/treatment strategies for perinatal ischemic brain injury.
Acknowledgments
The authors gratefully acknowledge the gift of the ovine IL-6 pQE30 vector and mouse monoclonal cell lines from Commonwealth Scientific and Industrial Research Organization, Livestock Industries (Victoria, Australia), that enabled the production of IL-6 protein and anti-IL-6 mAb, respectively. This work was supported by The U.S. National Institutes of Health, National Institute of General Medical Sciences (NIH NIGMS) Awards 1R01-HD-057100 and RI-INBRE P20RR016457-11; an Institutional Development Award from the NIH NIGMS Grants P20 RR018728 and P20GM103537; and a postdoctoral fellowship award from American Heart Association Grant 13POST16860015. The authors declare no conflicts of interest.
Glossary
- AIB
α-aminoisobutyric acid
- BBB
blood-brain barrier
- BSA
bovine albumin serum
- CP
cerebral palsy
- CSF
cerebrospinal fluid
- ECoG
electrocorticogram
- IC
internal control
- Ki
blood-to-brain transfer constant
- MMP
matrix metallopeptidase
- PLVAP
plasmalemma vesicle protein
- RBC
red blood cell
- ZO
zonula occludens
REFERENCES
- 1.Lai M. C., Yang S. N. (2011) Perinatal hypoxic-ischemic encephalopathy. J. Biomed. Biotechnol. 2011, 609813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Longo M., Hankins G. D. (2009) Defining cerebral palsy: pathogenesis, pathophysiology and new intervention. Minerva Ginecol. 61, 421–429 [PubMed] [Google Scholar]
- 3.Dickey E. J., Long S. N., Hunt R. W. (2011) Hypoxic ischemic encephalopathy—what can we learn from humans? J. Vet. Intern. Med. 25, 1231–1240 [DOI] [PubMed] [Google Scholar]
- 4.Ballabh P., Braun A., Nedergaard M. (2004) The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol. Dis. 16, 1–13 [DOI] [PubMed] [Google Scholar]
- 5.Abbott N. J., Patabendige A. A., Dolman D. E., Yusof S. R., Begley D. J. (2010) Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25 [DOI] [PubMed] [Google Scholar]
- 6.Abbott N. J., Rönnbäck L., Hansson E. (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat. Rev. Neurosci. 7, 41–53 [DOI] [PubMed] [Google Scholar]
- 7.Virgintino D., Robertson D., Errede M., Benagiano V., Tauer U., Roncali L., Bertossi M. (2002) Expression of caveolin-1 in human brain microvessels. Neuroscience 115, 145–152 [DOI] [PubMed] [Google Scholar]
- 8.Lakhan S. E., Kirchgessner A., Tepper D., Leonard A. (2013) Matrix metalloproteinases and blood-brain barrier disruption in acute ischemic stroke. Front Neurol 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hnasko R., Frank P. G., Ben-Jonathan N., Lisanti M. P. (2006) PV-1 is negatively regulated by VEGF in the lung of caveolin-1, but not caveolin-2, null mice. Cell Cycle 5, 2012–2020 [DOI] [PubMed] [Google Scholar]
- 10.Chen X., Threlkeld S. W., Cummings E. E., Juan I., Makeyev O., Besio W. G., Gaitanis J., Banks W. A., Sadowska G. B., Stonestreet B. S. (2012) Ischemia-reperfusion impairs blood-brain barrier function and alters tight junction protein expression in the ovine fetus. Neuroscience 226, 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bralet A. M., Beley A., Beley P., Bralet J. (1979) Brain edema and blood-brain barrier permeability following quantitative cerebral microembolism. Stroke 10, 34–38 [DOI] [PubMed] [Google Scholar]
- 12.Kaur C., Ling E. A. (2008) Blood brain barrier in hypoxic-ischemic conditions. Curr. Neurovasc. Res. 5, 71–81 [DOI] [PubMed] [Google Scholar]
- 13.Szaflarski J., Burtrum D., Silverstein F. S. (1995) Cerebral hypoxia-ischemia stimulates cytokine gene expression in perinatal rats. Stroke 26, 1093–1100 [DOI] [PubMed] [Google Scholar]
- 14.Martín-Ancel A., García-Alix A., Pascual-Salcedo D., Cabañas F., Valcarce M., Quero J. (1997) Interleukin-6 in the cerebrospinal fluid after perinatal asphyxia is related to early and late neurological manifestations. Pediatrics 100, 789–794 [DOI] [PubMed] [Google Scholar]
- 15.Cai Z., Hutchins J. B., Rhodes P. G. (1998) Intrauterine hypoxia-ischemia alters nitric oxide synthase expression and activity in fetal and neonatal rat brains. Brain Res. Dev. Brain Res. 109, 265–269 [DOI] [PubMed] [Google Scholar]
- 16.Chen X., Sadowska G. B., Zhang J., Kim J. E., Cummings E. E., Bodge C. A., Lim Y. P., Makeyev O., Besio W. G., Gaitanis J., Threlkeld S. W., Banks W. A., Stonestreet B. S. (2015) Neutralizing anti-interleukin-1beta antibodies modulate fetal blood-brain barrier function after ischemia. Neurobiol. Dis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lotz M., Jirik F., Kabouridis P., Tsoukas C., Hirano T., Kishimoto T., Carson D. A. (1988) B cell stimulating factor 2/interleukin 6 is a costimulant for human thymocytes and T lymphocytes. J. Exp. Med. 167, 1253–1258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iadecola C., Anrather J. (2011) The immunology of stroke: from mechanisms to translation. Nat. Med. 17, 796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Wagoner N. J., Oh J. W., Repovic P., Benveniste E. N. (1999) Interleukin-6 (IL-6) production by astrocytes: autocrine regulation by IL-6 and the soluble IL-6 receptor. J. Neurosci. 19, 5236–5244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ali C., Nicole O., Docagne F., Lesne S., MacKenzie E. T., Nouvelot A., Buisson A., Vivien D. (2000) Ischemia-induced interleukin-6 as a potential endogenous neuroprotective cytokine against NMDA receptor-mediated excitotoxicity in the brain. J. Cereb. Blood Flow Metab. 20, 956–966 [DOI] [PubMed] [Google Scholar]
- 21.Chiesa C., Pellegrini G., Panero A., Osborn J. F., Signore F., Assumma M., Pacifico L. (2003) C-reactive protein, interleukin-6, and procalcitonin in the immediate postnatal period: influence of illness severity, risk status, antenatal and perinatal complications, and infection. Clin. Chem. 49, 60–68 [DOI] [PubMed] [Google Scholar]
- 22.Pousset F. (1994) Developmental expression of cytokine genes in the cortex and hippocampus of the rat central nervous system. Brain Res. Dev. Brain Res. 81, 143–146 [DOI] [PubMed] [Google Scholar]
- 23.Chai Z., Gatti S., Toniatti C., Poli V., Bartfai T. (1996) Interleukin (IL)-6 gene expression in the central nervous system is necessary for fever response to lipopolysaccharide or IL-1 beta: a study on IL-6-deficient mice. J. Exp. Med. 183, 311–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pantoni L., Sarti C., Inzitari D. (1998) Cytokines and cell adhesion molecules in cerebral ischemia: experimental bases and therapeutic perspectives. Arterioscler. Thromb. Vasc. Biol. 18, 503–513 [DOI] [PubMed] [Google Scholar]
- 25.Hagberg H., Gilland E., Bona E., Hanson L. A., Hahin-Zoric M., Blennow M., Holst M., McRae A., Söder O. (1996) Enhanced expression of interleukin (IL)-1 and IL-6 messenger RNA and bioactive protein after hypoxia-ischemia in neonatal rats. Pediatr. Res. 40, 603–609 [DOI] [PubMed] [Google Scholar]
- 26.Shrivastava K., Llovera G., Recasens M., Chertoff M., Giménez-Llort L., Gonzalez B., Acarin L. (2013) Temporal expression of cytokines and signal transducer and activator of transcription factor 3 activation after neonatal hypoxia/ischemia in mice. Dev. Neurosci. 35, 212–225 [DOI] [PubMed] [Google Scholar]
- 27.Dammann O., Leviton A. (1997) Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatr. Res. 42, 1–8 [DOI] [PubMed] [Google Scholar]
- 28.Dammann O., Leviton A. (2000) Brain damage in preterm newborns: biological response modification as a strategy to reduce disabilities. J. Pediatr. 136, 433–438 [DOI] [PubMed] [Google Scholar]
- 29.Rezaie P., Dean A. (2002) Periventricular leukomalacia, inflammation and white matter lesions within the developing nervous system. Neuropathology 22, 106–132 [DOI] [PubMed] [Google Scholar]
- 30.Yoon B. H., Jun J. K., Romero R., Park K. H., Gomez R., Choi J. H., Kim I. O. (1997) Amniotic fluid inflammatory cytokines (interleukin-6, interleukin-1beta, and tumor necrosis factor-alpha), neonatal brain white matter lesions, and cerebral palsy. Am. J. Obstet. Gynecol. 177, 19–26 [DOI] [PubMed] [Google Scholar]
- 31.Yoon B. H., Romero R., Yang S. H., Jun J. K., Kim I. O., Choi J. H., Syn H. C. (1996) Interleukin-6 concentrations in umbilical cord plasma are elevated in neonates with white matter lesions associated with periventricular leukomalacia. Am. J. Obstet. Gynecol. 174, 1433–1440 [DOI] [PubMed] [Google Scholar]
- 32.Yoon B. H., Romero R., Kim C. J., Koo J. N., Choe G., Syn H. C., Chi J. G. (1997) High expression of tumor necrosis factor-alpha and interleukin-6 in periventricular leukomalacia. Am. J. Obstet. Gynecol. 177, 406–411 [DOI] [PubMed] [Google Scholar]
- 33.Desai T. R., Leeper N. J., Hynes K. L., Gewertz B. L. (2002) Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 104, 118–123 [DOI] [PubMed] [Google Scholar]
- 34.Cohen S. S., Min M., Cummings E. E., Chen X., Sadowska G. B., Sharma S., Stonestreet B. S. (2013) Effects of interleukin-6 on the expression of tight junction proteins in isolated cerebral microvessels from yearling and adult sheep. Neuroimmunomodulation 20, 264–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen X., Threlkeld S. W., Cummings E. E., Sadowska G. B., Lim Y. P., Padbury J. F., Sharma S., Stonestreet B. S. (2013) In-vitro validation of cytokine neutralizing antibodies by testing with ovine mononuclear splenocytes. J. Comp. Pathol. 148, 252–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gunn A. J., Gunn T. R., de Haan H. H., Williams C. E., Gluckman P. D. (1997) Dramatic neuronal rescue with prolonged selective head cooling after ischemia in fetal lambs. J. Clin. Invest. 99, 248–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bernhard C. G., Kolmodin G. M., Meyerson B. A. (1967) On the prenatal development of function and structure in the somesthetic cortex of the sheep. Prog. Brain Res. 26, 60–77 [DOI] [PubMed] [Google Scholar]
- 38.Back S. A., Riddle A., Hohimer A. R. (2006) Role of instrumented fetal sheep preparations in defining the pathogenesis of human periventricular white-matter injury. J. Child Neurol. 21, 582–589 [DOI] [PubMed] [Google Scholar]
- 39.Barlow R. M. (1969) The foetal sheep: morphogenesis of the nervous system and histochemical aspects of myelination. J. Comp. Neurol. 135, 249–262 [DOI] [PubMed] [Google Scholar]
- 40.Cook C. J., Gluckman P. D., Johnston B. M., Williams C. (1987) The development of the somatosensory evoked potential in the unanaesthetized fetal sheep. J. Dev. Physiol. 9, 441–455 [PubMed] [Google Scholar]
- 41.Cook C. J., Williams C., Gluckman P. D. (1987) Brainstem auditory evoked potentials in the fetal sheep, in utero. J. Dev. Physiol. 9, 429–439 [PubMed] [Google Scholar]
- 42.Petersson K. H., Pinar H., Stopa E. G., Faris R. A., Sadowska G. B., Hanumara R. C., Stonestreet B. S. (2002) White matter injury after cerebral ischemia in ovine fetuses. Pediatr. Res. 51, 768–776 [DOI] [PubMed] [Google Scholar]
- 43.Stonestreet B. S., Patlak C. S., Pettigrew K. D., Reilly C. B., Cserr H. F. (1996) Ontogeny of blood-brain barrier function in ovine fetuses, lambs, and adults. Am. J. Physiol. 271, R1594–R1601 [DOI] [PubMed] [Google Scholar]
- 44.Stonestreet B. S., Le E., Berard D. J. (1993) Circulatory and metabolic effects of beta-adrenergic blockade in the hyperinsulinemic ovine fetus. Am. J. Physiol. 265, H1098–H1106 [DOI] [PubMed] [Google Scholar]
- 45.Williams C. E., Gunn A. J., Mallard C., Gluckman P. D. (1992) Outcome after ischemia in the developing sheep brain: an electroencephalographic and histological study. Ann. Neurol. 31, 14–21 [DOI] [PubMed] [Google Scholar]
- 46.Mallard E. C., Williams C. E., Gunn A. J., Gunning M. I., Gluckman P. D. (1993) Frequent episodes of brief ischemia sensitize the fetal sheep brain to neuronal loss and induce striatal injury. Pediatr. Res. 33, 61–65 [DOI] [PubMed] [Google Scholar]
- 47.Seow H. F., Rothel J. S., Wood P. R. (1994) Expression and purification of recombinant ovine interleukin-1 beta from Escherichia coli. Vet. Immunol. Immunopathol. 41, 229–239 [DOI] [PubMed] [Google Scholar]
- 48.McWaters P., Hurst L., Chaplin P. J., Collins R. A., Wood P. R., Scheerlinck J. P. (2000) Characterisation of monoclonal antibodies to ovine interleukin-6 and the development of a sensitive capture ELISA. Vet. Immunol. Immunopathol. 73, 155–165 [DOI] [PubMed] [Google Scholar]
- 49.Wood P. R., Rothel J. S., McWaters P. G., Jones S. L. (1990) Production and characterization of monoclonal antibodies specific for bovine gamma-interferon. Vet. Immunol. Immunopathol. 25, 37–46 [DOI] [PubMed] [Google Scholar]
- 50.Banks W. A., Terrell B., Farr S. A., Robinson S. M., Nonaka N., Morley J. E. (2002) Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides 23, 2223–2226 [DOI] [PubMed] [Google Scholar]
- 51.Banks W. A., Farr S. A., Morley J. E., Wolf K. M., Geylis V., Steinitz M. (2007) Anti-amyloid beta protein antibody passage across the blood-brain barrier in the SAMP8 mouse model of Alzheimer’s disease: an age-related selective uptake with reversal of learning impairment. Exp. Neurol. 206, 248–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sadowska G. B., Threlkeld S. W., Flangini A., Sharma S., Stonestreet B. S. (2012) Ontogeny and the effects of in utero brain ischemia on interleukin-1β and interleukin-6 protein expression in ovine cerebral cortex and white matter. Int. J. Dev. Neurosci. 30, 457–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malaeb S. N., Sadowska G. B., Stonestreet B. S. (2007) Effects of maternal treatment with corticosteroids on tight junction protein expression in the cerebral cortex of the ovine fetus with and without exposure to in utero brain ischemia. Brain Res. 1160, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sadowska G. B., Malaeb S. N., Stonestreet B. S. (2010) Maternal glucocorticoid exposure alters tight junction protein expression in the brain of fetal sheep. Am. J. Physiol. Heart Circ. Physiol. 298, H179–H188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ron N. P., Kazianis J. A., Padbury J. F., Brown C. M., McGonnigal B. G., Sysyn G. D., Sadowska G. B., Stonestreet B. S. (2005) Ontogeny and the effects of corticosteroid pretreatment on aquaporin water channels in the ovine cerebral cortex. Reprod. Fertil. Dev. 17, 535–542 [DOI] [PubMed] [Google Scholar]
- 56.Kim C. R., Sadowska G. B., Petersson K. H., Merino M., Sysyn G. D., Padbury J. F., Stonestreet B. S. (2006) Effects of postnatal steroids on Na+/K+-ATPase activity and alpha1- and beta1-subunit protein expression in the cerebral cortex and renal cortex of newborn lambs. Reprod. Fertil. Dev. 18, 413–423 [DOI] [PubMed] [Google Scholar]
- 57.Stonestreet B. S., Sadowska G. B., McKnight A. J., Patlak C., Petersson K. H. (2000) Exogenous and endogenous corticosteroids modulate blood-brain barrier development in the ovine fetus. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R468–R477 [DOI] [PubMed] [Google Scholar]
- 58.Sadowska G. B., Patlak C. S., Petersson K. H., Stonestreet B. S. (2006) Effects of multiple courses of antenatal corticosteroids on blood-brain barrier permeability in the ovine fetus. J. Soc. Gynecol. Investig. 13, 248–255 [DOI] [PubMed] [Google Scholar]
- 59.Ohno K., Pettigrew K. D., Rapoport S. I. (1978) Lower limits of cerebrovascular permeability to nonelectrolytes in the conscious rat. Am. J. Physiol. 235, H299–H307 [DOI] [PubMed] [Google Scholar]
- 60.Cserr H. F., DePasquale M., Patlak C. S. (1987) Regulation of brain water and electrolytes during acute hyperosmolality in rats. Am. J. Physiol. 253, F522–F529 [DOI] [PubMed] [Google Scholar]
- 61.Hans V. H., Kossmann T., Joller H., Otto V., Morganti-Kossmann M. C. (1999) Interleukin-6 and its soluble receptor in serum and cerebrospinal fluid after cerebral trauma. Neuroreport 10, 409–412 [DOI] [PubMed] [Google Scholar]
- 62.Hergenroeder G. W., Moore A. N., McCoy J. P. Jr., Samsel L., Ward N. H. III, Clifton G. L., Dash P. K. (2010) Serum IL-6: a candidate biomarker for intracranial pressure elevation following isolated traumatic brain injury. J. Neuroinflammation 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McClain C., Cohen D., Phillips R., Ott L., Young B. (1991) Increased plasma and ventricular fluid interleukin-6 levels in patients with head injury. J. Lab. Clin. Med. 118, 225–231 [PubMed] [Google Scholar]
- 64.Suehiro E., Fujisawa H., Akimura T., Ishihara H., Kajiwara K., Kato S., Fujii M., Yamashita S., Maekawa T., Suzuki M. (2004) Increased matrix metalloproteinase-9 in blood in association with activation of interleukin-6 after traumatic brain injury: influence of hypothermic therapy. J. Neurotrauma 21, 1706–1711 [DOI] [PubMed] [Google Scholar]
- 65.Prout, A. P., Frasch, M. G., Veldhuizen, R. A., Hammond, R., Ross, M. G., Richardson, B. S. (2010) Systemic and cerebral inflammatory response to umbilical cord occlusions with worsening acidosis in the ovine fetus. Am. J. Obstet. Gynecol. 202, 82 [DOI] [PubMed]
- 66.Prout A. P., Frasch M. G., Veldhuizen R., Hammond R., Matushewski B., Richardson B. S. (2012) The impact of intermittent umbilical cord occlusions on the inflammatory response in pre-term fetal sheep. PLoS ONE 7, e39043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lavine S. D., Hofman F. M., Zlokovic B. V. (1998) Circulating antibody against tumor necrosis factor-alpha protects rat brain from reperfusion injury. J. Cereb. Blood Flow Metab. 18, 52–58 [DOI] [PubMed] [Google Scholar]
- 68.Stonestreet B. S., Goldstein M., Oh W., Widness J. A. (1989) Effects of prolonged hyperinsulinemia on erythropoiesis in fetal sheep. Am. J. Physiol. 257, R1199–R1204 [DOI] [PubMed] [Google Scholar]
- 69.Gibson K. J., Lumbers E. R. (1995) Extracellular volume and blood volume in chronically catheterized fetal sheep. J. Physiol. 485, 835–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Roelfsema V., Bennet L., George S., Wu D., Guan J., Veerman M., Gunn A. J. (2004) Window of opportunity of cerebral hypothermia for postischemic white matter injury in the near-term fetal sheep. J. Cereb. Blood Flow Metab. 24, 877–886 [DOI] [PubMed] [Google Scholar]
- 71.Shankaran S., Laptook A. R., Poole W. K.; Eunice Kennedy Shriver NICHD Neonatal Research Network (2010) Hypothermia for perinatal asphyxial encephalopathy. N. Engl. J. Med. 362, 1051–1052, author reply 1052 [DOI] [PubMed] [Google Scholar]
- 72.Sas A., Jones R., Tyor W. (2008) Intra-peritoneal injection of polyclonal anti-interferon alpha antibodies cross the blood brain barrier and neutralize interferon alpha. Neurochem. Res. 33, 2281–2287 [DOI] [PubMed] [Google Scholar]
- 73.Atwal J. K., Chen Y., Chiu C., Mortensen D. L., Meilandt W. J., Liu Y., Heise C. E., Hoyte K., Luk W., Lu Y., Peng K., Wu P., Rouge L., Zhang Y., Lazarus R. A., Scearce-Levie K., Wang W., Wu Y., Tessier-Lavigne M., Watts R. J. (2011) A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci. Transl. Med. 3, 84ra43. [DOI] [PubMed] [Google Scholar]
- 74.Bauman M. D., Iosif A. M., Ashwood P., Braunschweig D., Lee A., Schumann C. M., Van de Water J., Amaral D. G. (2013) Maternal antibodies from mothers of children with autism alter brain growth and social behavior development in the rhesus monkey. Transl. Psychiatr. 3, e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Brimberg L., Sadiq A., Gregersen P. K., Diamond B. (2013) Brain-reactive IgG correlates with autoimmunity in mothers of a child with an autism spectrum disorder. Mol. Psychiatry 18, 1171–1177 [DOI] [PubMed] [Google Scholar]
- 76.Zhang Y., Pardridge W. M. (2001) Mediated efflux of IgG molecules from brain to blood across the blood-brain barrier. J. Neuroimmunol. 114, 168–172 [DOI] [PubMed] [Google Scholar]
- 77.Schlachetzki F., Zhu C., Pardridge W. M. (2002) Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J. Neurochem. 81, 203–206 [DOI] [PubMed] [Google Scholar]
- 78.McCarthy K. M., Yoong Y., Simister N. E. (2000) Bidirectional transcytosis of IgG by the rat neonatal Fc receptor expressed in a rat kidney cell line: a system to study protein transport across epithelia. J. Cell Sci. 113, 1277–1285 [DOI] [PubMed] [Google Scholar]
- 79.del Zoppo G. J. (2009) Inflammation and the neurovascular unit in the setting of focal cerebral ischemia. Neuroscience 158, 972–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Abbruscato T. J., Davis T. P. (1999) Combination of hypoxia/aglycemia compromises in vitro blood-brain barrier integrity. J. Pharmacol. Exp. Ther. 289, 668–675 [PubMed] [Google Scholar]
- 81.Mark K. S., Davis T. P. (2002) Cerebral microvascular changes in permeability and tight junctions induced by hypoxia-reoxygenation. Am. J. Physiol. Heart Circ. Physiol. 282, H1485–H1494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ito U., Ohno K., Nakamura R., Suganuma F., Inaba Y. (1979) Brain edema during ischemia and after restoration of blood flow. Measurement of water, sodium, potassium content and plasma protein permeability. Stroke 10, 542–547 [DOI] [PubMed] [Google Scholar]
- 83.Nedelcu J., Klein M. A., Aguzzi A., Boesiger P., Martin E. (1999) Biphasic edema after hypoxic-ischemic brain injury in neonatal rats reflects early neuronal and late glial damage. Pediatr. Res. 46, 297–304 [DOI] [PubMed] [Google Scholar]
- 84.Fischer S., Clauss M., Wiesnet M., Renz D., Schaper W., Karliczek G. F. (1999) Hypoxia induces permeability in brain microvessel endothelial cells via VEGF and NO. Am. J. Physiol. 276, C812–C820 [DOI] [PubMed] [Google Scholar]
- 85.Kumar A., Mittal R., Khanna H. D., Basu S. (2008) Free radical injury and blood-brain barrier permeability in hypoxic-ischemic encephalopathy. Pediatrics 122, e722–e727 [DOI] [PubMed] [Google Scholar]
- 86.Brown R. C., Davis T. P. (2002) Calcium modulation of adherens and tight junction function: a potential mechanism for blood-brain barrier disruption after stroke. Stroke 33, 1706–1711 [DOI] [PubMed] [Google Scholar]
- 87.Huber J. D., Witt K. A., Hom S., Egleton R. D., Mark K. S., Davis T. P. (2001) Inflammatory pain alters blood-brain barrier permeability and tight junctional protein expression. Am. J. Physiol. Heart Circ. Physiol. 280, H1241–H1248 [DOI] [PubMed] [Google Scholar]
- 88.Stonestreet B. S., Sadowska G. B., Hanumara R. C., Petrache M., Petersson K. H., Patlak C. S. (2012) Comparative effects of glucose- and mannitol-induced osmolar stress on blood-brain barrier function in ovine fetuses and lambs. J. Cereb. Blood Flow Metab. 32, 115–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stonestreet B. S., Sadowska G. B., Leeman J., Hanumara R. C., Petersson K. H., Patlak C. S. (2006) Effects of acute hyperosmolality on blood-brain barrier function in ovine fetuses and lambs. Am. J. Physiol. Regul. Integr. Comp. Physiol. 291, R1031–R1039 [DOI] [PubMed] [Google Scholar]
- 90.Fujimoto M., Takagi Y., Aoki T., Hayase M., Marumo T., Gomi M., Nishimura M., Kataoka H., Hashimoto N., Nozaki K. (2008) Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 28, 1674–1685 [DOI] [PubMed] [Google Scholar]
- 91.Ellison V. J., Mocatta T. J., Winterbourn C. C., Darlow B. A., Volpe J. J., Inder T. E. (2005) The relationship of CSF and plasma cytokine levels to cerebral white matter injury in the premature newborn. Pediatr. Res. 57, 282–286 [DOI] [PubMed] [Google Scholar]
- 92.Chiesa C., Pellegrini G., Panero A., De Luca T., Assumma M., Signore F., Pacifico L. (2003) Umbilical cord interleukin-6 levels are elevated in term neonates with perinatal asphyxia. Eur. J. Clin. Invest. 33, 352–358 [DOI] [PubMed] [Google Scholar]
- 93.Maruo N., Morita I., Shirao M., Murota S. (1992) IL-6 increases endothelial permeability in vitro. Endocrinology 131, 710–714 [DOI] [PubMed] [Google Scholar]
- 94.Benjelloun, N., Renolleau, S., Represa, A., Ben-Ari, Y., Charriaut-Marlangue, C. (1999) Inflammatory responses in the cerebral cortex after ischemia in the P7 neonatal rat. Stroke. 30, 1916–1923 [DOI] [PubMed]
- 95.Stanimirovic D., Satoh K. (2000) Inflammatory mediators of cerebral endothelium: a role in ischemic brain inflammation. Brain Pathol. 10, 113–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fischer S., Wobben M., Marti H. H., Renz D., Schaper W. (2002) Hypoxia-induced hyperpermeability in brain microvessel endothelial cells involves VEGF-mediated changes in the expression of zonula occludens-1. Microvasc. Res. 63, 70–80 [DOI] [PubMed] [Google Scholar]
- 97.Zhang J., Takahashi H. K., Liu K., Wake H., Liu R., Maruo T., Date I., Yoshino T., Ohtsuka A., Mori S., Nishibori M. (2011) Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 42, 1420–1428 [DOI] [PubMed] [Google Scholar]
- 98.Gu Y., Zheng G., Xu M., Li Y., Chen X., Zhu W., Tong Y., Chung S. K., Liu K. J., Shen J. (2012) Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J. Neurochem. 120, 147–156 [DOI] [PubMed] [Google Scholar]
- 99.Gu Y., Dee C. M., Shen J. (2011) Interaction of free radicals, matrix metalloproteinases and caveolin-1 impacts blood-brain barrier permeability. Front Biosci (Schol Ed) 3, 1216–1231 [DOI] [PubMed] [Google Scholar]
- 100.Shue E. H., Carson-Walter E. B., Liu Y., Winans B. N., Ali Z. S., Chen J., Walter K. A. (2008) Plasmalemmal vesicle associated protein-1 (PV-1) is a marker of blood-brain barrier disruption in rodent models. BMC Neurosci. 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Goussev S., Hsu J. Y., Lin Y., Tjoa T., Maida N., Werb Z., Noble-Haeusslein L. J. (2003) Differential temporal expression of matrix metalloproteinases after spinal cord injury: relationship to revascularization and wound healing. J. Neurosurg. 99(2, Suppl)188–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Carson-Walter E. B., Hampton J., Shue E., Geynisman D. M., Pillai P. K., Sathanoori R., Madden S. L., Hamilton R. L., Walter K. A. (2005) Plasmalemmal vesicle associated protein-1 is a novel marker implicated in brain tumor angiogenesis. Clin. Cancer Res. 11, 7643–7650 [DOI] [PubMed] [Google Scholar]
- 103.Virgintino D., Girolamo F., Rizzi M., Ahmedli N., Sadowska G. B., Stopa E. G., Zhang J., Stonestreet B. S. (2014) Ischemia/Reperfusion-induced neovascularization in the cerebral cortex of the ovine fetus. J. Neuropathol. Exp. Neurol. 73, 495–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saito K., Suyama K., Nishida K., Sei Y., Basile A. S. (1996) Early increases in TNF-alpha, IL-6 and IL-1 beta levels following transient cerebral ischemia in gerbil brain. Neurosci. Lett. 206, 149–152 [DOI] [PubMed] [Google Scholar]
- 105.Suzuki S., Tanaka K., Suzuki N. (2009) Ambivalent aspects of interleukin-6 in cerebral ischemia: inflammatory versus neurotrophic aspects. J. Cereb. Blood Flow Metab. 29, 464–479 [DOI] [PubMed] [Google Scholar]
- 106.Thorns V., Walter G. F., Licastro F. (2002) Effects of IL6 and IL1beta on aFGF expression and excitotoxicity in NT2N cells. J. Neuroimmunol. 127, 22–29 [DOI] [PubMed] [Google Scholar]
- 107.Samland H., Huitron-Resendiz S., Masliah E., Criado J., Henriksen S. J., Campbell I. L. (2003) Profound increase in sensitivity to glutamatergic- but not cholinergic agonist-induced seizures in transgenic mice with astrocyte production of IL-6. J. Neurosci. Res. 73, 176–187 [DOI] [PubMed] [Google Scholar]
- 108.Loddick S. A., Turnbull A. V., Rothwell N. J. (1998) Cerebral interleukin-6 is neuroprotective during permanent focal cerebral ischemia in the rat. J. Cereb. Blood Flow Metab. 18, 176–179 [DOI] [PubMed] [Google Scholar]
- 109.Yamada M., Hatanaka H. (1994) Interleukin-6 protects cultured rat hippocampal neurons against glutamate-induced cell death. Brain Res. 643, 173–180 [DOI] [PubMed] [Google Scholar]
- 110.Toulmond S., Vige X., Fage D., Benavides J. (1992) Local infusion of interleukin-6 attenuates the neurotoxic effects of NMDA on rat striatal cholinergic neurons. Neurosci. Lett. 144, 49–52 [DOI] [PubMed] [Google Scholar]
- 111.Brunssen S. H., Moy S. S., Toews A. D., McPherson C. A., Harry G. J. (2013) Interleukin-6 (IL-6) receptor/IL-6 fusion protein (Hyper IL-6) effects on the neonatal mouse brain: possible role for IL-6 trans-signaling in brain development and functional neurobehavioral outcomes. Brain Behav. Immun. 27, 42–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tita A. T., Andrews W. W. (2010) Diagnosis and management of clinical chorioamnionitis. Clin. Perinatol. 37, 339–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Malaeb S., Dammann O. (2009) Fetal inflammatory response and brain injury in the preterm newborn. J. Child Neurol. 24, 1119–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leviton, A., Kuban, K., O'Shea, T. M., Paneth, N., Fichorova, R., Allred, E. N., Dammann, O. (2011) The relationship between early concentrations of 25 blood proteins and cerebral white matter injury in preterm newborns: the ELGAN study. J Pediatr. 158, 897–903 [DOI] [PubMed]
- 115.Quagliarello V. J., Wispelwey B., Long W. J. Jr., Scheld W. M. (1991) Recombinant human interleukin-1 induces meningitis and blood-brain barrier injury in the rat. Characterization and comparison with tumor necrosis factor. J. Clin. Invest. 87, 1360–1366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Abraham C. S., Deli M. A., Joo F., Megyeri P., Torpier G. (1996) Intracarotid tumor necrosis factor-alpha administration increases the blood-brain barrier permeability in cerebral cortex of the newborn pig: quantitative aspects of double-labelling studies and confocal laser scanning analysis. Neurosci. Lett. 208, 85–88 [DOI] [PubMed] [Google Scholar]
- 117.Megyeri P., Abrahám C. S., Temesvári P., Kovács J., Vas T., Speer C. P. (1992) Recombinant human tumor necrosis factor alpha constricts pial arterioles and increases blood-brain barrier permeability in newborn piglets. Neurosci. Lett. 148, 137–140 [DOI] [PubMed] [Google Scholar]