

Abstract
Mycobacterium tuberculosis requires the phosphate-sensing signal transduction system Pst/SenX3-RegX3 to resist host immune responses. A ΔpstA1 mutant lacking a Pst phosphate uptake system component is hypersensitive to diverse stress conditions in vitro and is attenuated in vivo due to constitutive expression of the phosphate starvation-responsive RegX3 regulon. Transcriptional profiling of the ΔpstA1 mutant revealed aberrant expression of certain pe and ppe genes. PE and PPE proteins, defined by conserved N-terminal domains containing Pro-Glu (PE) or Pro-Pro-Glu (PPE) motifs, account for a substantial fraction of the M. tuberculosis genome coding capacity, but their functions are largely uncharacterized. Because some PE and PPE proteins localize to the cell wall, we hypothesized that overexpression of these proteins sensitizes M. tuberculosis to stress by altering cell wall integrity. To test this idea, we deleted pe and ppe genes that were overexpressed by ΔpstA1 bacteria. Deletion of a single pe gene, pe19, suppressed hypersensitivity of the ΔpstA1 mutant to both detergent and reactive oxygen species. Ethidium bromide uptake assays revealed increased envelope permeability of the ΔpstA1 mutant that was dependent on PE19. The replication defect of the ΔpstA1 mutant in NOS2−/− mice was partially reversed by deletion of pe19, suggesting that increased membrane permeability due to PE19 overexpression sensitizes M. tuberculosis to host immunity. Our data indicate that PE19, which comprises only a 99-amino-acid PE domain, has a unique role in the permeability of the M. tuberculosis envelope that is regulated to resist stresses encountered in the host.
INTRODUCTION
Over 15 years ago, the novel PE and PPE protein families were identified in the complete genome sequence of Mycobacterium tuberculosis; together, these proteins represent over 7% of the genome coding capacity (1). Despite the attention placed on these protein families, their functions remain largely uncharacterized. PE and PPE proteins are defined by conserved N-terminal domains of ∼110 or ∼180 amino acids that contain Pro-Glu (PE) or Pro-Pro-Glu (PPE) sequence motifs, respectively (1). Although PE and PPE proteins can be identified in the genomes of all sequenced members of the Mycobacterium genus, their expansion into large multiprotein families is restricted to the slow-growing pathogenic mycobacterial species, including M. tuberculosis, and associated with the expansion of the ESX type VII secretion systems (2). There is evidence that some PE and PPE proteins are exported to the bacterial cell surface or extracellular milieu in an ESX-dependent manner (3–6). ESX-dependent export requires specific sequences within the PE or PPE domain (7, 8), including a recently described YxxxD/E ESX secretion targeting motif located near the C terminus of the ∼110-amino-acid PE domain (9).
The 99 PE proteins and 69 PPE proteins encoded by the M. tuberculosis H37Rv genome can be further divided into subfamilies based on C-terminal sequence motifs (2). The PE_PGRS (polymorphic GC-rich sequence) and PPE_MPTR (major polymorphic tandem repeat) subfamilies, which include 65 and 23 members, respectively, each have highly repetitive C-terminal sequences. The PPE-PPW (10 members) and PPE-SVP (24 members) subfamilies, in contrast, have C-terminal domains with well-conserved and nonrepetitive sequence motifs.
Given the variability of the PE and PPE C-terminal domains and their potential extracellular localization, some have speculated that PE or PPE protein antigen variation could enable the evasion of antigen-specific host immune responses (1). This theory has been fueled by observations that there is variation in certain pe_pgrs gene sequences among clinical isolates (10, 11) and that recombination leading to gene deletions can occur between different copies of the pe and ppe genes (12). There is, however, little evidence to support rapid alterations of pe and ppe gene sequences in vivo (13). In addition, a recent comparative analysis of pe and ppe sequences from 40 clinical isolates suggested the absence of evolutionary selective pressure on these genes, an observation incompatible with positive selective pressure from the immune response that drives antigenic variation (14). Finally, there is evidence from both human tuberculosis infection and animal models that the conserved PE and PPE domains are themselves antigenic, generating immune responses that can cross-react with other PE and PPE proteins of similar sequence (15–17).
Only a few PE or PPE proteins have been investigated either through the generation of M. tuberculosis genetic mutants or biochemical characterization of purified proteins. Genome-wide mutagenesis approaches indicate that most pe and ppe genes are not essential for M. tuberculosis replication either in laboratory culture or in an animal model (18–20), suggesting functional redundancy among members of the PE and PPE families. However, PE_PGRS30 has been implicated in M. tuberculosis virulence (21), and several pe and ppe transposon insertion mutants have been identified in screens for genes involved in phagosome maturation arrest (22, 23). In addition, several PE proteins have enzymatic activity. LipY (PE_PGRS63) is a triacylglycerol lipase that is targeted to the cell wall in a manner dependent on the PE domain (8, 24, 25). PE_PGRS11 exhibits phosphoglycerate mutase activity in vitro (26). Finally, a conserved serine hydrolase fold has been identified in several PE and PPE proteins (27). One of these proteins, PE16, has in vitro esterase activity against short-chain fatty acid esters (28). These observations suggest that some PE and PPE proteins have specific enzyme activities that could be important for M. tuberculosis metabolism and physiology. However, since the majority of PE and PPE proteins do not have conserved domains suggestive of enzymatic activity, this is not likely to be a general property of all PE or PPE proteins.
Knowledge of the physiological growth conditions that induce the expression of specific PE and PPE proteins could help to elucidate their function. The pe and ppe genes are differentially regulated at the transcriptional level in response to a variety of environmental signals (29) and are frequently found among the differentially expressed genes in regulatory mutants (30, 31). In addition, there is evidence that some PE_PGRS family members are expressed specifically in granulomatous lesions (32). Therefore, the differential expression of PE and PPE proteins may be an adaptive response by M. tuberculosis to specific environmental conditions that promotes bacterial survival.
The M. tuberculosis phosphate starvation-responsive signal transduction system Pst/SenX3-RegX3 is a transcriptional regulatory system that is required for resistance to host immune defenses (33). The SenX3-RegX3 two-component system normally is activated by inorganic phosphate (Pi) starvation. The activation of the DNA binding response regulator RegX3 is inhibited when Pi is abundant through a poorly understood interaction with the Pst (phosphate-specific transport) Pi uptake system. A ΔpstA1 mutant that lacks a membrane-spanning component of the Pst system exhibits constitutive activation of RegX3 and dysregulated expression of RegX3-dependent genes in Pi-rich medium. We previously demonstrated that the hypersensitivity of the ΔpstA1 mutant to various stress conditions in vitro and to host immune responses in vivo was attributable to aberrant activation of RegX3, because these hypersensitivity phenotypes were suppressed by deletion of regX3.
Our results suggested that a RegX3-regulated factor(s) is responsible for both sensitivity to stress conditions in vitro and sensitivity to host immune responses in vivo. The pleiotropic phenotypes of the ΔpstA1 mutant, particularly sensitivity to the cell wall-disrupting detergent sodium dodecyl sulfate (SDS), suggested a cell wall defect (33). As part of our previous studies, we performed transcriptional profiling to define the set of genes that are aberrantly expressed by ΔpstA1 bacteria in a RegX3-dependent manner (33). Among this set of genes, there were several that encode PE and PPE proteins. Of the PE and PPE proteins that have been characterized to date, many are localized to the mycobacterial cell wall (3, 7, 8, 34–36). We hypothesized that the overexpression of cell wall-localized PE and/or PPE proteins in the ΔpstA1 mutant sensitize these bacteria to stress by subtly altering the architecture or permeability of the cell wall. Here, we demonstrate that overexpression of the PE protein PE19 sensitizes the ΔpstA1 mutant to cell wall and oxidative stress conditions in vitro by increasing cell envelope permeability. We further demonstrate that the overexpression of PE19 is partially responsible for attenuation of the ΔpstA1 mutant in NOS2−/− mice. Our results suggest that the PE19 protein has a unique and important function in M. tuberculosis physiology and that its expression is carefully controlled to enable bacterial persistence.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
M. tuberculosis Erdman and derivative strains were grown at 37°C in Middlebrook 7H9 (Difco) liquid culture medium supplemented with 10% albumin-dextrose-saline (ADS), 0.5% glycerol, and 0.1% Tween 80 or on Middlebrook 7H10 (Difco) solid culture medium supplemented with 10% oleic acid-albumin-dextrose-catalase (OADC; BD Biosciences) and 0.5% glycerol. For cultivation of M. tuberculosis mc27000 (H37Rv ΔRD1, ΔpanCD) (37) and derivative strains, complete Middlebrook 7H9 or 7H10 medium was supplemented with 50 μg/ml pantothenic acid (Sigma). Frozen stocks were prepared by growing liquid cultures to mid-exponential phase (OD600 of 0.6 to 0.8), adding glycerol to 15% final concentration, and storing aliquots at −80°C. For phosphate-limiting (Pi-free) 7H9 broth, a 100× liquid stock of 7H9 base was reconstituted without the addition of the Pi-buffering components (Na2HPO4, KH2PO4). 1× Pi-free 7H9 was made with 0.5% glycerol, 10% ADS, 0.1% Tween 80, and 50 mM morpholinepropanesulfonic acid (MOPS) buffer, pH 6.6. Doubling times were calculated from daily OD600 measurements taken over the first 72 h of growth after dilution of mid-exponential-phase (OD600 of 0.5) cultures to an OD600 of 0.05 in complete 7H9 medium.
Cloning of deletion and complementation constructs.
Constructs for deletion of putative pe-ppe gene pairs or operons were generated in the allelic exchange vector pJG1100 (38), which contains the aph (kanamycin resistance), hyg (hygromycin resistance), and sacB (sucrose sensitivity) markers (see Table S1 in the supplemental material). Genomic regions of ∼800 bp 5′ and 3′ of the genes to be deleted were PCR amplified from M. tuberculosis Erdman genomic DNA using the oligonucleotides listed in Table S2. Reverse primers to amplify the 5′ regions were designed with an AvrII restriction site in-frame with the translation start codon. Forward primers to amplify the 3′ regions were designed with an AvrII restriction site in-frame with the stop codon. Resulting PCR products were cloned in pCR2.1-TOPO (Invitrogen) and sequenced. The 5′ and 3′ regions were removed from pCR2.1 by restriction with PacI/AvrII or AvrII/AscI, respectively, and ligated together in pJG1100 between the PacI and AscI sites to generate in-frame deletion constructs. Deletion constructs generated in pJG1100 and the genes they are designed to delete are listed in Table S1.
For complementation of the Δppe27-pe19 and Δpe19 deletions, the ppe27-pe19 locus or the full-length pe19 gene was PCR amplified from M. tuberculosis Erdman genomic DNA using the primers indicated in Table S2 in the supplemental material. For the complementation of the Δpe19 deletion with C-terminal His6 or hemagglutinin (HA) epitope-tagged versions of PE19, the full-length pe19 gene was PCR amplified from M. tuberculosis Erdman genomic DNA using primer 1791compF and the reverse primers indicated in Table S2. The resulting PCR products were cloned in pCR2.1-TOPO and sequenced. For ppe27-pe19 complementation, the insert was removed from pCR2.1 by digestion with ClaI and HpaI and cloned in similarly digested pMV261 to generate pMVppe27-pe19. For pe19 complementation, the inserts were removed from pCR2.1 by digestion with PstI and HindIII and cloned in similarly digested pMV261 to generate pMVpe19, pMVpe19His6, or pMVpe19HA.
M. tuberculosis strain construction.
M. tuberculosis strains harboring in-frame unmarked deletions were generated by a two-step homologous recombination allelic exchange method (39, 40). Deletions constructed and the vectors used to generate them are listed in Table S1 in the supplemental material. Plasmids were introduced into wild-type (WT) or ΔpstA1 mutant (33) M. tuberculosis Erdman by electroporation with 1 to 2 μg of purified plasmid DNA. Electrocompetent M. tuberculosis strains were prepared as previously described (33). Transformants were grown for 24 h in 7H9 broth before selecting recombinants containing chromosomally integrated pJG1100 on 7H10 agar containing kanamycin (15 μg/ml) and hygromycin (50 μg/ml). Kanr/Hygr colonies were picked and grown in 7H9 broth without antibiotics to mid-exponential phase. Integration of the constructs at the correct chromosomal locus was confirmed by PCR on heat-inactivated cell lysates using the primer pairs for detection of the 5′ and 3′ integrations (see Table S3). Clones that contained the allelic exchange vector integrated at the correct locus were serially diluted and plated on 7H10 agar containing 2% sucrose for counterselection of the pJG1100 vector. Sucrose-resistant clones were grown in 7H9 broth, and isolates in which the deletion replaced the wild-type allele were identified by PCR using the primers for detection of the gene deletion (see Table S3). The ΔpstA1 mutation was constructed in M. tuberculosis mc27000 as described previously (33). The Δpe19 deletion was constructed in mc27000 WT and ΔpstA1 backgrounds as described above. Analysis of phthiocerol dimycocerosate (PDIM) production was done by thin-layer chromatography of [14C]propionate-labeled lipid extracts as previously described (38).
For complementation of the Δpe19 mutation, the Δpe19 and ΔpstA1 Δpe19 strains were made electrocompetent as described above and electroporated with plasmid pMVpe19. Transformants were selected on 7H10 agar containing kanamycin (15 μg/ml). The presence of the complementing plasmid in Kanr isolates was confirmed by PCR on heat-inactivated cell lysates using primers pMVinsF/1791compR.
Southern hybridization.
Genomic DNA was extracted from WT M. tuberculosis Erdman, the ΔpstA1 mutant, and the indicated pe-ppe deletion mutants as described previously (41). Equivalent amounts of genomic DNA were digested overnight with the restriction enzymes BamHI, KpnI, or SalI. Restricted DNA was separated by electrophoresis on a Tris-acetate-EDTA (TAE) agarose gel and transferred to a Hybond N+ nylon membrane (Amersham). Probes were generated by PCR amplification from M. tuberculosis Erdman genomic DNA using primers 1787F/1787R, 3622F/3622R, or 1790F/1790R (see Table S2 in the supplemental material) and labeled using the ECL direct nucleic acid labeling kit (Amersham) according to the manufacturer's instructions. The blot was blocked with Amersham Gold hybridization buffer prepared with 250 mM NaCl and 5% (wt/vol) blocking reagent for 2.5 h at 42°C. The labeled probe was added and the blot was incubated overnight at 42°C. The membrane was washed according to the manufacturer's instructions, and the probe was detected with ECL detection reagents (Amersham). Autoradiographic films (Kodak) were exposed to the blots for 1 to 5 min and developed on an automatic film processor.
qRT-PCR.
For confirmation of the transcriptional profiling results, bacteria were grown to mid-exponential phase (OD600 of 0.5) in 7H9 broth. To assess the response to Pi starvation, bacteria were grown to mid-exponential phase in 7H9 broth and then washed twice and resuspended to an OD600 of 0.05 in Pi-free 7H9 broth. Cultures were incubated at 37°C, and bacteria were collected for RNA extraction at 0, 24, 48, 72, and 96 h. Cells were collected by centrifugation (4,700 × g, 10 min, 4°C), and RNA was extracted as described previously (33). Equivalent amounts of total RNA were treated with Turbo DNase (Ambion) and reverse transcribed to cDNA with the Transcriptor first-strand cDNA synthesis kit (Roche) using random hexamers for priming and the following cycling conditions: 10 min annealing at 25°C, 60 min extension at 50°C, and 5 min heat inactivation at 85°C. cDNA was stored at −20°C until real-time PCRs were performed. Quantitative real-time reverse transcription-PCRs (qRT-PCRs) were prepared with 2× LightCycler 480 Sybr green I master mix (Roche), 2 μl cDNA, and 0.3 μM primers and were run in absolute quantification mode on a LightCycler 480 (Roche). PCR cycling conditions were 95°C for 10 min; 45 cycles of 95°C for 10 s, 60°C for 20 s, and 72°C for 20 s with data collected once per cycle during the extension phase; and one cycle of 95°C for 5 s, 60°C for 1 min, and 97°C with a ramp rate of 0.11°C/s to generate melting curves for confirmation of product specificity. Mock reactions (no RT) were performed on each sample to confirm the absence of genomic DNA contamination. Cp values were converted to copy numbers using standard curves for each gene. Target cDNA was internally normalized to sigA cDNA (Pi-rich cultures) or 16S cDNA (Pi-starved cultures). Primers for qRT-PCR used in this study are listed in Table S2 in the supplemental material. Primers were designed using Primer Express software (Applied Biosystems) and were tested in standard PCRs using 100 M. tuberculosis genome equivalents as the template.
Cell wall and ROS stress.
Bacteria were grown to mid-exponential phase (OD600 of 0.5) in 7H9 broth, diluted to an OD600 of 0.05 in fresh 7H9 broth, and incubated at 37°C after addition of 0.125% SDS or 3 mM hydrogen peroxide (H2O2). CFU were enumerated at 0 and 24 h by plating serially diluted culture aliquots on 7H10 agar.
Reactive nitrogen species (RNS) stress.
Bacteria were grown to mid-exponential phase (OD600 of 0.5) in 7H9 broth and treated with the NO donor diethylenetriamine-NONOate (DETA-NO; Sigma) as described previously (42), except that complete 7H9 broth was used. DETA-NO was added at 0, 24, and 48 h. CFU were enumerated at 0 and 72 h by plating serially diluted culture aliquots on 7H10 agar.
Acidic pH stress.
Bacteria were grown to mid-exponential phase (OD600 of 0.5) in 7H9 broth, diluted to an OD600 of 0.05 in fresh 7H9 broth at pH 5.5, and incubated at 37°C. CFU were enumerated at 0, 2, 4, and 7 days by plating serially diluted culture aliquots on 7H10 agar.
Ethidium bromide uptake.
The accumulation of ethidium bromide was measured as previously described (43). M. tuberculosis mc27000 and derivative strains were grown to mid-exponential phase (OD600 of 0.6 to 1.0), pelleted by centrifugation at room temperature, and resuspended in ethidium bromide uptake buffer (50 mM KH2PO4 [pH 7.0], 5 mM MgSO4) to an OD600 of 0.5. Cells were preenergized with 25 mM glucose (Sigma) for 5 min. Ethidium bromide was added at a final concentration of 20 μM. Uptake was measured at room temperature using black, flat-bottom 96-well microplates (Corning Costar) and a SpectraMax M5 microplate reader (Molecular Devices, LLC) in top-reading mode with excitation at 530 nm and emission at 590 nm.
Mouse infections.
Male and female NOS2−/− and C57BL/6J mice 6 to 8 weeks of age were purchased from Jackson Laboratory. Irgm1−/− mice (44) were bred under specific-pathogen-free conditions at the EPFL Center of Phenogenomics or the University of Minnesota Research Animal Resources. All mouse strains were on the C57BL/6 background. Mice were infected via the aerosol route with ∼100 CFU using either a custom-built aerosol chamber as described previously (38) or an inhalation exposure system (GlasCol). For experiments using the inhalation exposure system, the nebulizer was loaded with a bacterial suspension in PBS containing 0.05% Tween 80 at an OD600 of 0.01, and mice were exposed to the aerosol for 20 min. Infected mice were euthanized by CO2 overdose. Bacterial CFU were enumerated by plating serially diluted lung homogenates on 7H10 agar and counting colonies after 3 to 4 weeks of incubation at 37°C. The animal protocols used in this study were reviewed and approved by the chief veterinarian of EPFL, the Service de la Consommation et des Affaires Vétérinaires of the Canton of Vaud, the Swiss Office Vétérinaire Fédéral, and the University of Minnesota Institutional Animal Care and Use Committee. All animal experiments were done in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (45) and the Swiss Law for the Protection of Animals.
Statistical analysis.
Student's unpaired t test (two-tailed) was used for pairwise comparisons between WT and mutant strains of M. tuberculosis. The Mantel-Cox log-rank test was used for comparison of Kaplan-Meier plots of mouse survival. P values were calculated using GraphPad Prism 5.0 software (GraphPad Software, Inc.). P < 0.05 was considered significant.
RESULTS
PstA1 negatively regulates expression of specific pe and ppe genes.
Using transcriptional profiling, we previously identified a subset of pe and ppe genes that were differentially expressed by ΔpstA1 bacteria relative to a wild-type (WT) control during growth in Pi-rich medium (Table 1; data are reproduced from reference 33). Each of the differentially expressed pe genes encodes only a PE domain. Each of the overexpressed ppe genes encodes a protein from the PPE-SVP subfamily, while the underexpressed ppe20 gene (rv1387) encodes a PPE-PPW subfamily protein (2). Transcript abundance for each of these genes was restored to the wild-type level by deletion of regX3 (Table 1) (33). These data suggested that some pe and ppe genes are regulated by the Pst/SenX3-RegX3 signal transduction system, and that PstA1 is required to negatively regulate their expression when Pi is readily available. To confirm the transcriptional profiling data, we examined transcript levels of several differentially expressed pe and ppe genes by quantitative real-time RT-PCR (qRT-PCR) in RNA extracted from bacteria grown in Pi-rich medium. Consistent with the transcriptional profiling data, we found that the ppe25, pe19, and ppe65 genes were significantly overexpressed by ΔpstA1 mutant bacteria in a RegX3-dependent manner (Fig. 1A). The fold increase in expression ranged from 4-fold for pe19 to 35-fold for ppe65. The aberrant gene expression pattern characteristic of the ΔpstA1 mutant was restored by complementation of the ΔregX3 deletion in trans (Fig. 1A). These data suggest that the expression of certain pe and ppe genes is controlled by the Pst/SenX3-RegX3 system.
TABLE 1.
pe and ppe genes differentially expressed by M. tuberculosis ΔpstA1 mutanta
| Gene | Rv numbering | Differential gene expression |
|||
|---|---|---|---|---|---|
| ΔpstA1 vs WT |
ΔpstA1 ΔregX3 vs WT |
||||
| Ratio | P value | Ratio | P value | ||
| ppe65 | rv3621 | 12.245 | 0.00438 | 1.004 | 0.987 |
| pe32 | rv3622 | 11.616 | 0.00362 | 0.847 | 0.405 |
| ppe25 | rv1787 | 9.775 | 0.000088 | 1.775 | 0.243 |
| ppe27 | rv1790 | 6.819 | 0.000234 | 1.284 | 0.652 |
| pe18 | rv1788 | 3.625 | 0.000952 | 1.006 | 0.99 |
| pe19b | rv1791 | 4.089 | 0.0645 | 1.260 | 0.706 |
| ppe19 | rv1361 | 3.176 | 0.0207 | 0.752 | 0.251 |
| pe15 | rv1386 | 0.0846 | 0.00111 | 1.364 | 0.705 |
| ppe20 | rv1387 | 0.0797 | 0.0273 | 2.022 | 0.15 |
| pe31 | rv3477 | 0.0237 | 0.00009 | 1.676 | 0.217 |
Expression ratios were determined by microarray analysis using RNA extracted from bacteria grown in Pi-rich 7H9 medium. Data are reproduced from reference 33.
Did not achieve P value cutoff of <0.05.
FIG 1.

Specific pe and ppe genes are regulated by Pst/SenX3-RegX3. (A) RNA was extracted from cultures of M. tuberculosis WT, ΔpstA1, ΔregX3, ΔpstA1ΔregX3, ΔregX3 pNDregX3, and ΔpstA1ΔregX3 pNDregX3 strains grown to mid-exponential phase (OD600 of 0.5) in Pi-rich 7H9 medium. The abundance of ppe25, pe19, and ppe65 transcripts relative to that of sigA was determined by real-time quantitative RT-PCR. Data shown are the means ± standard deviations from three independent experiments. Asterisks indicate statistically significant differences from the WT: *, P < 0.05; **, P < 0.001. (B) RNA was extracted from cultures of M. tuberculosis WT, ΔregX3, and ΔregX3 pNDregX3 strains at the indicated times after shifting to Pi-free 7H9 medium. pe19 transcript abundance relative to that of 16S rRNA was determined by real-time quantitative RT-PCR. Data shown are the means ± standard deviations from three independent experiments. Asterisks indicate statistically significant differences from both the respective 0-h time point and the ΔregX3 mutant: *, P < 0.005; **, P < 0.0005.
We previously demonstrated that transcription of ppe20 is repressed in a RegX3-dependent manner during Pi limitation and is constitutively repressed in the ΔpstA1 mutant (33). In contrast, we predicted that pe19 expression would be induced by Pi limitation in a RegX3-dependent manner. The abundance of the pe19 transcript was increased 6-fold in WT M. tuberculosis within 24 h of Pi limitation. This induction of pe19 transcription was statistically significant and required RegX3 (Fig. 1B). These data confirm that some pe and ppe genes are regulated by the Pst/SenX3-RegX3 system in response to available Pi.
Deletion of the ppe27-pe19 locus suppresses sensitivity to cell wall and reactive oxygen stress.
We previously showed that ΔpstA1 bacteria are hypersensitive to a variety of stress conditions in vitro, and that this stress sensitivity can be attributed to aberrant RegX3-dependent transcription (33). To test if overexpression of specific pe and/or ppe genes is responsible for stress hypersensitivity of the ΔpstA1 mutant, we constructed four deletions of gene pairs or putative operons encoding PE and PPE proteins that were highly overexpressed by ΔpstA1 bacteria (Δppe25-pe19, Δppe25-ppe26, Δppe27-pe19, and Δpe32-ppe65) (Fig. 2D and E). In-frame, unmarked deletions were constructed by two-step homologous recombination in both the WT and ΔpstA1 mutant strain backgrounds and were confirmed by PCR (data not shown) and Southern blotting (Fig. 2A and C). Notably, several of the deletions caused a growth defect in standard Pi-rich 7H9 medium. The Δppe25-pe19 and Δppe27-pe19 deletion mutations caused a significant increase in the doubling time, independent of the strain background in which the deletions were constructed (Table 2). The Δppe25-pe19 and Δppe27-pe19 mutants also formed smaller colonies on 7H10 solid agar medium independent of the strain background (data not shown). The growth defect of the Δppe27-pe19 mutants was only partially complemented by providing the ppe27-pe19 locus in trans (Table 2), suggesting polarity of the Δppe27-pe19 deletion on downstream gene expression. Nevertheless, these data indicate that the ppe27-pe19 locus plays an important role in M. tuberculosis physiology to enable optimal replication in vitro.
FIG 2.
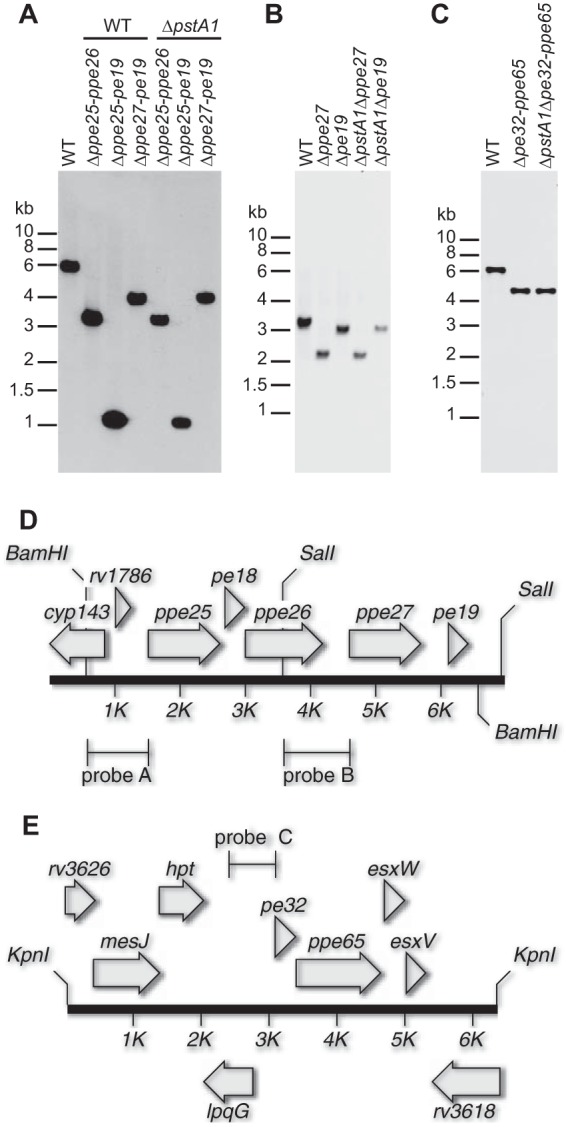
Southern blotting confirms pe-ppe deletions. (A to C) Southern blots. Genomic DNA from pe-ppe deletion strains and the M. tuberculosis wild type (WT) was digested with the indicated restriction enzymes. Bands that hybridized to a gene-specific probe were detected by enhanced chemiluminescence. Positions of molecular size markers are indicated. The pe-ppe deletions are expected to remove the following lengths of DNA sequence: Δppe25-pe19, 4.89 kbp; Δppe25-ppe26, 2.66 kbp; Δppe27-pe19, 1.80 kbp; Δppe27, 1.10 kbp; Δpe19, 0.30 kbp; Δpe32-ppe65, 2.65 kbp. (A) BamHI digest, probe A (primers 1787F/1787R); (B) SalI digest, probe B (primers 1790F/1790R); (C) KpnI digest, probe C (primers 3622F/3622R). (D and E) Maps of the mutated pe-ppe loci. Genes are indicated by gray arrows. Lines indicate locations of BamHI, SalI, and KpnI restriction enzyme sites and probes used for Southern blotting. (D) ppe25-pe19 locus. (E) pe32-ppe65 locus.
TABLE 2.
Doubling times of pe-ppe deletion mutants in Pi-rich 7H9 mediuma
| pe-ppe deletion | Doubling timeb (h), WT background | P value (vs WT) | Doubling timeb (h), ΔpstA1 background | P value (vs ΔpstA1) |
|---|---|---|---|---|
| None | 19.41 ± 1.59 | 19.71 ± 1.70 | ||
| Δpe32-ppe65 | 20.99 ± 0.50 | 0.1314 | 18.92 ± 0.92 | 0.4704 |
| Δppe25-pe19 | 27.63 ± 4.29 | 0.0004 | 27.73 ± 2.19 | <0.0001 |
| Δppe25-ppe26 | 20.87 ± 0.34 | 0.1568 | 19.50 ± 0.88 | 0.8509 |
| Δppe27-pe19 | 28.32 ± 2.55 | <0.0001 | 25.86 ± 2.19 | 0.0005 |
| Δppe27-pe19 pMVppe27-pe19 | 23.25 ± 0.98 | 0.0236 | 21.77 ± 1.13 | 0.1554 |
| Δppe27 | 18.54 ± 0.24 | 0.4018 | 18.15 ± 0.33 | 0.1937 |
| Δpe19 | 22.67 ± 0.87 | 0.003 | 22.95 ± 2.52 | 0.0137 |
| Δpe19 pMV-pe19 | 20.70 ± 0.93 | 0.2210 | 19.65 ± 0.61 | 0.9581 |
Doubling times were determined from optical density measurements taken during the first 72 h of growth after dilution to an OD600 of 0.05 in 7H9 medium.
Means ± standard deviations of doubling times from three independent cultures.
Because ΔpstA1 bacteria exhibit hypersensitivity to either the cell wall-disrupting detergent SDS or the reactive oxygen species H2O2, we first tested if any of the pe-ppe deletions altered resistance to these two stress conditions. Deletion of either pe32-ppe65 or ppe25-ppe26 in the ΔpstA1 mutant had no significant effect on sensitivity to SDS or H2O2 (Fig. 3A). In contrast, the Δppe25-pe19 and Δppe27-pe19 deletions both reversed the hypersensitivity of the ΔpstA1 mutant to SDS and H2O2. The ΔpstA1Δppe25-pe19 and ΔpstA1Δppe27-pe19 mutants exhibited no significant difference in resistance to either SDS or H2O2 compared to the WT control (Fig. 3A). The Δppe25-pe19 and Δppe27-pe19 deletions also significantly increased SDS resistance in the WT strain background, suggesting that basal expression of the ppe27-pe19 locus contributes to sensitivity to cell wall stress (Fig. 3A).
FIG 3.

Hypersensitivity of the ΔpstA1 mutant to cell wall and reactive oxygen stress is suppressed by deletion of pe19. The indicated M. tuberculosis strains were grown to mid-exponential phase (OD600 of 0.5) in 7H9 medium and diluted to an OD600 of 0.05 in fresh 7H9 medium. Sodium dodecyl sulfate (SDS) or hydrogen peroxide (H2O2) was added at 0.125% or 3 mM final concentration, respectively, and cultures were incubated at 37°C with shaking. CFU were enumerated at 0 and 24 h by plating serially diluted cultures on 7H10 agar medium and incubating at 37°C for 3 to 4 weeks. Percent survival was calculated as (CFU poststress)/(CFU prestress) × 100. Results presented are the means ± standard deviations from at least three independent experiments. (A) Deletions of putative pe-ppe operons Δpe32-ppe65, Δppe25-pe19, Δppe25-ppe26, and Δppe27-pe19 in the WT and ΔpstA1 parental strain backgrounds. (B) Individual Δppe27 and Δpe19 mutations in the WT and ΔpstA1 parental strain backgrounds. (C) Complementation of the Δpe19 deletion. Asterisks indicate statistically significant differences from the WT control: *, P < 0.05; **, P < 0.005.
Overexpression of pe19 sensitizes M. tuberculosis to cell wall and oxidative stress.
To further elucidate which genes are involved in sensitivity to cell wall or oxidative stress, we constructed independent in-frame unmarked deletions of the ppe27 and pe19 genes in both WT Erdman and the ΔpstA1 mutant. The Δppe27 and Δpe19 deletions were confirmed by PCR (data not shown) and Southern blotting (Fig. 2B). The Δppe27 mutation had no impact on the growth rate in either the WT or ΔpstA1 mutant backgrounds (Table 2). In contrast, the Δpe19 and ΔpstA1 Δpe19 mutants both had significantly reduced growth rates in standard Pi-rich 7H9 medium (Table 2 and Fig. 4). Both strains harboring the Δpe19 deletion also formed smaller colonies than the parental strain on 7H10 agar medium (data not shown). These growth phenotypes were fully complemented by providing a copy of the pe19 gene on the episomal plasmid pMV261 under the control of the strong constitutive hsp60 promoter (Table 2 and Fig. 4).
FIG 4.

pe19 deletion mutants have a growth defect in liquid culture. M. tuberculosis WT, Δpe19, Δpe19 pMVpe19, ΔpstA1, ΔpstA1 Δpe19, and ΔpstA1 Δpe19 pMVpe19 strains were grown to mid-exponential phase (OD600 of 0.5) in 7H9 medium, diluted to an OD600 of 0.05 in fresh 7H9 medium, and incubated with shaking at 37°C. Aliquots of each culture were taken at the indicated times for measurement of the OD600. Results are averages ± standard deviations from triplicate cultures and are representative of two independent experiments.
The Δppe27 and Δpe19 strains were tested for resistance to both SDS and H2O2. The Δppe27 mutation did not alter sensitivity to either stress condition (Fig. 3B). In contrast, the Δpe19 mutation fully suppressed sensitivity of the ΔpstA1 mutant to both SDS and H2O2 (Fig. 3B). Complementation of the Δpe19 mutation with the pMVpe19 plasmid restored the hypersensitivity to SDS and H2O2 characteristic of ΔpstA1 bacteria, confirming that pe19 is responsible for these stress sensitivity phenotypes (Fig. 3C). These data suggest that overexpression of pe19 causes sensitivity of the ΔpstA1 mutant to SDS and H2O2 stress. The pMVpe19 plasmid also caused increased sensitivity to SDS and H2O2 in both the WT and Δpe19 strain backgrounds, although the change in H2O2 sensitivity did not achieve statistical significance (Fig. 3C). To test if pe19 is overexpressed from the pMVpe19 plasmid, we examined the level of pe19 transcript by qRT-PCR in strains harboring the complementation plasmid. Although the difference was not statistically significant, the pe19 gene was expressed at a 2.5-fold higher level in the Δpe19 pMVpe19 strain than in the WT control (Fig. 5A). These data suggest that increased expression of pe19 is sufficient to cause hypersensitivity to cell wall and oxidative stress.
FIG 5.

Gene expression in pe19 deletion mutant and complemented strains. RNA was extracted from cultures of M. tuberculosis WT, Δpe19, Δpe19 pMVpe19, ΔpstA1, ΔpstA1 Δpe19, and ΔpstA1 Δpe19 pMVpe19 strains grown to mid-exponential phase (OD600 of 0.5) in Pi-rich 7H9 medium. The abundance of pe19 (A) and udgA, mgtA, and rv0784 (B) transcripts relative to that of sigA was determined by real-time quantitative RT-PCR. Data shown are the means ± standard deviations from three independent experiments. ND indicates no detectable transcript. Asterisks indicate statistically significant differences from the WT: *, P < 0.01; **, P < 0.005.
Although the data suggest that PE19 directly mediates changes in stress sensitivity, an alternative possibility is that PE19 normally participates in the Pst/SenX3-RegX3 signal transduction pathway. In this scenario, deletion of pe19 would cause global changes in the expression of genes that are controlled by the Pst/SenX3-RegX3 system. We tested the expression of several genes that we previously showed were overexpressed by ΔpstA1 bacteria in a RegX3-dependent manner (33). Deletion of pe19 did not alter transcription of any of these known targets of Pst/SenX3-RegX3 regulation (Fig. 5B). There were no significant differences in transcript levels between the WT and Δpe19 strains or between the ΔpstA1 and ΔpstA1 Δpe19 strains. These data suggest that PE19 is not involved in signal transduction and that the phenotypes caused by overexpression of pe19 are due to the function of PE19 itself.
Overexpression of pe19 does not cause sensitivity to acid or nitrosative stress.
We previously demonstrated that ΔpstA1 bacteria are more sensitive to nitrosative stress, and this stress sensitivity can be reversed by deletion of regX3 (33). These data suggested that a RegX3-dependent factor causes sensitivity to reactive nitrogen species. To test whether sensitivity to nitrosative stress also might be caused by overexpression of pe19, we analyzed the Δpe19 deletion mutants for resistance to the nitric oxide donor DETA-NO. The Δpe19 deletion did not cause any significant change in sensitivity to DETA-NO (Fig. 6A). These data suggest that sensitivity to nitrosative stress is caused by a RegX3-dependent factor other than PE19.
FIG 6.
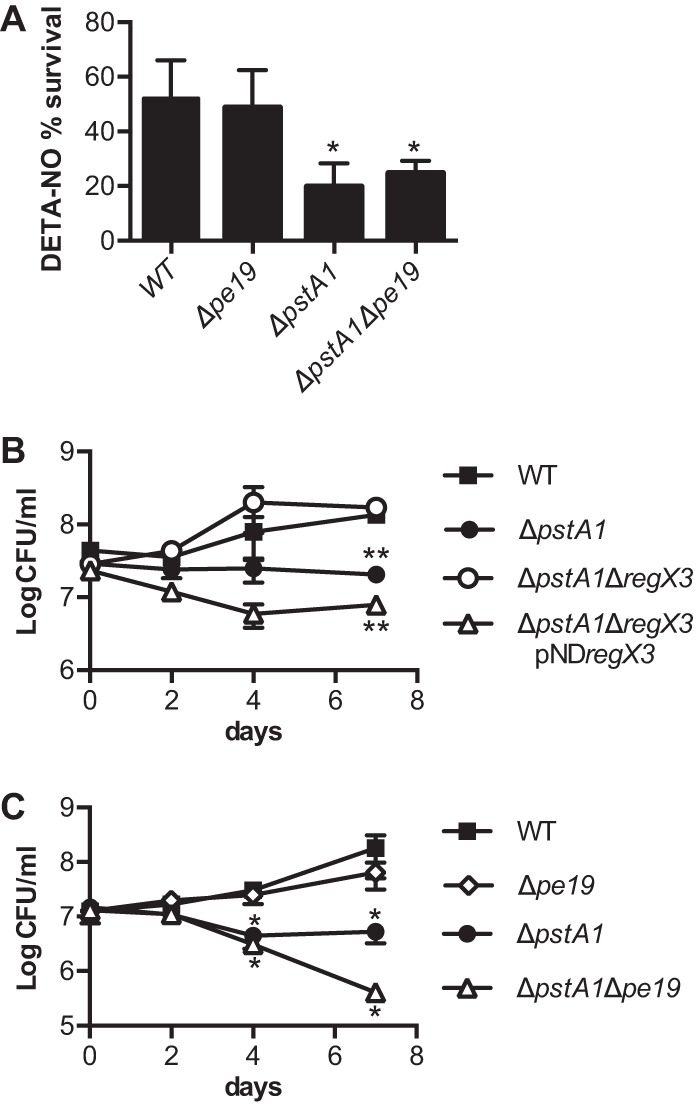
pe19 deletion does not suppress sensitivity of the ΔpstA1 mutant to reactive nitrogen or acidic pH stress. The indicated M. tuberculosis strains were grown to mid-exponential phase (OD600 of 0.5) in 7H9 medium before being subjected to reactive nitrogen or acidic pH stress conditions. CFU were enumerated by plating serially diluted cultures on 7H10 agar medium and incubating at 37°C for 3 to 4 weeks. Results presented are the averages ± standard deviations from triplicate cultures and are representative of two independent experiments. (A) Reactive nitrogen stress. Cultures were diluted to an OD600 of 0.05 in 7H9 medium and incubated at 37°C with shaking. DETA-NO was added to cultures at 0.2 mM final concentration at 0, 24, and 48 h. CFU were enumerated at 0 and 72 h. Percent survival was calculated at (CFU poststress)/(CFU prestress) × 100. (B and C) The indicated strains were diluted to an OD600 of 0.05 in 7H9 medium acidified to pH 5.5. CFU were enumerated at 0, 2, 4, and 7 days. Asterisks indicate statistically significant differences from the WT control: *, P < 0.05; **, P < 0.0005.
In preliminary experiments, we tested sensitivity to nitrosative stress using an acidified nitrate medium (46). Control experiments revealed that ΔpstA1 bacteria are hypersensitive to mild acid, pH 5.5 (Fig. 6B). Survival of the ΔpstA1 mutant was significantly reduced after 7 days of culture in 7H9, pH 5.5, compared to that of the WT control. Acid sensitivity of the ΔpstA1 mutant was attributable to aberrant gene expression mediated by RegX3, since the ΔpstA1 ΔregX3 mutant was as resistant to acidified 7H9 medium as the WT control (Fig. 6B). Complementation of the regX3 deletion by the introduction of plasmid-encoded regX3 restored acid sensitivity (Fig. 6B).
To determine if PE19 is the RegX3-dependent factor responsible for acid sensitivity, we examined sensitivity of the Δpe19 mutants to acidified 7H9 medium. The Δpe19 mutation did not significantly alter acid resistance in the WT background (Fig. 6C). In contrast, the ΔpstA1 Δpe19 mutant was significantly more sensitive than the ΔpstA1 parental strain to acidified medium (P = 0.0138) (Fig. 6C). These data suggest that a RegX3-regulated factor(s) other than PE19 causes acid sensitivity.
Sensitivity to acid is not mediated by other RegX3-regulated PE or PPE proteins.
Since acid sensitivity of the ΔpstA1 mutant was not attributable to the overexpression of pe19, we explored the possibility that the overexpression of other PE and PPE proteins contribute to acid sensitivity. We tested sensitivity of the ΔpstA1 Δppe25-ppe26 and ΔpstA1 Δpe32-ppe65 mutants to 7H9, pH 5.5. Both of these mutants remained significantly hypersensitive to acidic pH (Fig. 7A and B). Neither of these pe-ppe deletions significantly impacted replication in acidified medium in the WT background (Fig. 7A and B). These data suggest that RegX3-regulated factor(s) other than the PE and PPE proteins that we have deleted cause sensitivity of the ΔpstA1 mutant to mild acid.
FIG 7.
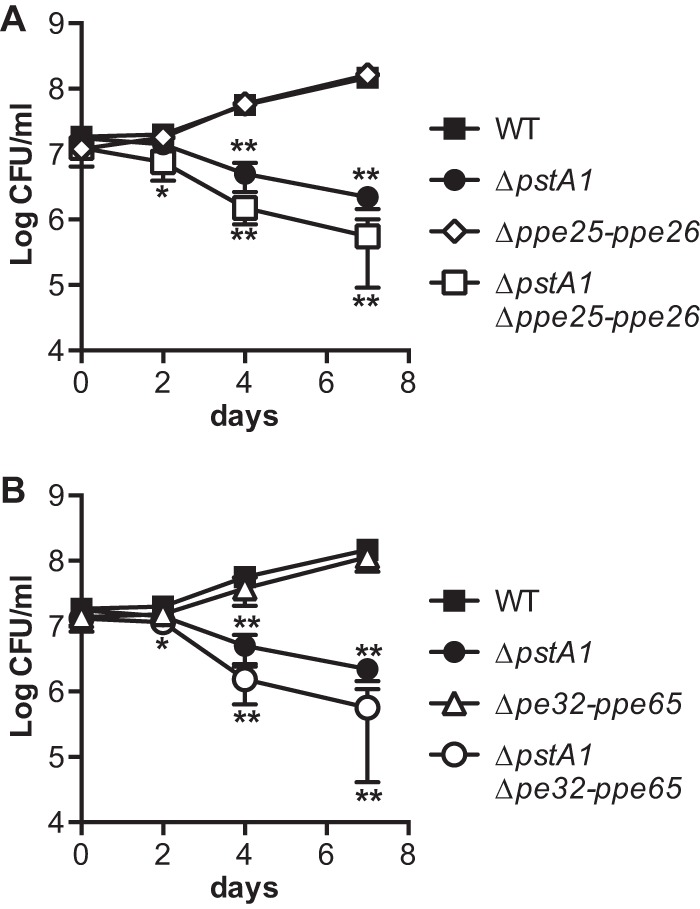
Δppe25-ppe26 and Δpe32-ppe65 deletions do not suppress sensitivity of the ΔpstA1 mutant to acidic pH stress. The indicated M. tuberculosis strains were grown to mid-exponential phase (OD600 of 0.5) in 7H9 medium, diluted to an OD600 of 0.05 in 7H9 medium acidified to pH 5.5, and incubated at 37°C with shaking. CFU were enumerated at 0, 2, 4, and 7 days by plating serially diluted cultures on 7H10 agar medium and incubating at 37°C for 3 to 4 weeks. Results presented are the averages ± standard deviations from three independent experiments. Asterisks indicate statistically significant differences from the WT control: *, P < 0.05; **, P < 0.001.
Overexpression of pe19 causes increased cell envelope permeability.
Since some PE and PPE proteins are known to localize to the mycobacterial cell wall (3, 7, 8, 34, 36), we predicted that overexpression of pe19 contributes to stress sensitivity by altering the permeability of the cell envelope. To test this hypothesis, we used an ethidium bromide uptake assay which measures the increase in fluorescence of ethidium bromide after uptake and binding to bacterial nucleic acids (43). To facilitate performing these assays, we reconstructed the ΔpstA1 and Δpe19 mutations in the attenuated M. tuberculosis strain mc27000 (H37Rv, ΔRD1, ΔpanCD [37]). In the mc27000 background. The ΔpstA1 mutation caused increased sensitivity to both SDS and H2O2, while the ΔpstA1 Δpe19 double mutant was as resistant to these stress conditions as the WT mc27000 control (see Fig. S1 in the supplemental material). The Δpe19 deletion also caused a significant decrease in growth rate of the mc27000 strain (see Table S4). These data indicate that pe19 is required for optimal replication and that pe19 overexpression causes sensitivity to cell wall and oxidative stress in the attenuated mc27000 background, like in the virulent Erdman strain.
In ethidium bromide uptake assays, we observed a statistically significant 2-fold increase in the uptake rate for the ΔpstA1 mutant (1.165 ± 0.1225 RFU/min; P = 0.002) compared with that of the WT mc27000 control (0.57 ± 0.0773 RFU/min) (Fig. 8). The Δpe19 mutant consistently exhibited a slower initial rate of ethidium bromide uptake, although the steady-state uptake rate (0.523 ± 0.0835 RFU/min) was only modestly slower than that of the WT mc27000 control (Fig. 8). The ΔpstA1 Δpe19 double mutant exhibited a rate of ethidium bromide uptake (0.489 ± 0.147 RFU/min) similar to that of the WT and Δpe19 strains (Fig. 8). These data indicate that overexpression of pe19 increases permeability of the mycobacterial envelope in the ΔpstA1 mutant.
FIG 8.
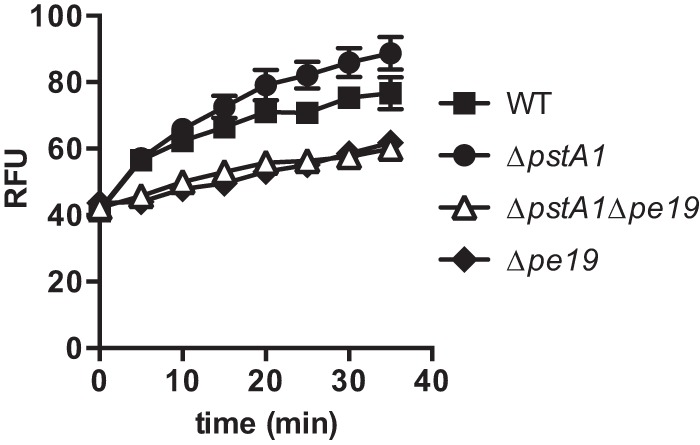
Increased envelope permeability of the ΔpstA1 mutant is caused by pe19. The indicated M. tuberculosis strains in the mc27000 background were incubated with 20 μM ethidium bromide, and uptake rates were determined by measuring emission at 590 nm upon excitation at 530 nm. Data presented are expressed as relative fluorescence units (RFU) and are the means ± standard deviations of triplicate samples from a single experiment. Data are representative of three independent experiments. Uptake rates presented in the text were determined using data between the 5- and 30-min time points and are the means ± standard deviations from three independent experiments.
To determine if PE19 directly influences cell envelope permeability by localizing to the cell wall, we generated versions of PE19 that were tagged at the C terminus with either the His6 or HA epitope. It was necessary to construct epitope-tagged PE19 for these experiments due to the high similarly of PE19 to other PE proteins (89.9% identical, 91.9% similar to PE18) that would preclude the specific detection of PE19 with antibodies to either the full-length PE19 or predicted antigenic peptides. C-terminal HA epitope tags previously have been used to determine the localization of the PE domain from other PE proteins (9, 35). The PE19-His6 and PE19-HA epitope-tagged proteins were functional, since they could complement the H2O2 resistance phenotypes of the ΔpstA1 Δpe19 mutant as effectively as untagged PE19 (see Fig. S2 in the supplemental material). However, we were unable to detect either PE19-His6 or PE19-HA in Western blot experiments performed on the whole-cell lysate and secreted protein fractions from these strains (data not shown).
Overexpression of pe19 causes attenuation of the ΔpstA1 mutant in NOS2−/− mice.
We previously demonstrated that ΔpstA1 bacteria are sensitive to host gamma interferon (IFN-γ)-dependent adaptive immune responses. The ΔpstA1 mutant is attenuated for replication and virulence in mice lacking either the IFN-γ-regulated GTPase Irgm1 or the IFN-γ-inducible nitric oxide synthase NOS2 (33). The attenuation of ΔpstA1 bacteria in NOS2−/− mice could be partially reversed by deletion of regX3, suggesting that the aberrant expression of RegX3-dependent gene(s) causes sensitivity to host immune responses (33). We sought to determine whether the overexpression of pe19 causes the in vivo attenuation of the ΔpstA1 mutant. Because we previously demonstrated that M. tuberculosis can become attenuated due to spontaneous mutations that eliminate the production of the complex cell wall lipid phthiocerol dimycocerosate (PDIM) (38), we first confirmed that the pe19 deletion did not alter PDIM synthesis (see Fig. S3 in the supplemental material). NOS2−/− mice were infected by the aerosol route with ∼100 CFU of the ΔpstA1 Δpe19 mutant, WT Erdman, or the ΔpstA1 mutant. Deletion of pe19 partially restored the ability of the ΔpstA1 mutant to replicate in the lungs of NOS2−/− mice (Fig. 9A). Bacterial loads were significantly higher in the lungs of ΔpstA1 Δpe19-infected mice than of ΔpstA1-infected mice at both 4 weeks (P = 0.0465) and 6 weeks (P = 0.0044) postinfection (Fig. 9A). Viable CFU recovered from the lungs of ΔpstA1 Δpe19-infected mice were not significantly different from those of the WT control at any time point. These data suggest that the overexpression of pe19 sensitizes the ΔpstA1 mutant to a host immune response that is independent of nitric oxide production by NOS2.
FIG 9.

Deletion of pe19 partially suppresses the replication defect of the ΔpstA1 mutant in NOS2−/− mice. NOS2−/− (A), Irgm1−/− (B), or C57BL/6J (C) mice were infected by the aerosol route with ∼100 CFU of the M. tuberculosis WT, ΔpstA1, ΔpstA1 Δpe19, or Δpe19 strain. Groups of infected mice (n = 4) were euthanized at the indicated time points. Bacterial CFU were enumerated by plating lung homogenates on 7H10 agar and incubating for 3 to 4 weeks at 37°C. Symbols represent means, and error bars indicate standard errors of the means. Results for the ΔpstA1 Δpe19 mutant are from a single experiment and are representative of two independent experiments. All other results are from a single experiment. Asterisks indicate statistically significant differences between ΔpstA1 and ΔpstA1 Δpe19 mutants (*, P < 0.05; **, P < 0.005) (A), between both the ΔpstA1 and ΔpstA1 Δpe19 mutants compared to the WT (*, P < 0.05; **, P < 0.007) (B), and between Δpe19 and WT strains (*, P < 0.05; **, P < 0.005; ***, P < 0.001) (C).
We previously observed that the deletion of regX3 did not suppress the replication or virulence defects of the ΔpstA1 mutant in Irgm1−/− mice (33). However, we were unable to rule out the possibility that a RegX3-regulated factor(s) contributes to these replication and virulence defects, because a ΔregX3 mutant was similarly attenuated (33). Since the Δpe19 deletion partially reversed the replication defect of the ΔpstA1 mutant in NOS2−/− mice, we tested if pe19 also was responsible for the attenuation of the ΔpstA1 mutant in Irgm1−/− mice. We infected Irgm1−/− mice by the aerosol route with ∼100 CFU of the ΔpstA1 Δpe19 mutant, WT Erdman, or the ΔpstA1 mutant. The ΔpstA1 Δpe19 mutant remained significantly attenuated for replication in the lungs of Irgm1−/− mice (Fig. 9B). Viable CFU recovered from the lungs of Irgm1−/− mice infected with the ΔpstA1 Δpe19 mutant were significantly reduced compared to those of the WT control at 2, 4, and 6 weeks postinfection (Fig. 9B). There was no significant difference in CFU recovered from Irgm1−/− mice infected with the ΔpstA1 or ΔpstA1 Δpe19 mutant at any time point. The ΔpstA1 Δpe19 mutant also was significantly attenuated for virulence in Irgm1−/− mice. While Irgm1−/− mice infected with 100 CFU of WT Erdman succumb rapidly to the infection, with a mean survival time of 38.5 days (33), all 8 mice infected with the ΔpstA1 Δpe19 mutant survived to 16 weeks postinfection, when the experiment was terminated (P < 0.0001) (data not shown). To determine if the Δpe19 mutation causes attenuation in Irgm1−/− mice, we infected Irgm1−/− mice with the Δpe19 mutant. The Δpe19 mutant replicated extensively in the lungs of Irgm1−/− mice, with kinetics similar to those of the WT control (Fig. 9B). Bacterial burden in the lungs of Δpe19-infected Irgm1−/− mice was not significantly different from that of the WT control at either the 4- or 6-week time point (Fig. 9B). These data suggest that a factor or factors other than the overexpression of pe19 cause attenuation of the ΔpstA1 mutant in Irgm1−/− mice.
Finally, we examined if PE19 is important for the replication or persistence of M. tuberculosis in wild-type mice. C57BL/6 mice were infected by the aerosol route with ∼100 CFU of WT Erdman or ∼500 CFU of the Δpe19 mutant. Despite a higher initial inoculum, the Δpe19 mutant exhibited a significant replication defect in the lungs over the first 2 weeks of infection (Fig. 9C). After 4 weeks of infection, the number of CFU recovered from the lungs of Δpe19-infected mice began to decline and remained significantly different from that of the WT control (Fig. 9C). The phenotype of the Δpe19 mutant in C57BL/6 mice strongly resembled the phenotypes of both the ΔregX3 and ΔpstA1 mutants, which also exhibited a persistence defect during the chronic phase of infection (33). These data indicate that PE19 is important for replication and persistence in the lungs of an immunocompetent mammalian host.
DISCUSSION
Although the PE and PPE proteins represent a large fraction of the coding capacity of the M. tuberculosis genome, their functions in mycobacterial physiology and virulence remain poorly understood. Since many PE and PPE proteins that have been characterized to date are localized to the cell wall (3, 7, 8, 34–36), we predicted that the aberrant overexpression of these proteins would sensitize M. tuberculosis to stress. We demonstrate that the overexpression of the PE protein PE19, which consists of only a 99-amino-acid PE domain, causes sensitivity to specific cell wall and oxidative stress conditions in vitro and to NOS2-independent host immune responses in vivo. These phenotypes likely are due to a general increase in the permeability of the mycobacterial envelope upon PE19 overexpression that we observed by ethidium bromide uptake assays. We further show that PE19 is necessary for optimal replication both in liquid culture and in the lungs of immunocompetent mice. Together, these data suggest that PE19 has an important and nonredundant role in M. tuberculosis physiology and virulence. To our knowledge, this is the first direct association of specific in vitro growth, stress sensitivity, and virulence phenotypes with the expression of a single PE protein consisting of only the PE domain in M. tuberculosis.
Our previous work demonstrated that an M. tuberculosis ΔpstA1 mutant is sensitive to a variety of stress conditions in vitro, including the cell wall-disrupting detergent SDS and the reactive oxygen species H2O2, due to constitutive activation of the RegX3 DNA-binding response regulator (33). These data suggested that the constitutive expression of a RegX3-regulated factor or factors causes sensitivity to stress. Because the pleiotropic stress sensitivity of the ΔpstA1 mutant was reminiscent of M. tuberculosis strains with cell wall defects (47), we targeted the RegX3-regulated PE and PPE proteins for our analysis. By deleting potential pe-ppe gene pairs or operons and ultimately making single gene deletions, we demonstrate that the overexpression of a single pe gene, pe19, is responsible for the sensitivity of the ΔpstA1 mutant to both SDS and H2O2. The overexpression of pe19 also is sufficient to cause sensitivity to these two stress conditions in WT M. tuberculosis. It is likely that the increased sensitivity to stress caused by pe19 overexpression is due to an increase in the permeability of the mycobacterial envelope, which we observed using ethidium bromide uptake assays. Although the detergent SDS and the reactive oxygen species H2O2 are quite different stressors that are predicted to have different effects on the cell, it is possible that the general increased permeability of the M. tuberculosis envelope in the ΔpstA1 mutant allows increased penetration of these compounds. Indeed, M. tuberculosis mutant strains with predicted defects in cell wall biogenesis were similarly sensitive to both SDS and H2O2 in vitro (47), suggesting that alterations to the cell wall can influence the ability to resist both of these stressors.
The overexpression of pe19 also contributes to the sensitivity of the ΔpstA1 mutant to host immune responses in vivo, since the deletion of pe19 partially restored the ability to replicate in the lungs of NOS2−/− mice. This suggests that the overexpression of PE19 causes sensitivity to an immune response other than nitric oxide produced by NOS2. It is possible that the increased expression of PE19 sensitizes ΔpstA1 mutant bacteria to reactive oxygen species produced by the phagocyte oxidase (phox), since the overexpression of pe19 caused sensitivity to H2O2 in vitro. However, pe19 is not the only factor that contributes to the attenuation of the ΔpstA1 mutant, since the ΔpstA1 Δpe19 double mutant is severely attenuated for replication in the lungs and virulence in Irgm1−/− mice. The ΔpstA1 Δpe19 bacteria might remain sensitive to either phagosome acidification or nitric oxide produced by NOS2 in Irgm1−/− mice, since the ΔpstA1 Δpe19 mutant remained hypersensitive to both acid and nitrosative stress in vitro. Together, our data indicate that the ΔpstA1 mutant is attenuated due to pleiotropic changes in gene expression caused by the constitutive activation of RegX3, of which PE19 overexpression is only one part. The aberrant expression of other RegX3-regulated factors likely contributes to sensitivity to acid, nitrosative stress, and host immunity.
In addition to causing a general alteration in the permeability of the M. tuberculosis envelope, PE19 also is necessary for optimal replication both in vitro and in vivo in the lungs of wild-type mice. PE19 may enhance the permeability of the envelope to specific nutrients that are necessary for M. tuberculosis replication in vitro and in vivo. However, the Δpe19 mutants had a relatively modest decrease in growth rate compared with that of either the Δppe27-pe19 or Δppe25-pe19 mutant. We were not able to fully complement the growth defect of the Δppe27-pe19 mutant by providing the ppe27-pe19 locus in trans, suggesting the polarity of this deletion on the expression of other genes. A large 426-bp intergenic region between ppe27 and pe19 that contains multiple transcription start sites (48) also is deleted in the Δppe27-pe19 mutant. We have evidence that this intergenic region contains a RegX3-dependent promoter that drives the expression of genes 3′ to pe19, which include conserved components and secreted substrates of the specialized type VII protein secretion system ESX-5 (S. R. Elliott and A. D. Tischler, unpublished data). We speculate that the more substantial growth defect of the Δppe25-pe19 and Δppe27-pe19 mutants is due to the decreased production of ESX-5 core components, which are essential for growth in vitro (49, 50).
Our data suggest that PE19 has a nonredundant function in M. tuberculosis physiology. This is surprising, since PE19 has high similarity to the PE18 protein that also is encoded by the ppe25-pe19 locus (89.9% identical, 91.9% similar). However, the deletion of pe18 (in the Δppe25-ppe26 mutant) did not influence either the growth rate of M. tuberculosis or the sensitivity of the ΔpstA1 mutant to stress. PE proteins are thought to form heterodimeric complexes with a partner PPE protein based on the crystal structure of the PE/PPE pair PE25 and PPE41 (51). It is possible that the sequence differences between PE19 and PE18 enable these proteins to form heterodimers with different PPE partner proteins that have different functions. However, it is unclear which PPE proteins might interact with PE19 to mediate its functions. Many pe genes are located adjacent to and coexpressed with a ppe gene (2, 52, 53). These co-operonic pe and ppe genes are assumed to form heterodimeric complexes like PE25/PPE41. However, we were unable to recapitulate the phenotypes associated with the deletion of pe19 by deleting the ppe genes located in the ppe25-pe19 locus. It is possible that PE19 can interact with several different PPE proteins to carry out its function. In this scenario, it would be necessary to delete all of the ppe genes that encode PE19 partners to observe changes in growth rate or stress sensitivity similar to those caused by the deletion of pe19.
The reduced growth rate of Δpe19 mutant bacteria and association of PE19 overexpression with sensitivity to cell wall stress and increased cell envelope permeability suggest that PE19 is localized to the cell wall and, along with its potential PPE partner(s), influences cell wall architecture or permeability. A recent structural study of EspB, an M. tuberculosis protein secreted by the ESX-1 type VII secretion system, revealed that this protein adopts a conformation similar to that of the PE25/PPE41 structure and forms ring-shaped multimers (54). PE19 and its PPE partner(s) may form similar multimers that localize to the cell wall and influence its permeability. Since pe19 expression is regulated by the Pi starvation-responsive Pst/SenX3-RegX3 signal transduction system, we speculate that PE19 is specifically involved in permeabilizing the cell wall to allow the uptake of nutrients that contain Pi. We attempted to determine the localization of PE19 by expressing C-terminal epitope-tagged versions of the protein. Although these epitope-tagged PE19 proteins apparently were functional, since they could complement stress sensitivity phenotypes associated with the Δpe19 mutation, we were unable to detect PE19 in Western blots. It is possible that PE19 is produced at a low level or is unstable, such that the protein is not present in sufficient quantities for detection by Western blotting. Alternatively, PE19 may be proteolytically processed at the C terminus, which would remove the C-terminal epitope tags we engineered. It may be necessary to use more sensitive or larger epitope tags (e.g., green fluorescent protein) or to N-terminally tag PE19 to determine its localization. Our future work will focus on further biochemical characterization of PE19 to determine its localization, interacting partners, and potential functions in nutrient acquisition.
Supplementary Material
ACKNOWLEDGMENTS
We thank Manisha Lotlikar and Laetitia Martin for expert technical assistance with animal experiments, John Hartzheim for constructing pMVpe19, Greg Taylor for providing Irgm1−/− mice, and Bill Jacobs for providing the mc27000 strain.
This work was supported by an Irvington Postdoctoral Fellowship of the Cancer Research Institute (A.D.T.), Swiss National Science Foundation grant 310030_156945 (J.D.M.), and institutional start-up funds from the University of Minnesota (A.D.T.).
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00942-15.
REFERENCES
- 1.Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CEI, Takala F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, Barrell BG. 1998. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 2.Gey van Pittius NC, Sampson SL, Lee H, Kim Y, van Helden PD, Warren RM. 2006. Evolution and expansion of the Mycobacterium tuberculosis PE and PPE multigene families and their association with the duplication of the ESAT-6 (esx) gene cluster regions. BMC Evol Biol 6:95. doi: 10.1186/1471-2148-6-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delogu G, Pusceddu C, Bua A, Fadda G, Brennan MJ, Zanetti S. 2004. Rv1818c-encoded PE_PGRS protein of Mycobacterium tuberculosis is surface exposed and influences bacterial cell structure. Mol Microbiol 52:725–733. doi: 10.1111/j.1365-2958.2004.04007.x. [DOI] [PubMed] [Google Scholar]
- 4.Abdallah AM, Verboom T, Hannes F, Safi M, Strong M, Eisenberg D, Musters RJ, Vandenbrouck-Grauls CM, Appelmelk BJ, Luirink J, Bitter W. 2006. A specific secretion system mediates PPE41 transport in pathogenic mycobacteria. Mol Microbiol 62:667–679. doi: 10.1111/j.1365-2958.2006.05409.x. [DOI] [PubMed] [Google Scholar]
- 5.Abdallah AM, Verboom T, Weerdenburg EM, Gey van Pittius NC, Mahasha PW, Jimenez C, Parra M, Cadieux N, Brennan MJ, Appelmelk BJ, Bitter W. 2009. PPE and PE_PGRS proteins of Mycobacterium marinum are transported via the type VII secretion system ESX-5. Mol Microbiol 73:329–340. doi: 10.1111/j.1365-2958.2009.06783.x. [DOI] [PubMed] [Google Scholar]
- 6.Bottai D, Di Luca M, Majlessi L, Frigui W, Simeone R, Sayes F, Bitter W, Brennan MJ, Leclerc C, Batoni G, Campa M, Brosch R, Esin S. 2012. Disruption of the ESX-5 system of Mycobacterium tuberculosis causes loss of PPE protein secretion, reduction of cell wall integrity and strong attenuation. Mol Microbiol 83:1195–1209. doi: 10.1111/j.1365-2958.2012.08001.x. [DOI] [PubMed] [Google Scholar]
- 7.Cascioferro A, Daleke MH, Ventura M, Dona V, Delogu G, Palú G, Bitter W, Manganelli R. 2011. Functional dissection of the PE domain responsible for translocation of PE_PGRS33 across the mycobacterial cell wall. PLoS One 6:e27713. doi: 10.1371/journal.pone.0027713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Daleke MH, Cascioferro A, de Punder K, Ummels R, Abdallah AM, van der Wel N, Peters PJ, Luirink J, Manganelli R, Bitter W. 2011. Conserved Pro-Glu (PE) and Pro-Pro-Glu (PPE) protein domains target LipY lipases of pathogenic mycobacteria to the cell surface via the ESX-5 pathway. J Biol Chem 286:19024–19034. doi: 10.1074/jbc.M110.204966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daleke MH, Ummels R, Bawono P, Heringa J, Vandenbrouck-Grauls CM, Luirink J, Bitter W. 2012. General secretion signal for the mycobacterial type VII secretion pathway. Proc Natl Acad Sci U S A 109:11342–11347. doi: 10.1073/pnas.1119453109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Talarico S, Cave MD, Marrs CF, Foxman B, Zhang L, Yang Z. 2005. Variation of the Mycobacterium tuberculosis PE_PGRS33 gene among clinical isolates. J Clin Microbiol 43:4954–4960. doi: 10.1128/JCM.43.10.4954-4960.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Talarico S, Zhang L, Marrs CF, Foxman B, Cave MD, Brennan MJ, Yang Z. 2008. Mycobacterium tuberculosis PE_PGRS16 and PE_PGRS26 genetic polymorphism among clinical isolates. Tuberculosis (Edinb) 88:283–294. doi: 10.1016/j.tube.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karboul A, Mazza A, Gey van Pittius NC, Ho JL, Brousseau R, Mardassi H. 2008. Frequent homologous recombination events in Mycobacterium tuberculosis PE/PPE multigene families: potential role in antigenic variability. J Bacteriol 190:7838–7846. doi: 10.1128/JB.00827-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, Flynn JL, Fortune SM. 2011. Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat Genet 43:482–486. doi: 10.1038/ng.811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McEvoy CRE, Cloete R, Müller B, Schürch AC, van Helden PD, Gagneux S, Warren RM, Gey van Pittius NC. 2012. Comparative analysis of Mycobacterium tuberculosis pe and ppe genes reveals high sequence variation and an apparent absence of selective constraints. PLoS One 7:e30593. doi: 10.1371/journal.pone.0030593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vordermeier HM, Hewinson RG, Wilkinson RJ, Wilkinson KA, Gideon HP, Young DB, Sampson SL. 2012. Conserved immune recognition hierarchy of mycobacterial PE/PPE proteins during infection in natural hosts. PLoS One 7:e40890. doi: 10.1371/journal.pone.0040890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sayes F, Sun L, Di Luca M, Simeone R, Degaiffier N, Fiette L, Esin S, Brosch R, Bottai D, Leclerc C, Majlessi L. 2012. Strong immunogenicity and cross-reactivity of Mycobacterium tuberculosis ESX-5 type VII secretion-encoded PE-PPE proteins predicts vaccine potential. Cell Host Microbe 11:352–363. doi: 10.1016/j.chom.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Arlehamn CSL, Gerasimova A, Mele F, Henderson R, Swann J, Greenbaum JA, Kim Y, Sidney J, James EA, Taplitz R, McKinney DM, Kwok WW, Grey H, Sallusto F, Peters B, Sette A. 2013. Memory T cells in latent Mycobacterium tuberculosis infection are directed against three antigenic islands and are largely contained in a CXCR3+CCR6+ Th1 subset. PLoS Pathog 9:e1003130. doi: 10.1371/journal.ppat.1003130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sassetti CM, Boyd DH, Rubin EJ. 2003. Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 19.Sassetti CM, Rubin EJ. 2003. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A 100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, Sassetti CM. 2011. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iantomasi R, Sali M, Cascioferro A, Palucci I, Zumbo A, Soldini S, Rocca S, Greco E, Maulucci G, De Spirito M, Fraziano M, Fadda G, Manganelli R, Delogu G. 2012. PE_PGRS30 is required for full virulence of Mycobacterium tuberculosis. Cell Microbiol 14:356–367. doi: 10.1111/j.1462-5822.2011.01721.x. [DOI] [PubMed] [Google Scholar]
- 22.Stewart GR, Patel J, Robertson BD, Rae A, Young DB. 2005. Mycobacterial mutants with defective control of phagosomal acidification. PLoS Pathog 1:269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brodin P, Poquet Y, Levillain F, Peguillet I, Lorrouy-Maumus G, Gilleron M, Ewann F, Christophe T, Jang J, Jang MS, Park SJ, Rauzier J, Carralot J-P, Shrimpton R, Genovesio A, Gonzalo-Asenio JA, Puzo G, Martin C, Brosch R, Stewart GR, Gicquel B, Neyrolles O. 2010. High content phenotypic cell-based visual screen identifies Mycobacterium tuberculosis acyltrehalose-containing glycolipids involved in phagosome remodeling. PLoS Pathog 9:e1001100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deb C, Daniel J, Sirakova TD, Abomoelak B, Dubey VS, Kolattukudy PE. 2006. A novel lipase belonging to the hormone-sensitive lipase family induced under starvation to utilize stored triacylglycerol in Mycobacterium tuberculosis. J Biol Chem 281:3866–3875. doi: 10.1074/jbc.M505556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mishra KC, de Chastellier C, Narayana Y, Bifani P, Brown AK, Besra GS, Katoch VM, Joshi B, Balaji KN, Kremer L. 2008. Functional role of the PE domain and immunogenicity of the Mycobacterium tuberculosis triacylglycerol hydrolase LipY. Infect Immun 76:127–140. doi: 10.1128/IAI.00410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chaturvedi R, Bansal K, Narayana Y, Kapoor N, Sukumar N, Togarsimalemath SK, Chandra N, Mishra S, Ajitkumar P, Joshi B, Katoch VM, Patil SA, Balaji KN. 2010. The multifunctional PE_PGRS11 protein from Mycobacterium tuberculosis plays a role in regulating resistance to oxidative stress. J Biol Chem 285:30389–30403. doi: 10.1074/jbc.M110.135251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sultana R, Tanneeru K, Guruprasad L. 2011. The PE-PPE domain in Mycobacterium reveals a serine α/β hydrolase fold and function: an in-silico analysis. PLoS One 6:e16745. doi: 10.1371/journal.pone.0016745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sultana R, Vemula MH, Banerjee S, Guruprasad L. 2013. The PE16 (Rv1430) of Mycobacterium tuberculosis is an esterase belonging to serine hydrolase superfamily of proteins. PLoS One 8:e55320. doi: 10.1371/journal.pone.0055320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Voskuil MI, Schnappinger D, Rutherford R, Liu Y, Schoolnik GK. 2004. Regulation of the Mycobacterium tuberculosis PE/PPE genes. Tuberculosis (Edinb) 84:256–262. doi: 10.1016/j.tube.2003.12.014. [DOI] [PubMed] [Google Scholar]
- 30.Sampson SL. 2011. Mycobacterial PE/PPE proteins at the host-pathogen interface. Clin Dev Immunol 2011:497203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fishbein S, van Wyk N, Warren RM, Sampson SL. 2015. Phylogeny to function: PE/PPE protein evolution and impact on Mycobacterium tuberculosis pathogenicity. Mol Microbiol 96:901–916. doi: 10.1111/mmi.12981. [DOI] [PubMed] [Google Scholar]
- 32.Ramakrishnan L, Federspiel NA, Falkow S. 2000. Granuloma-specific expression of Mycobacterium virulence proteins from the glycine-rich PE-PGRS family. Science 288:1436–1439. doi: 10.1126/science.288.5470.1436. [DOI] [PubMed] [Google Scholar]
- 33.Tischler AD, Leistikow RL, Kirksey MA, Voskuil MI, McKinney JD. 2013. Mycobacterium tuberculosis requires phosphate-responsive gene regulation to resist host immunity. Infect Immun 81:317–328. doi: 10.1128/IAI.01136-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sampson SL, Lukey P, Warren RM, van Helden PD, Richardson M, Everett MJ. 2001. Expression, characterization and subcellular localization of the Mycobacterium tuberculosis PPE gene Rv1917c. Tuberculosis (Edinb) 81:305–317. doi: 10.1054/tube.2001.0304. [DOI] [PubMed] [Google Scholar]
- 35.Cascioferro A, Delogu G, Colone M, Sali M, Stringaro A, Arancia G, Fadda G, Palu G, Manganelli R. 2007. PE is a functional domain responsible for protein translocation and localization on mycobacterial cell wall. Mol Microbiol 66:1536–1547. [DOI] [PubMed] [Google Scholar]
- 36.Dona V, Ventura M, Sali M, Cascioferro A, Provvedi R, Palu G, Delogu G, Manganelli R. 2013. The PPE domain of PPE17 is responsible for its surface localization and can be used to express heterologous proteins on the mycobacterial surface. PLoS One 8:e57517. doi: 10.1371/journal.pone.0057517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sambandamurthy VK, Derrick SC, Hsu T, Chen B, Larsen MH, Jalapathy KV, Chen M, Kim J, Porcelli SA, Chan J, Morris SL, Jacobs WR Jr. 2006. Mycobacterium tuberculosis ΔRD1 ΔpanCD: a safe and limited replicating mutant strain that protects immunocompetent and immunocompromised mice against experimental tuberculosis. Vaccine 24:6309–6320. doi: 10.1016/j.vaccine.2006.05.097. [DOI] [PubMed] [Google Scholar]
- 38.Kirksey MA, Tischler AD, Siméone R, Hisert KB, Uplekar S, Guilhot C, McKinney JD. 2011. Spontaneous phthiocerol dimycocerosate-deficient variants of Mycobacterium tuberculosis are susceptible to gamma interferon-mediated immunity. Infect Immun 79:2829–2838. doi: 10.1128/IAI.00097-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pavelka MS Jr, Jacobs WR Jr. 1999. Comparison of the construction of unmarked deletion mutations in Mycobacterium smegmatis, Mycobacterium bovis Bacillus Calmette-Guerin, and Mycobacterium tuberculosis H37Rv by allelic exchange. J Bacteriol 181:4780–4789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parish T, Stoker NG. 2000. Use of a flexible cassette method to generate a double unmarked Mycobacterium tuberculosis tlyA plcABC mutant by gene replacement. Microbiology 146:1969–1975. doi: 10.1099/00221287-146-8-1969. [DOI] [PubMed] [Google Scholar]
- 41.Larsen MH, Biermann K, Tandberg S, Hsu T, Jacobs WR Jr. 2007. Genetic manipulation of Mycobacterium tuberculosis. Curr Protoc Microbiol Chapter 10:Unit 10A.2. [DOI] [PubMed] [Google Scholar]
- 42.Vandal OH, Nathan CF, Ehrt S. 2009. Acid resistance in Mycobacterium tuberculosis. J Bacteriol 191:4714–4721. doi: 10.1128/JB.00305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danilchanka O, Mailaender C, Niederweis M. 2008. Identification of a novel multidrug efflux pump of Mycobacterium tuberculosis. Antimicrob Agents Chemother 52:2503–2511. doi: 10.1128/AAC.00298-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Collazo CM, Yap GS, Sempowski GD, Lusby KC, Tessarollo L, Vande Woude GF, Sher A, Taylor GA. 2001. Inactivation of LRG-47 and IRG-47 reveals a family of interferon γ-inducible genes with essential, pathogen-specific roles in resistance to infection. J Exp Med 194:181–187. doi: 10.1084/jem.194.2.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 46.Darwin KH, Ehrt S, Gutierrez-Ramos J-C, Weich N, Nathan CF. 2003. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 47.Vandal OH, Roberts JA, Odaira T, Schnappinger D, Nathan CF, Ehrt S. 2009. Acid-susceptible mutants of Mycobacterium tuberculosis share hypersusceptibility to cell wall and oxidative stress and to the host environment. J Bacteriol 191:625–631. doi: 10.1128/JB.00932-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shell SS, Wang J, Lapierre P, Mir M, Chase MR, Pyle MM, Gawande R, Ahmad R, Sarracino DA, Ioerger TR, Fortune SM, Derbyshire KM, Wade JT, Gray TA. 2015. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet 4:e1005641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Luca M, Bottai D, Batoni G, Orgeur M, Aulicino A, Counoupas C, Campa M, Brosch R, Esin S. 2012. The ESX-5 associated eccB5-eccC5 locus is essential for Mycobacterium tuberculosis viability. PLoS One 7:e52059. doi: 10.1371/journal.pone.0052059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ates LS, Ummels R, Commandeur S, van der Weerd R, Sparrius M, Weerdenburg E, Alber M, Kalscheuer R, Piersma SR, Abdallah AM, El Ghany MA, Abdel-Haleem AM, Pain A, Jiménez CR, Bitter W, Houben ENG. 2015. Essential role of the ESX-5 secretion system in outer membrane permeability of pathogenic mycobacteria. PLoS Genet 11:e1005190. doi: 10.1371/journal.pgen.1005190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. 2006. Toward the structural genomics of complexes: crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 103:8060–8065. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tundup S, Akhter Y, Thiagarajan D, Hasnain SE. 2006. Clusters of PE and PPE genes of Mycobacterium tuberculosis are organized in operons: evidence that PE Rv2431c is co-transcribed with PPE Rv2430c and their gene products interact with each other. FEBS Lett 580:1285–1293. doi: 10.1016/j.febslet.2006.01.042. [DOI] [PubMed] [Google Scholar]
- 53.Tiwari BM, Kannan N, Vemu L, Raghunand TR. 2012. The Mycobacterium tuberculosis PE proteins Rv0285 and Rv1386 modulate innate immunity and mediate bacillary survival in macrophages. PLoS One 7:e51686. doi: 10.1371/journal.pone.0051686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solomonson M, Setiaputra D, Makepeace KAT, Lameignere E, Petrotchenko EV, Conrady DG, Bergeron JR, Vuckovic M, DiMaio F, Borchers CH, Yip CK, Strynadka NCJ. 2015. Structure of EspB from the ESX-1 Type VII secretion system and insights into its export mechanism. Structure 23:571–583. doi: 10.1016/j.str.2015.01.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.