

Abstract
At a human/livestock/wildlife interface, Escherichia coli populations were used to assess the risk of bacterial and antibiotic resistance dissemination between hosts. We used phenotypic and genotypic characterization techniques to describe the structure and the level of antibiotic resistance of E. coli commensal populations and the resistant Enterobacteriaceae carriage of sympatric African buffalo (Syncerus caffer caffer) and cattle populations characterized by their contact patterns in the southern part of Hwange ecosystem in Zimbabwe. Our results (i) confirmed our assumption that buffalo and cattle share similar phylogroup profiles, dominated by B1 (44.5%) and E (29.0%) phylogroups, with some variability in A phylogroup presence (from 1.9 to 12%); (ii) identified a significant gradient of antibiotic resistance from isolated buffalo to buffalo in contact with cattle and cattle populations expressed as the Murray score among Enterobacteriaceae (0.146, 0.258, and 0.340, respectively) and as the presence of tetracycline-, trimethoprim-, and amoxicillin-resistant subdominant E. coli strains (0, 5.7, and 38%, respectively); (iii) evidenced the dissemination of tetracycline, trimethoprim, and amoxicillin resistance genes (tet, dfrA, and blaTEM-1) in 26 isolated subdominant E. coli strains between nearby buffalo and cattle populations, that led us (iv) to hypothesize the role of the human/animal interface in the dissemination of genetic material from human to cattle and toward wildlife. The study of antibiotic resistance dissemination in multihost systems and at anthropized/natural interface is necessary to better understand and mitigate its multiple threats. These results also contribute to attempts aiming at using E. coli as a tool for the identification of pathogen transmission pathway in multihost systems.
INTRODUCTION
As human activities increase, the pressure on natural ecosystems through land encroachment, unsustainable use of natural resources, and fragmentation of habitats tend to expand worldwide (1). This trend is exacerbated by human population growth and the need to access more land to feed all in developing countries (2, 3). In these contexts, the spread of pathogens and genetic material can represent a burden on wildlife, livestock, and human population health (4–6). Diseases significantly impact livestock productions, which are a key livelihood option in semiarid areas and can also threaten endangered wildlife species (7). The dissemination of antibiotic resistance (ABR) into remote, supposedly pristine, areas resulting from a high and inappropriate use of antibiotics in humans and domestic animals (in particular medicated feed) (8, 9), demonstrates how the most remote ecosystems are not exempt from a human footprint (10). The consequences of ABR diffusion in natural ecosystems are largely unknown. However, the evolution and selection of resistance genes in the wild could compromise the use of antibiotics (11), the main tool to fight infectious diseases in domestic animals and human (10). Moreover, the ABR pollution “in the wild” could threaten biodiversity (12).
The dynamics and processes of microorganism transmission between hosts and the environment should therefore be a focus of research at wildlife/livestock/human interfaces to provide management options to reduce or deal with their negative effects (i.e., impact on human health, livestock production, and biodiversity conservation). These interfaces represent complex multihost and multipathogen systems that have been so far little studied (13). Even if focusing on a single pathogen, the large diversity of hosts constrains the efficiency of past and current surveillance and control approaches. New frameworks are therefore needed that bridge biological fields (14, 15). Since pathogens have a limited number of transmission modes to infect a new host (e.g., direct, environmental such as waterborne, foodborne, or vector/insect-borne transmission), a framework trying to identify the transmission processes linking one host to different sources of pathogens could help identifying hotspots of pathogen transmission and predicting future microorganism transmission at a local level (16, 17).
The bacterium Escherichia coli is a good indicator of transmission pathways within multihost systems because E. coli is ubiquitous, shares the same niche as enteric pathogens and is transferred by the same route, and is one of the best-studied and best-known bacteria. E. coli diversity and population dynamics have been the focus of recent studies (17–20) investigating the relationship between E. coli populations and proxies of interhost contacts. For example, E. coli sharing between human, primates, and livestock increased with the frequency and intensity of interspecies contacts in Uganda (21). However, more studies are needed with different animal models, in different ecosystems and using the new available molecular tools to characterize bacterial diversity. The dissemination of ABR in pristine ecosystems can also be used to track directional genetic transfer from human and livestock toward wildlife (10, 22, 23).
A wealth of studies exists on the host, temporal stability, and geographical structure of E. coli associated with humans and domestic animals (see, for example, references 24, 25, and 26). The factors contributing to the sharing of E. coli between host populations include (i) feeding modes, (ii) phylogenetic relatedness, and (iii) host contact patterns related to bacterial transmission (27). Since it can be difficult to weigh each factor against each other, estimating the proportion of the E. coli population similarity related to the last factor could be difficult. However, a recent study (28) provided a semiexperimental setup that we used here. The animal model offers a good opportunity to investigate E. coli population sharing between hosts as the African buffalo (Syncerus caffer caffer) and cattle (Bos taurus/indicus) are bovids and therefore phylogenetically related, their diets overlap substantially and telemetry studies indicate that both populations can come into contact (28). Finally, ungulate population movements can be used to detect the degree of contacts between populations defining a contact variable that can be used to test hypotheses on E. coli population sharing.
The study was therefore initiated with a double objective: first, to increase the knowledge on the dissemination of ABR genes between hosts in these complex systems, so far little studied, in order to assess the risk associated with this anthropological threat on natural ecosystems; and second, to explore the processes of E. coli transmission between hosts as a model for pathogen transmission and potentially as a predictive tool. Hence, the genetic structures of commensal E. coli populations and their ABRs were explored simultaneously in sympatric ungulate hosts. We hypothesized that (i) the phylogenetic proximity and the diet overlap between cattle and buffalo in our study site would result in similar E. coli phylogroups' profiles but that (ii) ABR in buffalos should increase with the level of habitat sharing with domestic hosts, since the use of antibiotics is restricted to human and domestic populations.
MATERIALS AND METHODS
Study site and experimental setting.
The study was conducted in the Hwange district of Zimbabwe, Africa. The Hwange National Park and its periphery (including the Sikumi Forest and surrounding communal lands) are part of the Kavango-Zambezi Transfrontier Conservation Area (KAZA TFCA) (28) (Fig. 1). In southern Africa, TFCAs aim at combining sustainable development and biodiversity conservation through the promotion of the sustainable use of natural resources and agricultural production (29). The livelihoods of small-scale farmers rely heavily on basic livestock production (herd average n = 5), little or no agricultural input (fertilizer, antibiotic feeds) (28), and maize and sorghum cropping in a semiarid ecosystem (average rainfall, 600 mm per year).
FIG 1.

Study site, including home ranges (95% UD, 2012 to 2014) of adult female buffaloes were drawn in red and cattle drawn in green. Herd A (4 GPS collars, approximately 1,000 individuals) roamed in Hwange NP (dark gray) and herd B (4 GPS collars, approximately 500 individuals) remained in Sikumi Forest (gray) and privately owned safari areas (light gray). Three cattle home ranges drawn in green (95% UD, 2010 and 2011) are representative of cattle living in Magoli and Jwapi villages in Hwange Communal Area (white) and entering Sikumi Forest. No fence separates any of the land uses displayed. The map was created using Quantum GIS version 2.4.
The telemetry protocol presented previously (28) targeting sympatric buffalo and cattle populations was carried out on the same ungulate populations as of December 2012. Adult females were equipped with GPS collars since their movements are representative of the herd movements (30, 31). Annual home ranges for each individual/herd were calculated using the 95% utilization distribution method (32) and are displayed in Fig. 1. E. coli sampling protocols were implemented in three populations identified using the telemetry results: a distant buffalo population (A) whose home range does not overlap the other two populations (population size estimated at around 1,000 individuals), a neighboring buffalo population (B) (population size estimated also at around 500 individuals), and a cattle population (C) (several hundred individuals) sharing Sikumi Forest.
In this area, interviews with animal health technicians, farmers, and human health professionals revealed that antibiotics were used in cattle populations to treat tick-borne diseases and other infections (these individuals were asked to list by order of importance the antibiotic they use or prescribe). The antibiotics used most frequently in cattle were mainly tetracycline, followed by oxytetracycline, penicillin, and streptomycin (principally injected intramuscularly). There does not appear to be any preventive use of antibiotics in the area in cattle. In humans, antibiotics were mainly used to treat human tuberculosis (Mycobacterium tuberculosis), an infection with a high prevalence in the area (especially due to the high HIV burden). The main antibiotics used in humans were trimethoprim, co-trimoxazole (a combination of trimethoprim and sulfonamides), amoxicillin, and doxycycline.
Sample collection.
Fresh fecal samples of animals from the three populations A, B, and C were collected on the ground a few seconds or minutes after deposition between 31 October and 4 November 2012. For cattle, the protocol was implemented in two villages (i.e., Magoli and Jwapi), following cattle herds returning from their daily roaming in the Sikumi forest to the kraal (i.e., overnight enclosure located close to the homestead) before sunset. For buffalo populations (A and B), the herd was located using recent GPS positions transmitted by satellite and very high frequency devices. After visual contact was established with the buffalo herd, movements were monitored, and the samples were collected just after the herd moved out of an open area. This protocol ensured that the fecal material collected was obtained from the right host (population A, B, or C) and endeavored to minimize the sampling of fecal material from the same individuals by selecting distant dungs (>10 m) or dungs with clear dissimilarities in color and/or density. A sample size of around 50 (5 or 10%) was estimated from the population size (500 heads for each buffalo populations and several hundred heads for the cattle population) and practically to minimize double sampling of individuals and taking into account laboratory time and costs. Labeled with unique identifying numbers, transport swabs (Clinical Sciences Diagnostics containing Amies transport medium) were immersed in the fecal material and transported in a cool box with ice packs from the field to a deep freezer (in less than 6 h) in the research camp where they were then maintained at −20°C. During the same week, they were transported by car to Harare, capital of Zimbabwe, without defreezing and stored in another deep freezer until shipment by plane to the INSERM laboratory in France in March 2013. Once in the laboratory, each swab was then discharged in brain heart infusion broth with 20% glycerol and stored at −80°C until used.
Isolation of the dominant E. coli clone.
The stool-containing suspensions were plated onto Drigalski agar plates and incubated overnight at 37°C. Then, one yellow colony was randomly picked and confirmed by matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) analysis (MALDI Biotyper Microflex; Bruker) to belong to E. coli/Escherichia clade species. This colony was considered to represent the dominant E. coli/Escherichia clade clone as it has been recently shown (33, 34). The strain was tested for antibiotic susceptibility, phylotyped, and stored at −80°C. The nomenclature used for the designations of these strains was as follows: the letter of the population, the number of the individual, and “DOM” for dominant (e.g., B24DOM).
Antibiotic resistance.
Two protocols were used to analyze ABR. First, global ABR was analyzed in each sample by plating 100 μl of the glycerol dilution on Drigalski agar on which antibiotics disks containing amoxicillin (25 μg), kanamycin (30 IU), streptomycin (10 IU), tetracycline (30 IU), trimethoprim (5 μg), sulfonamides (200 μg), and chloramphenicol (30 μg) were plated, as described previously (35). Plates were incubated 24 h at 37°C and, if colonies were present within the zone of inhibition (as defined by the French Society for Microbiology [www.sfm-microbiologie.org/]), the sample was reported to be carrying resistant Enterobacteriaceae. A Murray score was calculated as described previously (36) using the following equation: Murray score = total number of resistances/total number of possible resistances for each individual sample. In addition, one randomly selected yellow colony falling within the zone of inhibition of tetracycline, amoxicillin, and trimethoprim was purified on Mueller-Hinton medium with the corresponding antibiotic disk each time it was present. The E. coli/Escherichia clade identification was confirmed by MALDI-TOF and stored at −80°C. These strains were then called tetracycline-, amoxicillin-, and trimethoprim-resistant strains, respectively, and labeled by the letter of the population, the number of the individual, and the abbreviation of the antibiotic (e.g., B24TET).
Second, classical antibiotic susceptibilities were determined using the disk diffusion method according to the 2012 recommendations of the French Society for Microbiology on the dominant and on the tetracycline-, amoxicillin-, and trimethoprim-resistant (see above) E. coli strains. The following antimicrobial agents were tested: amoxicillin (25 μg), amoxicillin + clavulanic acid (20 + 10 μg), ticarcillin (75 μg), cefoxitin (30 μg), cefepime (30 μg), cefotaxime (30 μg), ceftazidime (30 μg), streptomycin (10 IU), gentamicin (10 IU), kanamycin (30 IU), tetracycline (30 IU), trimethoprim (5 μg), sulfonamides (200 μg), chloramphenicol (30 μg), nalidixic acid (30 μg), and ofloxacin (5 μg).
Further characterization was performed on the subdominant antibiotic-resistant strains. Detection of tetracycline resistance efflux pump-encoding genes (tetA to tetE) by using a multiplex PCR (37) was performed on the tetracycline-resistant E. coli strains. β-Lactamase-encoding gene blaTEM was screened by PCR (38), followed by Sanger sequencing on the amoxicillin-resistant E. coli strains. Multiplex PCR detection of dihydrofolate reductase-encoding genes dfrA1, dfrA5-dfrA14, dfrA7-dfrA17, and dfrA12 was performed, followed by Sanger sequencing, on the trimethoprim-resistant E. coli strains. The choice of these genes was based on their prevalence in the E. coli genome database Mage (http://www.genoscope.cns.fr/agc/microscope/home/) (39). The primers for the dfrA PCR and the length of the PCR products were as follows: dhfr1.f (AACCAATGGCTGTTGGTTGG) and dhfr1.r (CTGAAACAATGACATGATCCG), 180 bp; dhfr5.f (CCACCAGACACTATAACGTG) and dhfr5.r (CATACCCTGGTCCGCGAAAG), 237 bp; dhfr7.f (TCAGAAAATGGCGTAATCGG) and dhfr7.r (ACGTGAACAGTAGACAAATG), 332 bp; and dhfr12.f (TGAGACAAGCTCGAATTCTG) and dhfr12.r (TGAACTCGGAATCAGTACGC), 430 bp. The PCR conditions were as described previously (40). Differentiation between the dfrA5 and dfrA14 genes on one hand and the dfrA7 and dfrA17 genes on the other hand was performed by sequencing.
E. coli phylogenetic grouping and strain relatedness.
Dominant and subdominant tetracycline-, amoxicillin-, and trimethoprim-resistant E. coli strains were assigned to one of the seven main phylogenetic phylogroups (A, B1, B2, C, D, E, and F) using the new Clermont quadruplex method (40) or to one of the five Escherichia clades (I to V) as described previously (40, 41). The subdominant tetracycline-resistant E. coli strain relatedness was assessed by repetitive extragenic palindromic PCR (rep-PCR) using a DiversiLab strain typing system (bioMérieux) as reporter earlier (42). Relatedness among the strains was also assessed by random amplification of polymorphic DNA (RAPD) using the 1254 primer (5′-CCGCAGCCAA-3′), as described previously (43).
Statistical analyses.
Using the R software (44), after checking for homogeneity of variance (no distribution was normally distributed), nonparametric tests (Kruskal-Wallis, Wilcoxon, chi-square, and Spearman rank correlation tests) were implemented to compare the ABR and the phylogroup population structure between the three host populations.
RESULTS
We collected 53 samples from isolated wild buffalo (population A), 52 samples from neighboring wild buffalo (population B), and 50 samples from domestic cattle (population C).
Phylogenetic group distribution and antibiotic resistance of the dominant E. coli/Escherichia clade strain.
E. coli/Escherichia clade dominant strains were detected in 152 of 155 samples (n = 52 for buffalo [A], n = 50 for neighboring buffalo [B], and n = 50 for cattle [C]). For three samples, the dominant Enterobacteriaceae did not belong to the Escherichia genus (two Klebsiella oxytoca in the B population and one Enterobacter cloacae in the A population).
The three host populations had similar patterns of E. coli phylogenetic group distribution (A and B: Spearman, P = 0.96; A and C: Spearman, P = 0.81; B and C: Spearman, P = 0.77) (Fig. 2). B1 was the main phylogroup detected in these three populations (36 to 54% of the detected dominant strains), followed by phylogroup E (24 to 34%). D phylogroup strains were present at more than 10% in population A. The A, B2, and C phylogroups were rarely detected (<6%). Three Escherichia clade I strains were isolated, all in buffalo (2 in the A population and 1 in the B population). No phylogroup F strain was observed.
FIG 2.
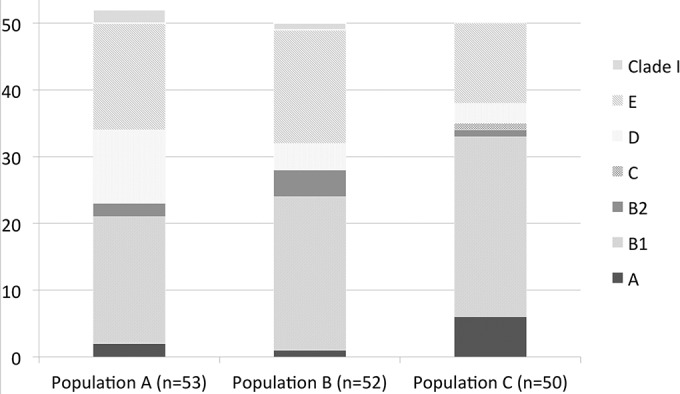
E. coli/Escherichia clade phylogenetic distribution of the dominant clones for each of the three ungulate populations: population A (buffalo not in contact, n = 53), population B (buffalo at the interface, n = 52), and population C (cattle, n = 50). Results for phylogroups A, B1, B2, C, D, and E and Escherichia clade I (Clade I) are displayed for each host population (no phylogroup F was observed).
ABR was found very rarely in the dominant strains, as only one B1-phylogroup E. coli from the buffalo population at the interface with cattle (B24DOM) was resistant streptomycin, tetracycline, and sulfonamides.
Global antibiotic resistance of fecal Enterobacteriaceae.
To have an overview of ABR in Enterobacteriaceae, the 155 fecal samples were tested for antibiotic-resistant Enterobacteriaceae by direct plating, gathering by this approach both dominant and subdominant strains (Table 1). A significant difference between the ABR patterns of the three populations was observed (Kruskal-Wallis test, P < 0.01) (Table 1). Buffalo with no contact with cattle (population A, average Murray score = 0.146) presented a lower Murray score than buffalo at the interface (population B, average Murray score = 0.258; Wilcoxon test, P < 0.01) and cattle (population C, average Murray score = 0.340; Wilcoxon test, P < 0.01). Cattle did not exhibit a significantly higher resistant score than buffalo at the interface (population B; Wilcoxon test, P = 0.21). Trends by antibiotics were quite consistent: for four of seven antibiotics (tetracycline, trimethoprim, sulfonamide, and chloramphenicol), we observed an increasing antibiotic resistance along the gradient A < B < C; for two of seven (streptomycin and amoxicillin), we observed an A ≪ C < B gradient, and for the remaining one (kanamycin), we noted an A = B < C gradient. Specifically, tetracycline resistance was significantly different between populations A and C (chi-square test, P < 0.01), between populations A and B (chi-square test, P = 0.04), and between populations B and C (chi-square test, P < 0.05). Amoxicillin resistance was significantly different between populations A and C (chi-square test, P < 0.01), between populations A and B (chi-square test, P < 0.01), and between populations B and C (chi-square test, P < 0.01). Trimethoprim resistance was significantly different between populations A and C (chi-square test, P < 0.01), not significant between populations A and B (chi-square test, P = 0.61), and between populations B and C (chi-square test, P < 0.01). In addition, buffalo (A and B, Murray score = 0.201) had significantly less ABR than cattle (C) (Wilcoxon test, P < 0.01) and populations in contact (B and C, Murray score = 0.298) had significantly more resistance than isolated population (A) (Wilcoxon test, P < 0.01).
TABLE 1.
Global antibiotic resistance prevalence of fecal Enterobacteriaceae for each ungulate population
| Antibiotic | No. of resistant samples (%)a |
||
|---|---|---|---|
| Host population A (n = 53) | Host population B (n = 52) | Host population C (n = 50) | |
| Streptomycin | 2 (3.8) | 9 (17.3) | 8 (16.0) |
| Tetracycline | 0 | 4 (7.7) | 17 (34.0) |
| Amoxicillin | 20 (37.7) | 45 (86.5) | 34 (68.0) |
| Trimethoprim | 9 (17.0) | 11 (21.2) | 23 (46.0) |
| Sulfonamide | 20 (37.7) | 20 (38.5) | 25 (50.0) |
| Kanamycin | 2 (3.8) | 2 (3.8) | 5 (10.0) |
| Chloramphenicol | 1 (1.9) | 3 (5.8) | 7 (14.0) |
Population A, a buffalo population not in contact with cattle; population B, a buffalo population in contact with cattle; population C, a cattle population. For each antibiotic, the first number represents the number of resistant samples, and the related percentage for the given host population is indicated in parentheses. Mean Murray scores ± the 95% confidence intervals were calculated for all antibiotics as described by Murray et al. (36) as follows: host population A, 0.146 ± 0.150; host population B, 0.258 ± 0.204; and host population C, 0.340 ± 0.275.
E. coli subdominant antibiotic-resistant strains.
Due to the veterinary and human medicine practices in Zimbabwe, we characterized further the presence of E. coli subdominant strains resistant to tetracycline, which was the most commonly used antibiotic in cattle, as well as resistance to amoxicillin and trimethoprim, which were largely used in humans. Furthermore, a markedly contrasting pattern of tetracycline-resistant Enterobacteriaceae and, to a lesser extent, of amoxicillin and trimethoprim resistance, among host populations was observed (Table 1). No antibiotic-resistant E. coli strain was identified in population A (buffalo with no contact with cattle), whereas 3 and 19 fecal samples yielded resistant E. coli strains in population B (buffalo in contact with cattle) and population C (cattle), respectively (significant difference between population C and the two other populations; chi-square test, P < 0.01 for C and A and for C and B) (Table 2). Using our strategy, we sometimes isolated several strains that were resistant to two or three of the tested antibiotics in a single sample. We considered that the strains were identical when they belonged to the same phylogroup, exhibited the same pattern of antibiotic resistance on the antibiogram, possessed the same resistance gene, and shared an identical RAPD profile. Thus, 3 and 23 subdominant resistant strains were identified in populations B and C, respectively (significant difference between population C and the two other populations; chi-square test, P < 0.01 for C and A and for C and B) (Table 2). Of note, the subdominant resistant strain isolated in the B24 sample (B24TET) was identical to the dominant strain resistant to antibiotics (B24DOM), as confirmed by RAPD analysis.
TABLE 2.
Antibiotic resistance phenotype and phylogenetic group belonging to each antibiotic-resistant subdominant E. coli strain detected in two of three host populations
| Isolatea | Antibiotic resistance phenotypeb | Gene |
E. coli phylogenetic groupc | ||
|---|---|---|---|---|---|
| tet | blaTEM-1 | dfr | |||
| Buffalo at the interface (B) | |||||
| B1TET | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA5 | C |
| B4TET | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA7 | D |
| B24TETd | TET, SMN, SUL | B | NDe | ND | E |
| Cattle (C) | |||||
| C1TET | TET | A | ND | ND | A |
| C2TMP | TMP, SUL | ND | ND | dfrA14 | B1 |
| C9TET | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA14 | A |
| C12TET | TET, SMN, AMX, TIC | B | + | ND | B1 |
| C18TET | TET | A | ND | ND | A |
| C18AMX | TET, AMX, SUL, TIC, AMC | A | + | ND | C |
| C25TET | TET, AMX, TMP, SUL, TIC, AMC | A | + | dfrA14 | B1 |
| C26TET | TET, SMN, AMX, TIC | B | + | ND | B1 |
| C26TMP | TMP, SUL | ND | ND | dfrA14 | A |
| C29TET | TET, SMN, AMX, TIC | B | + | ND | B1 |
| C31TMP | TET, TMP, SUL | B | ND | dfrA14 | A |
| C32TET | TET, SMN, AMX, TMP, SUL, KAN, TIC, AMC | B | + | dfrA1 | C |
| C36TET | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA1 | A |
| C36TMP | TET, AMX, TMP, SUL, TIC, AMC | A | + | dfrA14 | C |
| C37TET | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA7 | A |
| C38TMP | TMP, SUL | ND | ND | dfrA14 | B1 |
| C40TET | TET, AMX, TMP, SUL, TIC, AMC | A | + | dfrA14 | A |
| C42TMP | TMP, SUL | ND | ND | dfrA14 | B1 |
| C43TET | TET, TMP, SUL | B | ND | dfrA17 | E |
| C43TMP | TET, SMN, AMX, TMP, SUL, TIC | A | + | dfrA5 | B1 |
| C44TET | TET, TMP, SUL | A | ND | dfrA17 | A |
| C45TMP | TMP, SMN, AMX, SUL, TIC | ND | + | dfrA7 | B1 |
| C46TET | TET, SMN, AMX, TMP, SUL, GEN, TIC, AMC | C | + | dfrA17 | D |
The strains are labeled by the letter of the population, the number of the individual, and the abbreviation of the antibiotic on which they were isolated. When a strain was isolated on several antibiotics, only one is arbitrarily presented.
Abbreviations: amoxicillin (AMX), amoxicillin + acid clavulanic (AMC), ticarcillin (TIC), streptomycin (SMN), gentamicin (GEN), kanamycin (KAN), tetracycline (TET), trimethoprim (TMP), and sulfonamide (SUL).
This strain was identical to the dominant strain (B24DOM), as shown by RAPD.
ND, not determined.
In the isolated resistant strains, a high diversity of E. coli phylogenetic groups was observed with five phylogroups represented (Table 2). To document this heterogeneity further, we performed rep-PCR on the most frequently isolated subdominant tetracycline-resistant strains (Fig. 3). Only three B1 phylogroup strains from the cattle population (C12TET, C26TET, and C29TET) belong to the same clone. For the remaining strains, the rep-PCR did not reveal any identical strain between the buffalos at the interface and the cattle subdominant tetracycline-resistant strain population. Similarly, B1 phylogroup strains C2TMP, C38TMP, and C42TMP all produced a clear and distinct RAPD pattern. Of note, in the cattle population, the main phylogroups of the resistant subdominant strains were the A and B1 phylogroups (39.1% each), followed by the C phylogroup (13%), in contrast to the B1 and E phylogroups for the dominant strains (Fig. 2).
FIG 3.

Comparison of E. coli subdominant tetracycline-resistant strains by repetitive extragenic palindromic PCR using a DiversiLab strain typing system (bioMérieux, Marcy l'Etoile, France). The Clermont genotypes determined as described by Clermont et al. (40, 43) are indicated on the right of the figure.
The antibiotic-resistant strains were very rarely resistant to only one antibiotic (two strains resistant only to tetracycline) but were resistant to up to 8 of the tested antibiotics. The most common ABR was the tetracycline and sulfonamide resistance (21 strains, 80.7% of [all] the resistant strains), followed by trimethoprim (18 strains, 69.2%), amoxicillin/ticarcillin (16 strains, 61.5%), and streptomycin (13 strains, 50.0%) resistances (Table 2). Six strains were resistant to the association amoxicillin-clavulanic acid and one to kanamycin. A multiplex PCR assay of tetA to tetE genes responsible for tetracycline resistance (37) identified a tet gene in all of the tetracycline-resistant strains (Table 2). The genes were mainly tetA and tetB and found in both buffalo and cattle populations. Only one cattle strain had tetC. A multiplex PCR assay of the dfr genes involved in trimethoprim resistance identified a majority of dfrA14 genes but some dfrA1, dfrA5, dfrA7, and dfrA17 genes, the dfrA5 and dfrA7 genes being shared between buffalo and cattle populations. Lastly, we confirmed by PCR sequencing that the amoxicillin resistance found in both populations was due to narrow-spectrum β-lactamase TEM-1 (Table 2).
Altogether, these data indicate that diverse E. coli strains bearing antibiotic resistance genes (tet, dfrA, and blaTEM-1) are present in buffalo in contact with cattle and especially in cattle, but not in buffalo without contact with cattle.
DISCUSSION
We explored the structure and the level of antibiotic resistance of E. coli commensal populations and the resistant Enterobacteriaceae carriage of sympatric buffalo and cattle populations characterized by their contact patterns in a southern African ecosystem. Our results (i) identify an ABR gradient that we genetically characterized from cattle to buffalo, structured by host phylogeny and contact patterns, and (ii) confirm our initial assumptions that buffalo and cattle shared similar phylogroup profiles, albeit with some variability, that led us (iii) to hypothesize the role of the human/animal interface in the diffusion of genetic material from human to cattle and finally toward wildlife.
The main result of this study is the identification of an ABR gradient between sympatric domestic and wild ungulate populations in a tropical ecosystem. We detected this gradient at several levels. First, at the Enterobacteriaceae community level, the Murray score indicated that the cattle population had significantly more ABR than buffalo and that ungulate populations in contact (i.e., populations B and C with overlapping home ranges) shared more ABR than ungulate populations that were not in contact (population A) (Table 1, Fig. 1). Second, whereas almost no ABR was detected in dominant E. coli strains isolated from the three host populations, subdominant antibiotic-resistant E. coli strains were mainly present in cattle and, at a lower isolation ratio in the buffalo population in contact with the cattle population, whereas antibiotic-resistant E. coli strains were absent from the buffalo population that had no contact with the two other populations (Table 2). Finally, the molecular characterization of ABR associated with the observed various genetic backgrounds in the subdominant resistant E. coli strains found in populations B and C suggested that these strains rarely spread between individuals in contrast to the antibiotic resistance genes that are shared within the cattle population, as well as between buffalo and cattle at the interface. It can be hypothesized that strains can be transmitted at the interface rapidly, but that antibiotic resistance genes spread independently. This is facilitated by the fact that these genes are borne by mobile genetic structures. In E. coli, tet efflux genes are found in transposons inserted into diverse plasmids from a variety of incompatibility groups (45), and blaTEM-1 has been observed to disseminate on the Tn3 transposon (46). Similarly, dfr genes are often integron-borne genes (47). In these subdominant resistant strains, multiple resistance was observed (Table 2), which is mainly conferred by mobile genetic elements. Such a mechanism of selfish gene spread rather than strain or plasmid spread has recently been proposed to explain the dissemination of acquired resistance to β-lactams in small wild mammals in French Guiana pristine forest from an Amerindian village (48).
ABR in natural ecosystems can originate from two sources: (i) natural ABR emerging in the wild through natural selection processes or (ii) diffusion of genetic material or organisms harboring these ABR from an anthropological origin, i.e., through the use of antibiotics in domestic animals or in humans and their subsequent diffusion in the environment (10). We are confident that the gradient identified originated from the latter process (i) because the main ABR detected in the buffalo population matched the most frequently used antibiotics in domestic animal and human populations (tetracycline and streptomycin for domestic animal and trimethoprim and amoxicillin for humans), (ii) because ABR in cattle was also detected for antibiotics used in human populations, and (iii) because the resistance genes identified here have already been isolated in many different contexts, and their emergence is supposed to be a rare event. In addition, the buffalo population in contact with cattle had an intermediate degree of ABR both at the global and subdominant antibiotic-resistant E. coli strains, and all ABR found in wildlife was also found in cattle. The dominant clone is usually the clone with the best fitness in a given environment. Many drug resistances confer a fitness cost (49), and it is likely that antibiotic-resistant bacteria will be outcompeted in a low antibiotic pressure environment, such as protected areas. In this case, resistant clones will probably not be selected as dominant. However, several processes act to stabilize resistance (compensatory evolution) (50), and there is also evidence that the genetic adaptations to the costs of resistance can virtually preclude resistant E. coli lineages from reverting to sensitivity (51). This could explain why only one buffalo in contact with cattle had a dominant strain resistant to antibiotics (B24) (Table 2). Our data are in line with a worldwide study of commensal E. coli in wild and domestic animals that showed the anthropogenic origin of antibiotic resistance and integron, a molecular vector of resistance (8).
The profiles of E. coli populations between the three host populations shared a degree of similarity. The phylogenetic proximity of ungulate hosts and the fact that they seasonally use food and water resources in the same ecosystem (no supplementary feeding for cattle except for crop residues left in the fields in the study area) can explain these results. The dominant phylogroup for the three ungulate populations was B1, followed by phylogroup E (Fig. 2), in agreement with available knowledge for ruminant populations (52). However, the third phylogroup prevalence differed between populations A, B, and C. In cattle, the third most prevalent phylogroup was A, a dominant phylogroup for human populations (33), suggesting a transfer of strains between human and cattle that interact through frequent and close contacts (8). In buffalo, the third most prevalent phylogroups were D and B2, respectively, for populations A and B, indicating that different subdominant phylogroups dominate in different populations of the same species, as suggested for humans (52). Interestingly, the subdominant resistant strains of the cattle population were mainly of phylogroup A (9/26) with only two strains of phylogroup E, as opposed to the dominant clones, suggesting also a transfer of human origin (52). However, this result was not observed in the buffalo populations, from which only three strains were isolated (Table 2).
Although the mechanisms of genetic material transfer are not known, we demonstrate that the level of ABR varies according to the contact patterns between host populations. Sharing pasture and water points offers opportunities for direct and indirect transfer of organisms or genetic materials between wild and domestic ungulates. Close contacts between human and cattle occur regularly, especially when cattle are kept in the kraal every evening, where lactating female are milked, and the herders manipulate animals. Often, human and livestock share a unique water source. These behaviors can explain the presence of phylogroups of potential human origin (i.e., phylogroup A) and ABR against human antibiotics. It has been recently shown in the Amazonian forest that acquired ABR did not disseminate in the wild far (600 m) from the point of selective pressure represented by the village (48).
These results are important at two levels. First, they provide some information on the dissemination of bacteria and their resistance at wild/domestic/human interfaces, indicating that wildlife populations within a protected conservation area are not exempt from anthropological pollution, even in the most remote areas. The impacts of E. coli (and potentially other bacteria) and resistance genes transfers to wild populations are difficult to assess, but they could alter the microbiome structures in wildlife and affect their behavior and/or health (53, 54). This dissemination can also pose a threat to the domestic and human populations from which they originate, since resistance genes in different selective environments can evolve into more harmful variants when they are introduced back into domestic or human populations (55). Follow-up studies on the mechanisms of bacteria and gene diffusion in this ecosystem could be targeted at describing the human E. coli population structure and ABR and the role of other domestic and wild hosts and the environment.
Second, these results support the potential use of E. coli as an indicator of transmission pathways in multihost systems, as recently suggested (17). Dominant strains are shared between hosts in contact (e.g., phylogroup B1 and E between cattle and buffalo; potentially A between human and cattle) and offer a first level of variability to be used to assess transmission processes between hosts. If resistant subdominant strains were not shared between in-contact host populations, their ABR genes were, identifying a second level of exploitable variability and a directional transmission pathway from cattle to buffalo, with humans as the probable source population. The intensity, frequency, and directionality of these transmission events between hosts could be further investigated using new next-generation sequencing tools targeting specific genetic sequences and applied to time series of multihost sampling coupled with studies estimating proxies of interhost contacts. For example, Miguel et al. (28) indicated seasonal and interannual interhost contact patterns that could translate into pulses of ABR dissemination. The outcome would be a framework to identify “highways” of transmission between hosts, with potential spatial and temporal variability, giving a head-start to the surveillance of emerging disease spillover events.
ACKNOWLEDGMENTS
This study was implemented within the framework of the research Platform Conservation and Production in Partnership (www.rp-pcp.org) and in collaboration with CNRS within the framework of the Zone Atelier in the Hwange area.
We thank the ANR SAVARID (ANR-11-CEPL-003) project for supporting this study.
REFERENCES
- 1.Wittemyer G, Elsen P, Bean WT, Burton ACO, Brashares JS. 2008. Accelerated human population growth at protected area edges. Science 321:123–126. doi: 10.1126/science.1158900. [DOI] [PubMed] [Google Scholar]
- 2.Baudron F, Giller KE. 2014. Agriculture and nature: trouble and strife? Biol Conserv 170:232–245. doi: 10.1016/j.biocon.2013.12.009. [DOI] [Google Scholar]
- 3.Cumming GS, Buerkert A, Hoffmann EM, Schlecht E, von Cramon-Taubadel S, Tscharntke T. 2014. Implications of agricultural transitions and urbanization for ecosystem services. Nature 515:50–57. doi: 10.1038/nature13945. [DOI] [PubMed] [Google Scholar]
- 4.Caron A, Miguel E, Gomo C, Makaya P, Pfukenyi D, Hove T, Foggin C, de Garine-Wichatitksy M. 2013. Relationship between burden of infection in ungulate populations and wildlife/livestock interfaces. Epidemiol Infect 141:1522–1535. doi: 10.1017/S0950268813000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woolhouse ME. 2008. Emerging diseases go global. Nature 451:898–899. doi: 10.1038/451898a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Daszak P, Cunningham AA, Hyatt AD. 2000. Emerging infectious diseases of wildlife: threats to biodiversity and human health. Science 287:443–449. doi: 10.1126/science.287.5452.443. [DOI] [PubMed] [Google Scholar]
- 7.Kock R. 2005. What is this infamous “wildlife/livestock interface”?: a review of current knowledge, p xxxiii and 220 In Ososfsky S, Cleaveland S, Karesh WB, Kock MD, Nyphus PJ, Starr L, Yang A (ed), Conservation and development interventions at the wildlife/livestock interface: implications for wildlife, livestock and human health, vol 30 IUCN, Gland, Switzerland. [Google Scholar]
- 8.Skurnik D, Ruimy R, Andremont A, Amorin C, Rouquet P, Picard B, Denamur E. 2006. Effect of human vicinity on antimicrobial resistance and integrons in animal faecal Escherichia coli. J Antimicrob Chemother 57:1215–1219. doi: 10.1093/jac/dkl122. [DOI] [PubMed] [Google Scholar]
- 9.Cabello FC. 2006. Heavy use of prophylactic antibiotics in aquaculture: a growing problem for human and animal health and for the environment. Environ Microbiol 8:1137–1144. doi: 10.1111/j.1462-2920.2006.01054.x. [DOI] [PubMed] [Google Scholar]
- 10.Allen HK, Donato J, Wang HH, Cloud-Hansen KA, Davies J, Handelsman J. 2010. Call of the wild: antibiotic resistance genes in natural environments. Nat Rev Microbiol 8:251–259. doi: 10.1038/nrmicro2312. [DOI] [PubMed] [Google Scholar]
- 11.Wadman M. 2001. Group urges survey of antibiotics in animals. Nature 409:273–273. doi: 10.1038/35053297. [DOI] [PubMed] [Google Scholar]
- 12.Martinez JL. 2009. Environmental pollution by antibiotics and by antibiotic resistance determinants. Environ Pollut 157:2893–2902. doi: 10.1016/j.envpol.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 13.Viana M, Mancy R, Biek R, Cleaveland S, Cross PC, Lloyd-Smith JO, Haydon DT. 2014. Assembling evidence for identifying reservoirs of infection. Trends Ecol Evol 29:270–279. doi: 10.1016/j.tree.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caron A, Morand S, de Garine-Wichatitsky M. 2012. Epidemiological interaction at the wildlife/livestock/human interface: can we anticipate emerging infectious diseases in their hotspots? A framework for understanding emerging diseases processes in their hot spots, p 311–332. In Morand S, Beaudeau F, Cabaret J (ed), New frontiers of molecular epidemiology of infectious diseases. Springer, Dordrecht, Netherlands. [Google Scholar]
- 15.Daszak P, Zambrana-Torrelio C, Bogich TL, Fernandez M, Epstein JH, Murray KA, Hamilton H. 2012. Interdisciplinary approaches to understanding disease emergence: the past, present, and future drivers of Nipah virus emergence. Proc Natl Acad Sci U S A 110(Suppl 1):3681–3688. doi: 10.1073/pnas.1201243109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van den Broek PJ, Bernards AT, van der Reijden TJ, van Strijen B, Dijkshoorn L. 2009. Can Escherichia coli be used as an indicator organism for transmission events in hospitals? Eur J Clin Microbiol Infect Dis 28:169–173. doi: 10.1007/s10096-008-0597-0. [DOI] [PubMed] [Google Scholar]
- 17.VanderWaal KL, Atwill ER, Isbell LA, McCowan B. 2014. Quantifying microbe transmission networks for wild and domestic ungulates in Kenya. Biol Conserv 169:136–146. doi: 10.1016/j.biocon.2013.11.008. [DOI] [Google Scholar]
- 18.VanderWaal KL, Atwill ER, Isbell LA, McCowan B. 2013. Linking social and pathogen transmission networks using microbial genetics in giraffe (Giraffa camelopardalis). J Anim Ecol 83:406–414. doi: 10.1111/1365-2656.12137:n/a-n/a. [DOI] [PubMed] [Google Scholar]
- 19.Pesapane R, Ponder M, Alexander KA. 2013. Tracking pathogen transmission at the human-wildlife interface: banded mongoose and Escherichia coli. Ecohealth 10:115–128. doi: 10.1007/s10393-013-0838-2. [DOI] [PubMed] [Google Scholar]
- 20.Benavides JA, Godreuil S, Bodenham R, Ratiarison S, Devos C, Petretto MO, Raymond M, Escobar-Paramo P. 2012. No evidence for transmission of antibiotic-resistant Escherichia coli strains from humans to wild western lowland gorillas in Lope National Park, Gabon. Appl Environ Microbiol 78:4281–4287. doi: 10.1128/AEM.07593-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rwego IB, Gillespie TR, Isabirye-Basuta G, Goldberg TL. 2008. High rates of Escherichia coli transmission between livestock and humans in rural Uganda. J Clin Microbiol 46:3187–3191. doi: 10.1128/JCM.00285-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez JL, Fajardo A, Garmendia L, Hernandez A, Linares JF, Martinez-Solano L, Sanchez MB. 2009. A global view of antibiotic resistance. FEMS Microbiol Rev 33:44–65. doi: 10.1111/j.1574-6976.2008.00142.x. [DOI] [PubMed] [Google Scholar]
- 23.Levy SB, Marshall B. 2004. Antibacterial resistance worldwide: causes, challenges and responses. Nat Med 10:S122–S129. doi: 10.1038/nm1145. [DOI] [PubMed] [Google Scholar]
- 24.Gordon DM, Cowling A. 2003. The distribution and genetic structure of Escherichia coli in Australian vertebrates: host and geographic effects. Microbiology 149:3575–3586. doi: 10.1099/mic.0.26486-0. [DOI] [PubMed] [Google Scholar]
- 25.Duriez P, Clermont O, Bonacorsi S, Bingen E, Chaventre A, Elion J, Picard B, Denamur E. 2001. Commensal Escherichia coli isolates are phylogenetically distributed among geographically distinct human populations. Microbiology 147:1671–1676. doi: 10.1099/00221287-147-6-1671. [DOI] [PubMed] [Google Scholar]
- 26.Hancock DD, Besser TE, Rice DH, Herriott DE, Tarr PI. 1997. A longitudinal study of Escherichia coli O157 in fourteen cattle herds. Epidemiol Infect 118:193–195. doi: 10.1017/S0950268896007212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lescat M, Clermont O, Woerther PL, Glodt J, Dion S, Skurnik D, Djossou F, Dupont C, Perroz G, Picard B, Catzeflis F, Andremont A, Denamur E. 2013. Commensal Escherichia coli strains in Guiana reveal a high genetic diversity with host-dependent population structure. Environ Microbiol Rep 5:49–57. doi: 10.1111/j.1758-2229.2012.00374.x. [DOI] [PubMed] [Google Scholar]
- 28.Miguel E, Grosbois V, Caron A, Boulinier T, Fritz H, Cornélis D, Foggin C, Makaya PV, Tshabalala PT, de Garine-Wichatitsky M. 2013. Contacts and foot and mouth disease transmission from wild to domestic bovines in Africa. Ecosphere 4:art51. doi: 10.1890/ES12-00239.1. [DOI] [Google Scholar]
- 29.Cumming DHM. 2004. Sustaining animal health and ecosystem services in large landscapes, 2nd draft Wildlife Conservation Society, Bronx, NY: http://www.wcs-ahead.org/documents/gltfca_cumming.pdf. [Google Scholar]
- 30.Sinclair A. 1977. The African buffalo: a study of resource limitation by populations. University of Chicago Press, Chicago, IL. [Google Scholar]
- 31.Zengeya FM, Murwira A, De Garine-Wichatitsky M. 2014. Seasonal habitat selection and space use by a semi-free range herbivore in a heterogeneous savanna landscape. Austral Ecol 39:722–731. doi: 10.1111/aec.12137. [DOI] [Google Scholar]
- 32.Benhamou S. 2011. Dynamic approach to space and habitat use based on biased random bridges. PLoS One 6:e14592. doi: 10.1371/journal.pone.0014592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smati M, Clermont O, Le Gal F, Schichmanoff O, Jaureguy F, Eddi A, Denamur E, Picard B, Coliville G. 2013. Real-time PCR for quantitative analysis of human commensal Escherichia coli populations reveals a high frequency of subdominant phylogroups. Appl Environ Microbiol 79:5005–5012. doi: 10.1128/AEM.01423-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smati M, Clermont O, Bleibtreu A, Fourreau F, David A, Daubié A-S, Hignard C, Loison O, Picard B, Denamur E. 2015. Quantitative analysis of commensal Escherichia coli populations reveals host-specific enterotypes at the intra-species level. Microbiologyopen 4:604–615. doi: 10.1002/mbo3.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lester SC, del Pilar Pla M, Wang F, Perez Schael I, Jiang H, O'Brien TF. 1990. The carriage of Escherichia coli resistant to antimicrobial agents by healthy children in Boston, in Caracas, Venezuela, and in Qin Pu, China. N Engl J Med 323:285–289. doi: 10.1056/NEJM199008023230501. [DOI] [PubMed] [Google Scholar]
- 36.Murray BE, Mathewson JJ, DuPont HL, Ericsson CD, Reves RR. 1990. Emergence of resistant fecal Escherichia coli in travelers not taking prophylactic antimicrobial agents. Antimicrob Agents Chemother 34:515–518. doi: 10.1128/AAC.34.4.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nawaz M, Sung K, Khan SA, Khan AA, Steele R. 2006. Biochemical and molecular characterization of tetracycline-resistant Aeromonas veronii isolates from catfish. Appl Environ Microbiol 72:6461–6466. doi: 10.1128/AEM.00271-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitout JD, Thomson KS, Hanson ND, Ehrhardt AF, Coudron P, Sanders CC. 1998. Plasmid-mediated resistance to expanded-spectrum cephalosporins among Enterobacter aerogenes strains. Antimicrob Agents Chemother 42:596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vallenet D, Belda E, Calteau A, Cruveiller S, Engelen S, Lajus A, Le Fevre F, Longin C, Mornico D, Roche D, Rouy Z, Salvignol G, Scarpelli C, Thil Smith AA, Weiman M, Medigue C. 2013. MicroScope: an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data. Nucleic Acids Res 41:D636–D647. doi: 10.1093/nar/gks1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clermont O, Christenson JK, Denamur E, Gordon DM. 2013. The Clermont Escherichia coli phylo-typing method revisited: improvement of specificity and detection of new phylo-groups. Environ Microbiol Rep 5:58–65. doi: 10.1111/1758-2229.12019. [DOI] [PubMed] [Google Scholar]
- 41.Clermont O, Gordon DM, Brisse S, Walk ST, Denamur E. 2011. Characterization of the cryptic Escherichia lineages: rapid identification and prevalence. Environ Microbiol 13:2468–2477. doi: 10.1111/j.1462-2920.2011.02519.x. [DOI] [PubMed] [Google Scholar]
- 42.Woerther PL, Angebault C, Lescat M, Ruppe E, Skurnik D, Mniai AE, Clermont O, Jacquier H, Costa AD, Renard M, Bettinger RM, Epelboin L, Dupont C, Guillemot D, Rousset F, Arlet G, Denamur E, Djossou F, Andremont A. 2010. Emergence and dissemination of extended-spectrum beta-lactamase-producing Escherichia coli in the community: lessons from the study of a remote and controlled population. J Infect Dis 202:515–523. doi: 10.1086/654883. [DOI] [PubMed] [Google Scholar]
- 43.Clermont O, Glodt J, Burdet C, Pognard D, Lefort A, Branger C, Denamur E, Members CG. 2013. Complexity of Escherichia coli bacteremia pathophysiology evidenced by comparison of isolates from blood and portal of entry within single patients. Int J Med Microbiol 303:529–532. doi: 10.1016/j.ijmm.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 44.Development Core Team R. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 45.Chopra I, Roberts M. 2001. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev 65:232–260. doi: 10.1128/MMBR.65.2.232-260.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marcade G, Deschamps C, Boyd A, Gautier V, Picard B, Branger C, Denamur E, Arlet G. 2009. Replicon typing of plasmids in Escherichia coli producing extended-spectrum beta-lactamases. J Antimicrob Chemother 63:67–71. doi: 10.1093/jac/dkn428. [DOI] [PubMed] [Google Scholar]
- 47.Partridge SR, Tsafnat G, Coiera E, Iredell JR. 2009. Gene cassettes and cassette arrays in mobile resistance integrons. FEMS Microbiol Rev 33:757–784. doi: 10.1111/j.1574-6976.2009.00175.x. [DOI] [PubMed] [Google Scholar]
- 48.Grall N, Barraud O, Wieder I, Hua A, Perrier M, Babosan A, Gaschet M, Clermont O, Denamur E, Catzeflis F, Decre D, Ploy MC, Andremont A. 2015. Lack of dissemination of acquired resistance to beta-lactams in small wild mammals around an isolated village in the Amazonian forest. Environ Microbiol Rep 7:698–708. doi: 10.1111/1758-2229.12289. [DOI] [PubMed] [Google Scholar]
- 49.Andersson DI. 2003. Persistence of antibiotic resistant bacteria. Curr Opin Microbiol 6:452–456. doi: 10.1016/j.mib.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 50.Andersson DI. 2006. The biological cost of mutational antibiotic resistance: any practical conclusions? Curr Opin Microbiol 9:461–465. doi: 10.1016/j.mib.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 51.Schrag SJ, Perrot V, Levin BR. 1997. Adaptation to the fitness costs of antibiotic resistance in Escherichia coli. Proc R Soc Lond Ser B Biol Sci 264:1287–1291. doi: 10.1098/rspb.1997.0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tenaillon O, Skurnik D, Picard B, Denamur E. 2010. The population genetics of commensal Escherichia coli. Nat Rev Microbiol 8:207–217. doi: 10.1038/nrmicro2298. [DOI] [PubMed] [Google Scholar]
- 53.Taschuk R, Griebel PJ. 2012. Commensal microbiome effects on mucosal immune system development in the ruminant gastrointestinal tract. Anim Health Res Rev 13:129–141. doi: 10.1017/S1466252312000096. [DOI] [PubMed] [Google Scholar]
- 54.Power ML, Emery S, Gillings MR. 2013. Into the wild: dissemination of antibiotic resistance determinants via a species recovery program. PLoS One 8:e63017. doi: 10.1371/journal.pone.0063017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davies J, Davies D. 2010. Origins and evolution of antibiotic resistance. Microbiol Mol Biol Rev 74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]