

Abstract
Background
Rare genetic variants influence blood pressure (BP).
Methods and Results
Whole exome sequencing was performed on DNA samples from 17,956 individuals of European and African ancestry (14,497 first stage and 3,459 second stage discovery) to examine the impact of rare variants on hypertension and four BP traits: systolic and diastolic BP (SBP, DBP), pulse pressure (PP), and mean arterial pressure (MAP). Tests of ∼170,000 common variants (minor allele frequency, MAF, ≥1%, statistical significance P≤2.9×10-7) and gene-based tests of rare variants (MAF<1%, ∼17,000 genes, statistical significance P≤1.5×10-6) were evaluated for each trait and ancestry, followed by multiethnic meta-analyses. In the first stage discovery, rare coding variants (splicing, stop-gain, stop-loss, nonsynonymous variants, or indels) in CLCN6 were associated with lower DBP (cumulative MAF=1.3%, β=-3.20, P=4.1×10-6), and were independent of a nearby common variant (rs17367504) previously associated with BP. CLCN6 rare variants were also associated with lower SBP (β=-4.11, P=2.8×10-4), MAP (β=-3.50, P=8.9×10-6), and reduced hypertension risk (odds ratio=0.72, P=0.017). Meta-analysis of the two-stage discovery samples showed that CLCN6 was associated with lower DBP at exome-wide significance (cumulative MAF=1.1%, β=-3.30, P=5.0×10-7).
Conclusions
These findings implicate the effect of rare coding variants in CLCN6 in BP variation, and offer new insights into BP regulation.
Keywords: blood pressure, hypertension, rare variants, whole exome sequencing
Introduction
Blood pressure (BP) is a heritable quantitative trait influenced by both genetic and environmental stimuli.1, 2 Persistently elevated BP is a risk factor for cardiovascular disease and a major contributor to cardiovascular death.3, 4 Identifying genetic determinants of BP regulation may add novel insights into cardiovascular disease prevention, and may lead to more efficacious treatments. Large scale genome-wide association studies (GWAS) have reported common variants at approximately 60 loci that are associated with systolic (SBP) and diastolic BP (DBP) in individuals of European ancestry (EA), with effect sizes ranging from 0.4 to 1.2 mmHg for SBP and 0.2 to 0.7 mmHg for DBP per copy of the minor allele.5-7 Additional variants for pulse pressure (PP) and mean arterial pressure (MAP) have also been identified with effect sizes of similar magnitudes.8 A recent large BP GWAS demonstrated that BP variants identified in EAs may have effects in individuals of African ancestry, so an analysis of multiethnic samples has the potential to find novel genetic determinants of BP traits in this field.9 Despite the fact that numerous BP variants have been identified by GWAS, the proportion of explained variance in BP measures remains limited.
Studies have shown that rare coding mutations contribute to BP variation,10, 11 but a recent study involving targeted sequencing of six BP genes identified by GWAS did not reveal novel rare variants associated to the trait.12 In contrast, whole exome sequencing (WES), which captures both common and rare coding variation, has successfully been applied to identify rare coding variants contributing to multiple complex traits.13, 14 To date, no WES study has evaluated the association between rare coding variants and BP traits. To address this, we performed first stage WES on 9,950 EAs and 4,547 individuals of African-American ancestry (AA) from six large population-based cohort studies to examine the impact of rare coding variants on SBP, DBP, PP, MAP, and hypertension. The second stage WES was conducted in two EA cohorts, comprising 3,459 individuals.
Methods
Study Populations and Blood Pressure Measurements
The first stage discovery sample consisted of 10,403 individuals from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium15 and 4,094 individuals from the National Heart, Lung, and Blood Institute GO Exome Sequencing Project (ESP) with BP measures. Individuals from CHARGE were from three population-based cohorts including the Atherosclerosis Risk in Communities (ARIC) study (n=5,704 EAs and 2,792 AAs), Cardiovascular Health Study (CHS, n=680 EAs) and the Framingham Heart Study (FHS, n=1,227 EAs). Independent individuals from ESP were sampled from six population-based cohorts: ARIC (n=512 EAs and 323 AAs), CHS (n=144 EAs and 64 AAs), FHS (n=404 EAs), Jackson Heart Study (JHS, n=359 AAs), Multi-Ethnic Study of Atherosclerosis (MESA, n=247 EAs and 151 AAs), and the Women's Health Initiative (WHI, n=1,032 EAs and 858 AAs). The detailed sampling strategy for ESP is described in Supplemental Methods. The second stage discovery sample consisted of individuals from the Rotterdam Study (RS, n=2,205 EAs) and the Erasmus Rucphen Family (ERF) study (n=1,254 EAs). Detailed descriptions of each of the eight cohorts have been published elsewhere.16-24
For all cohorts in this study, BP values were measured at the first examination and anti-hypertensive medication use was recorded from the medication history or medication inventory at the same time. Detailed descriptions for BP measurements in each cohort are summarized in Supplemental Methods. For individuals taking anti-hypertensive medication, untreated BP values were imputed by adding 15 mm Hg to measured SBP and 10 mm Hg to measured DBP.25, 26 All participants provided written informed consent as approved by local institutional review committees.
Exome Sequencing and Variant Calling
For CHARGE, DNA samples were prepared using the Baylor College of Medicine Human Genome Sequencing Center VCRome 2.1 design 27 (42Mb, NimbleGen), and were sequenced and called together. For ESP, DNA samples were prepared using either Roche Nimblegen SeqCap EZ or Agilent SureSelect Human All Exon 50Mb. All samples were paired end sequenced using Illumina GAII or HiSeq instruments. Details on sequencing, variant calling and variant quality control are provided in Supplemental Methods.
Annotation of Whole Exome Sequence Variants
To facilitate meta-analysis between CHARGE and ESP, a combined variant annotation file was created to include all quality variants observed in either CHARGE or ESP. Variants were annotated from CHARGE and ESP separately using ANNOVAR 28 and dbNSFP v2.0 29 according to the reference genome GRCh37 and National Center for Biotechnology Information RefSeq. Coding variants were annotated to a unique gene as well as the following categories that were considered for inclusion in gene-based tests: splicing, stop-gain, stop-loss, nonsynonymous variants, and indels. The CHARGE and ESP annotated variant lists were merged into a joint file to ensure that a variant present in both studies had the same reference allele and annotation category.
Statistical Analyses
Individuals with untreated SBP<60 mmHg or untreated DBP<40 mmHg were excluded from analysis. PP was calculated by subtracting DBP from SBP, and MAP was defined as DBP plus PP/3. Hypertension was defined as individuals having SBP≥140mmHg, or DBP≥90mmHg, or use of BP lowering medication at the first examination. All four continuous traits were winsorized at the 99.9th percentile prior to the analysis by utilizing BP data available from the entire cohort. Cohort-level and ancestry-specific analyses were carried out using the R seqMeta package (http://cran.r-project.org/web/packages/seqMeta/index.html) adjusting for age, age-squared, sex, BMI, and principal components (PCs, generated by EIGENSTRAT30) or study site as needed within each cohort and ancestry stratum. Fixed effect inverse variance weighted meta-analyses of single variant and gene-based tests were then conducted using seqMeta to combine cohort-level and ancestry-specific summary results for multiethnic analyses. Only variants on autosomal chromosomes were analyzed in this study, and all analyses used additive genetic models.
Single variants (common variants, MAF≥1%) were tested for association with the four BP traits and hypertension. Single variant associations were considered to be significant if P≤2.9×10-7, reflecting Bonferroni correction for testing ∼170,000 variants. For gene-based analysis, we performed a T1 test for each gene, in which annotated coding variants with MAF≤1% within a gene were collapsed into a single gene-based burden score and then the score was analyzed using linear regression.31 We also implemented the Sequence Kernel Association Test (SKAT) using default beta weights,32 which analyzed annotated coding variants with a MAF≤1% and is more powerful when effects are both BP-raising and BP-lowering. For multiethnic meta-analyses, genes with cumulative MAF (cMAF) ≥0.1% were analyzed using both T1 and SKAT implemented by seqMeta, and an association was considered to be significant if P≤1.5×10-6 given a Bonferroni correction for ∼17,000 genes and two burden tests.
Second Stage Discovery
The top T1 gene-based association identified in this study was followed up in two independent sample sets, RS (n=2,205 EAs) and ERF (n=1,254 EAs). For ERF, sequencing was done using the Agilent version V4 capture kit on an Illumina Hiseq2000 sequencer. In the RS, individuals were sequenced using the Nimblegen SeqCap EZ V2 capture kit on an Illumina Hiseq2000 sequencer. Details on sequencing, variant calling and variant quality control are provided in Supplemental Methods. Coding variants included in the analyses were defined as splicing, stop-gain, stop-loss, nonsynonymous, and indels. A gene-based T1 test was conducted as described above, with the significance threshold set at P<0.05.
Results
Participant Characteristics
The study sample for this analysis consisted of 17,956 individuals, with 14,497 in the first stage discovery data set and 3,459 in the second stage discovery data set. In general, individuals from each cohort were middle aged, with a greater proportion of females than males. Compared to EAs, AAs had higher prevalence of hypertension, type 2 diabetes, and higher mean BMI and BP values. Ancestry-stratified characteristics of the two-stage discovery cohorts are summarized in Table S1.
Gene-based Test Results
For each BP trait, the first stage discovery results from T1 and SKAT gene-based tests at P<5×10-4 and rare coding variants in the identified genes are summarized in Tables S2-S3. The most significant association was for the chloride channel, voltage-sensitive 6 gene (CLCN6) with DBP, in the T1 test. There were 95 rare coding variants in CLCN6 present in CHARGE or ESP (cMAF=1.3%, annotated variant level results with DBP are shown in Table S4); 34 of which were not reported by the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org, accessed Mar 31st, 2015). The aggregation of rare coding variants in CLCN6 were associated with lower DBP (β =-3.20, P=4.1×10-6), SBP (β =-4.11, P=2.8×10-4) and MAP (β=-3.50, P=8.9×10-6), but were not associated with PP. Rare coding variants were seen in both ancestries with similar cMAF of 1.2% (Figure 1). The magnitude of the effect sizes were consistent between EA and AA, where each copy of a rare allele was associated with 3-4 mm Hg lower DBP (Table 1). There were 29 BP genetic loci, including 42 genes, previously reported by Ehret, et al., the largest BP GWAS thus far.6 Tables S5-S6 contains T1 and SKAT results for the 42 genes and the four BP traits. After accounting for multiple testing for the 42 genes (p < 0.001), only CLCN6 exceeded this significance threshold.
Figure 1.
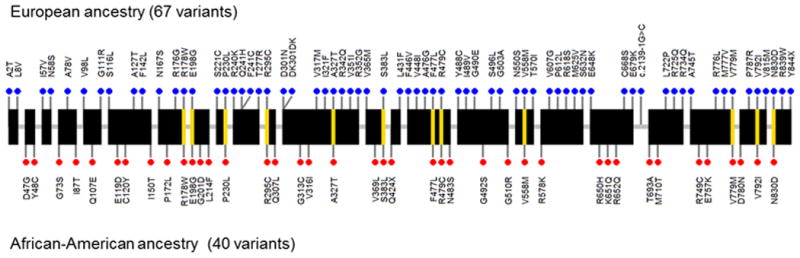
Chloride channel, voltage-sensitive 6 (CLCN6) rare coding mutations discovered in the first stage cohorts. Blue dots above and red dots below represent the mutations observed in European ancestry or African-American ancestry, respectively. Yellow lines across the gene connect the same mutation seen in both ancestries.
Table 1. T1 gene-based results for CLCN6 on four BP traits across two ancestries in CHARGE and ESP.
| EA (n=9,950, cMAF=0.01) |
AA (n=4,547, cMAF=0.01) |
EA & AA (n=14,497, cMAF=0.01) |
||||
|---|---|---|---|---|---|---|
| Beta (SE) | P | Beta (SE) | P | Beta (SE) | P | |
| SBP | -3.80 (1.26) | 2.60E-03 | -5.43 (2.58) | 0.03 | -4.11 (1.13) | 2.82E-04 |
| DBP | -3.12 (0.78) | 5.70E-05 | -3.44 (1.53) | 0.02 | -3.20 (0.69) | 4.10E-06 |
| MAP | -3.35 (0.88) | 1.44E-04 | -4.11 (1.76) | 0.02 | -3.50 (0.79) | 8.91E-06 |
| PP | -0.52 (0.85) | 0.54 | -1.90 (1.75) | 0.28 | -0.78 (0.76) | 0.31 |
Beta corresponds to mmHg per mutated allele for BP traits
EA - European ancestry; AA - African-American ancestry; cMAF - cumulative minor allele frequency; SE - standard error
In a T1 burden test for hypertension in the first stage discovery sample, rare coding variants in CLCN6 accounted for a 28% lower odds of hypertension (OR=0.72, 95% CI=0.55 to 0.94, P=0.017). CLCN6 is located in 1p36. A common intronic SNP, rs17367504 (MAF=14%), 3.4 kb upstream from CLCN6, was associated with reduced DBP in a previous GWAS.5 Therefore, we re-examined the association between CLCN6 and DBP in CHARGE EAs and AAs, adjusting for rs17367504. The results showed the observed effect size and significance of CLCN6 on DBP levels had the same magnitude as in the unconditional analyses (Table 2).
Table 2. T1 gene-based results for CLCN6 on DBP in CHARGE conditioning on rs17367504.
| Unconditional analysis | Condition on rs17367504 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cohort | Gene | cMAF | N | P | Beta (SE) | N | p | Beta (SE) |
| European ancestry | ||||||||
| ARIC | CLCN6 | 0.01 | 5704 | 0.002 | -2.88 (0.95) | 5639 | 0.002 | -2.92 (0.95) |
| CHS | CLCN6 | 0.01 | 683 | 0.22 | -4.10 (3.31) | 683 | 0.29 | -3.89 (3.68) |
| FHS | CLCN6 | 0.01 | 1227 | 0.03 | -3.92 (1.77) | 1216 | 0.04 | -3.89 (1.87) |
| African-America ancestry | ||||||||
| ARIC | CLCN6 | 0.007 | 2792 | 0.05 | -4.00 (2.07) | 2719 | 0.09 | -3.66 (2.13) |
| Multiethnic | ||||||||
| CHARGE | CLCN6 | 0.01 | 10406 | 1.39E-05 | -3.28 (0.76) | 10257 | 2.81E-05 | -3.22 (0.77) |
Beta corresponds to mmHg per mutated allele for DBP.
cMAF - cumulative minor allele frequency; SE - standard error
Corroborating Evidence
There are three sources of corroborating data for the observed first stage discovery findings: second stage discovery, previous GWAS, and animal model studies. When compared to the first stage discovery cohorts, the two EA second stage cohorts had a smaller cMAF (cMAF=0.3% vs 1.3%) for CLCN6 in the T1 test, but the direction of the effect was consistent. CLCN6 remained significantly associated with DBP in ERF (β=-7.25, P=0.04), but not in RS (β=-1.19, P=0.68) (variant level results are shown in Table S7, and T1 results for the other BP traits are shown in Table S8). After meta-analyzing the two-stage discovery samples, CLCN6 was exome-wide significantly related to lower DBP (cMAF = 1.1%, β = -3.30, P = 5.0 × 10-7, Figure 2). Second, CLCN6 is near a previous DBP GWAS locus5 that contains multiple candidate genes. Third, a knock-out homologue Clcn6 in the rat results in reduced BP levels and lower hypertension risk,33 supporting results similar to our observations.
Figure 2.
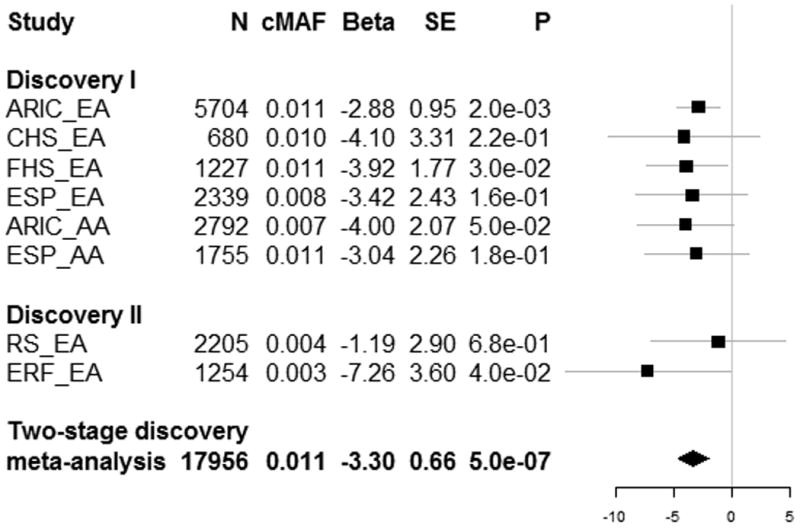
Cohort and ancestry specific effects of CLCN6 on DBP in two-stage discovery cohorts. cMAF indicates cumulative minor allele frequency; SE, standard error, EA, European ancestry and AA, African-American ancestry. Beta corresponds to mmHg per mutated allele for DBP.
Single Variant Test Results
Common variants (MAF≥1%) were analyzed in relation to BP traits using multiethnic meta-analyses. No single variant test reached our pre-defined significance threshold. The associations for each BP trait with P<5×10-5 are shown in Table S9. Four coding variants located in ULK4, SLC39A8, HFE and SH2B3 previously reported by Ehret, et al., the largest BP GWAS thus far,6 were captured in this study, and thus, were available for analysis. Our results showed consistent directional effects for the coded alleles with the GWAS findings, and the associations with DBP all had P<0.05 (Table S10).
Discussion
By analyzing exome sequence data from two large consortia (n=14,497) in relation to BP traits, we identified an aggregation of rare coding variants in CLCN6 that were associated with lower DBP among EAs and AAs. The association was corroborated in the second stage discovery cohorts, and a meta-analysis of two-stage discovery cohorts showed that CLCN6 was exome-wide significantly related to lower DBP (P=5.0×10-7). In addition to DBP, CLCN6 was related to lower levels of SBP and MAP as well as lower risk for hypertension. This indicates a potential role of CLCN6 in BP regulation, and positions this gene as an attractive therapeutic target for future studies.
We demonstrated that the effect of CLCN6 was independent of a previously reported common GWAS SNP in this region. CLCN6 is located in 1p36, a region with several BP candidate genes identified by GWAS, including AGTRAP, MTHFR, CLCN6, NPPA, and NPPB. A common SNP in this region is rs17367504, in the intron of MTHFR, with a modest BP effect size (<1 mm Hg) in BP.6, 34 We identified 95 rare coding variants in CLCN6, and these rare variants, in aggregate, were associated with decreased BP levels (3-4 mm Hg), independent of the tagging SNP, rs17367504. The effect size for the rare coding variants in CLCN6 was about four- to six-fold larger than previous common BP variants from GWAS.
CLCN6 belongs to the voltage-dependent chloride channel (ClC) family. The function of chloride channels range from ion homeostasis to cell migration and regulation of electrical excitability.35 However, the physiological role of CLCN6 is less well characterized. CLCN6 has four conserved domains, where ClC_6_like and CBS_pair_EriC_assoc_euk_bac are the most likely functional domains. Most rare variants identified in our study (80%) are located in these two domains. ClC_6_like belongs to the ClC superfamily. It shares the unique double-barreled architecture and voltage-dependent gating mechanism, though the function is not clear.35 CBS_pair_EriC_assoc_euk_bac, coexisting with other functional domains, contains two tandem repeats of the cystathionine beta-synthase (CBS pair) domains, and mutations within this domain are associated with Bartter syndrome.36 Interestingly, CLCNKA and CLCNKB, two other genes belonging to chloride channel family, share CBS_pair_EriC_assoc_euk_bac domain and are involved in blood pressure regulation. CLCNKA and CLCNKB play a key role in transporting chloride ions through ClC Ka and Kb, which is part of the mechanism of kidney reabsorption of sodium chloride to help maintain blood pressure.37 Studies have shown CLCNKA and CLCNKB harbored mutations associated with low blood pressure in Mendelian conditions, including Bartter's and Gitelman's syndromes.38 Rare independent mutations in other renal salt handling genes, including SLC12A3, SLC12A1 and KCNJ1, were reported to contribute to lower BP levels (e.g. -3.4 mm Hg for long-term average DBP) and reduced prevalence of hypertension in a community-based study as well.11 Our study showed that CLCN6 has a similar magnitude of effect on BP levels and hypertension. Consistent results were observed in both EAs and AAs (β = -3.12 for EAs and β = -3.44 for AAs), which would enhance the global understanding of genetic determinants for BP regulation.
Common variants associated with BP have been studied extensively in large-scale GWAS, and many variants have been reported with effects of about 0.5 to 1 mm Hg (per variant allele).5, 6, 8, 9, 39-41 In this study, in contrast with the rare variant result, we did not identify novel common variants that significantly influenced BP levels. We showed consistent results for four common BP SNPs, located in ULK4, SLC39A8, HFE, SH2B3, that were reported by the ICBP consortium6 and were captured in our whole exome sequencing. Large scale GWAS is a powerful approach to detect common variants associated with complex traits;42 we had limited power to detect novel common variants in this study given the sample size with whole exome sequence compared to GWAS.
To our knowledge, this study is the first and largest WES study for BP traits among EAs and AAs. We observed rare coding variants in CLCN6 that in aggregate have large effects on BP. Additional sequencing in larger samples will help demonstrate the robustness of our findings and further replication is warranted. Our study focused on BP measurements at baseline, and repeat measurements may provide a more precisely estimated phenotype to detect genetic determinants for BP variation.43 Therefore, future whole exome sequencing studies incorporating repeated BP measurements are justified.
In summary, by analyzing WES, we identified that an aggregation of rare coding variants in CLCN6 was associated with lower DBP and lower risk of hypertension among 13,409 EAs and 4,547 AAs from eight large population-based cohort studies. In addition, the effect sizes of CLCN6 were consistent across two ancestries. Our findings provide evidence for a functional role of CLCN6 in BP regulation and point toward this gene as a therapeutic target.
Supplementary Material
Clinical Perspective.
Genetic variants that are rare in the general population may influence blood pressure. Our study focused on the protein coding (exome) sequence from 17,956 individuals of European and African ancestry (14,497 first stage and 3,459 second stage discovery) and identified rare coding variants in CLCN6 significantly associated with lower diastolic blood pressure. The association persisted after conditioning on a nearby known blood pressure related common variant, rs17367504. CLCN6 was also shown to have effects on other blood pressure traits, including systolic blood pressure and mean arterial pressure, and decreased odds of hypertension. CLCN6 belongs to the voltage-dependent chloride channel family with a known domain that is involved in blood pressure regulation. Corroborating evidence comes from a separate study showing that a knock-out homologue Clcn6 in the rat reduced blood pressure levels and lowered hypertension risk. Our study showed that CLCN6 rare coding variants have a similar magnitude of effect on blood pressure levels and hypertension compared to common variants reported by genome wide association studies, and the effect was consistent between European ancestry and African ancestry. These findings implicate the roles of rare coding variants in explaining blood pressure variation, contributing to hypertension, and suggesting potential therapeutic interventions for cardiovascular diseases.
Acknowledgments
The authors thank the staff and participants of the ARIC study for their important contributions. The authors acknowledge the support of the National Heart, Lung, and Blood Institute (NHLBI) and the research institutions in creating this resource for biomedical research and thank the study investigators, field staff and study participants for their important contributions. A full list of principal CHS investigators and institutions can be found at CHS-NHLBI.org. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors thank the WHI investigators and staff for their dedication, and the study participants for making the program possible. A full listing of WHI investigators can be found at: http://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Short%20List.pdf. We are grateful to all study participants and their relatives, general practitioners and neurologists for their contributions and to P. Veraart for her help in genealogy, J. Vergeer for the supervision of the laboratory work, and P. Snijders for his help in data collection for the ERF study. We thank Mr. Pascal Arp, Ms. Mila Jhamai, Jeroen van Rooij, BSc, Mr. Marijn Verkerk, and Dr Robert Kraaij for their help in creating the RS-Exome Sequencing database. Finally, the authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists.
Funding Sources: Funding for GO ESP was provided by NHLBI grants RC2 HL-103010 (HeartGO), RC2 HL-102923 (LungGO) and RC2 HL-102924 (WHISP). The exome sequencing was performed through NHLBI grants RC2 HL-102925 (BroadGO) and RC2 HL-102926 (SeattleGO).
Funding support for “Building on GWAS for NHLBI-diseases: the U.S. CHARGE consortium” was provided by the NIH through the American Recovery and Reinvestment Act of 2009 (ARRA) (5RC2HL102419). Sequencing was carried out at the Baylor Genome Center (U54 HG003273).
The ARIC Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute (NHLBI) contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN2682011000010C, HHSN2682011000011C, and HHSN2682011000012C), R01HL087641, R01HL59367 and R01HL086694.
The Framingham Heart Study is conducted and supported by the NHLBI in collaboration with Boston University (Contract No. N01-HC-25195), and its contract with Affymetrix, Inc., for genome-wide genotyping services (Contract No. N02-HL-6-4278), for quality control by Framingham Heart Study investigators using genotypes in the SNP Health Association Resource (SHARe) project. A portion of this research was conducted using the Linux Cluster for Genetic Analysis (LinGA) computing resources at Boston University Medical Campus.
Cardiovascular Health Study: This CHS research was supported by NHLBI contracts HHSN268201200036C, HHSN268200800007C, N01HC55222N01HC55222, N01HC85079, N01HC85080, N01HC85081, N01HC85082, N01HC85083, N01HC85086; and NHLBI grants U01HL080295, R01HL087652, R01HL105756, R01HL103612, and R01HL120393 with additional contribution from the National Institute of Neurological Disorders and Stroke (NINDS). Additional support was provided through R01AG023629R01AG023629 from the National Institute on Aging (NIA).
The Jackson Heart Study is supported by contracts HHSN268201300046C, HHSN268201300047C, HHSN268201300048C, HHSN268201300049C, HHSN268201300050C from the National Heart, Lung, and Blood Institute and the National Institute on Minority Health and Health Disparities.
MESA: This research was supported by the Multi-Ethnic Study of Atherosclerosis (MESA) contracts N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, N01-HC-95169 and by grants UL1-TR-000040 and UL1-RR-025005 from NCRR. The provision of genotyping data was supported in part by the National Center for Advancing Translational Sciences, CTSI grant UL1TR000124, and the National Institute of Diabetes and Digestive and Kidney Disease Diabetes Research Center (DRC) grant DK063491 to the Southern California Diabetes Endocrinology Research Center. The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN268201100003C, HHSN268201100004C, and HHSN271201100004C.”
Erasmus Rucphen family study: The ERF study as a part of EUROSPAN (European Special Populations Research Network) was supported by European Commission FP6 STRP grant number 018947 (LSHG-CT-2006-01947) and also received funding from the European Community's Seventh Framework Programme (FP7/2007-2013)/grant agreement HEALTH-F4-2007-201413 by the European Commission under the programme “Quality of Life and Management of the Living Resources” of 5th Framework Programme (no. QLG2-CT-2002-01254). High-throughput analysis of the ERF data was supported by joint grant from Netherlands Organization for Scientific Research and the Russian Foundation for Basic Research (NWO-RFBR 047.017.043). Exome sequencing analysis in ERF was supported by the ZonMw grant (project 91111025). Najaf Amin is supported by the Hersenstichting Nederland (project number F2013(1)-28).
Rotterdam study: The generation and management of GWAS genotype data for the Rotterdam Study is supported by the Netherlands Organisation of Scientific Research NWO Investments (nr. 175.010.2005.011, 911-03-012). This study is funded by the Research Institute for Diseases in the Elderly (014-93-015; RIDE2), the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) project nr. 050-060-810 and Netherlands Consortium for Healthy Ageing (NCHA). The generation and management of the exome sequencing data for the Rotterdam Study was executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Netherlands. The Exome Sequencing data set was funded by the Netherlands Genomics Initiative (NGI)/Netherlands Organisation for Scientific Research (NWO) sponsored Netherlands Consortium for Healthy Aging (NCHA; project nr. 050-060-810), by the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, and by the and by a Complementation Project of the Biobanking and Biomolecular Research Infrastructure Netherlands (BBMRI-NL; www.bbmri.nl ; project number CP2010-41).
The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, Netherlands Organization for the Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam.
References
- 1.Fuentes RM, Notkola IL, Shemeikka S, Tuomilehto J, Nissinen A. Familial aggregation of blood pressure: a population-based family study in eastern Finland. J Hum Hypertens. 2000;14:441–445. doi: 10.1038/sj.jhh.1001049. [DOI] [PubMed] [Google Scholar]
- 2.Hong Y, de Faire U, Heller DA, McClearn GE, Pedersen N. Genetic and environmental influences on blood pressure in elderly twins. Hypertension. 1994;24:663–670. doi: 10.1161/01.hyp.24.6.663. [DOI] [PubMed] [Google Scholar]
- 3.Stokes J, 3rd, Kannel WB, Wolf PA, D'Agostino RB, Cupples LA. Blood pressure as a risk factor for cardiovascular disease. The Framingham Study--30 years of follow-up. Hypertension. 1989;13:I13–I18. doi: 10.1161/01.hyp.13.5_suppl.i13. [DOI] [PubMed] [Google Scholar]
- 4.Lewington S, Clarke R, Qizilbash N, Peto R, Collins R. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. 2002;360:1903–1913. doi: 10.1016/s0140-6736(02)11911-8. [DOI] [PubMed] [Google Scholar]
- 5.Newton-Cheh C, Johnson T, Gateva V, Tobin MD, Bochud M, Coin L, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41:666–676. doi: 10.1038/ng.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ehret GB, Munroe PB, Rice KM, Bochud M, Johnson AD, Chasman DI, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478:103–109. doi: 10.1038/nature10405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Levy D, Ehret GB, Rice K, Verwoert GC, Launer LJ, Dehghan A, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wain LV, Verwoert GC, O'Reilly PF, Shi G, Johnson T, Johnson AD, et al. Genome-wide association study identifies six new loci influencing pulse pressure and mean arterial pressure. Nat Genet. 2011;43:1005–1011. doi: 10.1038/ng.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franceschini N, Fox E, Zhang Z, Edwards TL, Nalls MA, Sung YJ, et al. Genome-wide association analysis of blood-pressure traits in African-ancestry individuals reveals common associated genes in African and non-African populations. Am J Hum Genet. 2013;93:545–554. doi: 10.1016/j.ajhg.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cruz DN, Simon DB, Nelson-Williams C, Farhi A, Finberg K, Burleson L, et al. Mutations in the Na-Cl cotransporter reduce blood pressure in humans. Hypertension. 2001;37:1458–1464. doi: 10.1161/01.hyp.37.6.1458. [DOI] [PubMed] [Google Scholar]
- 11.Ji W, Foo JN, O'Roak BJ, Zhao H, Larson MG, Simon DB, et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008;40:592–599. doi: 10.1038/ng.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morrison AC, Bis JC, Hwang SJ, Ehret GB, Lumley T, Rice K, et al. Sequence analysis of six blood pressure candidate regions in 4,178 individuals: the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) targeted sequencing study. PLoS One. 2014;9:e109155. doi: 10.1371/journal.pone.0109155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet. 2014;94:233–245. doi: 10.1016/j.ajhg.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schick UM, Auer PL, Bis JC, Lin H, Wei P, Pankratz N, et al. Association of exome sequences with plasma C-reactive protein levels in >9000 participants. Hum Mol Genet. 2015;24:559–571. doi: 10.1093/hmg/ddu450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Psaty BM, O'Donnell CJ, Gudnason V, Lunetta KL, Folsom AR, Rotter JI, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: Design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. The ARIC investigators. Am J Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 17.Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 18.Dawber TR, Meadors GF, Moore FE., Jr Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–281. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The Framingham Offspring Study. Design and preliminary data. Prev Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 20.Taylor HA, Jr, Wilson JG, Jones DW, Sarpong DF, Srinivasan A, Garrison RJ, et al. Toward resolution of cardiovascular health disparities in African Americans: design and methods of the Jackson Heart Study. Ethn Dis. 2005;15:S6–4. 17. [PubMed] [Google Scholar]
- 21.Design of the Women's Health Initiative clinical trial and observational study. The Women's Health Initiative Study Group. Control Clin Trials. 1998;19:61–109. doi: 10.1016/s0197-2456(97)00078-0. [DOI] [PubMed] [Google Scholar]
- 22.Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, et al. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 23.Hofman A, Breteler MM, van Duijn CM, Krestin GP, Pols HA, Stricker BH, et al. The Rotterdam Study: objectives and design update. Eur J Epidemiol. 2007;22:819–829. doi: 10.1007/s10654-007-9199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pardo LM, MacKay I, Oostra B, van Duijn CM, Aulchenko YS. The effect of genetic drift in a young genetically isolated population. Ann Hum Genet. 2005;69:288–295. doi: 10.1046/J.1469-1809.2005.00162.x. [DOI] [PubMed] [Google Scholar]
- 25.Tobin MD, Sheehan NA, Scurrah KJ, Burton PR. Adjusting for treatment effects in studies of quantitative traits: antihypertensive therapy and systolic blood pressure. Stat Med. 2005;24:2911–2935. doi: 10.1002/sim.2165. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Kraja AT, Oberman A, Lewis CE, Ellison RC, Arnett DK, et al. A summary of the effects of antihypertensive medications on measured blood pressure. Am J Hypertens. 2005;18:935–942. doi: 10.1016/j.amjhyper.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Bainbridge MN, Wang M, Wu Y, Newsham I, Muzny DM, Jefferies JL, et al. Targeted enrichment beyond the consensus coding DNA sequence exome reveals exons with higher variant densities. Genome Biol. 2011;12:R68. doi: 10.1186/gb-2011-12-7-r68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34:E2393–E2402. doi: 10.1002/humu.22376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 31.Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. doi: 10.1016/j.ajhg.2011.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flister MJ, Tsaih SW, O'Meara CC, Endres B, Hoffman MJ, Geurts AM, et al. Identifying multiple causative genes at a single GWAS locus. Genome Res. 2013;23:1996–2002. doi: 10.1101/gr.160283.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ehret GB, Caulfield MJ. Genes for blood pressure: an opportunity to understand hypertension. Eur Heart J. 2013;34:951–961. doi: 10.1093/eurheartj/ehs455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jentsch TJ, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 36.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, et al. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–284. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Estevez R, Boettger T, Stein V, Birkenhager R, Otto E, Hildebrandt F, et al. Barttin is a Cl- channel beta-subunit crucial for renal Cl- reabsorption and inner ear K+ secretion. Nature. 2001;414:558–561. doi: 10.1038/35107099. [DOI] [PubMed] [Google Scholar]
- 38.Simon DB, Bindra RS, Mansfield TA, Nelson-Williams C, Mendonca E, Stone R, et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter's syndrome type III. Nat Genet. 1997;17:171–178. doi: 10.1038/ng1097-171. [DOI] [PubMed] [Google Scholar]
- 39.Kato N, Takeuchi F, Tabara Y, Kelly TN, Go MJ, Sim X, et al. Meta-analysis of genome-wide association studies identifies common variants associated with blood pressure variation in east Asians. Nat Genet. 2011;43:531–538. doi: 10.1038/ng.834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kelly TN, Takeuchi F, Tabara Y, Edwards TL, Kim YJ, Chen P, et al. Genome-wide association study meta-analysis reveals transethnic replication of mean arterial and pulse pressure loci. Hypertension. 2013;62:853–859. doi: 10.1161/HYPERTENSIONAHA.113.01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang D, Pang Z, Li S, Jiang W, Wang S, Thomassen M, et al. Genome-wide linkage and association scans for pulse pressure in Chinese twins. Hypertens Res. 2012;35:1051–1057. doi: 10.1038/hr.2012.90. [DOI] [PubMed] [Google Scholar]
- 42.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 43.Ganesh SK, Chasman DI, Larson MG, Guo X, Verwoert G, Bis JC, et al. Effects of long-term averaging of quantitative blood pressure traits on the detection of genetic associations. Am J Hum Genet. 2014;95:49–65. doi: 10.1016/j.ajhg.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.