

Abstract
Stepwise deletions in the only plasmid in Thermus thermophilus HB27, megaplasmid pTT27, showed that two distantly located loci were important for maintenance of the plasmid. One is a minimum replicon including one gene, repT, coding a replication initiator, and the other encodes subunits of class I ribonucleotide reductase (RNR) for deoxynucleoside triphosphate (dNTP) synthesis. Since the initiator protein, RepT, bound to direct repeats downstream from its own gene, it was speculated that a more-downstream A+T-rich region, which was critical for replication ability, could be unwound for replication initiation. On the other hand, the class I RNR is not necessarily essential for cell growth, as evidenced by the generation of the plasmid-free strain by the loss of pTT27. However, the plasmid-free strain culture has fewer viable cells than the wild-type culture, probably due to a dNTP pool imbalance in the cell. This is because of the introduction of the class I RNR genes or the supplementation of 5′-deoxyadenosylcobalamin, which stimulated class II RNR encoded in the chromosome, resolved the decrease in the number of viable cells in the plasmid-free strain. Likewise, these treatments dramatically enhanced the efficiency of transformation by exogenous plasmids and the stability of the plasmids in the strain. Therefore, the class I RNR would enable the stable maintenance of plasmids, including pTT27, as a result of genome replication normalized by reversing the dNTP pool imbalance. The generation of this plasmid-free strain with great natural competence and its analysis in regard to exogenous plasmid maintenance will expand the availability of HB27 for thermophilic cell factories.
INTRODUCTION
Thermus spp. are extremely thermophilic bacteria that can grow at high temperatures from 50 to 82°C. Their thermophilic enzymes, e.g., DNA polymerase for PCR, have been used in industrial applications, and their potential as cell factories to produce enzymes from other thermophiles has been demonstrated (1). The bacteria themselves have also been extensively investigated as model organisms for systems biology including structural genomics (1, 2), and their genomic DNAs, with G+C contents as high as about 70%, are an interesting target for synthetic biology (3). Thermus-related studies have been encouraged by the existence of an established genetic engineering system based on natural competence (4–6), with thermostabilized drug resistance markers (7–10) and other thermostable genetic tools (1, 11–16).
Complete genomic sequences of T. scotoductus SA-01, T. oshimai JL-2, Thermus sp. strain CCB_US3_UF1, T. aquaticus Y51MC23, T. thermophilus strains HB8, HB27, SG0.5JP17-16, and JL-18, and others are available online. Thermus spp. often harbor a megaplasmid larger than 100 kbp, whereas in some strains, the megaplasmid genes seem to be transferred to the chromosome (17, 18). Similar megaplasmids have been observed in other bacteria of the Thermus-Deinococcus group showing resistance to extreme stresses such as high temperature, radiation, desiccation, and oxidation. It has not been reported whether these megaplasmids, carrying hundreds of genes, are essential for cell growth in the bacterial group. Furthermore, the replication initiation mechanism remains to be analyzed, in contrast to that of smaller Thermus plasmids such as pTT8 (9.3 kbp) (19, 20), a 16-kbp unnamed plasmid from strain ATCC 27737 (21), pTA103 (2.0 kbp) (22), and pVV8 (81 kbp) (23).
Megaplasmids are often found in bacteria such as Alphaproteobacteria, Streptococcaceae, and Planctomycetaceae. Among them, the replication origins of alphaproteobacterial megaplasmids have been the most extensively studied (24–26). The three contiguous genes repABC are responsible for plasmid replication and segregation, and repAB and repC encode partitioning proteins and an initiation factor, respectively (24–28). An A+T-rich region observed in and around the repABC genes was speculated to be unwound for replication initiation. In Paracoccus versutus, belonging to the Alphaproteobacteria, the pTAV3 megaplasmid curing has been reported to lead to a growth defect in minimal medium, raising the question of whether the replicon is a plasmid or a minichromosome (25). Similarly, a large replicon, pNRC100, in a halophilic archaeon has been reported to harbor essential genes, and the potential of the replicon to evolve into a new chromosome has been mentioned, based on its resistance to curing (29). On what criteria should the issue of megaplasmid versus minichromosome be judged?
pTT27, with a size of about 233 kbp, is the only plasmid in T. thermophilus HB27, and its complete sequence was the first released among the megaplasmids from Thermus spp. (30). Thermus-Deinococcus bacteria are usually pigmented yellow or red because of carotenoid production (31), and HB27 is no exception. The terminal steps of the synthesis of this yellow pigment are encoded on pTT27 (30). Likewise, pTT27 encodes terminal biosynthesis steps for cobalamin (30), which serves as a cofactor for cellular enzymes, as a ligand for RNA riboswitches in the control of gene expression (32), and as a light-sensing chromophore to mediate light-dependent gene regulation (33). The closely related strain HB8 possesses a similar, 24-kb-larger megaplasmid with the same name, but synteny between the two pTT27 megaplasmids is less conserved than that between two chromosomes (34). Many large plasmids, as described above, seem to be present at low copy numbers in each cell (24–26), while the copy number of pTT27 is estimated to be four or five per cell in HB8 (35). T. thermophilus is polyploid, and the pTT27 copy number is equal to the chromosomal one (35). We focused on the sole plasmid pTT27 in HB27 to survey its replication origin and to determine which genes are essential for cell growth. If the plasmid had been smaller than 100 kb, allowing it to be easily handled for plasmid preparation and transformation, fragments of digested pTT27 would have been subcloned in Escherichia coli to identify the replication origin and essential genes, as performed previously in pVV8, which can be replicated by a repV gene alone (23). At the start of the present study, however, preparation of the intact high-purity pTT27 from HB27 was very difficult because of insufficient skill in manipulating the megaplasmid. Therefore, a strategy of pTT27 miniaturization in the HB27 cell was employed repeatedly until a size permitting easy handling was obtained.
In this study, a minimum replicon of the megaplasmid pTT27 was identified as a 2.3-kb region including a single gene encoding an initiator protein. During the process, a plasmid-free substrain of HB27 was generated as a result of pTT27 curing. This plasmid-free strain, with superior natural competence, has great potential as a host strain for DNA engineering and thermophilic cell factories. Findings on the stability of exogenous plasmids in the strain as presented here will contribute greatly to the development of DNA manipulation with T. thermophilus HB27 as well as its plasmid-free substrain.
MATERIALS AND METHODS
Cells, plasmids, and materials.
T. thermophilus HB27 was described previously (36). The construction of an HB27 recA-null mutant and of plasmids pUC-Hm and pUC-Vmini, in which a hygromycin resistance (Hmr) gene was cloned at the NdeI site of pUC19 and the replication origin of the intrinsic plasmid pVV8 from T. thermophilus HB8 was inserted into pUC-Hm, was performed as described previously (23). Plasmid pUC-TT8, in which the replication origin of pTT8 from HB8 (19) was inserted into pUC-Hm, was constructed in this study. A kanamycin resistance (Kmr) gene (7) and the Hmr gene (9) for T. thermophilus were kindly donated by Seiki Kuramitsu (Osaka University) and Yoshinori Koyama (AIST), respectively, whereas a bleomycin resistance (Bmr) gene (10) was chemically synthesized by TaKaRa Bio. E. coli Rosetta2(DE3) and the plasmid pET-22b were purchased from Merck Millipore, while plasmid pSTV29 for E. coli was from TaKaRa Bio. Oligonucleotides with a 6-carboxyfluorescein (FAM) label at the 5′ ends were synthesized by Eurofins Genomics K.K. 5′-Deoxyadenosylcobalamin (AdoCbl) (coenzyme [vitamin] B12) was purchased from Sigma-Aldrich.
Growth conditions and transformation.
T. thermophilus was grown at 70°C in TR medium, and 1.5% gellan gum with 1.5 mM CaCl2 and 1.5 mM MgCl2 was added to the medium for the plates (6). Km at 500 μg/ml, Hm at 100 μg/ml, Bm at 20 μg/ml, and/or AdoCbl at 0.1 μg/ml was added to the medium when needed. Transformation was carried out as described previously (6). The recA-null mutant was grown at 58°C as described previously (37). Synthetic medium (38) was used as the minimal medium.
Initial deletion of pTT27 region in HB27.
Fragments of approximately 5 kbp for recombination with pTT27 in T. thermophilus HB27 cells were amplified by PCR with the primers listed in Table 1 and LA Taq with GC buffer (TaKaRa Bio). One fragment digested by EcoRI and NotI was inserted into an EcoRI-SalI region of pUC-Hm with another fragment digested by NotI and SalI (Fig. 1A, panel i). The resultant plasmid was linearized by NotI digestion and then transferred to HB27. When all colonies appeared white on the TR plate containing Hm, it was judged that the expected deletion of the pTT27 region had been achieved in the transformants. The deletion was checked by restriction enzyme digestion of the extracted plasmid in addition to colony PCR.
TABLE 1.
PCR primer sequences to amplify ca. 5-kbp regions for homologous recombination with pTT27
| Locus | Location on pTT27 (nt) | Length of region (bp) | Primer sequencea |
|---|---|---|---|
| 1 | 231441–3800 | 4,965 | 5′-AAGCGGCCGCGAATTCCTACCTGCCCCAGGACCTTCCCGATGGAAAGCGGCAGGAG |
| 5′-AAGCGGCCGCGTCGACTTGAGCTTGTGGCCATCGCCGGGGTCTACCTCGAGGGGTACGAGG | |||
| 2 | 59961–65394 | 5,434 | 5′-AAGCGGCCGCGAATTCGCAGATCTTCTCCTTTGAGCCCCTCCTCATCGGAGGGAAC |
| 5′-AAGCGGCCGCGTCGACGCCTTGGCCCACTTCACCGGCTTCAAGGCCACTTCCGACC | |||
| 3 | 119941–125680 | 5,740 | 5′-AAGCGGCCGCGAATTCGAGGCAACTTGCTGAGGTCCAGGACGTTCTGCCGGGAACC |
| 5′-AAGCGGCCGCGTCGACCCGCTTCATGGGTACCTCCAGCGCACCTCGGGGTCGGACATG | |||
| 4 | 179931–185270 | 5,340 | 5′-AAGCGGCCGCGAATTCTTCACGCCTCCGGTTTGACGACCGGGTACATGTATCTTGC |
| 5′-AAGCGGCCGCGTCGACTAAGGACGGCGACGATGTTGAGGTTCTCCGTGTAGTTCGG |
The underlined, boldface, and italic sequences indicate NotI, EcoRI, and SalI sites, respectively.
FIG 1.

Large-scale deletion in megaplasmid pTT27. (A) Deletion approaches used in this study. Panel i, two PCR-amplified 5-kbp fragments, represented by the arrows, were inserted into pUC-Hm as a homologous region for recombination (dotted lines) with pTT27. The plasmid constructed in E. coli was transferred to T. thermophilus HB27 after linearization by NotI digestion. The transformants were selected by Hm resistance from pUC-Hm. Black and white stars indicate essential gene(s) and the replication origin, respectively. Panel ii, similar 5-kbp fragments were inserted into pSTV29 with Kmr as indicated. The resultant plasmid linearized by XbaI was transferred to the Hm-resistant HB27 shown in panel i. The transformants were selected in the presence of Km. (B) Actual large-scale deletions in pTT27. Panel I, deletion trials are schematically shown. The top line indicates the intact pTT27 of 232,605 bp, and the numbers above are positions on the megaplasmid. Red arrows 1, 2, 3, and 4 and blue arrows a, b, c, and d show the loci for homologous recombination (Table 1) and the target regions for colony PCR in panel iii, respectively. The lower lines represent the putative residual region after deletion, and the numbers at both ends display the end locations of the region. The replication ability of each region is shown by (+) or (−) on the left side, and the region exhibiting the replication ability, as shown by (+), is emphasized by a bold line. The leftmost bold numbers match the lane numbers in panel iv and Fig. 2C. Panel ii, colonies on the Hm-containing plate. The deletion trial of the region from locus 4 to locus 2 resulted in all colonies being white, owing to the lack of the carotenoid biosynthesis-related genes on pTT27. Panel iii, the existence or nonexistence of the four regions shown by blue arrows in panel i in the colonies of panel ii was checked by colony PCR with the primers listed in Table 2. The deletion from locus 4 to locus 2 was supported by the lack of amplification of regions a and d. Lane M, size marker. Panel iv, the intact pTT27 (lane 1) and pUC27H2 (lane 2) prepared from HB27 were digested by both NdeI and SphI, followed by CHEF electrophoresis. Lane M, size marker of concatemeric λ DNA and the HindIII-digested one. Running conditions were 3 V/cm with a 12-s pulse time and 16-h running time at 15°C.
Two 5-kbp fragments were cloned into an EcoRI-SalI region of pSTV29 in a similar manner, and subsequently the Kmr gene was inserted into the NotI site between the fragments (Fig. 1A, panel ii). The resultant plasmid was linearized by XbaI and transferred to the pTT27-miniaturized HB27. The transformants were selected on a Km-containing TR plate.
Stepwise deletions of pTT27 region in E. coli.
The Kmr-labeled pTT27 derivative (pUC27H4) is a shuttle vector between T. thermophilus and E. coli with a size of about 40 kbp. Additional deletions of the pTT27 region were carried out by commonly used methods with E. coli. The replication ability of the reduced pTT27 regions was verified by transformation of the recA-null mutant of HB27 harboring the intact pTT27. When the region possesses replication ability in T. thermophilus, colonies on the selection plate containing Hm are white because of the loss of intact pTT27 resulting from plasmid incompatibility.
DNA preparation and gel electrophoresis.
The alkali-SDS method was meticulously applied to the preparation of the megaplasmid and its derivatives from T. thermophilus, followed by ultracentrifugation in a CsCl-ethidium bromide gradient (39) especially for large-scale preparation. Intact genomic DNA was prepared in an agarose plug by a method reported previously (40), followed by in-gel digestion with restriction enzymes. The plasmid or genomic DNA was analyzed (Fig. 1B, panel iv; see Fig. 3B) by contour-clamped homogeneous electric field (CHEF) gel electrophoresis in a 1.0% agarose gel in TBE buffer (50 mM Tris-borate [pH 8.0] and 1.0 mM EDTA) at 15°C. Gels were stained with ethidium bromide and visualized under UV light.
FIG 3.

Curing pTT27 from T. thermophilus HB27. (A) Strategy to cure the pTT27 megaplasmid from HB27. When HB27 was transformed by pUC27H11, the intact pTT27 was excluded due to plasmid incompatibility. The transformant lacking pTT27 but containing pUC27H11 was subsequently transformed by pUC27K11. After both plasmid-possessing transformants obtained in the presence of both Hm and Km were cultivated without drugs, their colonies on a drug-free plate were checked for drug sensitivity. One drug-sensitive strain without any plasmid was selected as the PFW substrain. (B) Confirmation of the absence of plasmids in the PFW strain. Intact genomic DNAs of the WT (lane 1) and PFW (lane 2) strains were prepared in gel plugs and analyzed by CHEF electrophoresis after in-gel digestion by both EcoT22I and MunI. Lane M, size marker of concatemeric λ DNA and a HindIII-digested one. Running conditions were 5 V/cm with a 30-s pulse time and 20-h running time at 15°C. The absence of a linearized megaplasmid in lane 2 is indicated by the arrowhead.
Curing of pTT27 from HB27.
In plasmid pUC27H11, three genes (the ribonucleotide reductase [RNR] α and β subunit genes and repT) were cloned into pUC-Hm. When the Kmr gene replaces the Hmr gene on the plasmid, the plasmid is referred to as pUC27K11. T. thermophilus HB27 was initially transformed by pUC27H11. Subsequently, the obtained Hm-resistant white HB27 was transformed by pUC27K11, and transformants were selected in the presence of both Km and Hm. After a transformant resistant to both drugs was grown in TR medium in the absence of the antibiotics for 3 days, the cells were spread on an antibiotic-free TR plate. Among 300 colonies analyzed for drug resistance, 13 exhibited sensitivity to both Km and Hm. Also, when transformed by pUC27K11 initially and by pUC27H11 secondarily, drug-sensitive colonies were obtained in a similar fashion. Genomic DNAs from these strains were prepared in agarose plugs and checked by CHEF gel electrophoresis.
Preparation of RepT recombinant protein.
The plasmid pET-RepT, for the overexpression of the C-terminally His6-tagged RepT protein, was constructed by ligating a DNA fragment containing the repT gene to the NdeI-NotI site of pET-22b. The DNA fragment was amplified by PCR using the pTT27 plasmid as a template. The primer sequences for PCR were 5′-TTTTCATATGGCCCGCAAGCGCAAGCAGGACGCCACCCCGCCCCTC-3′ for the 5′ primer and 5′-TTTTGCGGCCGCGCCGGGGAGCATCTTGCGGAAGCGTTCCAGCCAACC-3′ for the 3′ primer, where the underlined bases show the positions of the NdeI (5′ primer) and NotI (3′ primer) sites. Subsequently, plasmid construction, overproduction, and purification of the C-terminally His6-tagged RepT fused to the maltose-binding protein (MBP) were performed as described in a previous report (23). To separate the His-tagged RepT from MBP, the purified proteins were treated with PreScission protease (GE Healthcare Bio-Sciences) and applied to a column of HisTrap HP (GE Healthcare Bio-Sciences) as described previously (23). The eluted proteins were concentrated, dialyzed against 20 mM Tris-HCl (pH 8.0) containing 1 mM EDTA, 0.5 M NaCl, and 3 mM β-mercaptoethanol, and used for further analyses. The protein concentration was determined by the Bradford assay and by measuring UV absorption using a theoretical molar extinction coefficient of 1.13, which was calculated from ε values of 1,576 M−1cm−1 for Tyr and 5,225 M−1cm−1 for Trp at 280 nm (41).
Gel retardation assay.
The plasmid pUC27H11 carries repT and RNR genes on the pUC-Hm vector. A PvuII fragment containing the genes from the plasmid was digested by XhoI and NcoI, and the resultant fragments were used at 0.05 μM as a substrate in for the binding assay (see Fig. 6A) in a total volume of 20 μl. The binding reaction was carried out at 37°C for 30 min in 10 mM Tris-HCl (pH 8.0) containing 0.1 M NaCl, 1 mM dithiothreitol (DTT), 50 ng/μl acetylated bovine serum albumin (AcBSA), and 100 ng/μl λ DNA. The products were analyzed by ethidium bromide staining after electrophoresis with a 2.0% agarose gel. Substrates for Fig. 6C were amplified by PCR using the pUC27H11 plasmid as a template and used for the assay.
FIG 6.

Gel retardation assay to identify the RepT-binding region. (A) The fragmented pTT27 regions of pUC27H11 were incubated with the recombinant RepT protein at 0, 0.37, 0.74, and 1.5 μM (lanes 1 to 4, respectively), and the RepT-binding region was analyzed by gel retardation assay. Lanes M1 and M2, size markers of HindIII-digested λ DNA and HaeIII-digested ϕX174, respectively. The upper scheme presents the pTT27 regions of pUC27H11, and the numbers indicate the size of each fragment. (B) Nucleotide sequence of the 699-bp RepT-binding fragment. Because the full sequence of XhoI at both ends is included, a 705-bp sequence is displayed. The numbers are the nucleotide positions initiated from the first C of the first XhoI sequence. Part of the repT gene and the next gene, parA, are boxed in gray. Nine direct repeat sequences and the A+T-rich region are shown in red and blue, respectively. The two regions boxed in orange are the exact same sequence. (C) Parts of the 699-bp fragment were amplified by PCR and used as substrates for gel retardation assay. They were incubated with the RepT protein at 0, 0.37, 0.74, and 1.5 μM (lanes 1 to 4, respectively). Lane M, size marker of HaeIII-digested ϕX174. The numbers along the right-hand side represent the corresponding regions in the 699-bp fragment (nt 1 to 705).
DNase I footprinting.
A region from nucleotide (nt) 1 to 420 of a 699-bp XhoI fragment (see Fig. 6B) was amplified by PCR, for which one of the two primers was labeled with fluorescein at its 5′ end, and used at 0.5 μM as a substrate for footprinting analysis. After the binding reaction in a total volume of 20 μl with 100 ng/μl poly(dI-dC)/poly(dI-dC) (Sigma) in exchange for λ DNA, the reaction mixture was incubated with DNase I (final concentration, 20 ng/μl) (TaKaRa Bio) for 5 min at room temperature. The reaction was stopped by the addition of an equal volume of DNase I stop solution (20 mM EDTA, 8 M urea, 1% SDS, and 125 ng/μl yeast tRNA), followed by phenol extraction and ethanol precipitation. The resultant DNAs were analyzed by 7 and 11% PAGE with 8 M urea by using a Molecular Imager FX (Bio-Rad). Parts of the XhoI fragment were amplified by PCR and used as markers to identify the binding sites.
RESULTS
Stepwise deletions in the pTT27 megaplasmid.
Deletion of half of the pTT27 megaplasmid in T. thermophilus HB27 was attempted in order to identify the replication origin and the genes essential for cell growth. As shown in Fig. 1 and Table 1, four almost equally spaced loci (loci 1, 2, 3, and 4) on pTT27 were randomly selected. An approximately 5-kbp region in each locus was cloned for homologous recombination with the megaplasmid as described in Materials and Methods. Deletions of the regions from locus 3 to locus 1, from locus 4 to locus 2, from locus 1 to locus 3, and from locus 2 to locus 4 correspond to the remains of the regions from locus 1 to locus 3 (127 kb), locus 2 to locus 4 (125 kb), locus 3 to locus 1 (114 kb), and locus 4 to locus 2 (118 kb), respectively. These deletion trials were performed as shown in Fig. 1A, panel i. Although we had guessed that only the transformant in which the expected deletion was established would form colonies on the selection plate, colonies were observed in all four trials. In the case of the deletion from locus 3 to locus 1, all colonies on the plate were as yellow as those of the wild-type (WT) HB27 in Fig. 1B, panel ii. On the other hand, all of the colonies with the deletion from locus 4 to locus 2 were white. With the deletions from locus 1 to locus 3 and from locus 2 to locus 4, several colonies were white, but a large portion of the colonies were yellow. The yellow colonies link to the carotenoid biosynthesis-related genes on pTT27, and these genes are located upstream of locus 2 (Fig. 1B, panel i). This means that the deletions from locus 4 to locus 2 and from locus 1 to locus 3 must produce white colonies on the selection plate, and the result for the deletion from locus 4 to locus 2 suggests that the expected deletion was completed. The colonies from each deletion trial were exposed to colony PCR with the primers listed in Table 2 to check the deletion of the area sandwiched by the two 5-kb regions for homologous recombination. The results in Fig. 1B, panel iii, suggest that only the deletion from locus 4 to locus 2 could be established in a predictable way. This was supported by restriction enzyme digestion of the extracted plasmid (Fig. 1B, panel iv). As the largest two fragments from the intact pTT27 digested by both NdeI and SphI contain the region for homologous recombination, they were not observed in the plasmid with the deletion from locus 4 to locus 2. Instead, two truncated fragments, each of about 16 to 17 kbp, would appear. In addition, three fragments of about 13 to 17 kbp, as well as one 10.5-kbp fragment, one 3.3-kbp fragment, and two 2.3-kbp fragments, were lost in the deletion plasmid, but one 2.4-kbp fragment derived from pUC19 was added. The fragments around 13 to 17 kbp were too intricate to assess, while the other changes were confirmed. Unlike the results for the deletion from locus 4 to locus 2, the expected digestion patterns were not observed in the other three deletion trials (data not shown). This halved pTT27 remaining from locus 2 to locus 4 is referred to as pUC27H2.
TABLE 2.
Primer sets for colony PCR to check deletions in pTT27
| Target region | Position on pTT27 (nt) | Amplified length (bp) | Primer sequence |
|---|---|---|---|
| a | 32391–33052 | 662 | 5′-GAGAGGACGAGCTTGGGGCCATGGTGGAGGAAAGGCTCCG |
| 5′-GAGGAGGTTGGCGAGGCGAAGCCTTTCCTCCACCGGTAGC | |||
| b | 92451–92950 | 490 | 5′-CCCATGGGAAAGCGTCTCTATGCCGTGGCGTACGACATTC |
| 5′-AAAGAGCCCGGCATGCTCCCGGAAGAAGTGGGCAGTGTAG | |||
| c | 162064–162504 | 441 | 5′-GCTTTTGGGCCTGGAGGAGGCCTTTGGTCAGGCCCTG |
| 5′-CTGTGGCCCACCAGGGCAGGCTCCCCCGCGGAGAAGAG | |||
| d | 210037–210487 | 451 | 5′-CCCACTTCCAGCACGAAGTCCACTTCCTGG |
| 5′-TGTGGCTCCGCGCCTACCTGCAAACCTACC |
Why was colony formation observed in all of the deletion trials? It was speculated that homologous recombination between the transferred DNA and pTT27 occurred and that the expected deletion was completed. The deletion would have applied to some, but not all, copies of pTT27 because its copy number is four or five per cell (35). If the Hmr-labeled deleted version of pTT27 contained its replication factors, it would replace the intact pTT27 through plasmid incompatibility in the presence of Hm, as shown in the white colonies caused by the deletion of locus 4 to locus 2. If the Hmr-labeled deleted version possessed no replication factors, on the other hand, it would never replicate in a cell. However, the deleted one must be incorporated into the intact pTT27 by a homologous recombination in the remaining region somewhere, since it has been implied that recombination occurs with great frequency in T. thermophilus cells (35, 37). This is why the Hm-resistant colonies were formed in all the deletion trials. Thus, the results of the colony PCR for the deletion from locus 4 to locus 2 were homogeneous, while those for the other deletions were heterogeneous, probably because of recombination at unspecified regions. These heterogeneous results were also shown in restriction enzyme digestion analysis of the extracted plasmids (data not shown). At least, the deletion of the region from locus 4 to locus 2 is established, and the four randomly selected loci fortunately encourage screening of the replication factors of pTT27 by the appearance of the white colonies due to the loss of the carotenoid biosynthesis genes.
Further deletion trials, as indicated in Fig. 1B, panel i, showed that the remainder of the region from nt 75911 to 170970 in pTT27 made all the colonies white. The resultant plasmid, as shown in lane 3 of Fig. 2C, is referred to as pUC27H3. When the pTT27 region of the plasmid was shortened at one end by about 4 to 5 kbp (Fig. 1B, panel i), most of the colonies were yellow. This suggests that both ends are important for pTT27 replication. Therefore, a center portion of the pTT27 region in pUC27H3 was replaced by the Kmr gene, as shown in Fig. 1A, panel ii, and B, panel i. The resultant smaller plasmid (pUC27H4) was confirmed (Fig. 2C, lane 4), showing that two loci positioned distantly on pTT27 were important for plasmid maintenance.
FIG 2.

Minimization of pTT27 in a stepwise manner. (A) Scheme for the stepwise deletions. The plasmids were constructed in E. coli, and replication ability was confirmed by transformation of the recA-null mutant of HB27. The top line indicates the pUC27H4 from Fig. 1B, panel i, and the numbers are positions on the intact pTT27. The lower lines represent the residual region, and their replication abilities are indicated in a manner similar to that in Fig. 1B. The V shape of the dotted line indicates a linkage between the two ends. In the bottom image, the two pTT27 regions included in pUC27H10 (of 10) are schematically shown. (B) Involvement of three genes, TT_P0160, TT_P0161 (RNR-β), and TT_P0162 (RNR-α), in pTT27 replication. Deletion of TT_P0161 or TT_P0162, which would compose the class I RNR enzyme, resulted in no transformant of the recA-null mutant, having a transformation efficiency below 1.0 × 100 CFU/μg. (C) Stepwise deletion in pTT27 shown by normal agarose gel electrophoresis of the pTT27 derivatives extracted from HB27 without digestion. The arrowheads indicate the pTT27 derivatives. Lane M, size marker of HindIII-digested λ DNA. The other lane numbers correspond to the leftmost boldface ones in Fig. 1B (panel i) and 2A. In the small-scale plasmid preparation from T. thermophilus by the alkali-SDS method, the shattered genomic DNA is often observed, at around 20 kbp.
The pUC27H4 plasmid is a shuttle vector between Thermus and E. coli with a size of about 40 kbp (Fig. 2A), which can be easily manipulated in E. coli. When the T. thermophilus HB27 recA-null mutant, which formed yellow colonies, was transformed by pUC27H4, all transformants on the selection plate were white as a result of the loss of intact pTT27 resulting from plasmid incompatibility with pUC27H4. This implies that all transformants of the recA-null mutant made by the pUC27H4 derivative and exhibiting replication ability would form white colonies on the selection plate. Therefore, additional shortening of the region required for pTT27 replication was performed in E. coli, and its replication ability was checked with the HB27 recA-null mutant in a fashion similar to that for pUC27H4. As shown in Fig. 2A, deletions of the pTT27 region were repeated in a stepwise fashion; finally, pUC27H10, which exhibited replication ability, was obtained (Fig. 2C, lane 10). The plasmid possesses two pTT27 regions of about 2.6 and 3.6 kbp, in which the single open reading frame (ORF) TT_P0085 and the three ORFs TT_P0160, TT_P0161, and TT_P0162 are carried, respectively. To judge the necessity of the three operon-like genes TT_P0160, TT_P0161, and TT_P0162 for pTT27 replication, a null mutant plasmid for each gene was constructed to maintain the polycistronic transcription (Fig. 2B). When the recA-null mutant was transformed by these plasmids, the plasmid with the loss of TT_P0160 showed replication ability (Fig. 2C, lane 11), but that with the loss of TT_P0161 or TT_P0162 did not. These ORFs are registered in the database as encoding the β subunit of RNR (RNR-β) for TT_P0161 and the α subunit of RNR (RNR-α) for TT_P0162. RNR, which converts ribonucleotides to deoxyribonucleotides, is a key enzyme for the de novo synthesis of deoxyribonucleotides. The RNR complex consisting of α and β subunits seems to be important for pTT27 replication. On the other hand, TT_P0085 is registered as encoding a replication initiation protein. Here, the TT_P0085 protein and its gene are referred to as RepT, the replication initiation protein for pTT27, and the repT gene, respectively. The TT_P0160-deleted version of pUC27H10 (Fig. 2C, lane 11) is called pUC27H11.
HB27 plasmid-free substrain.
In a previous study (36), we cured a 9.3-kbp plasmid, pTT8, in T. thermophilus HB8. In a similar way, pTT27 curing from HB27 was attempted in order to obtain a strain without a plasmid, as described in Materials and Methods and Fig. 3A. The absence of the plasmid in the obtained candidate strains was confirmed by the digestion of genomic DNA with both EcoT22I and MunI. There are four EcoT22I cleavage sites (but two sites are as close as 12 bases apart) and four MunI sites in the chromosome of HB27 and no EcoT22I sites and one MunI site in pTT27. As shown in Fig. 3B, the WT strain shows seven chromosome-derived bands and one for the linearized pTT27 with a size of 233 kbp, whereas the candidate lacks pTT27. For additional confirmation, the nonexistence of repT and RNR genes was checked by PCR (data not shown). One of the obtained plasmid-free strains with white colonies, named PFW, was used after this step. Generation of the plasmid-free substrain means that all the megaplasmid-borne genes would be nonessential under this growth condition, and even the pTT27-borne RNR gene is no exception.
Minimum replicon of pTT27.
As shown in Fig. 4, the megaplasmid-borne RNR genes were cloned in the chromosome of PFW. To determine whether the repT region is a replicon, a recA-null mutant of the resultant PFW I-RNR strain was transformed by pUC27H12, in which the RNR gene region of pUC27H11 was deleted and only the repT gene is present (Fig. 4). The transformants were obtained, and the plasmid from them was observed (Fig. 4, lane 12). The repT-containing region was found to be the true replication origin of pTT27, and it was shown that the RNR genes could serve in trans against the replicon. When the parental PFW was transformed by pUC27H12, a few colonies were formed on the selection plate with a remarkably low transformation efficiency of 2.0 × 100 CFU/μg, as shown in Table 3. However, the plasmid was not extracted from cultured cells of the colonies (data not shown). At this point, therefore, the RNR genes were thought to be an indispensable factor for pTT27 replication.
FIG 4.

RNR can function in pTT27 replication in trans. To test whether the RNR acts in the megaplasmid replication in trans, the transposase-encoding TT_C0665 gene on the chromosome of the PFW strain was replaced by the RNR genes with Bmr for the selection marker, followed by recA deletion to avoid incorporation of pUC27H11 as a control plasmid into the chromosome through homologous recombination. Substitutions of all the chromosomes in the polyploid were confirmed by PCR and Southern blotting (data not shown). The resultant strain was transformed by pUC27H12, in which the RNR part of pUC27H11 (lane 11) was lost. The transformants were obtained in the presence of Hm, and the plasmid was confirmed from them as shown in lane 12. Arrowheads indicate the undigested plasmids. Lanes 11 and 13, pUC27H11 and pUC27H13, respectively, prepared in the same fashion; lane M, size marker of HindIII-digested λ DNA.
TABLE 3.
Transformation efficiency
| Strain | AdoCbla | Transformation efficiency (CFU/μg) with plasmidb: |
||
|---|---|---|---|---|
| pUC27H12 | pUC-Vmini | pUC-TT8 | ||
| WT | − | 1.2 × 103 | 7.3 × 103 | |
| + | 3.3 × 103 | 6.8 × 103 | ||
| PFW I-RNR | − | 1.2 × 105 | 2.3 × 104 | 5.2 × 104 |
| + | 7.8 × 104 | 9.8 × 103 | 8.3 × 104 | |
| PFW | − | 2.0 × 100 | 1.0 × 100 | 6.0 × 100 |
| + | 7.5 × 104 | 4.4 × 104 | 5.4 × 104 | |
+, T. thermophilus was cultured in liquid medium with supplementation with 0.1 μg/ml AdoCbl, mixed with the plasmid DNA, and then spread on an AdoCbl-supplemented plate. −, AdoCbl was added to neither the liquid medium nor the plate.
The plasmid DNAs prepared from PFW I-RNR were used for transformation. When plasmids prepared from E. coli were used, their transformation efficiencies were approximately 100-fold lower than those with the plasmids from PFW I-RNR. This is probably because of the chromosome-encoded restriction enzyme TthHB27I, which constitutes the restriction-modification system in T. thermophilus HB27.
Effect of AdoCbl on PFW growth.
The plasmid-free strain PFW grew in nutrient-rich TR medium, although its growth was clearly slower than that of the WT (Fig. 5A), showing that the megaplasmid was dispensable for growth in that medium. In synthetic medium, on the other hand, the PFW strain never grew, while the WT did (Fig. 5A), indicating the necessity of some plasmid factors for growth under this condition. Since the pTT27 megaplasmid carries cobalamin biosynthesis-related genes in the region from locus 1 to 2 (Fig. 1B, panel i) (30), AdoCbl at 0.1 μg/ml was added to the synthetic medium. This addition reversed the lack of growth of the PFW strain in the medium (Fig. 5A), confirming that the cobalamin biosynthesis genes on pTT27 were essential for the growth of T. thermophilus HB27 in the synthetic medium. In the TR medium, the addition of AdoCbl also improved the poor growth rates of PFW and PFW I-RNR but did not change that of the WT (Fig. 5A). When cultured in the TR medium with or without the AdoCbl supplementation, the number of viable cells at an optical density at 600 nm (OD600) of 0.15 was estimated by counting colonies formed on the TR plate. Although PFW had fewer viable cells than the WT, AdoCbl supplementation or the existence of pTT27-derived RNR seems to resolve the decrease in the number of viable cells in PFW (Fig. 5B).
FIG 5.

Effect of the class I RNR genes or AdoCbl supplementation on growth. (A) Growth of the WT (circles), PFW (squares), and PFW I-RNR (triangles) strains in TR medium or synthetic medium with (filled symbols) or without (open symbols) supplementation with 0.1 μg/ml AdoCbl. A preculture with an OD600 of around 1.1 was diluted 1:100 into fresh medium and incubated with shaking at 70°C. (B) Count of viable cells at the same OD600 of culture. Each strain was cultured in TR medium with (solid bars) or without (hatched bars) AdoCbl supplementation and was spread on a TR plate when the OD600 reached 0.15.
Stability of exogenous plasmid in the PFW strain.
The AdoCbl supplementation improved the poor growth rate of the PFW strain in TR medium (Fig. 5A). Therefore, transformation of the strain by pUC27H12 was tried by culturing in AdoCbl-supplemented TR medium. The AdoCbl supplementation encouraged colony formation, with much higher transformation efficiency than that without AdoCbl (Table 3), and the plasmid was observed in extract from the transformant cultured in the AdoCbl-supplemented liquid medium. When the same transformant was cultured without the AdoCbl supplementation, however, the plasmid was not observed (data not shown). These results suggest that the addition of AdoCbl would be required in order to maintain the pUC27H12 plasmid in the cell. When the above-mentioned few colonies obtained from the transformation without AdoCbl (Table 3) were actually cultured in the AdoCbl-supplemented liquid medium, the plasmid was observed, unlike the case in the normal medium (data not shown). In the WT and PFW I-RNR, on the other hand, the pUC27H12 plasmid was stably maintained without AdoCbl (data not shown), and further, the addition of AdoCbl had little effect on transformation efficiency (Table 3). The presence of the pTT27-originated RNR seems to have the same effect on plasmid maintenance and transformation as the AdoCbl supplementation.
As shown in Table 3, similar results were observed with other plasmids, pUC-Vmini and pUC-TT8, in which the minimum replicons of pVV8 and pTT8 from T. thermophilus HB8 were each cloned in pUC-Hm. In these transformants of PFW, the plasmid also was stable and unstable with and without the AdoCbl supplementation, respectively (data not shown). The plasmid instability and the effect of AdoCbl on plasmid maintenance in PFW seem to be not specific to the pTT27 derivatives but rather to be applicable to other exogenous plasmids.
RepT-binding site.
It was speculated that, like other replication initiation mechanisms, the replication of pTT27 would be initiated by the binding of some proteins as initiators to a specific region of the plasmid as the replicator. As the RepT protein, encoded by the repT gene was thought to be an initiator, the recombinant protein was prepared as described in Materials and Methods. The replicator must be located in the pTT27 region included in pUC27H12, but that in pUC27H11 was used for the substrate for the RepT-binding assay. In the gel retardation assay (Fig. 6A), the disappearance/retardation of the 699-bp XhoI fragment was observed with the increase of RepT. When the region downstream from repT was deleted from the EcoRV site (Fig. 2A), its replication ability was lost. It is likely that the RepT binding to the site downstream from the repT gene is important for megaplasmid replication.
The sequence of the 699-bp fragment is shown in Fig. 6B. Two characteristics of the sequence are that nine direct repeats of 5′-TCGGAAGACTTGGGG-3′ lie downstream from the repT gene and that the second to ninth repeats seem to appear at almost the same intervals. In addition, the sequence from nt 188 to 262 framed by the orange lines is just the same as that from nt 301 to 375. Further downstream from these repeats, an A+T-rich region with a G+C content of 32.7% is located over a wide range from nt 448 to 545, while the T. thermophilus HB27 genome shows a much higher G+C content of about 70% (30). To examine the effect of the repeats on RepT binding, additional gel retardation assays were performed. In the left gel of Fig. 6C, RepT shows sufficient binding to the first seven repeats but no binding to only the first three. On the right, on the other hand, the protein exhibits sufficient binding to the last six repeats but no binding to only the last two. Because the bindings are also observed significantly in the first five and last four repeats only, the number of repeats rather than the region from the fourth to seventh repeats is likely to be important for RepT binding. DNase I footprinting analysis was performed using the fragment from nt 1 to 420 as a substrate. Protection of the repeat regions from the DNase I digestion is indicated in Fig. 7, and especially that of the fifth and sixth repeats is easy to follow. This means that the RepT protein would bind to multiple repeat sequences of 5′-TCGGAAGACTTGGGG-3′. Although a gel mobility shift assay was carried out with a fragment containing the fifth and/or sixth repeat sequences, no binding with RepT was detected (data not shown), supporting the interpretation of Fig. 6C that RepT requires multiple repeats for binding.
FIG 7.
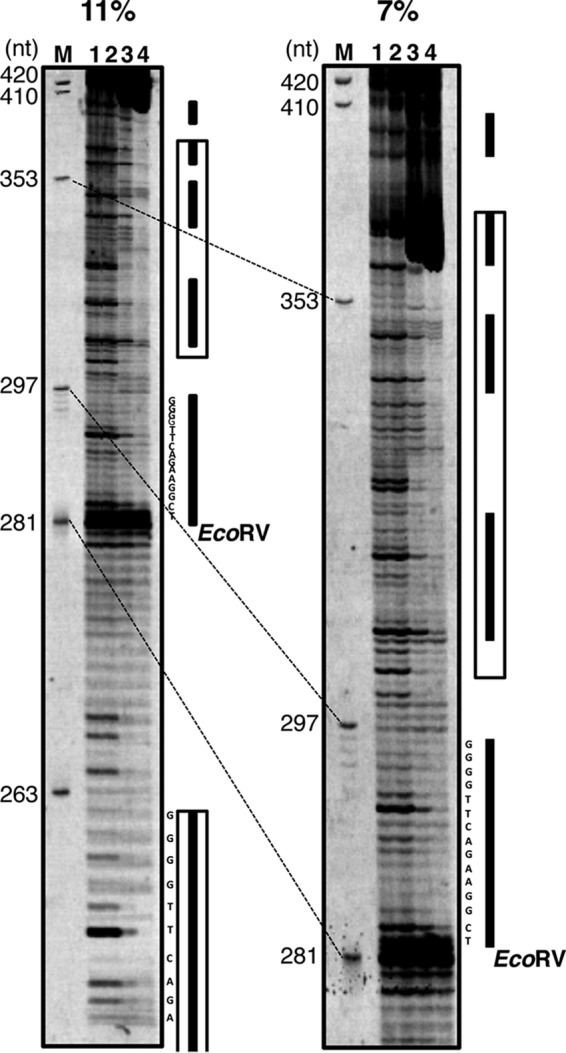
DNase I footprinting analysis of the RepT-binding region. The DNA fragment corresponding to nt 1 to 420 of the 699-bp fragment was prepared as a fluorescein-labeled substrate by PCR. The substrate was incubated with RepT at 0, 3.7, 11, and 33 μM (lanes 1 to 4, respectively) and subjected to DNase I digestion. The digests were analyzed on 11% and 7% polyacrylamide gels. The numbers along the left sides represent the nucleotide positions in the 699-bp fragment. Bold lines and boxes at the right sides indicate the direct repeat sequence and the long repeat boxed in orange in Fig. 6B, respectively.
The DNA duplex of the A+T-rich region downstream from the direct repeats was speculated to be unwound when RepT was bound to the direct repeats. Therefore, the necessity of the A+T-rich region was confirmed in vivo. The region downstream from the repT gene was deleted to the 5′ or 3′ side of the A+T-rich region in pUC27H12 (nt 445 or 560 in Fig. 6B), and the resultant plasmids were transferred to the PFW I-RNR. The plasmid including the A+T-rich region (pUC27H13) resulted in colonies on the selection plate and was extracted from the culture (Fig. 4, lane 13), whereas the plasmid without that region caused no colony formation. Hence, the A+T-rich region is indispensable for pTT27 replication.
DISCUSSION
Replication initiation of megaplasmid pTT27.
The repT gene and its adjacent regions, as in pUC27H13, would be the true minimum replicon for megaplasmid pTT27. The replication initiation protein RepT binds to the direct repeat sequences of 5′-TCGGAAGACTTGGGG-3′ downstream from its own gene repT, and the DNA duplex of the further-downstream A+T-rich region is likely to be unwound. Exclusion of the A+T-rich region from pUC27H13 caused a replication deficiency, strongly indicating the importance of the region. Eight repeats (the second to the ninth) are placed in a regular manner as shown in Fig. 6B, leading to the speculation that an oligomeric complex of the RepT proteins is coiled by the DNA duplex with the direct repeat sequences.
Plasmid maintenance in PFW.
Both the remarkably low efficiency of transformation of PFW by pUC27H12 (Table 3) and the lack of confirmation of the plasmid prepared from the resultant transformant culture had made us presume that the pTT27-borne RNR genes were indispensable for megaplasmid replication. However, this speculation was ruled out by the addition of AdoCbl, which caused the transformation efficiency to increase dramatically and encouraged stable maintenance of pUC27H12 in PFW (Table 3). Since RNR generally catalyzes the formation of deoxyribonucleotides from ribonucleotides, it is indispensable for DNA replication and repair and is essential for cell growth (42). The megaplasmid-borne RNR would be a class I enzyme using only NDP under aerobic conditions, while in T. thermophilus HB27, a probable class II RNR that uses both NDP and NTP as a substrate under aerobic and anaerobic conditions is encoded by TT_C1930 on the chromosome. Owing to the presence of another RNR, the absence of the megaplasmid-encoded enzyme in the PFW strain would not eliminate growth in TR medium. However, the decrease in viable cells in TR medium without AdoCbl (Fig. 5B) suggests the failure of some cellular mechanisms in PFW. The decrease in viable cells could be improved by AdoCbl supplementation or by the introduction of the pTT27-derived class I RNR genes into the chromosome, as shown in Fig. 5B. These results suggest that AdoCbl would work on the class II RNR, which requires AdoCbl as a cofactor involved in radical generation (42). The PFW cells probably would be under dNTP-deficient conditions in the normal TR medium, in which the amount of AdoCbl might be insufficient to stimulate the class II RNR completely, because the PFW strain loses both the class I RNR and AdoCbl biosynthesis ability as a result of pTT27 curing. The dNTP shortage or the imbalanced dNTP pool in PFW would lead to noncompletion or infidelity of chromosome and plasmid replication. Chromosome replication especially is crucial to the survival of the cells. In addition, the polyploidy of T. thermophilus and the high G+C content of its genome might make the situation serious. Introduction of the class I RNR or stimulation of the class II enzyme by the addition of AdoCbl resolves the dNTP shortage or the imbalanced dNTP pool in a cell, and then the completion or improved fidelity of chromosomal replication would result in an increase of viable PFW cells (Fig. 5B). For the same reason, the addition of AdoCbl would enable the stable maintenance of pUC27H12 as well as of other exogenous plasmids in PFW, and consequently, transformation efficiency would be improved also (Table 3). Replication failure would occur equally in the chromosome and plasmid, but failure of chromosome replication must be directly linked to death. As a result, the population of cells lacking the plasmid would increase with growth.
It is probable that in place of the AdoCbl supplementation, the presence of cobalamin biosynthesis-related genes would enable stable maintenance of the plasmid in PFW. Whereas our stepwise deletions in pTT27 showed that the combination of the repT and class I RNR genes was important in megaplasmid maintenance (Fig. 2), combination of repT and the cobalamin biosynthesis-related genes can be also assumed.
Necessity of megaplasmid pTT27 for growth.
First, it should be mentioned that the megaplasmid pTT27 must be highly beneficial for maintenance of genome stability in T. thermophilus HB27 because it carries the class I RNR genes and the biosynthesis genes for AdoCbl stimulating the chromosomal class II RNR gene. It is implied that this megaplasmid replication might be indirectly required for chromosomal replication.
Although many Thermus spp. harbor a megaplasmid, megaplasmid curing has yet to be reported. The megaplasmids have tended to be considered a basic need for cell growth because of their large size, carrying many genes. Indeed, physiologically important genes are located on the megaplasmid, such as genes encoding the hyperthermophile-specific reverse gyrase in T. thermophilus HB8, JL-18, and SG0.5JP17-16 (but not in HB27), the CRISPR-Cas system as a prokaryotic immune system, Argonaute for host defense from foreign DNA, the carotenoid and cobalamin biosynthesis transcriptional regulators, etc. The megaplasmid also carries some toxin-antitoxin system genes, implying its stable maintenance in a cell. Because the miniaturized pTT27 derivatives such as pUC27H11 and pUC27K11 lack all the toxin-antitoxin systems, curing pTT27 from HB27 might be achieved rather easily in the way described in Materials and Methods. The PFW strain can grow in nutrient-rich TR medium (Fig. 5A), suggesting that all megaplasmid-borne genes would be nonessential for cell growth under this condition.
On the other hand, the growth of the PFW strain in synthetic medium such as minimal medium requires AdoCbl supplementation (Fig. 5A). This means that cobalamin biosynthesis-related genes carried in megaplasmid pTT27 are essential for the growth of HB27 in synthetic medium. As described above, AdoCbl would work on the chromosomal class II RNR. However, the growth defect of PFW I-RNR, in which the class I RNR is active, in synthetic medium without AdoCbl (Fig. 5A) indicates that AdoCbl would work not only on RNR but also on some other cellular functions required for growth in synthetic medium. In T. thermophilus, AdoCbl also must have many important intracellular functions, as described in the introduction. Therefore, even in the TR medium, AdoCbl supplementation encourages growth of PFW I-RNR (Fig. 5A). Under the AdoCbl-supplemented condition, the presence of the pTT27-derived class I RNR has an inhibitory influence on the growth of PFW in synthetic medium, unlike in TR medium (Fig. 5A). During preculture in the AdoCbl-supplemented synthetic medium for Fig. 5A, however, the PFW strain exhibited an approximately 3-fold-longer lag phase than PFW I-RNR (data not shown). In Streptomyces and Pseudomonas aeruginosa, the transcription of class I RNR genes has been reported to increase at the beginning of the exponential phase (43, 44). In T. thermophilus, the class I genes might be transcribed in a similar pattern. In this case, the lag phase of PFW I-RNR might be shorter than that of PFW. Furthermore, AdoCbl has been reported to reduce the transcription of the class I RNR via a riboswitch mechanism in Streptomyces (45). For instance, a similar mechanism might be involved in the inhibitory effect of class I RNR on growth in the AdoCbl-supplemented synthetic medium, but the underlying mechanism remains to be identified in T. thermophilus.
A growth defect in minimal medium was observed also when the pTAV3 megaplasmid was lost in P. versutus (25). It has been discussed that the megaplasmid required for growth in minimal medium might carry housekeeping genes and thus not fit the strict definition of a plasmid (25). It remains to be judged whether this kind of DNA replicon, which is required only for growth in minimal medium, is a megaplasmid or a minichromosome.
DNA engineering in T. thermophilus.
In this study, an ∼110-kb region on the megaplasmid was deleted by replacement by the drug resistance gene, as shown in Fig. 1B. This means that two regions with sufficient length for homologous recombination enable the replacement of an intervening part as long as 100 kb without a nonessential gene. The longer the prepared homologous regions for recombination, the broader the part in the genome that might be substituted or deleted. Such a dynamic would introduce the possibility of genomic engineering in T. thermophilus. Incompatibility between the substituted and parental plasmids is implied to have priority over homologous recombination between them, according to a comparison of the results in Fig. 1B for the deletion from locus 4 to locus 2 and for the other three trials. In T. thermophilus, a polyploid bacterium, the nonessential genes on all genomic copies were replaced easily, whereas at least one copy had to remain intact in the case of the essential gene (35). The easy establishment of the deletion from locus 4 to locus 2 might have been caused by the same undefined mechanism that is involved in the replacement of the nonessential gene.
The PFW strain obtained in this study lost its pTT27-derived yellow pigment. This plasmid-free white strain with natural competence might have great potential as a host strain for DNA engineering in T. thermophilus. The absence of a plasmid in the host cell is attractive for plasmid transformation, and white colonies are suitable for color-based selection, such as yellow caused by carotenoid production (13) or blue caused by X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) and β-galactosidase (15). In this aspect, the PFW I-RNR strain, in which the megaplasmid-originated class I RNR genes were carried in the chromosome, would be better for use as the host cells, because it improves growth and stably maintains exogenous plasmids (Fig. 5A). Understandably, the class I RNR genes can also be cloned in the plasmid vector, as in the case of pUC27H11 (Fig. 2B). Alternatively, it is possible to clone the pTT27-derived cobalamin biosynthesis-related genes into the chromosome of PFW, but AdoCbl supplementation to the medium will make it easy. Another advantage of PFW is its higher transformation efficiency than WT, as shown in Table 3. pTT27 encodes Argonaute protein, which acts as a barrier for the uptake and propagation of foreign DNA. Deletion of the Argonaute gene has been reported to increase the transformation efficiency, and the transformation efficiency of the deletion mutant is 10-fold higher than that of the WT (46). In addition, the PFW host enables the plasmid-borne genes to be transiently kept, as with using a conditionally replicable vector. When it is not needed, cancellation of the AdoCbl supplementation would lead to lack of the genes with the plasmid in PFW. This can make a significant contribution to experiments involving verification of an essential gene in PFW.
When PFW is the host strain, the repT-dependent plasmid can be used as a cloning vector. The pUC27H13 plasmid, in which a 2.3-kb region with only repT is contained as the minimum replicon of pTT27, is a compact E. coli-Thermus shuttle vector. The repT vector can be expected to be suitable for large-scale cloning, because it was the origin of the 233-kb megaplasmid. This pTT27 replicon study, as well as the generation of the plasmid-free strain and analysis of exogenous plasmid maintenance in the resultant PFW strain, will serve as a foundation for the next wave of large-scale Thermus genetic/genomic engineering research, in addition to ensuring the availability of the HB27 (PFW) strain for thermophilic cell factories.
Funding Statement
This work was supported by research funds from the Yamagata Prefectural Government and Tsuruoka City, Japan.
REFERENCES
- 1.Cava F, Hidalgo A, Berenguer J. 2009. Thermus thermophilus as biological model. Extremophiles 13:213–231. doi: 10.1007/s00792-009-0226-6. [DOI] [PubMed] [Google Scholar]
- 2.Yokoyama S, Hirota H, Kigawa T, Yabuki T, Shirouzu M, Terada T, Ito Y, Matsuo Y, Kuroda Y, Nishimura Y, Kyogoku Y, Miki K, Masui R, Kuramitsu S. 2000. Structural genomics projects in Japan. Nat Struct Biol 7:943–945. doi: 10.1038/80712. [DOI] [PubMed] [Google Scholar]
- 3.Ohtani N, Hasegawa M, Sato M, Tomita M, Kaneko S, Itaya M. 2012. Serial assembly of Thermus megaplasmid DNA in the genome of Bacillus subtilis 168: a BAC-based domino method applied to DNA with high GC content. Biotechnol J 7:867–876. doi: 10.1002/biot.201100396. [DOI] [PubMed] [Google Scholar]
- 4.Koyama Y, Hoshino T, Tomizuka N, Furukawa K. 1986. Genetic transformation of the extreme thermophile Thermus thermophilus and of other Thermus spp. J Bacteriol 166:338–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hidaka Y, Hasegawa M, Nakahara T, Hoshino T. 1994. The entire population of Thermus thermophilus cells is always competent at any growth phase. Biosci Biotechnol Biochem 58:1338–1339. doi: 10.1271/bbb.58.1338. [DOI] [PubMed] [Google Scholar]
- 6.Hashimoto Y, Yano T, Kuramitsu S, Kagamiyama H. 2001. Disruption of Thermus thermophilus genes by homologous recombination using a thermostable kanamycin-resistant marker. FEBS Lett 506:231–234. doi: 10.1016/S0014-5793(01)02926-X. [DOI] [PubMed] [Google Scholar]
- 7.Hoseki J, Yano T, Koyama Y, Kuramitsu S, Kagamiyama H. 1999. Directed evolution of thermostable kanamycin-resistance gene: a convenient selection marker for Thermus thermophilus. J Biochem 126:951–956. doi: 10.1093/oxfordjournals.jbchem.a022539. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura A, Takakura Y, Kobayashi H, Hoshino T. 2005. In vivo directed evolution for thermostabilization of Escherichia coli hygromycin B phosphotransferase and the use of the gene as a selection marker in the host-vector system of Thermus thermophilus. J Biosci Bioeng 100:158–163. doi: 10.1263/jbb.100.158. [DOI] [PubMed] [Google Scholar]
- 9.Fujita A, Misumi Y, Koyama Y. 2012. Two versatile shuttle vectors for Thermus thermophilus-Escherichia coli containing multiple cloning sites, lacZα gene and kanamycin or hygromycin resistance marker. Plasmid 67:272–275. doi: 10.1016/j.plasmid.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 10.Brouns SJ, Wu H, Akerboom J, Turnbull AP, de Vos WM, van der Oost J. 2005. Engineering a selectable marker for hyperthermophiles. J Biol Chem 280:11422–11431. doi: 10.1074/jbc.M413623200. [DOI] [PubMed] [Google Scholar]
- 11.Koyama Y, Okamoto S, Furukawa K. 1990. Cloning of α- and β-galactosidase genes from an extreme thermophile, Thermus strain T2, and their expresion in Thermus thermophilus. Appl Environ Microbiol 56:2251–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ohta T, Tokishita SI, Imazuka R, Mori I, Okamura J, Yamagata H. 2006. β-Glucosidase as a reporter for the gene expression studies in Thermus thermophilus and constitutive expression of DNA repair genes. Mutagenesis 21:255–260. doi: 10.1093/mutage/gel025. [DOI] [PubMed] [Google Scholar]
- 13.Fujita A, Misumi Y, Honda S, Sato T, Koyama Y. 2013. Construction of new cloning vectors that employ the phytoene synthase encoding gene for color screening of cloned DNA inserts in Thermus thermophilus. Gene 527:655–662. doi: 10.1016/j.gene.2013.06.069. [DOI] [PubMed] [Google Scholar]
- 14.Blas-Galindo E, Cava F, Lopez-Vinas E, Mendieta J, Berenguer J. 2007. Use of a dominant rpsL allele conferring streptomycin dependence for positive and negative selection in Thermus thermophilus. Appl Environ Microbiol 73:5138–5145. doi: 10.1128/AEM.00751-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carr JF, Danziger ME, Huang AL, Dahlberg AE, Gregory ST. 2015. Engineering the genome of Thermus thermophilus using a counterselectable marker. J Bacteriol 197:1135–1144. doi: 10.1128/JB.02384-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cava F, de Pedro MA, Blas-Galindo E, Waldo GS, Westblade LF, Berenguer J. 2008. Expression and use of superfolder green fluorescent protein at high temperatures in vivo: a tool to study extreme thermophile biology. Environ Microbiol 10:605–613. doi: 10.1111/j.1462-2920.2007.01482.x. [DOI] [PubMed] [Google Scholar]
- 17.Gounder K, Brzuszkiewicz E, Liesegang H, Wollherr A, Daniel R, Gottschalk G, Reva O, Kumwenda B, Srivastava M, Bricio C, Berenguer J, van Heerden E, Litthauer D. 2011. Sequence of the hyperplastic genome of the naturally competent Thermus scotoductus SA-01. BMC Genomics 12:577. doi: 10.1186/1471-2164-12-577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumwenda B, Litthauer D, Reva O. 2014. Analysis of genomic rearrangements, horizontal gene transfer and role of plasmids in the evolution of industrial important Thermus species. BMC Genomics 15:813. doi: 10.1186/1471-2164-15-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki K, Itoh T. 2007. Characterization of the ColE2-like replicon of plasmid pTT8 from Thermus thermophilus. Biochem Biophys Res Commun 353:1028–1033. doi: 10.1016/j.bbrc.2006.12.150. [DOI] [PubMed] [Google Scholar]
- 20.Takayama G, Kosuge T, Maseda H, Nakamura A, Hoshino T. 2004. Nucleotide sequence of the cryptic plasmid pTT8 from Thermus thermophilus HB8 and isolation and characterization of its high-copy-number mutant. Plasmid 51:227–237. doi: 10.1016/j.plasmid.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 21.de Grado M, Lasa I, Berenguer J. 1998. Characterization of a plasmid replicative origin from an extreme thermophile. FEMS Microbiol Lett 165:51–57. doi: 10.1111/j.1574-6968.1998.tb13126.x. [DOI] [PubMed] [Google Scholar]
- 22.Chu SF, Shu HY, Lin LC, Chen MY, Tsay SS, Lin GH. 2006. Characterization of a rolling-circle replication plasmid from Thermus aquaticus NTU103. Plasmid 56:46–52. doi: 10.1016/j.plasmid.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Ohtani N, Tomita M, Itaya M. 2013. Identification of a replication initiation protein of the pVV8 plasmid from Thermus thermophilus HB8. Extremophiles 17:15–28. doi: 10.1007/s00792-012-0489-1. [DOI] [PubMed] [Google Scholar]
- 24.MacLellan SR, Smallbone LA, Sibley CD, Finan TM. 2005. The expression of a novel antisense gene mediates incompatibility within the large repABC family of alpha-proteobacterial plasmids. Mol Microbiol 55:611–623. [DOI] [PubMed] [Google Scholar]
- 25.Bartosik D, Baj J, Bartosik AA, Wlodarczyk M. 2002. Characterization of the replicator region of megaplasmid pTAV3 of Paracoccus versutus and search for plasmid-encoded traits. Microbiology 148:871–881. doi: 10.1099/00221287-148-3-871. [DOI] [PubMed] [Google Scholar]
- 26.Cervantes-Rivera R, Pedraza-López F, Pérez-Segura G, Cevallos MA. 2011. The replication origin of a repABC plasmid. BMC Microbiol 11:158. doi: 10.1186/1471-2180-11-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cevallos MA, Cervantes-Rivera R, Gutiérrez-Ríos RM. 2008. The repABC plasmid family. Plasmid 60:19–37. doi: 10.1016/j.plasmid.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 28.Pinto UM, Pappas KM, Winans SC. 2012. The ABCs of plasmid replication and segregation. Nat Rev Microbiol 10:755–765. doi: 10.1038/nrmicro2882. [DOI] [PubMed] [Google Scholar]
- 29.Ng WV, Ciufo SA, Smith TM, Bumgarner RE, Baskin D, Faust J, Hall B, Loretz C, Seto J, Slagel J, Hood L, DasSarma S. 1998. Snapshot of a large dynamic replicon in a halophilic archaeon: megaplasmid or minichromosome? Genome Res 8:1131–1141. [DOI] [PubMed] [Google Scholar]
- 30.Henne A, Brüggemann H, Raasch C, Wiezer A, Hartsch T, Liesegang H, Johann A, Lienard T, Gohl O, Martinez-Arias R, Jacobi C, Starkuviene V, Schlenczeck S, Dencker S, Huber R, Klenk HP, Kramer W, Merkl R, Gottschalk G, Fritz HJ. 2004. The genome sequence of the extreme thermophile Thermus thermophilus. Nat Biotechnol 22:547–553. doi: 10.1038/nbt956. [DOI] [PubMed] [Google Scholar]
- 31.Tian B, Hua Y. 2010. Carotenoid biosynthesis in extremophilic Deinococcus-Thermus bacteria. Trends Microbiol 18:512–520. doi: 10.1016/j.tim.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 32.Nahvi A, Barrick JE, Breaker RR. 2004. Coenzyme B12 riboswitches are widespread genetic control elements in prokaryotes. Nucleic Acids Res 32:143–150. doi: 10.1093/nar/gkh167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jost M, Fernández-Zapata J, Polanco MC, Ortiz-Guerrero JM, Chen PY, Kang G, Padmanabhan S, Elías-Arnanz M, Drennan CL. 2015. Structural basis for gene regulation by a B12-dependent photoreceptor. Nature 526:536–541. doi: 10.1038/nature14950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brüggemann H, Chen C. 2006. Comparative genomics of Thermus thermophilus: plasticity of the megaplasmid and its contribution to a thermophilic lifestyle. J Biotechnol 124:654–661. doi: 10.1016/j.jbiotec.2006.03.043. [DOI] [PubMed] [Google Scholar]
- 35.Ohtani N, Tomita M, Itaya M. 2010. An extreme thermophile, Thermus thermophilus, is a polyploid bacterium. J Bacteriol 192:5499–5505. doi: 10.1128/JB.00662-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ohtani N, Tomita M, Itaya M. 2012. The third plasmid pVV8 from Thermus thermophilus HB8: isolation, characterization, and sequence determination. Extremophiles 16:237–244. doi: 10.1007/s00792-011-0424-x. [DOI] [PubMed] [Google Scholar]
- 37.Castán P, Casares L, Barbé J, Berenguer J. 2003. Temperature-dependent hypermutational phenotype in recA mutants of Thermus thermophilus HB27. J Bacteriol 185:4901–4907. doi: 10.1128/JB.185.16.4901-4907.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka T, Kawano N, Oshima T. 1981. Cloning of 3-isopropylmalate dehydrogenase gene of an extreme thermophile and partial purification of the gene product. J Biochem 89:677–682. [DOI] [PubMed] [Google Scholar]
- 39.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 40.Itaya M, Tanaka T. 1991. Complete physical map of the Bacillus subtilis 168 chromosome constructed by a gene-directed mutagenesis method. J Mol Biol 220:631–648. doi: 10.1016/0022-2836(91)90106-G. [DOI] [PubMed] [Google Scholar]
- 41.Goodwin TW, Morton RA. 1946. The spectrophotometric determination of tyrosine and tryptophan in proteins. Biochem J 40:628–632. doi: 10.1042/bj0400628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Torrents E. 2014. Ribonucleotide reductases: essential enzymes for bacterial life. Front Cell Infect Microbiol 4:52. doi: 10.3389/fcimb.2014.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Torrents E, Poplawski A, Sjöberg BM. 2005. Two proteins mediate class II ribonucleotide reductase activity in Pseudomonas aeruginosa: expression and transcriptional analysis of the aerobic enzymes. J Biol Chem 280:16571–16578. doi: 10.1074/jbc.M501322200. [DOI] [PubMed] [Google Scholar]
- 44.Borovok I, Kreisberg-Zakarin R, Yanko M, Schreiber R, Myslovati M, Aslund F, Holmgren A, Cohen G, Aharonowitz Y. 2002. Streptomyces spp. contain class Ia and class II ribonucleotide reductases: expression analysis of the genes in vegetative growth. Microbiology 148:391–404. doi: 10.1099/00221287-148-2-391. [DOI] [PubMed] [Google Scholar]
- 45.Borovok I, Gorovitz B, Schreiber R, Aharonowitz Y, Cohen G. 2006. Coenzyme B12 controls transcription of the Streptomyces class Ia ribonucleotide reductase nrdABS operon via a riboswitch mechanism. J Bacteriol 188:2512–2520. doi: 10.1128/JB.188.7.2512-2520.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swarts DC, Jore MM, Westra ER, Zhu Y, Janssen JH, Snijders AP, Wang Y, Patel DJ, Berenguer J, Brouns SJ, van der Oost J. 2014. DNA-guided DNA interference by a prokaryotic Argonaute. Nature 507:258–261. doi: 10.1038/nature12971. [DOI] [PMC free article] [PubMed] [Google Scholar]