

Abstract
Pathogen identification and microbial source tracking (MST) to identify sources of fecal pollution improve evaluation of water quality. They contribute to improved assessment of human health risks and remediation of pollution sources. An MST microarray was used to simultaneously detect genes for multiple pathogens and indicators of fecal pollution in freshwater, marine water, sewage-contaminated freshwater and marine water, and treated wastewater. Dead-end ultrafiltration (DEUF) was used to concentrate organisms from water samples, yielding a recovery efficiency of >95% for Escherichia coli and human polyomavirus. Whole-genome amplification (WGA) increased gene copies from ultrafiltered samples and increased the sensitivity of the microarray. Viruses (adenovirus, bocavirus, hepatitis A virus, and human polyomaviruses) were detected in sewage-contaminated samples. Pathogens such as Legionella pneumophila, Shigella flexneri, and Campylobacter fetus were detected along with genes conferring resistance to aminoglycosides, beta-lactams, and tetracycline. Nonmetric dimensional analysis of MST marker genes grouped sewage-spiked freshwater and marine samples with sewage and apart from other fecal sources. The sensitivity (percent true positives) of the microarray probes for gene targets anticipated in sewage was 51 to 57% and was lower than the specificity (percent true negatives; 79 to 81%). A linear relationship between gene copies determined by quantitative PCR and microarray fluorescence was found, indicating the semiquantitative nature of the MST microarray. These results indicate that ultrafiltration coupled with WGA provides sufficient nucleic acids for detection of viruses, bacteria, protozoa, and antibiotic resistance genes by the microarray in applications ranging from beach monitoring to risk assessment.
INTRODUCTION
Waterborne pathogens pose a health risk to recreational water users (1), in drinking water systems (2), and in aquatic organisms such as shellfish that are consumed by humans (3). These waterborne pathogens include more than 40 different groups or genera, including viruses, bacteria, protozoa, cyanobacteria, and helminths (4). Additional waterborne pathogens will doubtless emerge over time due to increased proportions of sensitive populations, globalization of commerce, microbial evolution, and use of reclaimed water as drinking water (5). Many waterborne pathogens originate from fecal pollution in storm water runoff from agricultural and urban surfaces (6) or direct release of untreated sewage to surface water (7). Additional sources of waterborne fecal pathogens include wildlife and domesticated animals such as deer, dogs, raccoons, cats, and wild avian species (8). Still other waterborne pathogens, such as Vibrio spp., are autochthonous to aquatic environments (9).
The microbiological safety of surface water has been assessed for over a century by enumeration of fecal indicator bacteria (FIB) (10). Other monitoring techniques such as microbial source tracking (MST) are advantageous compared to enumeration of FIB because microorganisms or genes targeted via MST methods have an exclusive or preferential association with the gastrointestinal tract of a particular host species. These host-associated microorganisms are shed in feces, which may then be detected in water bodies. MST has been shown to be a useful method for determining the relationship between human health risk, water quality, and total maximum daily load (TMDL) (11). While there are currently over 100 different microbial source tracking marker genes proposed for use in water quality monitoring (12), it is impractical to monitor for all these microorganisms using quantitative PCR (qPCR) methods. However, as has been shown previously (13), microarrays, wherein thousands to hundreds of thousands of gene targets can be assayed at one time, allow for detection of multiple targets simultaneously. When whole-genome amplification (WGA) is used to amplify nucleic acids from environmental samples prior to microarray analysis, it is possible to simultaneously assay a sample for thousands of different organisms and multiple gene targets (e.g., virulence genes, 16S rRNA, antibiotic resistance genes, and mitochondrial DNA [mtDNA]).
One limitation to the monitoring of surface water via molecular methods is the low abundance of pathogens typically present in water; however, even low concentrations pose a health risk (14). Concentration methods such as hollow-fiber ultrafiltration (HFUF) (15–17) or a modification of this method, dead-end HFUF (DEUF) (18, 19), can help to overcome the dilution issue. Both methods have a high rate of recovery of microbes from large volumes of water (e.g., 100 liters). Here, we report on the use of ultrafiltration methods, WGA, and a novel MST microarray in order to detect waterborne pathogens and MST marker genes in surface water (freshwater and marine water), surface water spiked with sewage, and wastewater treatment plant (WWTP) effluent. The MST microarray combined with ultrafiltration methods could help regulators and researchers alike make informed decisions about water reuse for irrigation, in monitoring recreational and drinking water quality, and in tracking fecal pollution sources for remediation purposes.
MATERIALS AND METHODS
Microarray design.
The design of the microarray has been previously reported (13). Each array consisted of 411 distinct probes and associated controls (see below), which were replicated eight times on one slide. The probes included on each array targeted one or more of the following groups: (i) bacterial, eukaryotic, and viral waterborne pathogens; (ii) fecal indicator bacteria; (iii) previously published MST marker genes and mitochondrial DNA (mtDNA) genes; (iv) antibiotic resistance genes; (v) universal bacterial probes and enteric bacterial probes; and (vi) positive and negative controls. The distribution of gene probes was 43% rRNA (5S, 16S, 18S, or 23S of 157 different organisms by 174 probes), 16% viruses (17 different viruses by 69 probes), 14% mtDNA (28 different organisms by 54 probes), 20% pathogen virulence or housekeeping genes (77 different genes by 80 probes), 6% antibiotic resistance genes (3 different antibiotic groups by 25 probes), and 2% control probes (3 positive-control probes and 6 nonsense probes). A total of 174 probes were considered to be MST marker probes, although some probes were considered to belong in both the pathogen and MST marker gene categories. For example, human-associated adenovirus (20) was considered to be both an MST marker gene and a pathogen. All probes were 60 bases in length with a melting temperature of 65 to 82°C. Probes were included on the array if they were, first, previously validated probes (lengthened or shortened to 60-mers); second, previously published qPCR primers and/or probes that could be lengthened to 60-mers; or third, probes designed using CommOligo 2.0 (21), targeting microorganisms or genes listed above. The microarrays were printed by Agilent (Custom CGH, 8 × 15K platform; Agilent, Santa Clara, CA). Only two of the three positive-control probes on the arrays were used in this work.
Ultrafiltration methods.
All hollow-fiber ultrafiltration (HFUF) of samples were conducted on Rexeed-25S hemodialyzer filters (Asahi Kaei Medical America, Inc., Memphis, TN, or Dial Medical Supply, Chester Springs, PA), using previously published methods for dead-end ultrafiltration (DEUF) (19) or HFUF (22). The treated wastewater samples were concentrated using DEUF (at West Virginia University), while the sewage and surface water samples were concentrated using HFUF (at the University of South Florida). The only change from previously published methods was that DEUF retentate was eluted using sterile 1× phosphate-buffered saline (PBS) at pH 9.0 in order to enhance the efficiency of virus recovery.
In order to determine the recovery efficiency of the DEUF method, two tests were conducted. First, a known abundance of Escherichia coli (ATCC 9637) cultured to late exponential phase in LB broth was resuspended in 2 liters of 1× PBS and concentrated via DEUF to 75 ml. The abundance of the uidA gene of E. coli in 1 ml of the DEUF retentate was determined via qPCR. Second, the abundance of human polyomavirus was quantified in two samples: (i) 160 ml of raw sewage (Star City, WV) and (ii) 160 ml of raw sewage diluted in 1,440 ml of 1× PBS. The diluted sewage sample was concentrated via DEUF to 160 ml and then centrifuged at 16,000 × g for 1.5 h. Then, nucleic acids from 0.5 g of concentrate from centrifugation were extracted via the manual coextraction method described below.
In order to evaluate the effect of disinfection on targeted microorganisms in wastewater, 11.4 liters of secondarily treated wastewater was collected immediately before chlorination, and 11.4 liters of effluent was collected immediately postdechlorination from the Star City, WV, wastewater treatment plant. The Star City, WV, treatment plant treats up to 12 million gallons per day with primary treatment through sedimentation and secondary treatment with activated sludge or a rotating biological contactor followed by disinfection with chlorine gas and dechlorination with sodium bisulfite. The water samples were transported to the laboratory immediately after sampling on ice and were mixed thoroughly by shaking prior to ultrafiltration using the DEUF method. The prechlorination effluent was concentrated from 11.4 liters to 225 ml, and the dechlorinated effluent was concentrated from 11.4 liters to 210 ml. One milliliter of the concentrate was used for nucleic acid extraction.
To evaluate the potential for the microarray to detect pathogens in fresh and marine water, five samples were tested: (i) sewage from the Falkenburg Road Advanced Wastewater Treatment Plant, Tampa, FL; (ii) freshwater from the Hillsborough River, Tampa, FL; (iii) marine water from the Intracoastal Waterway, St. Petersburg, FL; (iv) freshwater spiked with sewage in a 1:50 ratio; and (v) marine water spiked with sewage in a 1:50 ratio. Forty liters of freshwater was collected from the Hillsborough River, Tampa, FL (N28 4.436, W82 22.526), and 40 liters of marine water was collected from the Intracoastal Waterway, St. Petersburg, FL (N27 48.0003, W82 46.004). Twenty liters of each water sample was concentrated via HFUF to obtain a final volume of 200 ml. This was further concentrated to 10 ml by centrifugation at 4°C in 15-ml centrifugal filter units with a 50,000-molecular-weight cutoff (50K MWCO) (Amicon Ultra-15; Millipore, Darmstadt, Germany). Another 20 liters of each water sample was spiked with 400 ml sewage influent collected from the Falkenburg Road Advanced Wastewater Treatment Plant, Tampa, FL, and concentrated to 200 ml using hollow-fiber filtration (22). The sewage-spiked river water and spiked marine water were further concentrated in centrifugal filters to 50 ml and 25 ml, respectively. One liter of the sewage influent was also concentrated to 35 ml as described above.
Nucleic acid extraction and handling.
Nucleic acids (DNA and RNA) from the concentrated treatment plant effluent samples were extracted using a manual coextraction method (23). cDNA was synthesized from DNase (Thermo Scientific, Pittsburgh, PA)-treated RNA by the Maxima first-strand synthesis kit (Thermo Scientific) according to the manufacturer's instructions. The DNA and RNA were coextracted from 250 μl of the final concentrated volumes for river, spiked river, marine, spiked marine, and sewage samples using the MoBio PowerWater RNA isolation kit (Carlsbad, CA). Then, 8 μl of the extracted mixture was converted to cDNA using the GoScript reverse transcription system (Promega, Madison, WI). The purified DNA and cDNA samples were shipped on ice to West Virginia University for testing via the microarray.
WGA and microarray handling.
The DNA and cDNA from each of the seven samples were amplified separately by the Illustra Genomiphi V2 DNA amplification kit (GE Healthcare, Pittsburgh, PA) according to the manufacturer's instructions. The general microarray sample handling has been reported previously (13) and can be summarized as follows: (i) total DNA or cDNA was amplified separately by WGA and then combined, (ii) restriction enzymes (PvuII, RsrII, SgrAI, and Nb.BbvcI; New England BioLabs, Ipswich, MA) were used to shorten WGA nucleic acids to less than 3,000 bp, (iii) known concentrations of positive controls were added, (iv) nucleic acids were labeled with Cy3 (SureTaq DNA labeling kit; Agilent, Santa Clara, CA), (v) labeled nucleic acids were hybridized to the microarray and then the unbound labeled nucleic acids were washed off, and (vi) the array was scanned and data were normalized before analyzing the results.
To normalize results and to minimize false-positive detections, the fluorescence levels of the perfect-match probes (PM; identical to the target genes) were compared against mismatch probes (MM; 3 to 9 nucleotides different from the corresponding PM probes). There were five replicates for each PM probe on the array. Twenty-seven MM probes were designed for each PM probe by replacing 1, 2, or 3 nucleotides at three probe regions at equally distant locations along the 60-mer probes (13). All the fluorescence unit (FU) signals were log transformed and background subtracted according to Agilent data normalization protocols. Then, the mean log FU of the negative-control and nonsense control probes for each microarray was subtracted from all PM and MM probe values. After sample labeling with Cy3, the microarray, labeled samples, filters, and backing slide were shipped to the Duke University Microarray Facility for sample hybridization to the microarray, washing, and scanning using an Agilent C scanner.
qPCR.
The abundance of several genes pre- and post-WGA was determined by quantitative PCR (qPCR) to evaluate WGA efficiency. Further, these gene abundances were used to correlate gene abundance via qPCR with the microarray fluorescence and to confirm the presence of various pathogens or markers of interest in environmental samples. Both SYBR green and TaqMan qPCR were applied in this study, and the primer and probe information can be found in Table S1 in the supplemental material. The qPCR primer and probe concentrations, thermocycler conditions, and positive and negative controls have been reported previously (13, 24). SYBR green-based detection was used for human Bacteroidales, human norovirus, and human polyomaviruses, while TaqMan-based detection was used for the remaining genes (see Table S1).
Data normalization and statistical analysis.
Log FU of PM signals was normalized to that of added standards by the following approach. Two positive-control genes (cloned into plasmids) which are not expected to be in sewage or surface waters, the 5.8S rRNA gene of Oncorhynchus mykiss and the 16S rRNA gene of Dehalococcoides mccartyi, were added to each sample following WGA and before Cy3 labeling. A range of control concentrations at seven increments, ranging from 0.2 to 1.4 ng DNA, was added at one concentration per sample. The resulting linear regression of nanograms of positive-control DNA added versus log FU for seven separate arrays is presented in Fig. 1. The standard curve was used to normalize all PM probe fluorescence among samples on a given set of arrays based on the log FU of positive controls added to each array. If the log FU of either positive control for any array diverged from the linear regression (e.g., gray squares in Fig. 1 for the array to which the sewage sample was hybridized), then the log FU of the PM probes on the same array was normalized by the correction required to fit the positive control to the linear regression (e.g., log FU of sewage sample PM probes was reduced by a scaling factor until it fitted the regression [Fig. 1]). The applied scaling factors ranged from 0.94 to 1.04. All further references to log FU are understood to be the normalized values. The log FU was used to generate a heat map showing detection and relative signal intensity for pathogen and antibiotic resistance genes.
FIG 1.
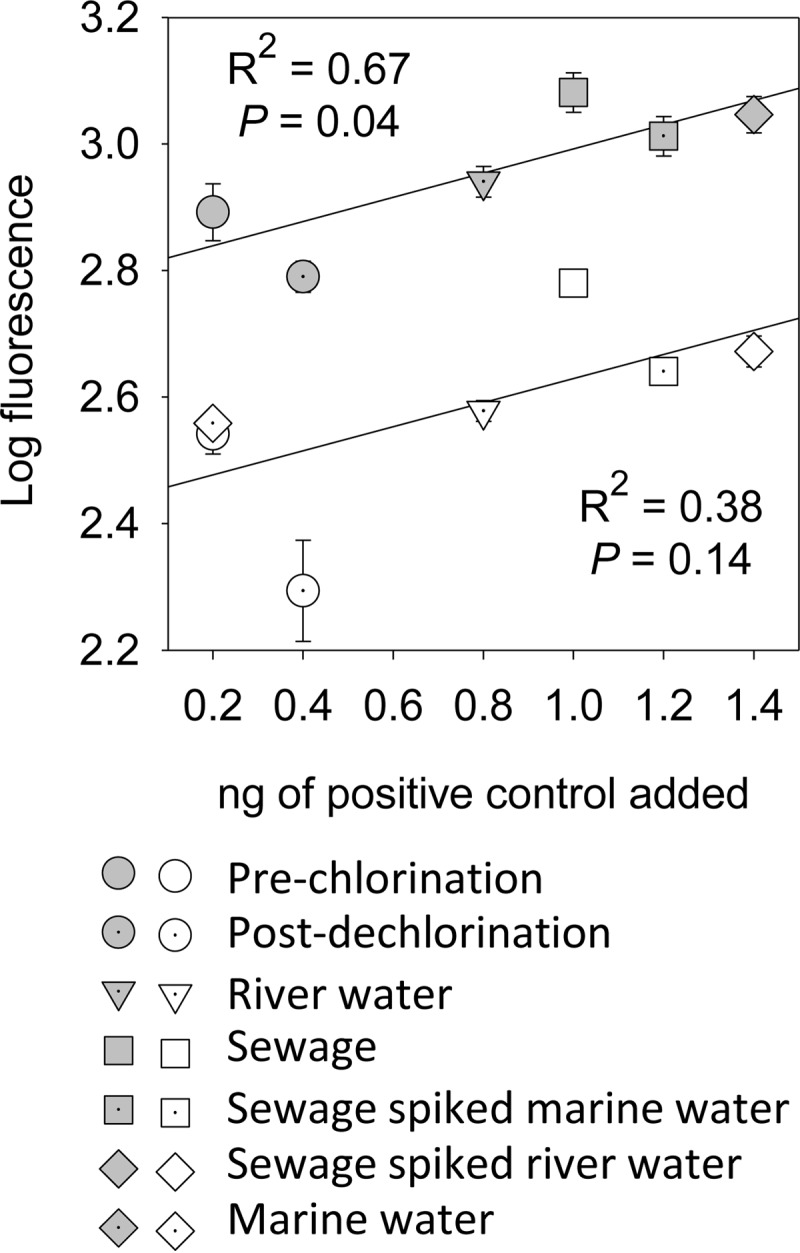
Log FU of two positive controls compared to nanograms of DNA added for each microarray. Shaded symbols represent probes for 5.8S rRNA of Oncorhynchus mykiss; white symbols represent probes for 16S rRNA of Dehalococcoides mccartyi. Error bars represent the standard deviations of the log FU from the 5 replicate PM probes on each array.
Nonmetric multidimensional scaling (NMDS) of microarray data from rRNA probes was used to discriminate the pollution sources of the seven microarray samples in the current study and the previously reported results using the same microarray design on different fecal samples (13). Microarray data from previously reported studies are labeled 1 and 2 here, while the microarray data from this study are labeled 3. All PM probes with normalized log FU for the PM probes exceeding 1.3 times the average signal for MM probes were considered a detection and assigned a “1”; otherwise, they were considered a nondetection and assigned a “0.” The NMDS plots were generated using PROC MDS of SAS (ver. 9.4; SAS Institute, Inc., Cary, NC) on a Bray-Curtis distance matrix. Twenty replicate plots were generated, and the plot with the least stress was selected. Typically, NMDS plots with stresses of less than 0.1 are considered to have ideal ordination with little likelihood of misinterpretation (25). Venn diagrams were generated using all PM probes on the microarrays to show probes detected in common between sample types.
Two methods were used to evaluate the microarray's ability to discriminate among fecal sources (Fig. 2). First, sensitivity and specificity based on individual probe performance were calculated, as they would be for a qPCR method with a singular gene target (26). Probes known to have human sewage association (i.e., MST markers or host-specific pathogens) and those considered to be general animal markers (e.g., Bacteroidales, present in the feces of most animals) by methods previously reported (see references in Table S2 in the supplemental material) were included in the calculation of sensitivity. Only MST markers and pathogens associated with specific animals other than humans were included in the calculation of specificity. An extensive review of the primary citation and subsequent literature detailing the host association of MST probes or primers included on the array was conducted to determine the true host association of the microarray MST probes. For this study, any MST probe found in greater than 50% of host fecal samples tested (as reported in the primary citation or subsequent literature) was deemed to be consistently associated with that host organism, and only these genes were included in calculation of sensitivity and specificity. A true positive (TP) was assumed to be the detection of an MST marker gene (e.g., human-associated Bacteroides) in a sample contaminated with fecal material in which the marker gene should be present (e.g., sewage). Furthermore, a false positive (FP) was the detection of an MST marker gene for a nontarget organism (e.g., swine feces marker) in a sewage-containing sample. True positive and false positive were used for calculating positive predictive value [TP/(TP + FP)], and true negative (TN) and false negative (FN) were used to calculate negative predictive value [TN/(FN + TN)]. Sensitivity (percent true positive) was calculated as TP/(TP + FN). Specificity was calculated as the percentage of true negatives, TN/(FP + TN), and the percentage of false positives was calculated as the number of FPs detected on a microarray (e.g., a cattle marker detected in the swine feces sample) divided by the total number of host-associated MST probes on the microarray, FP/(TP + TN + FP + FN) (27, 28).
FIG 2.

Methods for determining predictive accuracy of microarray for MST. Numbers of probes are indicated in parentheses. General fecal markers, e.g., general Bacteroidales, were considered true positives in the calculations. MST probes are those which have a host association, here assumed to be detected in greater than 50% of the fecal samples from that host or source organism. ARG, antibiotic resistance genes. Footnotes: a, probes indicated with an asterisk were included in further analysis; b, host-specific probes are found in >50% host feces tested and have a minimum of 75% specificity to host; c, source identifier probes were those detected in ≥50% of host feces (frequency from the literature); d, some MST probes were not considered if there was no dominant host association reported in the literature; e, multiple species, detected in two or more fecal types but not all animal feces.
The second method to evaluate the microarray's ability to discriminate among fecal sources contaminating a water sample was based on source identifier groups (29) and their abundance in samples (30) (Fig. 2). Source identifier groups are defined as operational taxonomic units (OTUs) (29) or probes for the microarray here, which are associated with a particular fecal source. A fecal source was assumed to contaminate a sample if >20% of the OTUs or probes for that source were detected in a sample. If two or more source identifiers met the >20% threshold, then the source with the highest percentage of positive probes was considered the true source. Furthermore, the source identifier probe intensity (30) (log FU) was also evaluated, and the source identifiers with the greatest overall and average FU were assumed to identify the true source.
RESULTS
DEUF method validation and WGA efficiency.
The uidA gene of E. coli was quantified by qPCR pre- and post-DEUF. Pre- and postfiltration concentrations were 5.87 and 8.45 log gene copies liter−1, respectively. The recovery efficiency was 95% for E. coli. The quantity of human polyomavirus in the 160-ml sewage sample was 2.62 log gene copies liter−1. The equivalent 160-ml sample, which was diluted and concentrated by DEUF, contained 2.69 log gene copes liter−1, indicating complete recovery of the human polyomaviruses from the filtration.
An average 46-fold increase in nucleic acid concentration by WGA for all the tested samples was observed (Table 1). Based on qPCR results, WGA increased the average abundance of all genes targeted from 3.1 to 4.9 log gene copies liter−1. However, some concentrations decreased between the pre- and post-WGA, such as norovirus from the WWTP effluent, S. aureus from the sewage-spiked river sample, Bacteroidales from the sewage-spiked marine sample, and E. coli from sewage-spiked river and marine water samples.
TABLE 1.
Whole-genome amplification efficiency evaluated by qPCR
| Source | Time of measurement and log change | Concn, log10 gene copies/liter for species (gene) |
Avg (SD) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| E. coli (uidA) | Enterococcus spp. (23S rRNA) | Bacteroidales (16S rRNA) | S. aureus (sec) | S. enterica (invA) | Human polyomavirus (T antigen) | Human norovirus (RNA polymerase) | |||
| WWTP effluent avga | Pre-WGA | 0.80g | 2.88 | 6.99 | 2.22 | 1.79 | 3.20 | 1.73 | |
| Post-WGA | 3.81 | 3.47 | 7.75 | 2.48 | 4.79 | 6.56 | 0.54 | ||
| Log change | 3.01 | 0.59 | 0.76 | 0.26 | 3.00 | 3.36 | −1.19 | 1.40 (1.74) | |
| Sewageb | Pre-WGA | 5.33 | 4.79 | 11.24 | 1.11g | 0.91g | 6.43 | 1.00 | |
| Post-WGA | 5.78 | 6.52 | 11.61 | 1.11g | 6.61 | 11.22 | 1.28 | ||
| Log change | 0.45 | 1.73 | 0.37 | BDLh | 5.72 | 4.79 | 0.28 | 2.22 (2.42) | |
| Riverc | Pre-WGA | 3.61 | 1.25 | 4.20 | 2.80 | 0.91g | 0.96g | 0.55 | |
| Post-WGA | 3.24 | 2.27 | 7.61 | 4.84 | 4.74 | 3.63 | 2.15 | ||
| Log change | −0.37 | 1.02 | 3.41 | 2.04 | 3.84 | 2.03 | 1.60 | 1.93 (1.42) | |
| Marined | Pre-WGA | 1.64 | −0.16 | 5.33 | 1.11g | 2.60 | 0.96g | 0.79 | |
| Post-WGA | 3.74 | 3.33 | 5.93 | 4.21 | 4.82 | 6.21 | 0.83 | ||
| Log change | 2.10 | 3.49 | 0.60 | 3.10 | 2.22 | 4.61 | 0.04 | 2.31 (1.60) | |
| River + sewagee | Pre-WGA | 3.31 | 1.76 | 9.05 | 4.73 | 3.56 | 5.46 | 0.99 | |
| Post-WGA | 4.47 | 4.19 | 9.09 | 1.11g | 5.34 | 9.64 | 2.02 | ||
| Log change | 1.16 | 2.43 | 0.04 | −3.62 | 1.78 | 4.18 | 1.03 | 1.00 (2.42) | |
| Marine + sewagef | Pre-WGA | 4.72 | 3.89 | 10.11 | 1.11g | 0.91g | 5.52 | 1.00 | |
| Post-WGA | 3.88 | 4.22 | 9.98 | 4.94 | 5.22 | 7.74 | 1.28 | ||
| Log change | −0.84 | 0.33 | −0.13 | 3.83 | 4.33 | 2.22 | 0.28 | 1.43 (2.04) | |
Star City wastewater treatment plant effluent, Morgantown, WV.
Falkenburg Road Advanced Wastewater Treatment Plant, Tampa, FL.
Hillsborough River, Tampa, FL.
Marine site, Intracoastal Waterway, St. Petersburg, FL.
Hillsborough River water spiked with sewage.
Marine site water, Intracoastal Waterway, St. Petersburg, FL, spiked with sewage.
Marker concentrations were below analytical detection limits; therefore, one-half the analytical detection limit was substituted.
BDL, below analytical detection limits.
Pathogen detection in environmental samples.
Detections of all PM probes associated with microorganisms and gene targets on the microarray are reported in Table S2 in the supplemental material. An abbreviated list of pathogens and antibiotic resistance genes detected in the samples tested here as well as the normalized log FU detected in each sample is presented in a heat map (Table 2). Four human virus genes were detected via the microarray, with the highest FU found in sewage and sewage-spiked samples. The presence of polyomavirus and adenovirus genes in positive microarray samples was confirmed via qPCR. Norovirus was not detected via the microarray but was detected via qPCR, although at lower abundances pre-WGA (1.0 ± 0.4 log gene copies liter−1) than those of polyomavirus (3.9 ± 2.1 log gene copies liter−1) and adenovirus (4.1 ± 2.0 log gene copies liter−1) (data not shown). The post-WGA abundance of norovirus by qPCR was 1.4 ± 0.6 log gene copies liter−1. In total, 78% of samples and gene targets (n = 7 samples and 8 gene targets [see Table S1]) tested via qPCR were also detected via the microarray.
TABLE 2.
Heat map indicating pathogens and antibiotic resistance genes detected on the microarrays and the probes' normalized relative fluorescence (numbers)a

Where multiple probes for an organism or target gene were detected in a sample, the average of the normalized fluorescence is displayed in the table. The probe FU colors are based on four arbitrary increments, in order of increasing color intensity: 0.01 to 0.65, >0.65 to 1.30, >1.30 to 1.95, and >1.95.
b —, the probe was not detected in that sample.
c WW, wastewater.
Other potentially pathogenic microorganisms, such as Campylobacter fetus, Clostridium spp., E. coli, Enterococcus faecalis, Legionella pneumophila, Staphylococcus aureus, Salmonella enterica and Shigella flexneri, were detected in most of the water samples via the microarray (Table 2). Furthermore, the presence of S. aureus, S. enterica, E. coli, and Enterococcus faecalis was confirmed via qPCR. The antibiotic resistance genes for aminoglycosides, beta-lactams, and tetracycline were mainly detected in sewage, river, spiked river, and spiked marine water samples via the microarray (Table 2).
In most cases, the addition of sewage to river or marine samples resulted in higher normalized log FU of microarray probes for sewage-associated microorganisms (Table 2). Of the 365 probes detected in the river, marine, and spiked water samples tested, only in 36 cases did the log FU of probes detected in the unamended water samples (river or marine) exceed those in sewage-spiked samples. Possible reasons for the greater FU of certain probes in water samples than in sewage-spiked samples include the following: (i) genes were in low concentrations in both sewage and natural waters and were near the detection limit for the microarray (e.g., bocavirus and antibiotic resistance genes), (ii) the sewage sample contained lower concentrations of the target microorganism than did the surface water samples (e.g., Clostridium perfringens, commonly found in sediments), and (iii) averaging of multiple probes for one gene target may artificially decrease the log FU (e.g., four Enterococcus faecalis 23S rRNA probes have different numbers of fluorescently labeled U nucleotides).
This correlation between pathogen concentration in a sample and normalized microarray FU is supported by our qPCR analysis for various pathogens. For example, the regression between qPCR gene copies liter−1 and microarray-normalized log FU for probes targeting Enterococcus spp., adenovirus, E. coli, and Bacteroidales (Fig. 3) indicates an overall significant linear correlation. The best individual correlation was for Enterococcus spp. (n = 4, R2 = 0.59, P = 0.04). Further evidence for the semiquantitative nature of the microarray is shown by the correlation between the mass of positive-control DNA hybridized to the array and observed FU (Fig. 1).
FIG 3.

Linear regression of qPCR log gene copies liter−1 versus average and standard deviation of normalized microarray probe log fluorescence. Circles, Enterococcus spp. (6 different probes in n = 4 samples, R2 = 0.59, P = 0.04); diamonds, Bacteroidales (7 different probes in n = 6 samples, R2 = 0.70, P = 0.23); triangles, adenovirus (7 different probes in n = 4 samples, R2 = 0.58, P = 0.24); inverted triangles, E. coli (3 different probes in n = 3 samples, R2 = 0.92, P = 0.18). The thick black line represents the overall regression line (R2 = 0.3, P < 0.012). Error bars represent the standard deviations of the log FU for all probes present on the array specific to a particular order, family, or genus.
Venn diagrams show the overlap in the probes detected in each related sample type (Fig. 4). Seventy-one microorganisms were found in common among the river, spiked river water, and sewage samples (Fig. 4A), while 60 microorganisms were found in common among the marine, spiked marine water, and sewage samples (Fig. 4B). Thirty-seven probes were detected in both the prechlorination and postdechlorination samples of treated wastewater (Fig. 4C). Relatively few organisms were detected only in one particular water type or sewage. Probes for five genes were detected only in sewage: a Bacteroides sp. strain (closely related to Bacteroides vulgatus Human 3, accession number JQ317268) (31), an E. coli strain, the iron-oxidizing bacterium Leptospirillum ferriphilum, human polyomavirus, and a tetracycline resistance gene. Similarly, the fish-associated Photobacterium phosphoreum and Tenacibaculum maritimum were found only in the marine sample, and Edwardsiella ictaluri was detected in both marine and river samples. One of the probes for Giardia intestinalis was found only in the prechlorination wastewater sample, while another G. intestinalis gene was found in sewage and a spiked river sample.
FIG 4.

Venn diagrams indicating perfect-match probes found in common among samples tested via the microarray. (A) Sewage/river water; (B) sewage/marine water; (C) effluent prechlorination and postdechlorination. River, Hillsborough River; sewage, Falkenburg Road, Tampa, FL, advanced wastewater treatment plant; spike river, water from the Hillsborough River, FL, spiked with sewage; marine, St. Petersburg, FL, marine water; spike marine, St. Petersburg, FL, marine water spiked with sewage; prechlorination, Star City, WV, wastewater treatment plant postsecondary treatment prior to chlorination; postdechlorination, Star City, WV, wastewater treatment plant postdechlorination immediately prior to discharge.
Utility of microarray for microbial source tracking.
Clustering of different sample types based on NMDS analysis is shown in Fig. 5 for the samples tested here and those reported previously (13). In general, related samples (e.g., marine, spiked marine, spiked river, river, and sewage) tended to cluster together and could be differentiated from samples from other sources (e.g., swine or bird feces). When the sensitivity and specificity of the microarray were calculated using methods originally intended for evaluation of single assays (i.e., qPCR for a single MST marker gene), the microarray's sensitivity was 51 to 57% and specificity was 79 to 81%.
FIG 5.

Overview of the separation of the spiked river and marine water samples, fecal samples, and treated wastewater by NMDS using rRNA gene data. Only rRNA genes detected on the current microarray (labeled 3) or those microarrays (labeled 1 and 2) previously published (13) are shown. Plot stress, 0.099.
The source identifier classification method (Table 3) and the total and average normalized FU of source identifier probes indicated that animal- as well as human- and sewage-associated probes predominated in the sewage sample, while animal-, human-, and avian-associated probes dominated the sewage-spiked surface water samples. Specifically, the data indicated that all samples contained feces from warm-blooded animals (100% of animal probes detected). The next most abundant probes in the sewage sample after animal-associated probes were human- and sewage-associated probes (42% of human- and sewage-associated probes detected) at high total (7.3 log FU) and average (0.8 ± 0.3 log FU of all human- and sewage-associated probes detected) probe intensities (Fig. 6). In fact, all other MST probes detected in the sewage sample, not including animals, were only 5.3 log FU total and had significantly lower intensities than did human-associated probes, averaging 0.6 ± 0.3 log FU (Fig. 6). Finally, the source identifier analysis of the microarray indicated that sewage-spiked marine and river water samples contained animal, human, and avian fecal inputs based on the percentage of source identifier probes detected (36 to 38% of human probes and 29% of avian probes detected in both samples), the probe category total intensity (Table 3), and average probe intensity (Fig. 6).
TABLE 3.
Predictive accuracy of microarray for the MST markers in sewage-containing samplesa
| Parameter | Sewage influent | Sewage-spiked marine water | Sewage-spiked river water |
|---|---|---|---|
| Performance based on individual probe results (%) | |||
| Sensitivity | 57 | 51 | 54 |
| Positive predictive value | 68 | 68 | 67 |
| False positive | 12 | 11 | 12 |
| Specificity | 79 | 81 | 79 |
| Negative predictive value | 70 | 68 | 69 |
| Performance based on aggregate source identifier resultsb | |||
| Animal (n = 6) | 6.3 (6) | 5.1 (6) | 7.0 (6) |
| Human and sewage (n = 36) | 7.3 (15) | 2.5 (13) | 3.6 (14) |
| Cow (n = 13) | 1.1 (2) | 0.5 (2) | 0.8 (2) |
| Avian (n = 17) | 3.3 (5) | 2.8 (5) | 3.0 (5) |
| Pig (n = 19) | 0.9 (3) | 0.3 (3) | 0.8 (3) |
| Sheep (n = 7) | 0 (0) | 0 (0) | 0 (0) |
| Source identifier | Human, animal | Animal, avian, human | Animal, human, avian |
General fecal markers, e.g., general Bacteroidales, were considered true positives in the calculations and are included in the animal category in Fig. 2. Boldface for “Source identifier” indicates the primary fecal source.
Sum of log FU of MST marker categories (number of probes detected).
FIG 6.

Average log FU of source identifier probes detected in sewage and sewage-spiked marine and river water. Means with a different letter are significantly different at an alpha level of 0.05 (analysis of variance, least significant difference) across treatments. The box represents 50% of the data, the dotted line represents the mean, the horizontal line within the box represents the median, and the whiskers above and below the box represent the 90th and 10th percentiles, respectively, while outliers are represented by circles.
Four human-associated Bacteroidales 16S rRNA probes (i.e., HF183 and HF134f) were detected only in sewage and sewage-spiked samples but not in either marine or river samples. Furthermore, these human feces-associated Bacteroidales probes had the greatest normalized log FU of all host-associated MST marker probes included on the microarray. The general probe targeting 16S rRNA for all enteric bacteria was detected in all samples except the postchlorination, wastewater effluent sample. In fact, the normalized log FU of the enteric bacterium probe was significantly higher (Student's t test, P = 0.043) in the sewage and sewage-spiked samples (1.064 ± 0.280 log FU) than in the river, marine, and treated wastewater samples (0.408 ± 0.432 log FU). Similarly, there were more overall bacteria based on the universal bacterial probe in sewage influent and spiked samples (1.927 ± 0.057 log FU) than in the river, marine, and other wastewater samples (1.248 ± 0.654 log FU), but not significantly more (Student's t test, P = 0.14). Significantly more (Student's t test, P < 0.001) Gram-negative organisms (1.893 ± 0.065 log FU) than Gram-positive organisms (1.271 ± 0.077 log FU) were detected in sewage and sewage-spiked samples, which is similar to previous findings (32).
DISCUSSION
Due to the small pore size of the ultrafilter (∼29 to 47 kDa), both DEUF and HFUF can be used for recovering diverse microorganisms, including all classes of microbial waterborne pathogens. The DEUF method, a modification of HFUF, has been used as a simple and portable technique for field concentration of bacteria, viruses, protozoa, and parasites from large water volumes (19, 33, 34). Two advantages to the DEUF method are that it is less likely to clog than other ultrafiltration methods and it is able to concentrate cells in the field followed by elution in the laboratory. The high recovery rates of 95% for E. coli and 100% for polyomavirus from water samples suggest that accurate determination of abundances of pathogens and detection limits in water samples is achievable.
WGA efficiency varied for human polyomaviruses and norovirus, E. coli, Enterococcus spp., Bacteroidales, Staphylococcus aureus, and Salmonella enterica. For instance, human polyomavirus (DNA virus) and norovirus (RNA virus) nucleic acid were not amplified as much as the bacteria (E. coli, Enterococcus spp., Bacteroidales, Staphylococcus aureus, and Salmonella enterica). Even for the five bacterial targets included in the evaluation, the increases in nucleotide concentrations are quite different from one another. The inefficient WGA amplification observed for some targets could be explained by either mechanistic issues with WGA in mixed environmental samples (35, 36) or bias in qPCR measurements. For example, factors affecting WGA efficiency could include the shorter genome sequences of the viruses, low initial nucleic acid concentrations not being amplified as efficiently as higher concentrations (i.e., “swamping out”), or the higher GC contents reducing amplification of the targeted nucleotides. Ongoing studies in our laboratory are attempting to determine the mechanisms behind the variation in WGA efficiency in mixed environmental samples.
Microarray probes for Vibrio cholerae rRNA genes (16S and 23S) were detected in all the water samples. This is surprising since nontoxigenic (ctx-negative) V. cholerae should be found primarily in estuarine environments, and toxigenic V. cholerae is rather rare in the United States. Unlike V. cholerae, Vibrio fluvialis and Vibrio parahaemolyticus (also in sewage) were found only in marine and spiked marine water samples. V. parahaemolyticus infections are much more common in the United States than V. cholerae infections; therefore, it is not as surprising to find the bacterium in sewage, and it is common in estuarine and coastal waters (37, 38). The most probable reason for the aberrant detection of V. cholerae by microarray is false-positive detection due to low specificity of the microarray probe. Comparison of the V. cholerae probe with the NCBI BLAST database showed a 100% match with Vibrio parahaemolyticus (accession number CP006008.1), Vibrio mimicus (KJ604709), and Vibrio navarrensis (isolated from wastewater; accession number AJ294423.1), which were not present in the database or were overlooked when the probe was originally designed. Similar nonspecificity for the detected Vibrio fluvialis probe was observed. Therefore, ongoing evaluation of the specificity of any microarray probe design against nucleotide databases (e.g., NCBI BLAST database) should be part of standard operating procedures. In the case of the pathogenic Vibrio spp., future microarray designs will include probes for toxin genes.
The quantitative nature of DNA microarrays has been studied since 2009; however, there are still arguments against the use of microarray for quantitative purposes. For example, Chen et al. (39) found a very low correlation between microarray fluorescence and qPCR. In contrast, Paliy et al. (40) developed a high-throughput phylogenetic microarray for microbe identifications in human intestines. Nicholson et al. (41) and Mehnert et al. (42) have used and designed automated tissue microarrays for clinical sample research. Finally, a quantitative liposome microarray has also been reported (43). In our study, two separate lines of evidence suggest that the microarray may be used semiquantitatively for detection of pathogens, namely, the decrease in probe log FU in the spiked samples compared to sewage samples and the correlation of log FU with qPCR results. The development of quantitative or semiquantitative microarrays has the potential to overcome the drawbacks to qPCR methods, including cost, time, and complexity of assays required. Based on qPCR concentrations pre-WGA for those genes not detected on the microarray, the following minimum detection limits (gene copies liter−1) were determined for the combination of the WGA and microarray process: 654 ± 38 of uidA gene of E. coli, 135 ± 8 of polyomavirus, 22 ± 4 of norovirus, and 186 of Enterococcus spp. Further, the relatively low detection limits found here of a few hundred gene copies liter−1 of water due to the combined methods of ultrafiltration, WGA, and MST microarray support the use of multiple-target microarrays for microbiological water quality monitoring.
The microarray's sensitivity is less than that previously reported for many qPCR assays with a single target (30, 44). It is greater than the sensitivity previously reported for some microarrays (13, 44) but less than that for other reported arrays which used the source identifier method (29, 30). In this case, the calculated sensitivity was influenced by the inclusion of over 37 different MST marker genes, many of which belong to pathogens that are relatively rare targets compared to indicator groups such as enterococci. The sensitivity of molecular assays for human-specific pathogens, such as viruses, varies by geographic location and species. For example, Ahmed et al. detected human adenoviruses in 73% of sewage samples in Australia (45), while Wolf et al. detected adenovirus F in 100% of sewage samples in New Zealand (46) but found adenovirus C in only 36% of sewage samples. A multilaboratory study found that viruses were highly specific indicators of sewage in water, but the sensitivity of nine different qPCR methods for human virus detection was very poor, ranging from 0% to 13.2% (47). The sensitivity of the microarray was estimated using only those probes for which we have sufficient evidence in the literature to assign consistent “host association.” In this study, we deemed that marker genes present in greater than 50% of host fecal samples tested (as reported in the literature) were consistently host associated. Sensitivity was then calculated as the number of correctly detected positive probes on the array divided by the number of probes on the array for that particular host. For the sewage sample, we found that 57% of the human-associated or animal-associated probes were detected in the samples. This is a rather high sensitivity when one considers that some of these probes we know a priori are likely to be in low concentrations in sewage samples (e.g., human mitochondrial DNA, which was not detected with the array) and some are likely to be in high concentration in sewage samples (e.g., Bacteroides HF183 and HF134f, which were detected with the array).
Overall, the results of these studies show that (i) the MST microarray can be used to discriminate between sources of fecal pathogens in surface water samples, (ii) microarray is correlated with qPCR-based enumeration of certain microbial gene targets, and (iii) common waterborne pathogen gene targets can be detected via microarray in surface water and reclaimed water samples. It is important to note that the microarray when used in combination with WGA will detect pathogen genes in infectious pathogens as well as in dead pathogens, and we have noted apparent cross-reactions of some probes (e.g., V. cholerae). Therefore, additional verification methods such as culture or qPCR may be required to assess the relationship between human health outcomes and pathogen gene detection via a microarray.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Duke Microarray Core facility for their technical support, microarray data management, and feedback on the generation of the microarray data reported in this work. We are grateful to Greg Shellito for providing water testing results from the Star City, WV, treatment plant. We appreciate the insightful comments of the independent peer reviewers and editorial board, which strengthened the manuscript.
Partial funding for this project was provided by the NSF ADVANCE IT Program (award HRD-1007978).
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.02583-15.
REFERENCES
- 1.Boehm AB, Ashbolt NJ, Colford JM Jr, Dunbar LE, Fleming LE, Gold MA, Hansel JA, Hunter PR, Ichida AM, McGee CD, Soller JA, Weisberg SB. 2009. A sea change ahead for recreational water quality criteria. J Water Health 7:9–20. doi: 10.2166/wh.2009.122. [DOI] [PubMed] [Google Scholar]
- 2.Hrudey SE, Hrudey EJ. 2007. Published case studies of waterborne disease outbreaks—evidence of a recurrent threat. Water Environ Res 79:233–245. doi: 10.2175/106143006X95483. [DOI] [PubMed] [Google Scholar]
- 3.Stewart JR, Gast RJ, Fujioka RS, Solo-Gabriele HM, Meschke JS, Amaral-Zettler LA, Del Castillo E, Polz MF, Collier TK, Strom MS, Sinigalliano CD, Moeller PD, Holland AF. 2008. The coastal environment and human health: microbial indicators, pathogens, sentinels and reservoirs. Environ Health 7(Suppl 2):S3. doi: 10.1186/1476-069X-7-S2-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Straub TM, Chandler DP. 2003. Towards a unified system for detecting waterborne pathogens. J Microbiol Methods 53:185–197. doi: 10.1016/S0167-7012(03)00023-X. [DOI] [PubMed] [Google Scholar]
- 5.Nwachcuku N, Gerba CP. 2004. Emerging waterborne pathogens: can we kill them all? Curr Opin Biotechnol 15:175–180. doi: 10.1016/j.copbio.2004.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sercu B, Van De Werfhorst LC, Murray JL, Holden PA. 2011. Terrestrial sources homogenize bacterial water quality during rainfall in two urbanized watersheds in Santa Barbara, CA. Microb Ecol 62:574–583. doi: 10.1007/s00248-011-9874-z. [DOI] [PubMed] [Google Scholar]
- 7.Passerat J, Ouattara NK, Mouchel JM, Rocher V, Servais P. 2011. Impact of an intense combined sewer overflow event on the microbiological water quality of the Seine River. Water Res 45:893–903. doi: 10.1016/j.watres.2010.09.024. [DOI] [PubMed] [Google Scholar]
- 8.Lu J, Ryu H, Hill S, Schoen M, Ashbolt N, Edge TA, Santo Domingo J. 2011. Distribution and potential significance of a gull fecal marker in urban coastal and riverine areas of southern Ontario, Canada. Water Res 45:3960–3968. doi: 10.1016/j.watres.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 9.Takemura AF, Chien DM, Polz MF. 2014. Associations and dynamics of Vibrionaceae in the environment, from the genus to the population level. Front Microbiol 5:38. doi: 10.3389/fmicb.2014.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leclerc H, Mossel DA, Edberg SC, Struijk CB. 2001. Advances in the bacteriology of the coliform group: their suitability as markers of microbial water safety. Annu Rev Microbiol 55:201–234. doi: 10.1146/annurev.micro.55.1.201. [DOI] [PubMed] [Google Scholar]
- 11.Harwood VJ, Staley C, Badgley BD, Borges K, Korajkic A. 2014. Microbial source tracking markers for detection of fecal contamination in environmental waters: relationships between pathogens and human health outcomes. FEMS Microbiol Rev 38:1–40. doi: 10.1111/1574-6976.12031. [DOI] [PubMed] [Google Scholar]
- 12.Roslev P, Bukh AS. 2011. State of the art molecular markers for fecal pollution source tracking in water. Appl Microbiol Biotechnol 89:1341–1355. doi: 10.1007/s00253-010-3080-7. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Harwood VJ, Nayak B, Staley C, Sadowsky MJ, Weidhaas J. 2015. A novel microbial source tracking microarray for pathogen detection and fecal source identification in environmental systems. Environ Sci Technol 49:7319–7329. doi: 10.1021/acs.est.5b00980. [DOI] [PubMed] [Google Scholar]
- 14.Leclerc H, Schwartzbrod L, Dei-Cas E. 2002. Microbial agents associated with waterborne diseases. Crit Rev Microbiol 28:371–409. doi: 10.1080/1040-840291046768. [DOI] [PubMed] [Google Scholar]
- 15.Hill VR, Kahler AM, Jothikumar N, Johnson TB, Hahn D, Cromeans TL. 2007. Multistate evaluation of an ultrafiltration-based procedure for simultaneous recovery of enteric microbes in 100-liter tap water samples. Appl Environ Microbiol 73:4218–4225. doi: 10.1128/AEM.02713-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hill VR, Polaczyk AL, Hahn D, Narayanan J, Cromeans TL, Roberts JM, Amburgey JE. 2005. Development of a rapid method for simultaneous recovery of diverse microbes in drinking water by ultrafiltration with sodium polyphosphate and surfactants. Appl Environ Microbiol 71:6878–6884. doi: 10.1128/AEM.71.11.6878-6884.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polaczyk AL, Narayanan J, Cromeans TL, Hahn D, Roberts JM, Amburgey JE, Hill VR. 2008. Ultrafiltration-based techniques for rapid and simultaneous concentration of multiple microbe classes from 100-L tap water samples. J Microbiol Methods 73:92–99. doi: 10.1016/j.mimet.2008.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Kearns EA, Magana S, Lim DV. 2008. Automated concentration and recovery of micro-organisms from drinking water using dead-end ultrafiltration. J Appl Microbiol 105:432–442. doi: 10.1111/j.1365-2672.2008.03757.x. [DOI] [PubMed] [Google Scholar]
- 19.Smith CM, Hill VR. 2009. Dead-end hollow-fiber ultrafiltration for recovery of diverse microbes from water. Appl Environ Microbiol 75:5284–5289. doi: 10.1128/AEM.00456-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hundesa A, Maluquer de Motes C, Bofill-Mas S, Albinana-Gimenez N, Girones R. 2006. Identification of human and animal adenoviruses and polyomaviruses for determination of sources of fecal contamination in the environment. Appl Environ Microbiol 72:7886–7893. doi: 10.1128/AEM.01090-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Z, Gentry TJ, Schadt CW, Wu L, Liebich J, Chong SC, Huang Z, Wu W, Gu B, Jardine P, Criddle C, Zhou J. 2007. GeoChip: a comprehensive microarray for investigating biogeochemical, ecological and environmental processes. ISME J 1:67–77. doi: 10.1038/ismej.2007.2. [DOI] [PubMed] [Google Scholar]
- 22.Rhodes ER, Hamilton DW, See MJ, Wymer L. 2011. Evaluation of hollow-fiber ultrafiltration primary concentration of pathogens and secondary concentration of viruses from water. J Virol Methods 176:38–45. doi: 10.1016/j.jviromet.2011.05.031. [DOI] [PubMed] [Google Scholar]
- 23.Griffiths RI, Whiteley AS, O'Donnell AG, Bailey MJ. 2000. Rapid method for coextraction of DNA and RNA from natural environments for analysis of ribosomal DNA- and rRNA-based microbial community composition. Appl Environ Microbiol 66:5488–5491. doi: 10.1128/AEM.66.12.5488-5491.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jothikumar N, Cromeans TL, Hill VR, Lu X, Sobsey MD, Erdman DD. 2005. Quantitative real-time PCR assays for detection of human adenoviruses and identification of serotypes 40 and 41. Appl Environ Microbiol 71:3131–3136. doi: 10.1128/AEM.71.6.3131-3136.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clarke KR. 1993. Non-parametric multivariate analyses of changes in community structure. Aust J Ecol 18:117–143. doi: 10.1111/j.1442-9993.1993.tb00438.x. [DOI] [Google Scholar]
- 26.Kildare BJ, Leutenegger CM, McSwain BS, Bambic DG, Rajal VB, Wuertz S. 2007. 16S rRNA-based assays for quantitative detection of universal, human-, cow-, and dog-specific fecal Bacteroidales: a Bayesian approach. Water Res 41:3701–3715. doi: 10.1016/j.watres.2007.06.037. [DOI] [PubMed] [Google Scholar]
- 27.Harwood VJ, Wiggins B, Hagedorn C, Ellender RD, Gooch J, Kern J, Samadpour M, Chapman AC, Robinson BJ, Thompson BC. 2003. Phenotypic library-based microbial source tracking methods: efficacy in the California collaborative study. J Water Health 1:153–166. [PubMed] [Google Scholar]
- 28.Myoda SP, Carson CA, Fuhrmann JJ, Hahm BK, Hartel PG, Yampara-Lquise H, Johnson L, Kuntz RL, Nakatsu CH, Sadowsky MJ, Samadpour M. 2003. Comparison of genotypic-based microbial source tracking methods requiring a host origin database. J Water Health 1:167–180. [PubMed] [Google Scholar]
- 29.Dubinsky EA, Esmaili L, Hulls JR, Cao Y, Griffith JF, Andersen GL. 2012. Application of phylogenetic microarray analysis to discriminate sources of fecal pollution. Environ Sci Technol 46:4340–4347. doi: 10.1021/es2040366. [DOI] [PubMed] [Google Scholar]
- 30.Cao Y, Van De Werfhorst LC, Dubinsky EA, Badgley BD, Sadowsky MJ, Andersen GL, Griffith JF, Holden PA. 2013. Evaluation of molecular community analysis methods for discerning fecal sources and human waste. Water Res 47:6862–6872. doi: 10.1016/j.watres.2013.02.061. [DOI] [PubMed] [Google Scholar]
- 31.Kabiri L, Alum A, Rock C, McLain JE, Abbaszadegan M. 2013. Isolation of Bacteroides from fish and human fecal samples for identification of unique molecular markers. Can J Microbiol 59:771–777. doi: 10.1139/cjm-2013-0518. [DOI] [PubMed] [Google Scholar]
- 32.Forster S, Snape JR, Lappin-Scott HM, Porter J. 2002. Simultaneous fluorescent gram staining and activity assessment of activated sludge bacteria. Appl Environ Microbiol 68:4772–4779. doi: 10.1128/AEM.68.10.4772-4779.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Knappett PS, Layton A, McKay LD, Williams D, Mailloux BJ, Huq MR, Alam MJ, Ahmed KM, Akita Y, Serre ML, Sayler GS, van Geen A. 2011. Efficacy of hollow-fiber ultrafiltration for microbial sampling in groundwater. Ground Water 49:53–65. doi: 10.1111/j.1745-6584.2010.00712.x. [DOI] [PubMed] [Google Scholar]
- 34.Leskinen SD, Brownell M, Lim DV, Harwood VJ. 2010. Hollow-fiber ultrafiltration and PCR detection of human-associated genetic markers from various types of surface water in Florida. Appl Environ Microbiol 76:4116–4117. doi: 10.1128/AEM.00025-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Binga E, Lasken R, Neufeld J. 2008. Something from (almost) nothing: the impact of multiple displacement amplification on microbial ecology. ISME J 2:233–241. doi: 10.1038/ismej.2008.10. [DOI] [PubMed] [Google Scholar]
- 36.Arriola E, Lambros MBK, Jones C, Dexter T, Mackay A, Tan DSP, Tamber N, Fenwick K, Ashworth A, Dowsett M, Reis-Filho JS. 2007. Evaluation of Phi29-based whole-genome amplification for microarray-based comparative genomic hybridisation. Lab Invest 87:75–83. doi: 10.1038/labinvest.3700495. [DOI] [PubMed] [Google Scholar]
- 37.Turner JW, Malayil L, Guadagnoli D, Cole D, Lipp EK. 2014. Detection of Vibrio parahaemolyticus, Vibrio vulnificus and Vibrio cholerae with respect to seasonal fluctuations in temperature and plankton abundance. Environ Microbiol 16:1019–1028. doi: 10.1111/1462-2920.12246. [DOI] [PubMed] [Google Scholar]
- 38.Johnson CN, Flowers AR, Noriea NF, Zimmerman AM, Bowers JC, DePaola A, Grimes DJ. 2010. Relationships between environmental factors and pathogenic vibrios in the northern Gulf of Mexico. Appl Environ Microbiol 76:7076–7084. doi: 10.1128/AEM.00697-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, Gelfond JA, McManus LM, Shireman PK. 2009. Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genomics 10:407. doi: 10.1186/1471-2164-10-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paliy O, Kenche H, Abernathy F, Michail S. 2009. High-throughput quantitative analysis of the human intestinal microbiota with a phylogenetic microarray. Appl Environ Microbiol 75:3572–3579. doi: 10.1128/AEM.02764-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nicholson AD, Guo X, Sullivan CA, Cha CH. 2014. Automated quantitative analysis of tissue microarray of 443 patients with colorectal adenocarcinoma: low expression of Bcl-2 predicts poor survival. J Am Coll Surg 219:977–987. doi: 10.1016/j.jamcollsurg.2014.07.007. [DOI] [PubMed] [Google Scholar]
- 42.Mehnert JM, McCarthy MM, Jilaveanu L, Flaherty KT, Aziz S, Camp RL, Rimm DL, Kluger HM. 2010. Quantitative expression of VEGF, VEGF-R1, VEGF-R2, and VEGF-R3 in melanoma tissue microarrays. Hum Pathol 41:375–384. doi: 10.1016/j.humpath.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saliba AE, Vonkova I, Ceschia S, Findlay GM, Maeda K, Tischer C, Deghou S, van Noort V, Bork P, Pawson T, Ellenberg J, Gavin AC. 2014. A quantitative liposome microarray to systematically characterize protein-lipid interactions. Nat Methods 11:47–50. doi: 10.1038/nmeth.2734. [DOI] [PubMed] [Google Scholar]
- 44.Boehm AB, Van De Werfhorst LC, Griffith JF, Holden PA, Jay JA, Shanks OC, Wang D, Weisberg SB. 2013. Performance of forty-one microbial source tracking methods: a twenty-seven lab evaluation study. Water Res 47:6812–6828. doi: 10.1016/j.watres.2012.12.046. [DOI] [PubMed] [Google Scholar]
- 45.Ahmed W, Goonetilleke A, Gardner T. 2010. Human and bovine adenoviruses for the detection of source-specific fecal pollution in coastal waters in Australia. Water Res 44:4662–4673. doi: 10.1016/j.watres.2010.05.017. [DOI] [PubMed] [Google Scholar]
- 46.Wolf S, Hewitt J, Greening GE. 2010. Viral multiplex quantitative PCR assays for tracking sources of fecal contamination. Appl Environ Microbiol 76:1388–1394. doi: 10.1128/AEM.02249-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harwood VJ, Boehm AB, Sassoubre LM, Vijayavel K, Stewart JR, Fong T-T, Caprais M-P, Converse RR, Diston D, Ebdon J, Fuhrman JA, Gourmelon M, Gentry-Shields J, Griffith JF, Kashian DR, Noble RT, Taylor H, Wicki M. 2013. Performance of viruses and bacteriophages for fecal source determination in a multi-laboratory, comparative study. Water Res 47:6929–6943. doi: 10.1016/j.watres.2013.04.064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.