

Abstract
The synthesis of all 20 common natural proteinogenic and 4 otherα-amino acid-isosteric α-amino tetrazoles has been accomplished, whereby the carboxyl group is replaced by the isosteric 5-tetrazolyl group. The short process involves the use of the key Ugi tetrazole reaction followed by deprotection chemistries. The tetrazole group is bioisosteric to the carboxylic acid and is widely used in medicinal chemistry and drug design. Surprisingly, several of the common α-amino acid-isosteric α-amino tetrazoles are unknown up to now. Therefore a rapid synthetic access to this compound class and non-natural derivatives is of high interest to advance the field.
Keywords: amino acids, bioisosterism, multicomponent reactions, synthetic methods, tetrazoles
Introduction
α-Amino acids are among the most important known classes of organic compounds, with multiple applications in nutrition, medicine, materials, chemistry, and biochemistry.[1] Sometimes, however, the physicochemical properties of α-amino acids are not suitable for a specific application and isosteric derivatives can be more suitable. For example some α-amino acids have a very high metabolic turnover or a low solubility at a specific pH. Well established isosteres of the carboxylic acid are hydroxamic, phosphonic and sulfonic acids, sulfonamides, acylsul-fonamides, and tetrazoles, to name just a few.[2] A specialized bioisostere database mentions >2000 COOH bioisosteres.[3] Among COOH isosteres, the tetrazole group is of special interest since it has a comparable pKa, a similar size, a similar spatial arrangement of the heteroatom lone pairs, and a similar molecular electrostatic potential, and therefore often undergoes a very similar receptor–ligand interaction (Figure 1).[4] However, the tetrazole group often exhibits a prolonged half-life, owing to enhanced metabolic stability,[5] enhanced spatial delocalization of the negative charge, and better membrane penetration resulting from increased lipophilicity.[6] Therefore, tetrazole represents the first-choice bioisosteric group if the corresponding COOH has issues in medicinal chemistry projects.[7]
Figure 1.
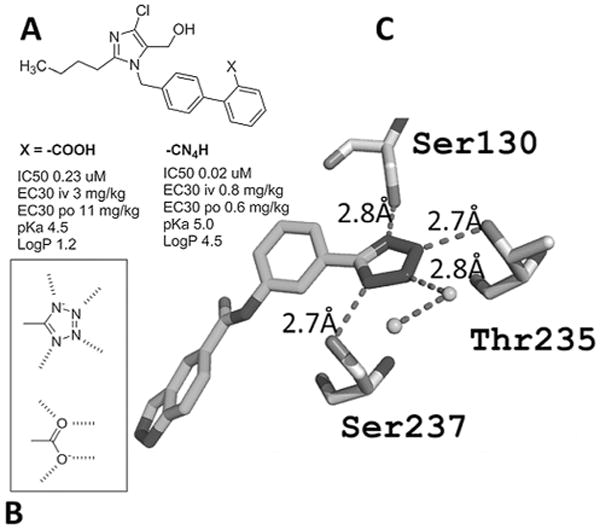
A) The bioisosteric carboxylic acid and tetrazolyl moieties in two Losartan derivatives;[8] B) the four lone pairs of the four nitrogen atoms of tetrazole and the two oxygen atoms of the carboxylic acid point into similar regions in space; C) receptor ligand interactions around a tetrazolate moiety in a β-lactamase (PDB ID:4DE1) showing the involvement of all four nitrogen lone pairs.[9]
In 1959, McManus and Herbst first reported two independent methods to prepare several α-amino acid tetrazole analogues.[10] Later, in 2002, Demko and Sharpless disclosed a novel [3+2] cycloaddition of azide onto nitrile or nitrilium to synthesize several endogenous α-amino acid-bioisosteric α-amino tetrazoles.[11] However, these approaches possess several disadvantages, such as high reaction temperatures, use of toxic reagents, metal catalyst utilization, and failure to synthesize all 20 natural proteinogenic α-amino acid-bioisosteric tetrazoles (Scheme 1).
Scheme 1.

Known syntheses of α-amino acid-bioisosteric α-amino tetrazoles and our approach.
To our knowledge, the preparation of all 20 natural proteinogenic α-amino acid-bioisosteric tetrazoles by one synthetic strategy has not been reported to date. Multicomponent reactions (MCRs) are a powerful way to afford the desired target compounds with great structural diversity in a short reaction sequences.[12] Isocyanide-based multicomponent reactions (IMCRs) are of special interest, owing to the variety of starting materials available and a plethora of transformations reported.[13] Ugi and Meyr reported the first application of IMCRs to synthesize tetrazole derivatives in 1961.[14] They employed NaN3 to replace the carboxylic acid in the 3-component Passerini reaction to give 1,5-disubstituted α-hydroxymethyl tetrazole derivatives. Afterwards, numerous tetrazole-fused structure-varied derivatives were synthesized by IMCRs to yield libraries of screening compounds applied in high-throughput screening.[15]
In this study, we instead employed the IMCR Ugi tetrazole synthesis with suitable starting materials aldehyde 1, cleavable amine 2, and isocyanide 4 in the presence of the azide source TMSN3 3 to access α-amino acid-bioisosteric α-amino tetrazoles 6 in a convergent way and to synthesize, for the first time, all the 20 endogenous α-amino acids isosteres (Scheme 1). Although the Ugi tetrazole synthesis is well established and has found multiple applications in the synthesis of different scaffolds, there has, to our knowledge, been no report on the use of this reaction to afford α-amino acid-bioisosteric α-amino tetrazoles. Therefore, we describe herein the synthesis of all 20 natural proteinogenic and 4 other amino acid analogues through the TMSN3 IMCR process.
Results and Discussion
In this study, we aimed to establish a novel method based on IMCRs to conveniently synthesize α-amino acid isosteric tetrazoles. Four protecting-group strategies were devised to account for compatibility with the amino acid side chains (Scheme 2): trityl/tert-octyl strategy, benzyl/benzyl strategy, mixed strategy, and Schiff base insertion strategy.
Scheme 2.

Four different protecting-group strategies to target α-amino acid-bioisosteric α-amino tetrazoles.
It is well established that ammonia is a poor amine component in most IMCRs, leading to multiple side products and overall poor yields.[16] Therefore an ammonia surrogate must be used in our MCR strategy. Based on our previous work, tritylamine 7 could be used as such in the Ugi reaction, giving moderate to high yields after mild cleavage of the trityl protecting group.[17] The combination of 7 and Walborsky's isocyanide (tert-octylisocyanide) 8 allowed for simultaneous deprotection of the Ugi products to give the α-amino tetrazoles in just one simple step (Scheme 2).
We therefore mixed the commercially available aldehydes and tritylamine 7 in the presence of methanol and heated in a microwave at 100°C for 15 min to obtain the Schiff base and then added TMSN3 3 and tert-octyl isocyanides 8, again under microwave irradiation, to yield the Ugi products containing simple aliphatic or aromatic side chains (5a–l). Notably, the high reaction temperature was not necessary for the Ugi ring closure but to force the Schiff base formation, as reported previously.[17] The final Ugi tetrazoles 5a–l were isolated in moderate to excellent yields by column chromatography on silica (Table 1). Next, the trityl and tert-octyl groups were simultaneously removed in ethanolic HCl (1.25 m) by heating at reflux overnight to give the α-amino acid-isosteric tetrazoles 6a–l in moderate to excellent yields (Table 1). For the amino acids Glu and Lys, the aldehydes are not commercially available and had to be synthesized. Aldehyde 10 for Glu Ugi product 5d could be conveniently synthesized from the commercial mixed acid chloride methyl ester 9 (Scheme 4).[18] Glu analogue 6d could be obtained by simultaneous removal of the trityl group and the tert-octyl group, as well as hydrolysis of the methyl ester by heating the Ugi product 5d at reflux in ethanolic 1.25 m HCl (Scheme 3). Aldehyde 13, for the Lys Ugi product 5e, was synthesized by N-Boc protection of commercial alcohol 11, followed by Swern oxidation at −78°C (Scheme 4).[19] Lys analogue 6e could be obtained by simultaneous removal of the Boc, trityl and tert-octyl groups by heating the Ugi product 5e at reflux in ethanolic 1.25 m HCl (Scheme 3). Racemic and commercial 2-methylbutanal led to the Ile Ugi product 5h and the Ile analogue 6h as a mixture of two diastereomers in 1:1 ratio (Table 1, 5h and 6h). This lack of stereoselectivity is a well-established feature of Ugi reactions.[20]
Table 1.
Trityl/tert-octyl strategy. Yields of the Ugi products (5a–l) and α-amino acid tetrazoles (6a–l).

|
Purification was carried out on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3.
Scheme 4.

Synthesis of aldehydes 10 and 13 for Ugi products 5d (Glu) and 5e (Lys).
Scheme 3.

Simultaneous deprotection of Ugi products 5d and 5e.
For side-chain-functionalized amino acid derivatives, the trityl/tert-octyl strategy did not work because of the lack of Schiff base formation, presumably due to the harsh reaction conditions required for trityl-Schiff base formation. Therefore, in several cases, a benzyl/benzyl strategy was employed. In contrast to the trityl/tert-octyl strategy, the benzyl amine 14 and benzylisocyanide 15 were used, which reacted smoothly with the aldehydes and TMSN3 3 at room temperature to give the Ugi tetrazoles in moderate to excellent yields (Table 2). Notably, for the Thr isostere 5r, a simultaneous ester hydrolysis took place during the Ugi reaction (Scheme 5). Similar to Ile precursor 5h, the race-mic aldehyde 21 led to the Thr precursor 5r and Ile analogue 6r as a mixture of two diastereomers in 1:1 ratio (Table 2, 5r and 6r). For the analogous Tyr, Thr, and Trp Ugi products, the aldehydes are also not commercially available and had to be synthesized. Commercial 16 was first esterified in re-fluxing methanolic HCl, yielding methylester 17; subsequent reduction with LiAlH4 and then Parikh–Doering oxidation were performed to give aldehyde 18.[21] Aldehyde 21, for the Thr Ugi product 5q, was synthesized by using a previously reported procedure.[22] Firstly, 4,4-dimethoxybutan-2-one 19 was reduced by NaBH4 at low temperature. The obtained alcohol was acetylated by acetic anhydride in the presence of trimethylamine to form acetyl acetal 20. Subsequently, compound 20 was deprotected by using cation-exchange resin Dowex 50 to give 4-ox-obutan-2-yl acetate 21. Aldehyde 24 was prepared by a similar method to aldehyde 18. Commercial acetic acid 22 was first reduced to alcohol 23 by using LiAlH4 in THF followed by Parikh–Doering oxidation (Scheme 6). It is important to note that aldehydes 18, 21, and 24 are unstable and cannot be stored and should therefore be used immediately after synthesis.
Table 2.
Benzyl/benzyl strategy. Yields of the Ugi products (5p-s) and α-amino acid tetrazoles (6p-s).

|
Purification was carried out on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3.
Scheme 5.

The Ugi reaction and an unexpected spontaneous hydrolysis to form Thr Ugi product 5r.
Scheme 6.

The preparation of aldehydes 18, 21 and 24 for Ugi products 5q (Tyr), 5r (Thr), and 5s (Trp).
For the other side-chain-functionalized amino derivatives (5m, 5n, 5t, 5u, and 5x), a mixed strategy was performed (Table 3). The trityl/tert-octyl strategy could not be employed for the Gln Ugi product 5m because the amide side chain is destroyed upon refluxing in an acidic solution required for the tert-octyl cleavage. Moreover, the benzyl/benzyl strategy was also not suitable, owing to the spontaneous formation of γ-lactam 5m″ (Scheme 7). Therefore we applied a trityl/benzyl strategy to produce Gln Ugi product 5m. The bulky tritylamine group in 5m avoided the spontaneous γ-lactamization that occurred for the benzyl analogue 5m″. To afford Gln precursor 5m′, the methyl ester in Ugi product 5m was hydrolyzed by LiOH. The formation of amide 5m′ was accomplished by a coupling step with 1-hydroxybenzotriazole (HOBt), 1-ethyl-3-(3-di-methylaminopropyl)carbodiimide (EDCI), N,N-diisopropylethyl-amine (DIEA), and (NH4)2CO3 as the ammonia source.[23] The de-protection of amide 5m′ to yield Gln analogue 6m was carried out in two more steps; trityl removal under mild acidic condition with trifluoroacetic acid (TFA) in CH2Cl2 at room temperature and hydrogenolytic benzyl deprotection by H2 and Pd on charcoal. Aldehyde 10, for Gln, was also used for Glu (Scheme 4). Similarly the tritylamine strategy is not compatible with Asp or Asn Ugi products. Although the Schiff base 25 was clearly formed, the subsequent Ugi reaction did not proceed, even under prolonged reaction times, presumably due to the bulkiness and enolic character of 25 (Scheme 7) blocking the attack of the isocyanide 41. Alternatively, we employed the benzyl/benzyl strategy to prepare the Ugi product 5n. Deprotection to 6n was accomplished by first ester hydrolysis using aqueous LiOH, followed by double debenzylation with H2 and Pd on charcoal. Notably, the benzyl/benzyl strategy also facilitated the transformation of carboxylic acid to amide without any loss of amide under the mild deprotection conditions for benzyl groups (Scheme 7). Likewise, for amide 5o the same coupling reaction as for amide 5m′ was carried out (Scheme 7). For the His Ugi product 5t, the trityl-protected imidazole aldehyde 30 had to be synthesized starting from commercial imidazole acetic acid 28, by a sequence of esterification, tritylation, and reduction by using diisobutylaluminium hydride (DIBAL) at low temperature (Scheme 8).[24] Aldehyde 30 was used in the Ugi reaction directly without further purification to afford the Ugi product 5t. His analogue 6t was obtained by TFA-mediated hydrolysis using triisopropylsilane (ITS) as a cation scavenger in medium yield (57%; Scheme 7).[25] For Arg Ugi product 5u, a more complicated synthetic strategy was required based on the demanding guanidine side chain. We utilized the benzyl/benzyl strategy to obtain Ugi product 5u. Before forming the guanidine group, the tailed NH2 group was deprotected under mild acidic conditions and was neutralized to the free amine. Next, di-tert-butyl thiourea with Mu-kaiyama's reagent as a coupling reagent was added in the presence of triethylamine to form di-Boc-protected guanidine-containing tetrazole derivative 5u′ (Scheme 7).[26] Di-tert-butyl thiourea 32 was prepared as described previously.[27] The Boc groups in the guanidine moiety in 5u′ were removed by using trii-sopropylsilane as a cation scavenger with TFA, followed by hydrogenolytical benzyl group removal leading to Arg analogue 6u in good yield (47%, two steps). Ornithine is another non-proteinogenic endogenous α-amino acid of great importance, being the metabolic product of l-arginine formed by the enzyme arginase. Thus we also synthesized Orn analogue 6x by simply removing the Boc group and benzyl groups of the Ugi product 5x (Scheme 7). The non-commercial aldehyde 35, for 5x, was prepared by using a procedure similar to that for aldehyde 13 (Scheme 8).
Table 3.
Mixed strategy: Yields of the Ugi products (5m–o and 5t–x) and α-amino acid tetrazoles (6m–o and 6t–x).

|
Purification was carried out on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3.
Scheme 7.
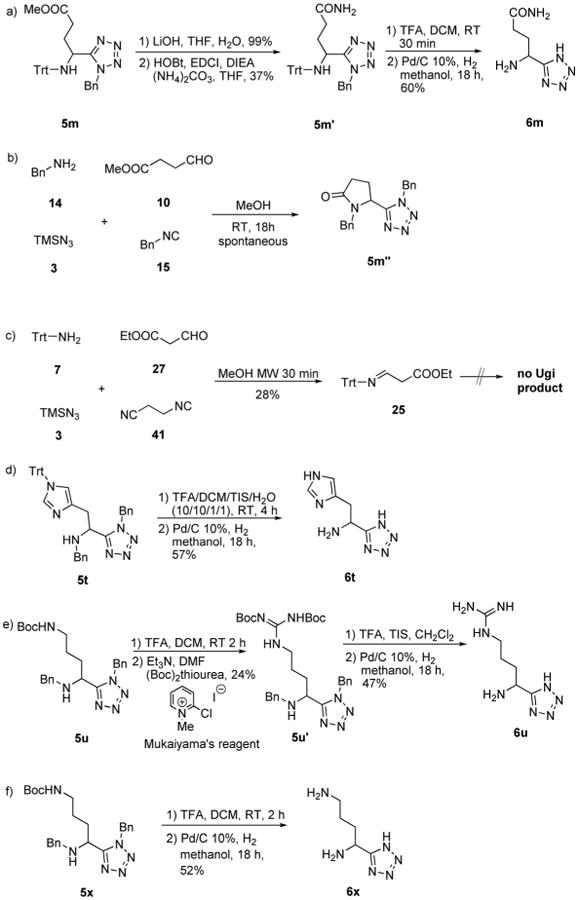
a) Synthesis of compound 5m′ and multistep deprotection to afford 6m; b) unexpected spontaneous cyclization to afford compound 5m″ by using the benzyl/benzyl strategy; c) Schiff base 25 formed by using tritylamine as an amino source; d) de-protection of 5t to give His analogue 6t; e) insertion of guanidine group to lead to compound 5u′ and deprotection to afford Arg analogue 6u; f) deprotection of 5x to give Orn analogue 6x.
Scheme 8.

Synthesis of aldehydes 27, 30, and 35 for Ugi products 5n, 5s and 5u and preparation of di-tert-butyl sulfinyldicarbamate 32.
For the amino acid analogues of Pro and Cys, yet another strategy had to be devised. For the Cys analogue, the harsh reaction conditions in the trityl/tert-octyl strategy could not afford the desired Ugi product either with mercaptoacetaldehyde dimer 39 or with chloroacetaldehyde 42 (Scheme 10). By using the benzyl/benzyl strategy, 42 worked well in the Ugi reaction and 43 could be isolated in 63% yield. From there, we envisioned the introduction of the mercapto side chain by nucleophilic substitution with NaSH or Na-SOCCH3. However, the attempted deprotection of the two Bn groups in the presence of H2 and Pd/C was not successful. Thus we turned to a different strategy and used a Ugi-ready protected form of mercaptoacetaldehyde: 2,2-dimethylthiazoli-dine 40. Thiazolidine 40 is a bench-stable liquid, which is well known to work as a component in the classical Ugi-4CR.[28] Moreover, it can be readily synthesized in good yields in one pot by another MCR, the Asinger-3CR of acetone, ammonia and mercaptoacetaldehyde dimer 39 (Scheme 9).[28, 29] The simple reaction of Walborsky's isocyanide 8 with TMSN3 and 40 yielded 5w in moderate yields. Heating the Ugi product 5w at reflux in ethanolic HCl provided the Cys analogue 6w in good yield by N,S-acetal and tetrazole deprotection (Scheme 10 and Table 4). For the Pro analogue, pyrrolidine 36 was oxidized to the cyclic Schiff base Δ1-pyrroline 37, which exists in equilibrium between cyclo trimer 38, oligomers, and the monomer (Scheme 9). The oxidation was accomplished with sodium persulfate with catalytic amounts of AgNO3 in water (Scheme 9).[30] Surprisingly, simply mixing Schiff base Δ1-pyrroline 37 with TMSN3 3 and β-cyanoethylisocyanide 41 in methanol did not lead to the Ugi product 5v. However upon addition of the Lewis acid ZnCl2 the Ugi tetrazole 5v was formed in good yields from cleavable β-cyanoethylisocyanide 41 (Scheme 10 and Table 4).
Scheme 10.
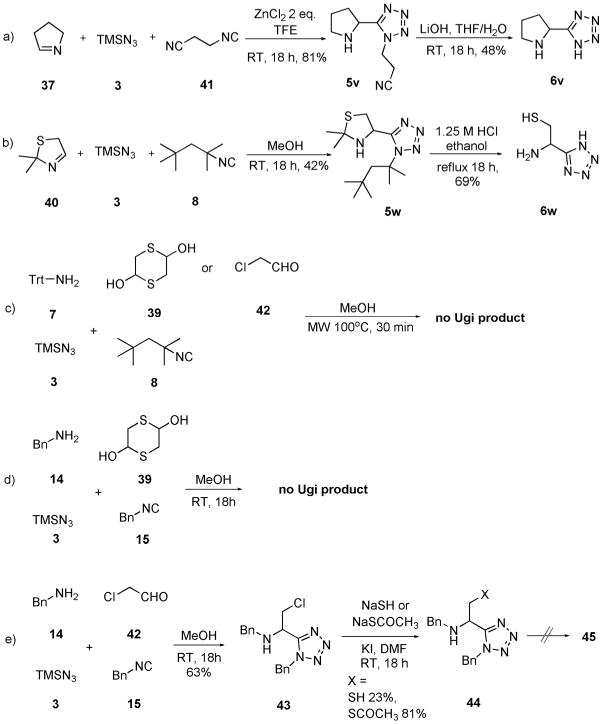
a) Synthesis of Ugi products 5v and 5w; b) final deprotection to produce 6v and 6w; c,d) failure to use trityl/tert-octyl strategy or benzyl/benzyl strategy in the preparation of Cys Ugi product; e) another route to obtain the precursor 44 for Cys analogue 6w.
Scheme 9.

Synthesis of Schiff bases 37 and 40 for Ugi products 6v and 6w.
Table 4.
Yields of the Ugi products 5v and 5w and α-amino acid tetrazoles 6v and 6w.

|
Purification was carried out on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3.
It is worth noting here that the Ugi products generally crystallized well and can be used to study the solid-state conformation and intermolecular contacts, as shown for three typical highly crowded examples in Figure 2.[23]
Figure 2.
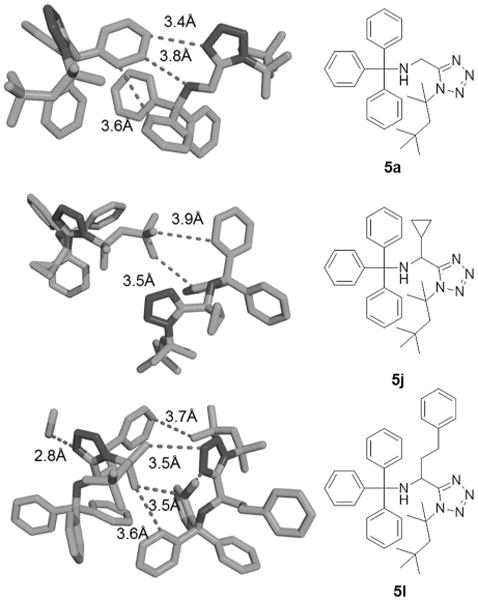
Molecular structures of compounds 5a, 5j, and 5j in the solid state illustrating the crowdedness of the trityl-protected products and showing some short intermolecular atom–atom contacts. The crystal contacts are mostly governed by hydrophobic interactions. In 5l, a co-crystallized metha-nol molecule forms a hydrogen bond to N4 of the tetrazole moiety. Thermal ellipsoids are drawn at the 50% probability level.
Conclusions
In summary, we have developed a concise and rapid synthetic route to α-amino acid-bioisosteric α-amino tetrazoles, exemplified in the synthesis of all 20 natural proteinogenic amino acids and 4 others by using a key azido-Ugi reaction. To our knowledge, this is the first report of the synthesis of all endogenous natural proteinogenic α-amino acid-bioisosteric α-amino tetrazoles by one general synthetic pathway. Surprisingly, the tetrazole derivatives of the α-amino acids Thr (6r), His (6t), Arg (6u), and Cys (6w) are, to our knowledge, hitherto unknown and reported herein for the first time. All compounds were synthesized as racemic mixtures and 6h and 6r as diastereomeric mixtures. Investigation of the biological activity of the endogenous natural proteinogenic α-amino acid-bioisosteric α-amino tetrazoles including applications in drug design are ongoing and will be reported in due course.
Experimental Section
General methods
Nuclear magnetic resonance spectra were recorded on a Bruker Advance 500 spectrometer [1H NMR (500 MHz), 13C NMR (126 MHz)], a Varian Inova 400 MHz spectrometer [1H NMR (400 MHz), 13C NMR (100 MHz)] or a Varian VXR 300 MHz spectrometer [1H NMR (300 MHz), 13C NMR (75 MHz)]. Chemical shifts δ for 1H NMR were reported in ppm and coupling constants were in Hz. The following abbreviations were used for spin multiplicity: s=singlet, d=doublet, t=triplet, dd=double doublet, m=multiplet. Chemical shifts δ for 13C NMR reported in ppm relative to the solvent peak. Mass spectra (HRMS) were recorded on an Orbitrap XL (Thermo Fisher Scientific; ESI pos. mode, resolution of 60000 m/z 400). Electrospray ionization mass spectra (ESI-MS) were recorded on a Waters Investigator Semi-prep 15 SFC-MS instrument. Mass spectra for deprotected α-amino acid-isosteric α-amino tetrazoles were measured on an API 3000 triple-quadrupole mass spectrometer (Applied Biosystems/MDS Sciex) via a TurboIonSpray source. Thin-layer chromatography was performed on precoated silica gel plates (TLC Silica gel 60 F254). Flash chromatography was performed on a Teledyne ISCO Combiflash Rf, using RediSep Rf Normal-phase Silica Flash Columns (Silica Gel 60 Å, 230–400 mesh). Reagents were available from commercial suppliers and used without any purification unless otherwise noted.
Synthetic procedure following Ugi reaction of trityl/tert-octyl and trityl/benzyl strategy
Aldehyde (1 mmol) and tritylamine (1 mmol) were suspended in MeOH (1 mL) in a sealed vial with a magnetic stirring bar. The reaction was heated at 100°C for 15 min by using microwave irradiation. Isocyanide (1 mmol) and azidotrimethylsilane (1 mmol) were then added into the reaction mixture and further heated at 100°C for 15 min by using microwave irradiation. The solvent was removed under reduced pressure and the residue was purified by using flash column chromatography.
1,1,1-Triphenyl-N-((1-(2,4,4-trimethylpentan-2-yl)-1 H-tetrazol-5-yl)methyl)methanamine (5a)
The product was obtained as a white solid (254 mg, 56%). Rf=0.43 (Petroleum ether:EtOAc 3:1); 1H NMR (500 MHz, CDCl3): δ=7.56–7.54 (m, 6H), 7.32–7.29 (m, 6H), 7.26–7.20 (m, 3H), 3.73 (dd, J=7.3, 2H), 1.66 (d, J=1.4 Hz, 2H), 1.60 (d, J=1.4 Hz, 6H), 0.65 ppm (d, J=1.5 Hz, 9H); 13C NMR (126 MHz, CDCl3): δ=153.8, 145.1, 128.8, 128.3, 127.0, 71.4, 64.3, 53.0, 40.3, 31.7, 30.7, 30.1 ppm; MS (ESI) m/z calculated [M+Na]+: 476.28; found [M+Na]+: 476.38.
Synthetic procedure following Ugi reaction of benzyl/benzyl strategy
Aldehyde (1 mmol) and benzylamine (1 mmol) were mixed in MeOH (1 mL) in a vial with a magnetic stirring bar. The reaction mixture stirred for 5 min at room temperature. Isocyanide (1 mmol) and azidotrimethylsilane (1 mmol) were then added to the reaction mixture and it was stirred overnight at room temperature. The solvent was removed under reduced pressure and the residue was purified by using flash column chromatography to obtain the desired product.
1-Benzyl-5-(1-benzyl-1 H-tetrazol-5-yl)pyrrolidin-2-one (5m″)
The product was obtained as a colorless solid (60 mg, 18%). Rf=0.49 (Petroleum ether:EtOAc = 2:1); 1H NMR (500 MHz, CDCl3): δ = 738–7.24 (m, 6H), 6.97–6.95 (m, 4H), 5.40 (d, J=15.5 Hz, 1 H), 5.04 (d, J=14.9 Hz, 1 H), 4.78 (d, J=15.6 Hz, 1 H), 4.62 (dd, J = 8.6, 5.0 Hz, 1 H), 3.72–3.61 (m, 1 H), 2.68–2.58 (m, 1 H), 2.47–2.41 (m, 1 H), 2.07–1.97 (m, 1 H), 1.74–1.67 ppm (m, 1 H); 13C NMR (126 MHz, CDCl3): δ = 174.6, 154.5, 135.1, 133.0, 129.5, 129.4, 129.2, 128.5, 128.3, 127.4, 51.1, 50.9, 45.3, 29.5, 24.4 ppm; MS (ESI) m/z calculated [M+Na] + : 356.15; found [M+Na]+: 356.30.
Synthetic procedure following Ugi reaction of mixed strategy
Methyl 4-(1-benzyl-1 H-tetrazol-5-yl)-4-(tritylamino)butanoate (5m)
The product was obtained as a light yellow or colorless solid (239 mg, 45%). Rf=032 (Petroleum ether:EtOAc 3:1); 1H NMR (500 MHz, CDCl3): δ = 7.38–7.23 (m, 10H), 7.22–7.10 (m, 8H), 7.08–7.01 (m, 2 H), 5.21 (d, J = 15.3 Hz, 1 H), 4.78 (d, J = 15.3 Hz, 1 H), 4.18 (d, J=5.6Hz, 1 H), 3.58 (s, 3H), 2.23–2.17 (m, 1 H), 2.04–1.98 (m, 1 H), 1.92–1.87 (m, 1 H), 1.59–1.52 ppm (m, 1 H); 13C NMR (126 MHz, CDCl3): δ = 173.4, 157.1, 145.2, 133.2, 129.2, 129.0, 128.9, 128.5, 128.1, 127.9, 126.8, 71.4, 51.8, 50.9, 47.0, 31.5, 28.9 ppm; MS (ESI) m/z calculated [M+Na] + : 540.24; found [M+Na]+: 540.21.
4-(1-Benzyl-1H-tetrazol-5-yl)-4-(tritylamino)butanamide (5m′)
Methyl 4-(1-benzyl-1H-tetrazol-5-yl)-4-(tritylamino)butanoate (5m; 0.2 g, 0.5 mmol) was dissolved in THF/H2O = 1/1 (2 mL) containing lithium hydroxide (48 mg, 1 mmol). The reaction mixture was stirred overnight at room temperature. The solvent was removed and the reaction mixture was acidified to pH 3 with 2 M HCl aqueous solution. Then it was extracted with CH2Cl2 (10 mL × 3). The organic layer was collected and concentrated to give the product as a white solid with a quantitative yield. It was used directly in the next step without further purification. 4-(1-benzyl-1H-tetrazol-5-yl)-4-(tritylamino)butanoic acid (0.2 g, 0.5 mmol) was dissolved in THF (4 mL) and the resultant solution was degassed with N2. Then DIEA (0.3 mL, 2.3 mmol), HOBt (67 mg, 0.55 mmol), and EDCI (88 μL, 0.055 mmol) were added. The reaction mixture was stirred for 10 min at room temperature. Ammonium carbonate (173 mg, 1.8 mmol) was added. The reaction mixture was further stirred overnight at room temperature. The solvent was removed and CH2Cl2 (7 mL) and water (7 mL) were added. The aqueous phase was separated and extracted with CH2Cl2 (20 mL × 3). The organic phases were combined and purified with a combiflash machine to give the product as a colorless solid (82 mg, 37%). Rf =0.49 (EtOAc); 1H NMR (500 MHz, CDCl3): δ=7.39–7.25 (m, 9H), 7.23–7.12 (m, 9H), 7.08–7.06 (m, 2H), 5.28 (d, J=15.2 Hz, 1H), 4.80 (d, J = 15.3 Hz, 1H), 4.19–4.16 (m, 1H), 2.23–2.17 (m, 1H), 2.02–1.98 (m, 1H), 1.92–1.87 (m, 1H), 1.59–1.54 ppm (m, 1H); 13C NMR (126 MHz, CDCl3): δ=174.1, 157.0, 145.2, 133.5, 129.2, 129.0, 128.6, 128.2, 128.1, 126.9, 71.7, 51.0, 47.5, 31.5, 30.1 ppm; MS (ESI) m/z calculated [M+Na]+: 525.24; found [M+Na]+: 525.41.
Synthetic procedure following Ugi reaction of Schiff base insertion strategy
3-(5-(Pyrrolidin-2-yl)-1H-tetrazol-1-yl)propanenitrile (5v)
3,4-Di-hydro-2H-pyrrole (37; 70 mg, 1 mmol) was dissolved in trifluoro-ethanol (1 mL) and zinc chloride (273 mg, 2 mmol) was added. The reaction mixture stirred for 5 min at room temperature. Then β-cyanoethylisocyanide (41; 81 mg, 1 mmol) was added. The reaction mixture was further stirred for 45 min at room temperature. Then 10 drops of aqueous ammonia solution (35%) was added. The reaction mixture was then stirred overnight at room temperature. The reaction was monitored by thin layer chromatography (TLC). When the reaction was completed, the reaction mixture was concentrated under reduced pressure and the residue was purified by using flash column chromatography. The product was a yellow solid (156 mg, 81%). Rf =0.42 (CH2Cl2:MeOH 8:2); 1H NMR (500 MHz, CDCl3): δ=5.03–4.94 (m, 1H), 4.85–4.78 (m, 1H), 4.70–4.64 (m, 1H), 3.15–3.06 (m, 3H), 3.01–2.92 (m, 1H), 2.37–2.22 (m, 2H), 2.04–1.84 ppm (m, 2H); 13C NMR (126 MHz, CDCl3): δ=157.1, 116.3, 53.0, 47.2, 43.3, 31.7, 25.9, 18.9 ppm; MS (ESI) m/z calculated [M+H]+: 193.12; found [M+H]+: 193.3.
Deprotecting procedure A using 1.25 m HCl in ethanol
Trityl group protected α-amino tetrazoles (5a–l and 5w; 0.5 mmol) was dissolved in ethanol containing 1.25 m HCl (7.6 mL) in a round-bottomed flask with a magnetic stir bar. The reaction mixture was heated at reflux overnight. The solvent was evaporated under reduced pressure and the crude product was purified by column chromatography on silica eluting with CH2Cl2 (6a–c, 6e–l) and methanol or on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3 (6d, 6w).
(1H-Tetrazol-5-yl)methanamine hydrogen chloride (6a)
The product was obtained as a light yellow oil (35 mg, 51%). Rf = 0.41 (CH2Cl2:MeOH 1:1); 1H NMR (500 MHz, [D4]methanol): δ=4.40 ppm (d, J=3.4, 2H); 13C NMR (126 MHz, [D4]methanol): δ=155.9, 34.7 ppm; MS (ESI) m/z calculated [M+H]+: 100.06; found [M+H]+: 100.1.
Deprotecting procedure B using 10% Pd on charcoal
Benzyl group protected α-amino tetrazoles (5n, 5o′, 5p–s; 0.5 mmol) was dissolved in methanol (2 mL) containing 10% Pd on charcoal (500 mg) in a round-bottomed flask with a magnetic stir bar. The reaction mixture was stirred overnight at room temperature. The solid was filtered and the filtrate was concentrated under reduced pressure. The residue was purified on a Dowex 50 cation-exchange column eluted with 2n aqueous NH3.
3-Amino-3-(1H-tetrazol-5-yl)propanoic acid (6n)
The product was obtained as a light yellow solid (20 mg, 26%). Rf=0.89 (CH2Cl2: MeOH 1:1); 1H NMR (300 MHz, D2O): δ = 4.98 (dd, J = 8.6, 5.3 Hz, 1 H), 3.04–2.88 ppm (m, 2H); 13C NMR (75 MHz, D2O): δ = 176.3, 44.8, 38.0, 25.8 ppm; MS (ESI) m/z calculated [M+H] + : 171.09; found [M+H]+: 171.2.
Deprotecting procedure using multiple deprotection
4-Amino-4-(1H-tetrazol-5-yl)butanamide (6m)
4-(1-Benzyl-1H-tetrazol-5-yl)-4-(tritylamino)butanamide (5m′; 70 mg, 0.14 mmol) was dissolved in methanol (0.5 mL). Then TFA (20 μL, 0.28 mmol) was added. The reaction mixture stirred for 30 min at room temperature. When the reaction was completed, 10% Pd on charcoal (139 mg) was added to the reaction mixture, which was then stirred overnight at room temperature under H2. The reaction mixture was then concentrated under reduced pressure and the residue was purified by using a Dowex 50 cation-exchange column eluted with 2n aqueous NH3. The product was white solid (14 mg, 60%). Rf = 0.5 (CH2Cl2:MeOH 1:1); 1H NMR (300 MHz, D2O): δ = 5.21–5.11 (m, 0.5 H), 4.93–4.83 (m, 0.5 H), 2.76–2.50 (m, 1 H), 2.42–2.20 (m, 2H), 2.15–1.95 ppm (m, 1 H); 13C NMR (126 MHz, D2O): δ = 181.5, 175.6, 158.2, 155.9, 48.6, 29.0, 26.6 ppm; MS (ESI) m/z calculated [M+H] + : 171.09; found [M+H] + : 171.2.
Supplementary Material
Acknowledgments
The work was financially supported by the NIH (1R01M097082-01) and by the Innovative Medicines Initiative (grant agreement no. 115489). T.Z. acknowledges a CSC PhD fellowship.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201504520.
References
- 1.Jakubke HD, Sewald N. Peptides from A-Z. Wiley-VCH; Weinheim, Germany: 2008. pp. 1–41. [Google Scholar]
- 2.Ballatore C, Huryn DM, Smith AB. ChemMedChem. 2013;8:385–395. doi: 10.1002/cmdc.201200585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wirth M, Zoete V, Michielin O, Sauer WH. Nucleic Acids Res. 2013;41:D1137–1143. doi: 10.1093/nar/gks1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hansch C, Leo A, Hoekman DH. Exploring Qsar: Fundamentals and Applications in Chemistry and Biology. American Chemical Society; 1995. [Google Scholar]
- 5.a) Holland GF, Pereira JN. J Med Chem. 1967;10:149–154. doi: 10.1021/jm00314a004. [DOI] [PubMed] [Google Scholar]; b) Figdor SK, von Wittenau MS. J Med Chem. 1967;10:1158–1159. doi: 10.1021/jm00318a038. [DOI] [PubMed] [Google Scholar]; c) Esplin DW, Woodbury DM. J Pharmacol Exp Ther. 1956;118:129–138. [PubMed] [Google Scholar]
- 6.a) Kubo K, Kohara Y, Yoshimura Y, Inada Y, Shibouta Y, Furukawa Y, Kato T, Nishikawa K, Naka T. J Med Chem. 1993;36:2343–2349. doi: 10.1021/jm00068a011. [DOI] [PubMed] [Google Scholar]; b) Kraus JL. Pharmacol Res Commun. 1983;15:183–189. doi: 10.1016/s0031-6989(83)80060-5. [DOI] [PubMed] [Google Scholar]; c) Obermeier MT, Chong S, Dando SA, Marino AM, Ryono DE, Starrett-Arroyo A, DiDonato GC, Warrack BM, White RE, Morrison RA. J Pharm Sci. 1996;85:828–833. doi: 10.1021/js9600282. [DOI] [PubMed] [Google Scholar]
- 7.Herr RJ. Bioorg Med Chem. 2002;10:3379–3393. doi: 10.1016/s0968-0896(02)00239-0. [DOI] [PubMed] [Google Scholar]
- 8.a) Wexler RR, Greenlee WJ, Irvin JD, Goldberg MR, Prendergast K, Smith RD, Timmermans PBMWM. J Med Chem. 1996;39:625–656. doi: 10.1021/jm9504722. [DOI] [PubMed] [Google Scholar]; b) Duncia JV, Carini DJ, Chiu AT, Johnson AL, Price WA, Wong PC, Wexler RR, Timmermans PB. Med Res Rev. 1992;12:149–191. doi: 10.1002/med.2610120203. [DOI] [PubMed] [Google Scholar]; c) Carini DJ, Duncia JV, Aldrich PE, Chiu AT, Johnson AL, Pierce ME, Price WA, Santella JB, Wells GJ. J Med Chem. 1991;34:2525–2547. doi: 10.1021/jm00112a031. [DOI] [PubMed] [Google Scholar]; d) Duncia JV, Pierce ME, Santella JB. J Org Chem. 1991;56:2395–2400. [Google Scholar]; e) Larsen RD, King AO, Chen CY, Corley EG, Foster BS, Roberts FE, Yang C, Lieberman DR, Reamer RA. J Org Chem. 1994;59:6391–6394. [Google Scholar]; f) Griffiths GJ, Hauck MB, Imwinkelried R, Kohr J, Roten CA, Stucky GC, Gosteli J. J Org Chem. 1999;64:8084–8089. doi: 10.1021/jo9824910. [DOI] [PubMed] [Google Scholar]; g) Segarra V, Crespo MI, Pujol F, Beleta J, Domenech T, Miralpeix M, Palacios JM, Castro A, Martinez A. Bioorg Med Chem Lett. 1998;8:505–510. doi: 10.1016/s0960-894x(98)00058-4. [DOI] [PubMed] [Google Scholar]
- 9.Nichols DA, Jaishankar P, Larson W, Smith E, Liu G, Beyrouthy R, Bonnet R, Renslo AR, Chen Y. J Med Chem. 2012;55:2163–2172. doi: 10.1021/jm2014138. [DOI] [PubMed] [Google Scholar]
- 10.McManus JM, Herbst RM. J Org Chem. 1959;24:1643–1649. [Google Scholar]
- 11.Demko ZP, Sharpless KB. Org Lett. 2002;4:2525–2527. doi: 10.1021/ol020096x. [DOI] [PubMed] [Google Scholar]
- 12.a) Hulme C, Gore V. Curr Med Chem. 2003;10:51–80. doi: 10.2174/0929867033368600. [DOI] [PubMed] [Google Scholar]; b) Ruijter E, Scheffelaar R, Orru RVA. ngew Chem Int Ed. 2011;50:6234–6246. doi: 10.1002/anie.201006515. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2011;123:6358–6371. [Google Scholar]
- 13.a) Dömling A. Chem Rev. 2006;106:17–89. doi: 10.1021/cr0505728. [DOI] [PubMed] [Google Scholar]; b) Dömling A, Wang W, Wang K. Chem Rev. 2012;112:3083–3135. doi: 10.1021/cr100233r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ugi I, Meyr R. Chem Ber. 1961;94:2229–2233. [Google Scholar]
- 15.a) Zarganes-Tzitzikas T, Patil P, Khoury K, Herdtweck E, Dömling A. Eur J Org Chem 2015. :51–55. doi: 10.1002/ejoc.201403401. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Patil P, Khoury K, Herdtweck E, Dömling A. Bioorg Med Chem. 2015;23:2699–2715. doi: 10.1016/j.bmc.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gunawan S, Ayaz M, De Moliner F, Frett B, Kaiser C, Patrick N, Xu Z, Hulme C. Tetrahedron. 2012;68:5606–5611. doi: 10.1016/j.tet.2012.04.068. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Gunawan S, Petit J, Hulme C. ACS Comb Sci. 2012;14:160–163. doi: 10.1021/co200209a. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Nixey T, Hulme C. Tetrahedron Lett. 2002;43:6833–6835. [Google Scholar]
- 16.a) Ugi I. Intra-Sci Chem Rep. 1971;5:229–261. [Google Scholar]; b Waki M, Meienhofer J. J Am Chem Soc. 1977;99:6075–6082. doi: 10.1021/ja00460a039. [DOI] [PubMed] [Google Scholar]; c Cao X, Moran EJ, Siev D, Lio A, Ohashi C, Mjalli AMM. Bioorg Med Chem Lett. 1995;5:2953–2958. [Google Scholar]
- 17.Zhao T, Boltjes A, Herdtweck E, Domling A. Org Lett. 2013;15:639–641. doi: 10.1021/ol303348m. [DOI] [PubMed] [Google Scholar]
- 18.C. Tao, Q. Wang, N. P. Desai, P. Soon-Shiong (Aventis Pharma S.A., Antony), US7517881 B2, 2010.
- 19.Mattingly PG. Synthesis. 1990:366–368.; b) Z. M. Van, A. Golebiowski, M. K. Ji, D. Whitehouse, T. Ryder, P. Beckett (Mars, Incorporated, McLean), WO2013059437A1, 2011.
- 20.Brown AL, Churches QI, Hutton CA. J Org Chem. 2015;80:9831–9837. doi: 10.1021/acs.joc.5b01519. [DOI] [PubMed] [Google Scholar]
- 21.Parikh JR, Doering WvE. J Am Chem Soc. 1967;89:5505–5507. [Google Scholar]
- 22.B. N. Naidu, K. Peese, I. B. Dicker, C. Li, M. Patel, N. A. Meanwell, M. A. Walker (Bristol-Myers Squibb, Princeton), WO2011046873 A1, 2011.
- 23.Y. Bergman, N. Choi, R. C. Foitzik, D. Ganame, C. F. Hemley, I. P. Holmes, R. Lessene, G. E. Lunniss, M. Nikac, S. R. Walker (Cancer Therapeutics CRC Pty Ltd., Bundoora), WO2012110773 A1, 2012.
- 24.a) F. Yang, B. Zhang, N. Wicnienski, J. Pfefferkorn, M. Greene, K. Chen, R. Nugent, M. Reding, R. Kelly, M. Mitchell (Pharmacia & Upjohn Company, La Jolla), US20050154056 A1, 2005; Browne LJ, Gude C, Rodriguez H, Steele RE, Bhatnager A. J Med Chem. 1991;34:725–736. doi: 10.1021/jm00106a038.
- 25.Shimamoto Y, Hattori Y, Kobayashi K, Teruya K, Sanjoh A, Nakaga-wa A, Yamashita E, Akaji K. Bioorg Med Chem. 2015;23:876–890. doi: 10.1016/j.bmc.2014.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yong YF, Kowalski JA, Lipton MA. J Org Chem. 1997;62:1540–1542. [Google Scholar]
- 27.Iwanowicz EJ, Poss MA, Lin J. Synth Commun. 1993;23:1443–1445. [Google Scholar]
- 28.Vorbruggen H. Helv Chim Acta. 1991;74:297–303. [Google Scholar]
- 29.Thiel M, Asinger F, Schmiedel K. Justus Liebigs Ann Chem. 1958;611:121–131. [Google Scholar]
- 30.Nomura Y, Ogawa K, Takeuchi Y, Tomoda S. Chem Lett. 1977:693–696. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.