

Abstract
Acute lung injury (ALI) as a serious diseases with high mortality and is considered a threat to human health and life. A number of studies have focused on the treatment and prevention of lung injury. However, the molecular mechanisms responsible for the development of lung injury are not yet fully understood, and this has impeded the development of effective drugs and treatment strategies. Hence, in the present study, mice with lipopolysaccharide (LPS)-induced ALI were used as a model to investigate the crosstalk between the CX3CL1-CX3CR1 axis and the nuclear factor (NF)-κB signaling pathway in the process of lung injury. CX3CL1-knockout (CX3CL1-KO or CX3CL1−/−) mice were used to examine the role of the CX3CL1-CX3CR1 axis in LPS-induced lung injury. We used baicalin, a natural product, to investigate its anti-inflammatory effects and its protective effects against lung injury. Western blot analysis, reverse transcription-quantitavie PCR (RT-qPCR), immunohistochemistry, enzyme-linked immunosorbent assay (ELISA) and the analysis of biochemical indicators were used to determine the key signaling pathway involved in the development of lung injury. The results indicated that, on the one hand, baicalin exerted potent anti-inflammatory effects by inhibiting the activation of the CX3CL1-CX3CR1 axis and NF-κB, thus preventing the the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway. In addition, the phosphorylation of AKT, which was significantly induced by LPS-induced ALI through the CX3CL1-CX3CR1 axis, was inhibited by treatment with baicalin. On the other hand, we further investigated the role of the CX3CL1-CX3CR1 axis in lung injury. We determined the diffrences in the expression levels of CX3CR1 between wild-type (WT) and CX3CL1−/− mice in order to establish its association with lung injury. Our results indicated that CX3CL1 may be the central and major indicator in the process of lung injury, which mediates the CX3CR1 receptor to activate AKT and further promote NF-κB activation. These findings demonstrate that the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB signaling pathway plays a direct role in LPS-induced lung injury. The inhibition of the activation of the CX3CL1-CX3CR1 axis may thus suppress the development of ALI. In addition, baicalin inhibited the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway in mice with LPS-induced ALI. Thus, treatment with baicalin may be a potential therapeutic strategy for ALI.
Keywords: acute lung injury, baicalin, CX3CL1-CX3CR1 axis, nuclear factor-κB, inflammation, CX3CL1-knockout
Introduction
Acute lung injury (ALI) is a distinct form of acute respiratory failure characterized by diffuse pulmonary infiltrates, progressive hypoxemia, reduced lung compliance and abnormal hydrostatic pressure (1). Over the past 30 or so years, this syndrome has come to be one of the central concerns of intensive care: lung injury arising from a variety of different etiologies, each characterized by bilateral diffuse infiltrates on X-ray, hypoxemia and non-cardiogenic pulmonary edema (2–4). Actually, ALI is caused by any stimulus of local or systemic inflammation, principally sepsis (5). It is most often observed as part of a systemic inflammatory process, particularly systemic sepsis, where the lung manifestations parallel those of other tissues, including widespread destruction of the capillary endothelium, extravasations of protein rich fluid and interstitial edema (6–8). In addition, the alveolar basement membrane is damaged, and fluid seeps into the airspaces, stiffening the lungs and causing ventilation-perfusion mismatch (9,10). According to the clinical diagnosis, there are two major stages: i) the acute phase characterized by the disruption of the alveolar-capillary interface, leakage of protein rich fluid into the interstitium and alveolar space and the excessive release of cytokines and the migration of neutrophils (11–14); and ii) the later reparative phase which is characterized by fibroproliferation, and the organization of lung tissue. ALI is a disease with a high mortality rate (30–50%) (15). However, there are still no effective therapeutic strategies or specific drugs to treat it. Therefore, the development of novel therapies for ALI is urgently needed.
Baicalin, a type of flavonoid, can be found and isolated from several species in the genus Scutellaria, including Scutellaria baicalensis and Scutellaria lateriflora, and has been reported to exert anti-hypertensive, antitumor and anti-inflammatory effects (16–18). As previously demonstrated, ALI involves a typical inflammatory response, and the nuclear factor (NF)-κB signaling pathway is directly involved in the developmental process of lung injury (19,20). Thus, the inhibition of the activation of the NF-κB pathway may prove to be key to the effective treatment of ALI. In addition, the CX3CL1-CX3CR1 axis has also been shown to play an important role in inflammation-related disorders, including hepatitis and fibrosis, nerve damage and tumor development (21–25). The CX3CL1-CX3CR1 axis has the ability to enhance the levels of phosphorylated NF-κB; in turn, the activation of NF-κB continually promotes the CX3CL1-CX3CR1 interaction (26). The crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway plays a critical role in inflammation (27–29). Hence, in this study, we wished to determine whether baicalin inhibits lipopolysaccharide (LPS)-induced ALI by interfering with the activation of the CX3CL1-CX3CR1 axis and NF-κB pathway. Furthermore, we used CX3CL1-knockout (CX3CL1-KO or CX3CL1−/−) mice as a model with which to evaluate the effects of CX3CL1 on inflammatory responses.
Materials and methods
Reagents and animals
Baicalin (CAS: 21967-41-9, HPLC analysis ≥98%, C21H18O11, MW: 446.36102) was purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China) (chemical strucure shown in Fig. 1). LPS was obtained from Sigma-Aldrich China, Inc. (Shanghai, China) and prepared in phosphate-buffered saline (PBS) at the concentration of 100 µg/ml. Anti-CX3CL1 (rabbit; ab25088) and anti-CX3CR1 (rabbit; ab8021) antibodies were obtained from Abcam (Shanghai, China). Anti-AKT (rabbit; 4961), anti-phosphorylated (p-) AKT (rabbit; 9614), anti-IκB (rabbit; 4812), anti-p-IκB (rabbit; 5209), anti-NF-κB (rabbit; 8242), anti-p-NF-κB (rabbit; 3033), anti-cyclooxygenase-2 (COX-2; rabbit; 12282) and anti-p-IKKβ (rabbit; 14938) antibodies were all obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). The tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6 and transforming growth factor (TGF)-β ELISA kits were obtained from R&D Systems, Inc., (Minneapolis, MN, USA). The IL-18 kit was from BioLegend (San Diego, CA, USA). Male BALB/c mice (n=120; 6–8 weeks old, weighing 20–25 g) were obtained from Charles River Laboratories (Wilmington, MA, USA) and housed in a temperature and humidity-controlled environment (25±2°C and 50±10% humidity) with a standard 12-h light/dark cycle with food and water provided in the cages. This study was approved by the Ethics Committee on Animal Research at the Capital Medical University, Beijing, China. All mice were randomly divided into 5 groups as follows: i) the control group; ii) the group treated with 3 mg/kg LPS; iii) the group treated with 3 mg/kg LPS + 50 mg/kg baicalin; iv) the group treated with 3 mg/kg LPS + 100 mg/kg baicalin; and v) the group treated with 3 mg/kg LPS + 200 mg/kg baicalin. The mice were orally administered with the various concentrations of baicalin for 7 days prior to the administration of LPS. LPS was administered to the mice via intraperitoneal injection for 48 h. All animal experiments were performed in accordance with the guide for the Care and Use of Laboratory Animals established by the US National Institutes of Health. In addition, 70 male C57BL/6 mice (6–8 weeks old, weighing 20–25 g) lacking the CX3CL1 gene (CX3CL1-KO or CX3CL1−/−) and wild-type (WT) mice (6–8 weeks old, weighing 20–25 g) were purchased from Beijing Biocytogen Co., Ltd. (Beijing, China). The C57BL/6 mice and WT mice were administered LPS in the same manner as the BALB/c mice.
Figure 1.
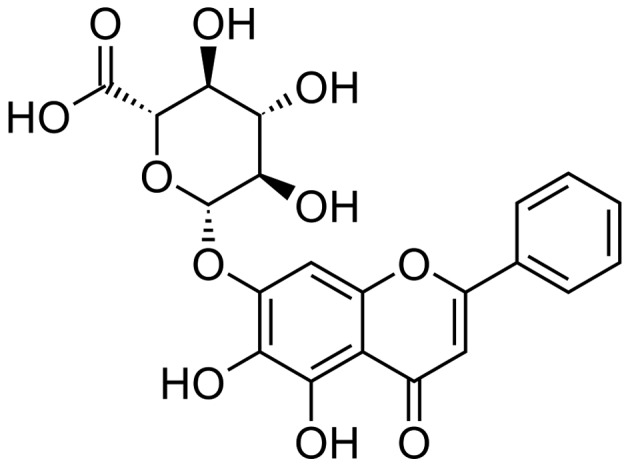
Chemical structure of baicalin.
Isolation and culture of lung epithelial cells
The immune adhesion method was used to isolate lung epithelial cells. In brief, after euthanizing the mice by ether anesthesia, the lungs were excised and the lung tissues of the CX3CL1-KO mice and WT mice were carefully isolated and mantained in Hanks' buffer. Subsequently, 0.25% trypsin (0.5 ml/5 g; Sigma-Aldrich China, Inc.) was added to homogenize the lung tissues at 4°C overnight. The tissues were then treated with an equal volume of trypsin and passed through a stainless steel mesh (size 60;Sigma-Aldrich China, Inc.), and suspended in DMEM/F12 medium (Gibco/Thermo Fisher Scientific, Inc., Waltham, MA, USA). The cell suspensions were centrifuged at 500 rpm for 5 min to remove the debris and impurities. The suspensions were collected and washed with Hanks' buffer 3 times. Subsequently, the cells at 3×106 ml were cultured in IgG-coated 6-wells plate at 37°C in 5% CO2 for 2.5 h. The unattached cells were removed to a new tube and collected by centrifugation at 800 rpm for 4 min. The isolated cells were then re-cultured in 6-wells plates. Cells at passage 3 were used in our experiments. The cells were serum-starved fro 36 h and then stimulated with TNF-α (50 ng/ml) for various periods of time (0, 2, 5, 10 and 15 min). The cells were then subjected to western blot analysis, RT-qPCR and the analysis of NF-κB activity.
Lung wet-to-dry weight ratio
After euthanizing the mice by ether anesthesia, the lungs were excised, blotted dry, weighed to obtain the 'wet' weight, and then placed in an oven at 80°C for 48 h to obtain the 'dry' weight. The ratio of the wet lung to dry lung was calculated to assess tissue edema.
Inflammatory cell counts in bronchoalveolar lavage fluid (BALF)
After the mice were euthanized, the total lung tissues (containing the trachea) of 8–10 mice were collected and subjected to perfusion with Hanks' buffer (0.3 ml, 3 times). The irrigation solution was collected and centrifuged (4°C, 3,000 rpm for 10 min) to pellet the cells The BALF samples were centrifuged (4°C, 3,000 rpm for 10 min) to pellet the cells. The cell pellets were resuspended in PBS for the total cell counts using a hemocytometer, and cytospins were prepared for differential cell counts by staining using the Wright-Giemsa staining method.
Histopathological examination of lung tissues
Histopathological evaluation was performed on the lungs of 8–10 mice that were not subjected to BALF collection. The lungs were fixed with 10% buffered formalin, imbedded in paraffin and sliced. Following hematoxylin and eosin (H&E) staining, the pathological changes of the lung tissues were observed under a light microscope. Some samples were also was subjected to immunohistochemical (IHC) staining (CX3CL1 and IκBα antibody) according to the instructions of the manufacturer (Cell Signaling Technology, Inc.) and were performed by Shanghai Zhenda Biotechnology, Co., Ltd. (Sanghai, China).
Western blot analysis and reverse transcription-quantitative PCR (RT-qPCR)
Six hours after the injection of LPS, the lung tissues were harvested and frozen in liquid nitrogen immediately until homogenization. Proteins were extracted from the lung tissue using T-PER Tissue Protein Extraction Reagent kit (Thermo Fisher Scientific) according to the manufacturer's instructions. Protein concentrations were determined using the BCA protein assay kit, and equal amounts of protein were loaded per well on a 10% sodium dodecyl sulphatepolyacrylamide gel. Subsequently, the proteins were transferred onto polyvinylidene difluoride membranes. The resulting membranes were blocked with Tris-buffered saline containing 0.05% Tween-20 (TBS-T), supplemented with 5% skim milk (Sigma-Aldrich China, Inc.) at room temperature for 2 h on a rotary shaker, followed by washing with TBS-T. The specific primary antibody, diluted in TBST, was incubated with the membrane at 4°C overnight. Subsequently, the membranes were washed with TBS-T followed by incubation with the peroxidase-conjugated secondary antibody at room temperature for 1 h. The immunoactive proteins were detected by using an enhanced chemiluminescence western blotting detection kit. The bands were observed using an ECL western blotting analysis system (GE Healthcare, Pittsburgh, PA, USA) and exposed to Kodak X-ray film.
RT-qPCR, was performed as previously described (30). Total RNA was extracted from tissues and cells using TRIzol® reagent (Invitrogen/Thermo Fisher Scientific, Inc.) and 1 µg total RNA was reverse transcribed using the M MLV RT system (Promega Corp., Madison, WI, USA). This was performed at 42°C for 1 h and terminated by the deactivation of the enzyme at 70°C for 10 min. qPCR was conducted using SYBR®-Green (Bio-Rad Laboratories, Inc., Hercules, CA, USA) in an ABI PRISM 7900HT detection system (Applied Biosystems; Thermo Fisher Scientific, Inc.). All the primers were produced by Thermo Fisher Scientific, Inc. The amplification of the pre-denatured products was conducted at 94°C for 60 sec; followed by 45 cycles at 95°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec; followed by 95°C for 10 sec, 65°C for 45 sec, and 40°C for 60 sec. Fold induction values were calculated using the to 2−ΔΔCq method, where ΔCq represents the differences in cycle threshold number between the target gene and GAPDH, and ΔΔCq represents the relative change in the differences between the control and treatment groups.
Two-dimensional gel electrophoresis
Each gel was loaded with 100 µg protein and mixed with water solution (7 mol/l urea, 2 M thiourea, 4% CHAPS and 0.2% ampholyte). The total sample volume was 290 µl. After hydration of the IPG adhesive strip (24 cm, pH 3–10, NL), the mineral oil was then covered. The isoelectric focusing procedure was set as 30 V/6 h→60 V/6 h→500 V/1 h→1,000 V/1 h→8,000 V/10 h, and the total voltage time product was 64,000 Vh. After isoelectric focusing, the IPG strip was preserved in the SDS equilibrium solution (6 M urea, 2% SDS, 0.02% bromophenol blue liquid reserves) for 15 min. After completion of the equilibrium, the IPG strip was placed above the 12.5% uniform polyacrylamide gel and 10 µl protein marker was added on the SDS gel. The air bubbles were removed and sealed using anhydrous ethanol. Subsequently, two-dimensional gel electrophoresis was performed with bromophenol blue tracking dye until the dye had move to the front of the rubber edge with a total duration of 20 h. Cy3 and Cy5 fluorescence staining was then applied. The protein expression of the TLR4/MyD88/NF-κB signaling-related indicators, AKT, CX3CR1, TNF-α, IL-6, was analyzed in this part. and the image was then subjected to intensity correction, point detection, background subtraction, homogenization and matching processing using DeCyder 2D 6.5 software.
Measurement of cytokine levels by ELISA
The levels of major inflammatory cytokines, such as TNF-α, IL-1β, IL-6, TGF-β and IL-18 levels in the cells or serum from mice were determined using respective ELISA kits, following the manufacturer's instructions. Blood samples were collected from the mice by cardiac puncture following diethyl ether anesthesia. Serum was obtained following centrifugation at 2,000 rpm for 15 min and used for ELISA. Briefly, polyclonal rat anti-mouse cytokine antibodies were used as capturing antibodies and biotinylated polyclonal rat anti-mouse cytokine antibodies for detection, and the standard curve of inflammatory cytokines was created. Color changes were determined at 450 nm.
Statistical analysis
Data are expressed as the means ± SEM. The treated cells, tissues and corresponding controls were compared using GraphPad PRISM (version 6.0; GraphPad Software, Inc., La Jolla, CA, USA) by a one-way ANOVA with Dunn's least significant difference tests. Differences between groups were considered significant at p<0.05.
Results
Baicalin inhibits LPS-induced inflammatory responses in mice
We examined the protective effects of baicalin against LPS-induced lung injury. As shown in Table I, the levels of major inflammatory cytokines in serum, such as TNF-α, interleukin (IL)-1β, IL-6, TGF-β and IL-18 were significantly increased following the administration of LPS. By contrast, in the baicalin-treated mice, the levels of these cytokines were decreased compared with the mice with ALI not treated with baicalin. Of note, the inhibitory effects of baicalin on LPS-induced inflammation in mice occurred in a dose-dependent manner.
Table I.
Effects of baicalin on the expression levels of inflammatory cytokines following LPS-induced lung injury.
| Events (pg/ml) | Control | 3 mg/kg LPS | 3 mg/kg LPS
|
||
|---|---|---|---|---|---|
| 50 mg/kg baicalin | 100 mg/kg baicalin | 200 mg/kg baicalin | |||
| TNF-α | 151.4±8.1 | 801.5±16.2c | 644.3±21.6e | 352.4±11.9e | 218.7±18.5f |
| IL-1β | 23.4±6.2 | 88.3±5.4c | 39.1±6.7e | 42.2±11.7e | 31.8±10.9f |
| IL-6 | 67.3±11.1 | 204.5±12.7c | 168.4 ± 5.2d | 116.4±17.2e | 121.8±14.3e |
| TGF-β | 98.5±5.7 | 235.8±6.7c | 187.6±10.7d | 143.8±7.5e | 132.9±9.9f |
| IL-18 | 195.2±8.3 | 507.2±18.4c | 327.2±14.8e | 298.2±14.7e | 265.5±11.1f |
Mean ± SEM. (n=8–10).
P<0.05,
P<0.01 and
P<0.001 vs. control mice.
P<0.05,
P<0.01 and
P<0.001 vs. LPS group. LPS, lipopolysaccharide. TNF-α, tumor necrosis factor α; IL, interleukin' TGF-β, transforming growth factor β.
Effects of baicalin on lung wet-to-dry weight ratio and inflammatory cell counts in BALF
We wished to determine whether the administration of LPS affects the ratio of lung wet-to-dry weight. As shown in Fig. 2A, the administration of LPS induced a significant increase in the wet-to-dry ratio, compared to the untreated control group. But in baicalin treatment, it has ability to decrease the lung wet-to-dry ratio. However, baicalin inhibited the LPS-induced increase in the lung wet-to-dry weight in a dose-dependent manner. Furthermore, we also examined the effects of baicalin on inflammatory cell counts in BALF. As shown in Fig. 2B–D, treatment with various concentrations of baicalin decreased the number of inflammatory cells in BALF, including neutrophils and macrophages, which was increased following the administration of LPS. Baicalin decreased the number of inflammatory cells in BALF in a dose-dependent manner.
Figure 2.

Effects of baicalin on lipopolysaccharide (LPS)-induced lung injury and inflammatory responses. (A) Wet-to-dry (W/D) ratio of lung tissue. (B–D) Inflammation-related cells counts in bronchoalveolar lavage fluid (BALF). (E) Hematoxylin and eosin (H&E) staining of lung tissue at different time points during LPS-induced lung injury. Data are expressed as the means ± SEM. +P<0.05 and +++P<0.001 vs. control; #P<0.05, ##P<0.01 and ###P<0.001 vs. LPS-challenged group.
Effects of baicalin on CX3CL1 protein expression and NF-κB activation following LPS-induced lung injury
In order to further examine the harmful effects of LPS on lung tissues, LPS-induced lung injury was examined at different time points (3, 6 and 12 h). As shown in Fig. 2E, the administration of LPS significantly enhanced the injury levels to the lungs (histopathological examination revealed blood capillary congestion, necrosis and exfoliation epithelium of the pulmonary alveoli, and the extravasation of fluid in lungs) compared to the control group in a time-dependent manner. In addition, at the 6-h time point (6 h post-LPS administration), we further examined whether treatment with baicalin would reduce the injury levels. As shown in Fig. 3A, treatment with baicalin inhibited the LPS-induced lung injury in a dose-dependent manner. Furthermore, we examined CX3CL1 expression in the lung tissues of mice with LPS-induced ALI (Figs. 3B and 4). The activation of the CX3CL1-CX3CR1 axis was observed in the LPS-treated group, but was inhibited in the baicalin-treated groups. As shown in Fig. 5, the levels of phosphorylated AKT, NF-κB and COX-2 were significantly upregulated following the administration of LPS, but were suppressed following treatment with various concentrations of baicalin.
Figure 3.

Hematoxylin and eosin (H&E) staining of lung tissues following lipopolysaccharide (LPS)-induced lung injury. (A) Protective effects of baicalin against LPS-induced lung injury at various concentrations. A histopathological examination revealed blood capillary congestion, necrosis and exfoliation epithelium of the pulmonary alveoli, and the extravasation of fluid in lungs. (B) Immunohistochemical analysis of CX3CL1 expression in lung tissue following LPS-induced lung injury.
Figure 4.

Effects of baicalin on the CX3CL1-CX3CR1 axis in mice with lipopolysaccharide (LPS)-induced lung injury. (A) Relative CX3CL1 protein expression in LPS-induced lung injury. (B) Relative CX3CR1 protein expression in LPS-induced lung injury. Data are expressed as the means ± SEM. ++P<0.01 and +++P<0.001 vs. control; #P<0.05, ##P<0.01 and ###P<0.001 vs. LPS group.
Figure 5.

Effects of baicalin on NF-κB and AKT pathways in lipopolysaccharide (LPS)-induced lung injury. (A) Phosphorylated AKT protein expression. (B) Phosphorylated IκBα protein expression. (C) Phosphorylated NF-κB protein expression. (D) COX-2 protein experssion. Data are expressed as the means ± SEM. ++P<0.01 and +++P<0.001 vs. control; #P<0.05, ##P<0.01 and ###P<0.001 vs. LPS group.
Inflammatory responses in CX3CL1−/− mice with LPS-induced lung injury
In order to determine the role of CX3CL1 in the pathogenesis of inflammation, WT and CX3CL1−/− mice were sensitized and challenged with LPS. BALF was collected 48 h after the final challenge and analyzed for the infiltration of inflammatory cells. The administration of LPS to the WT mice induced a significant increase in the number of inflammatory cells, including eosinophils, dendritic cells, and lymphocytes and macrophages, compared with non-LPS-challenged mice (Fig. 6). Among the inflammatory cell populations, eosinophils were the most dominant, followed by macrophages, T-cells, dendritic cells and B-cells. The number of inflammatory cells, including the number of eosinophils and lymphocytes in the LPS-challenged CX3CL1−/− mice was significantly lower than in the LPS-challenged WT mice. In addition, the increase in the number of inflammatory cytokines in the LPS-challenged CX3CL1−/− mice was significantly inhibited, compared with the WT mice (Fig. 7). These results demonstrate that a deficiency of CX3CL1 results in reduced inflammatory cell infiltration in BALF following challenge with LPS. We also performed IHC analysis (Fig. 8) to examine the changes in the expression levels of IκBα and CX3CR1 receptor following challenge with LPS. The CX3CL1−/− mice exhibited lower expression levels of IκBα and CX3CR1 than the WT mice. Of note, the decrease in the expression of IκBα and CX3CR1 was observed in a time-dependent manner. This illustrated that a deficiency of CX3CL1 indirectly or directly affects the activation of IκBα and CX3CR1 expression.
Figure 6.

Inflammation-related cell count in wild-type (WT) and CX3CL1−/− mice. (A–E) Relative number of eosinolhis, dendritic, B-cells, T-cells and macrophages in WT and CX3CL1−/− mice. Data are expressed as the means ± SEM. +++P<0.001 vs. PBS WT mice; In LPS group, ##P<0.01 vs. CX3CL1−/− mice. ns, not significant.
Figure 7.

Enzyme-linked immunosorbent assay (ELISA) of inflammatory cytokines in wild-type (WT) and CX3CL1−/− mice. (A–I) ELISA was used to analyze the levels of inflammatory cytokines in WT and CX3CL1−/− mice. Data are expressed as the means ± SEM. ++P<0.01 and +++P<0.001 vs. PBS WT mice; in LPS group, ##P<0.01 and ###P<0.001 vs. CX3CL1−/− mice. ns, not significant.
Figure 8.

Immunohistochemical analysis of IκBα and CX3CR1 expression in wild-type (WT) and CX3CL1−/− mice. (A) Immunohistochemical analysis of IκBα protein expression in WT and CX3CL1−/− mice at different time points. (B) Immunohistochemical analysis of CX3CR1 protein expression in WT and CX3CL1−/− mice at different time points.
AKT-induced activation of the NF-κB signaling pathway in CX3CL1−/− mice challenged with LPS
As shown in Figs. 9, 10 and 11, IHC analysis demonstrated that the AKT pathway was significantly activated following the administration of LPS. In the CX3CL1−/− mice, the phosphorylation levels of AKT were lower than those of the WT mice, and the inhibition of the AKT pathway in the CX3CL1−/− mice was observed in a time-dependent manner. In addition, the results of western blot analysis indicated that the AKT-induced activation of the TLR4/MyD88/NF-κB signaling pathway was significantly inhibited in the CX3CL1−/− mice compared with the WT mice. These findings suggest that the AKT pathway plays a role in the development of LPS-induced lung inflammation, which also directly involves the activation of the CX3CL1-CX3CR1 axis. The AKT-induced activation of the NF-κB signaling pathway was significantly inhibited in the CX3CL1−/− mice challenged with LPS.
Figure 9.

Immunohistochemical analysis of AKT expression in wild-type (WT) and CX3CL1−/− mice. Immunohistochemical analysis of AKT protein expression in WT and CX3CL1−/− mice at different time points.
Figure 10.

Western blot analysis of AKT and NF-κB protein in wild-type (WT) and CX3CL1−/− mice. (A) Phosphorylated AKT protein expression. (B) Phosphorylated IκB protein expression. (C) Phosphorylated IKKβ protein expression. (D) Phosphorylated NF-κB protein expression. Data are expressed as the means ± SEM. +P<0.05, ++P<0.01 and +++P<0.001 vs. CX3CL1−/− mice.
Figure 11.

Two-dimensional gel electrophoresis of AKT, CX3CR1 and NF-κB-related protein expression. Arrows indicate protein imprinting.
Deficiency of CX3CL1 inhibits the inflammatory response through the NF-κB pathway in lung epithelial cells
It has been demonstrated that the activation of the NF-κB pathway is involved in lung-related diseases both in experimental models and in humans (31). Moreover, the CX3CL1-CX3CR1 axis has been shown to be activated in lung inflammation and to regulate the NF-κB pathway in multiple cell systems (32). Thus, in this study, we wished to examine the effects of the CX3CL1-CX3CR1 axis on the NF-κB pathway and on the induction of inflammatory responses following the administration of LPS. As shown in Fig. 12A, the IκBα phosphorylation levels were significantly decreased in the CX3CL1-KO mice compared with the WT mice. The increase in the IκBα phosphorylation level resulted in the activation of the NF-κB pathway in the WT mice compared to the CX3CL1-KO mice (Fig. 12B). Of note, in the epithelial cells in which CX3CL1 was silenced, the levels of inflammatory cytokines were a lower than the WT group (Fig. 12C). Thus, these findings demonstrate that CX3CL1 plays a key role in LPS-induced inflammatory responses.
Figure 12.

The deficiency of CX3CL1 (CX3CL1−/−) inhibits tumor necrosis factor (TNF)-α induced inflammatory responses in lung epithelial cells. (A) Western blot analysis of phosphorylated IκB protein expression in TNF-α stimulated lung epithelial cells from wild-type (WT) and CX3CL1-knockout (KO) mice. (B) Relative nuclear factor (NF)-κB activity in cells from WT and CX3CL1-KO mice. (C) RTq-PCR analysis of inflammatory cytokine expression in TNF-α stimulated lung epithelial cells from WT and CX3CL1-KO mice. Data are expressed as the means ± SEM. +P<0.05 and ++P<0.01 vs. CX3CL1−/− mice; #P<0.05 vs. CX3CL1−/− mice in non-TNF-α administration.
Discussion
ALI as a critical illness consisting of hypoxemic respiratory failure with bilateral pulmonary infiltrates, and poses a serious threat to the health and lives populations worldwide (1,11). Moreover, as with acute respiratory distress syndrome, the formation and development of ALI are associated with high morbidity and mortality. As previously demonstated, some natural products have been used in animal experiments and in clinical studies in an aim to prevent ALI, with some promising results (2,4). However, the detailed underlying molecular mechanisms controlling lung injury are not yet fully understood. Effective treatments for lung-related diseases have yet to be developed. Baicalin, as a type of flavonoid, can be found and extracted from natural plants. It has been shown to exert anti-hypertensive, antitumor and anti-inflammatory effects (17,18). Thus, in this regard, we wished to determine whether baicalin has the ability to suppress LPS-induced ALI and to elucidate the molecular mechanisms through which it inhibits lung damage.
Firstly, we used BALB/c mice to construct a model of LPS-induced ALI in order to examine the protective effects of baicalin. Our findings indicated that treatment with baicalin inhibited the release of inflammatory cytokines, including TNF-α, IL-1β, IL-6, TGF-β and IL-18 in serum following the administration of LPS. Moreover, the inflammatory cell counts in BALF obtained from mice with LPS-induced lung injury were significantly reduced following treatment with various concentrations of baicalin. Treatment with baicalin also restored the lung wet-to-dry weight ratio. Furthermore, we examined the crucial role of the CX3CL1/CX3CR1 axis in LPS-induced lung injury. The results revealed that the CX3CL1/CX3CR1 axis is involved in the process of lung injury and is activated by LPS. The activation of the CX3CL1/CX3CR1 axis further increased the levels of NF-κB phosphorylation. As previously reported, the activation of the NF-κB pathway leads to inflammatory responses and cytokine production (33). In this study, the results of western blot analysis provide evidence of this. The protective effects of baicalin against LPS-induced lung injury occurred in a concentration-dependent manner. Some researchers have also indicated that the activation of the CX3CL1/CX3CR1 axis is often associated with the activation of the AKT pathway (34). Thus, we examined the association of the of AKT pathway with the CX3CL1/CX3CR1 axis. Our findings demonstrated that the administration of LPS to mice activated the AKT pathway and increased the phosphorylation levels of AKT. Phosphorylated AKT significantly activated the CX3CL1/CX3CR1 axis and the NF-κB pathway. Treatment with various concentrations of baicalin inhibited the activation of the NF-κB pathway, the CX3CL1/CX3CR1 crosstalk with other proteins, as well as the phosphorylation of AKT.
In addition, in order to examine the possible role of the CX3CL1/CX3CR1 axis in LPS-induced lung injury in mice, we used CX3CL1-KO mice as a model to examine the effects of the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway. IHC analysis of IκB at different time points (2, 4 and 8 h) of LPS administration indicated that IκB was involved in the development of LPS-induced lung injury in a time-dependent manner. Compared with the WT mice, the CX3CL1−/− mice exhibited a decreased in the levels of phosphorylated IκBα in a time-dependent manner. As the receptor of CX3CL1, we examined CX3CR1 expression between the WT and CX3CL1−/− mice by IHC analysis. The results indicated that the knockdown of CX3CL1 directly affects the expression of CX3CR1. The high expression of CX3CL1 induces the expression of CX3CR1. These data are consistent with those of another published study (35). As we mentioned above, the AKT signaling pathway may directly or indirectly be associated with LPS-induced lung injury. In the knockout mice, the expression of phosphorylated AKT was determined to be involved in the process of injury. These data indicated that AKT (upstream of the CX3CL1-CX3CR1 axis) may affect CX3CL1-mediated inflammatory responses. The inhibition of the activation of AKT may also suppress CX3CL1-CX3CR1 activation.
In conclusion, ALI as a serious diseases with a high mortality and poses a threat to human health and life. Nowadays, chemoprophylaxis using natural products isolated from plants and flora is a safe way of preventing a number of diseases, including hepatitis, cancer, diabetes, cardiac-cerebral vascular disease, kidney injury and lung injury (36,37). As regards lung injury, a number of drugs have been shown to exert anti-inflammatory effects (37); however, the molecular mechanisms responsible for the development of lung injury are not yet fully understood. Thus, in this regard, we used a mouse model of LPS-induced lung injury to determine the role of the CX3CL1-CX3CR1 axis and its related indicators in the development of lung injury, as well as the anti-inflammatory effects of baicalin. We found that baicalin, as a natural product isolated from the genus Scutellaria, exerts potent anti-inflammatory effects by inhibiting the activation of the CX3CL1-CX3CR1 axis and NF-κB, thus preventing the crosstalk between the CX3CL1-CX3CR1 axis and NF-κB pathway. In addition, the phosphorylation of AKT was significantly increased following LPS-induced lung injury through the CX3CL1-CX3CR1 axis, and was inhibited by treatment with baicalin. We also examined the role of the CX3CL1-CX3CR1 axis in the development of lung injury. We examined the differences in the expression of CX3CR1 between WT and CX3CL1−/− mice with LPS-induced lung injury. Our results indicated that CX3CL1 may be the central and major indicator in the process of lung injury, which mediates the CX3CR1 receptor and activates the AKT pathway and further promotes the activation of NF-κB.
References
- 1.Phua J, Badia JR, Adhikari NK, Friedrich JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, et al. Has mortality from acute respiratory distress syndrome decreased over time?: a systematic review. Am J Respir Crit Care Med. 2009;179:220–227. doi: 10.1164/rccm.200805-722OC. [DOI] [PubMed] [Google Scholar]
- 2.Rubenfeld GD, Caldwell E, Peabody E, Weaver J, Martin DP, Neff M, Stern EJ, Hudson LD. Incidence and outcomes of acute lung injury. N Engl J Med. 2005;353:1685–1693. doi: 10.1056/NEJMoa050333. [DOI] [PubMed] [Google Scholar]
- 3.Muñoz NM, Meliton AY, Meliton LN, Dudek SM, Leff AR. Secretory group V phospholipase A2 regulates acute lung injury and neutrophilic inflammation caused by LPS in mice. Am J Physiol Lung Cell Mol Physiol. 2009;296:L879–L887. doi: 10.1152/ajplung.90580.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu XL, Xie QM, Shen YH, Jiang JJ, Chen YY, Yao HY, Zhou JY. Mannose prevents lipopolysaccharide-induced acute lung injury in rats. Inflamm Res. 2008;57:104–110. doi: 10.1007/s00011-007-7037-y. [DOI] [PubMed] [Google Scholar]
- 5.Matthay MA, Zemans RL. The acute respiratory distress syndrome: Pathogenesis and treatment. Annu Rev Pathol. 2011;6:147–163. doi: 10.1146/annurev-pathol-011110-130158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 7.Li B, Yang J, Huang Q, Zhang Y, Peng C, Zhang Y, He Y, Shi J, Li W, Hu J, et al. Biodistribution and pulmonary toxicity of intratracheally instilled graphene oxide in mice. NPG Asia Mater. 2013;5:e44. doi: 10.1038/am.2013.7. [DOI] [Google Scholar]
- 8.Spragg RG, Bernard GR, Checkley W, Curtis JR, Gajic O, Guyatt G, Hall J, Israel E, Jain M, Needham DM, et al. Beyond mortality: future clinical research in acute lung injury. Am J Respir Crit Care Med. 2010;181:1121–1127. doi: 10.1164/rccm.201001-0024WS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci. 2004;77:126–134. doi: 10.1093/toxsci/kfg243. [DOI] [PubMed] [Google Scholar]
- 10.Song Y, Fukuda N, Bai C, Ma T, Matthay MA, Verkman AS. Role of aquaporins in alveolar fluid clearance in neonatal and adult lung, and in oedema formation following acute lung injury: studies in transgenic aquaporin null mice. J Physiol. 2000;525:771–779. doi: 10.1111/j.1469-7793.2000.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin TR, Matute-Bello G. Experimental models and emerging hypotheses for acute lung injury. Crit Care Clin. 2011;27:735–752. doi: 10.1016/j.ccc.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reiss LK, Uhlig U, Uhlig S. Models and mechanisms of acute lung injury caused by direct insults. Eur J Cell Biol. 2012;91:590–601. doi: 10.1016/j.ejcb.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Liu L, Gao Z, Xia C, Xu Y, Ma Z, Dong C, Li B. Comparative study of trans-oral and trans-tracheal intratracheal instillations in a murine model of acute lung injury. Anat Rec (Hoboken) 2012;295:1513–1519. doi: 10.1002/ar.22531. [DOI] [PubMed] [Google Scholar]
- 14.Su X, Bai C, Hong Q, Zhu D, He L, Wu J, Ding F, Fang X, Matthay MA. Effect of continuous hemofiltration on hemodynamics, lung inflammation and pulmonary edema in a canine model of acute lung injury. Intensive Care Med. 2003;29:2034–2042. doi: 10.1007/s00134-003-2017-3. [DOI] [PubMed] [Google Scholar]
- 15.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest. 2012;122:2731–2740. doi: 10.1172/JCI60331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evers DL, Chao CF, Wang X, Zhang Z, Huong SM, Huang ES. Human cytomegalovirus-inhibitory flavonoids: studies on antiviral activity and mechanism of action. Antiviral Res. 2005;68:124–134. doi: 10.1016/j.antiviral.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wan QF, Gu LG, Yin SJ, Qi GJ, Zhang S, Li GM, Ge DY. Protection effect of baicalin on lung injury of mice infected with influenza FM1. Chin J Tradit Chin Med Pharm. 2011:2848–2851. In Chinese. [Google Scholar]
- 18.Wan QF, Gu LG, Yin SJ, Ge DY, Li GM. Effect of baicalin on cell apoptosis FAS/FAS-L system of pneumonia mice lung tissue infected with FM1. Chin Pharmacol Bull. 2011;27:1555–1559. In Chinese. [Google Scholar]
- 19.Vergadi E, Vaporidi K, Theodorakis EE, Doxaki C, Lagoudaki E, Ieronymaki E, Alexaki VI, Helms M, Kondili E, Soennichsen B, et al. Akt2 deficiency protects from acute lung injury via alternative macrophage activation and miR-146a induction in mice. J Immunol. 2014;192:394–406. doi: 10.4049/jimmunol.1300959. [DOI] [PubMed] [Google Scholar]
- 20.Balakrishna S, Song W, Achanta S, Doran SF, Liu B, Kaelberer MM, Yu Z, Sui A, Cheung M, Leishman E, et al. TRPV4 inhibition counteracts edema and inflammation and improves pulmonary function and oxygen saturation in chemically induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2014;307:L158–L172. doi: 10.1152/ajplung.00065.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wasmuth HE, Zaldivar MM, Berres ML, Werth A, Scholten D, Hillebrandt S, Tacke F, Schmitz P, Dahl E, Wiederholt T, et al. The fractalkine receptor CX3CR1 is involved in liver fibrosis due to chronic hepatitis C infection. J Hepatol. 2008;48:208–215. doi: 10.1016/j.jhep.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Aoyama T, Inokuchi S, Brenner DA, Seki E. CX3CL1-CX3CR1 interaction prevents carbon tetrachloride-induced liver inflammation and fibrosis in mice. Hepatology. 2010;52:1390–1400. doi: 10.1002/hep.23795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Isse K, Harada K, Zen Y, Kamihira T, Shimoda S, Harada M, Nakanuma Y. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology. 2005;41:506–516. doi: 10.1002/hep.20582. [DOI] [PubMed] [Google Scholar]
- 24.Zhuang ZY, Kawasaki Y, Tan PH, Wen YR, Huang J, Ji RR. Role of the CX3CR1/p38 MAPK pathway in spinal microglia for the development of neuropathic pain following nerve injury-induced cleavage of fractalkine. Brain Behav Immun. 2007;21:642–651. doi: 10.1016/j.bbi.2006.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee KM, Jeon SM, Cho HJ. Interleukin-6 induces microglial CX3CR1 expression in the spinal cord after peripheral nerve injury through the activation of p38 MAPK. Eur J Pain. 2010;14:682.e1–12. doi: 10.1016/j.ejpain.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 26.Nishiyori A, Minami M, Ohtani Y, Takami S, Yamamoto J, Kawaguchi N, Kume T, Akaike A, Satoh M. Localization of fractalkine and CX3CR1 mRNAs in rat brain: does fractalkine play a role in signaling from neuron to microglia? FEBS Lett. 1998;429:167–172. doi: 10.1016/S0014-5793(98)00583-3. [DOI] [PubMed] [Google Scholar]
- 27.Andréasson U, Ek S, Merz H, Rosenquist R, Andersen N, Jerkeman M, Dictor M, Borrebaeck CA. B cell lymphomas express CX3CR1 a non-B cell lineage adhesion molecule. Cancer Lett. 2008;259:138–145. doi: 10.1016/j.canlet.2007.10.031. [DOI] [PubMed] [Google Scholar]
- 28.Erreni M, Solinas G, Brescia P, Osti D, Zunino F, Colombo P, Destro A, Roncalli M, Mantovani A, Draghi R, et al. Human glioblastoma tumours and neural cancer stem cells express the chemokine CX3CL1 and its receptor CX3CR1. Eur J Cancer. 2010;46:3383–3392. doi: 10.1016/j.ejca.2010.07.022. [DOI] [PubMed] [Google Scholar]
- 29.Rodero M, Marie Y, Coudert M, Blondet E, Mokhtari K, Rousseau A, Raoul W, Carpentier C, Sennlaub F, Deterre P, et al. Polymorphism in the microglial cell-mobilizing CX3CR1 gene is associated with survival in patients with glioblastoma. J Clin Oncol. 2008;26:5957–5964. doi: 10.1200/JCO.2008.17.2833. [DOI] [PubMed] [Google Scholar]
- 30.Babar IA, Czochor J, Steinmetz A, Weidhaas JB, Glazer PM, Slack FJ. Inhibition of hypoxia-induced miR-155 radiosensitizes hypoxic lung cancer cells. Cancer Biol Ther. 2011;12:908–914. doi: 10.4161/cbt.12.10.17681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dolcet X, Llobet D, Pallares J, Matias-Guiu X. NF-kB in development and progression of human cancer. Virchows Arch. 2005;446:475–482. doi: 10.1007/s00428-005-1264-9. [DOI] [PubMed] [Google Scholar]
- 32.McComb JG, Ranganathan M, Liu XH, Pilewski JM, Ray P, Watkins SC, Choi AM, Lee JS. CX3CL1 up-regulation is associated with recruitment of CX3CR1+ mononuclear phagocytes and T lymphocytes in the lungs during cigarette smoke-induced emphysema. Am J Pathol. 2008;173:949–961. doi: 10.2353/ajpath.2008.071034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valdivia-Silva JE, Franco-Barraza J, Silva AL, Pont GD, Soldevila G, Meza I, García-Zepeda EA. Effect of pro-inflammatory cytokine stimulation on human breast cancer: implications of chemokine receptor expression in cancer metastasis. Cancer Lett. 2009;283:176–185. doi: 10.1016/j.canlet.2009.03.040. [DOI] [PubMed] [Google Scholar]
- 34.Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004;64:4693–4698. doi: 10.1158/0008-5472.CAN-03-3437. [DOI] [PubMed] [Google Scholar]
- 35.Jamieson WL, Shimizu S, D'Ambrosio JA, Meucci O, Fatatis A. CX3CR1 is expressed by prostate epithelial cells and androgens regulate the levels of CX3CL1/fractalkine in the bone marrow: potential role in prostate cancer bone tropism. Cancer Res. 2008;68:1715–1722. doi: 10.1158/0008-5472.CAN-07-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cragg GM, Newman DJ. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta. 2013;1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]