

Abstract
Kupffer cells are resident liver macrophages and play a critical role in maintaining liver functions. Under physiological conditions, they are the first innate immune cells and protect the liver from bacterial infections. Under pathological conditions, they are activated by different components and can differentiate into M1-like (classical) or M2-like (alternative) macrophages. The metabolism of classical or alternative activated Kupffer cells will determine their functions in liver damage. Special functions and metabolism of Kupffer cells suggest that they are an attractive target for therapy of liver inflammation and related diseases, including cancer and infectious diseases. Here we review the different types of Kupffer cells and their metabolism and functions in physiological and pathological conditions.
Keywords: Kupffer cells, Liver, Metabolism, Macrophages
Introduction
The liver is the one of the largest organs in the body and has endocrine and exocrine properties. It is composed of 60% parenchymal cells, i.e., hepatocytes, and 30% to 35% non-parenchymal cells, i.e., Kupffer cells (KCs), hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells (LSECs) [1]. Kupffer cells were first identified by Karl Wilhelm von Kupffer in 1876 using a gold chloride-staining method and were named “Sternzelle” (stellate cells) [2]. Initially, KCs were associated to the family of perivascular cells of the connective tissues or to the adventitial cells (pericytes). Finally, after fundamental research by Tadeusz Browicz, KCs were identified as macrophages [3]. Kupffer cells are liver resident macrophages that localize within the lumen of the liver sinusoids and are adherent to the endothelial cells that compose the blood vessel walls. KCs are the first immune cells in the liver that come in contact with the gut bacteria and gut bacterial endotoxins and microbial debris derived from the gastrointestinal tract that have been transported to the liver via the portal vein [4]. They also play an essential role in the host defense [5,6] and participate in the metabolism of multiple compounds such as protein complexes, small particles, and lipids, and in removing apoptotic cells from the circulation [7,8]. Consequently, modifications or alterations of KC functions are associated with various liver diseases: viral hepatitis, steatohepatitis, alcoholic liver disease, intrahepatic cholestasis, activation or rejection of the liver during liver transplantation [9] and liver fibrosis [10]. Here we review the different type of KCs and their metabolism and functions in physiological and pathological conditions.
Ontogeny and Different Populations of Kupffer Cells
Ontogeny of Kupffer cells
KCs are liver resident macrophages and appear for the first time in the yolk sac during embryonic development in mammals [11]. Macrophages first migrate into the fetal liver via the umbilical veins and the left vitelline vein. The F4/80-positive macrophages are detected in the hepatic sinusoid at 11 days of gestation in mouse embryos, and their number increases with fetal age. At day 17, F4/80-positive macrophages exhibit peroxidase activity in the nuclear envelope and rough endoplasmic reticulum as observed in mouse adult liver KCs [12]. They proliferate quickly and differentiate into KCs in the late stage of embryonic development and after birth [13].
Life span and renewal of Kupffer cells in liver
Little is known concerning the life span and the renewal mechanisms of KCs. The calculated life span of mammalian KCs was determined to be 3.8 days [14]; however, experimental data showed a longer life span. Bouwens and collaborators [15] have shown in rats that the life span of KCs stretched from several weeks to 14 months. Moreover, in transplanted human livers, donor KCs persisted for up to one year [16].
The mechanisms of KC renewal have still remained elusive. Two hypotheses were put forward: The classical dogma assumes that KCs are not able to self-renew and come from bone marrow-derived monocytes [17,18], whereas the second hypothesis supports that KCs are a self-renewing population and can proliferate as mature cells, or they come from local intrahepatic progenitors [19–23]. To support this second hypothesis, Varol’s group treated mice with acetaminophen after an adoptive transfer experiment. Their data showed that monocytes characterized as Ly6ChighCD11bhighMHCIIneg were massively recruited and infiltrated into the damaged liver after 24 hours of treatment; at the same time, the number of KCs in the injured liver was decreased. These infiltrating monocytes differentiated into Ly6ClowF4/80high macrophages in the injured liver and became the predominant population at 72 hours following acetaminophen treatment before disappearing completely after 96 hours. These macrophages negatively regulated the recruitment of neutrophils in the injured liver. After 120 hours of treatment, KCs became the major macrophage population in the liver, and this repopulation of KCs was due to the self-renewal of differentiated KCs present in the liver [22]. Compared to bone marrow-derived macrophages, KCs exhibited a positive function on the recruitment of neutrophils and also protected hepatocytes from bacterial infection [24]. In order to maintain the constant number of KCs in liver, some data showed that KCs are able to migrate from the liver to the portal areas and into hepatic lymph nodes [25]. However, other hypotheses suggest that KCs can undergo apoptosis, and the apoptotic cells are recognized and phagocytized by adjacent KCs [14].
Subsets of mouse Kupffer cells
KCs are derived from monocytes and differentiate into liver resident macrophages. Because of their origin, macrophage surface markers were used for their identification; for example, F4/80, CD11b and CD68 are commonly used in mice [26]. F4/80 is a stable antigen of mononuclear phagocytes and does not present in other types of leukocytes [27,28]. CD11b antigen is present on the monocyte/macrophage, granulocyte and natural killer cytoplasmic surface [29], and CD68 antigen is usually used as a surface marker of macrophages and activated KCs [30]. Based on these surface markers, four populations of KCs were identified by Seki’s group [31]: F4/80+CD11b−, F4/80+CD11b+, F4/80+CD68− and F4/80+CD68+. F4/80+CD68+ and F4/80+CD11b− cells presented a higher phagocytic activity and showed significant reactive oxygen species (ROS) production after lipopolysaccharide (LPS) or adenosine triphosphate (ATP) stimulation, whereas the F4/80+CD11b+ and F4/80+CD68− cells showed a strong intensity of intracellular tumor necrosis factor (TNF) and interleukin 12 (IL-12) after LPS stimulation.
At the same time, Klein and collaborators irradiated mice and performed an adoptive transfer experiment. They identified two populations of KCs: the first one derived from the bone marrow and the second one was sessile KCs [21]. These two populations of KCs shared the same morphology and phagocytic activity. However, only the first one was implicated in inflammatory responses since they were recruited to the inflammatory foci after the generation of a liver inflammatory environment. Varol’s group used other surface markers to identify different populations of macrophages present in the injured liver caused by acetaminophen treatment [22]. As described above, they identified Ly6ChighCD11bhighMHCIIneg monocytes, which were able to differentiate into Ly6ClowF4/80high macrophages, and resident KCs. Furthermore, they analyzed the molecular signature of these three populations of liver macrophages and observed that: 1) KCs present in liver after 72 hours of acetaminophen treatment expressed the same gene profile as KCs in normal liver; 2) Ly6Chigh monocytes shared only one gene with KCs in normal liver and 667 genes with Ly6ClowF4/80 macrophages; and 3) the latter did not express any common gene with the KCs in the normal liver.
Together, data obtained from different groups show the presence of different populations of macrophages in injured liver, and moreover they express distinct gene expression profiles and are associated with specific functions to repair liver damage.
Metabolism and Functions of Activated Kupffer Cells
In the normal liver, KCs and other non-parenchymal cells represent from 30% to 35% of total liver cells. Thanks to their strategic position in the liver, KCs are the first ones that are in contact with materials absorbed from the gastrointestinal tract. The liver can be damaged by different injuries such as bacterial LPS [32], chemical substances, toxins and pharmacological agents [33,34] such as carbon tetrachloride (CCl4) [35], endotoxin [36], galactosamine [37], acetaminophen [38] and diethylnitrosamine (DEN) [39]. The immediate resulting effects of liver injuries are increased hepatocellular necrosis, which is one of the principal sources of KC activators [10]. Once they are activated, KCs display the ability to differentiate into M1-like macrophages (classical) or M2-like macrophages (alternative) depending on the signals they receive from their environment [40]. The term macrophage activation (classical activation) was used for the first time by Mackaness in the 1960s in an infection context to describe the antigen-dependent microbicidal activity of macrophages towards bacillus Calmette-Guerin and Listeria [41]. It was only in the 1990s, that Stein, Doyle and co-workers demonstrated the existence of an alternative activation phenotype of macrophages induced by IL-4 and IL-13 [42,43]. M1 and M2 macrophage populations differ from their capacity to respond to different stimuli and the repertoire of chemokines/cytokines and receptors they express after their activation [44]. However, both of them become active macrophages with high synthesis and secretion of inflammatory mediators including cytokines, superoxide, nitric oxide, eicosanoids, chemokines, and lysosomal and proteolytic enzymes [10]. Moreover, they exhibit high phagocytic and secretory activities.
M1-like macrophages
Interferon gamma (IFN-γ), alone or with microbial products such as LPS or inflammatory cytokines, such as TNF, can induce macrophage differentiation to the M1 population [45]. Interaction between IFN-γ and its receptor on macrophages activates STAT1 (signal transducers and activators of transcription1) and interferon regulatory factors (IRF) [46]. These classical macrophages are characterized by a high capacity to present antigen, high expression and production of IL-12, IL-23 [47] and IRF-5 [45], high production of nitric oxide (NO), and production of ROS [44,48]. Classical macrophages are IL- 12high and IL-10low. The activated M1 macrophages express opsonic receptors such as FcγRIII and exhibit a high level of arginine metabolism. This metabolism consists of the transformation of arginine to nitric oxide and citrulline by nitric oxide synthase (iNOS; NOS2) [49]. Furthermore, they display an important glycolytic activity by inducing the expression of the pro- glycolytic PFKFB3 isoform (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase), which gives them an energetic advantage in hypoxic regions. In response to different stimuli, classical M1 macrophages up-regulate the expression and secretion of IL-1β, and they increase the succinylation of metabolic proteins and the expression of immunoresponsive genes (IRGs) such as IRG1, which exerts an anti-microbial activity during bacterial infections. Consequently, M1 macrophages are associated with antimicrobial killing and skewing T cell responses toward T helper cells type 1 (TH1) [50].
M2-like macrophages
Activation of macrophages to the M2 population can be induced by IL-4, IL-10, IL-13, IL-33, transforming growth factor (TGF-β), and granulocyte colony-stimulating factor (G-CSF). These activated macrophages are also divided by Mantovani and colleagues into different subtypes [41], because of the broad range of activities they perform: M2a (where “a” stands for alternative), induced by IL4 or IL-13; M2b, induced by exposure to immune complex and agonists of Toll-like receptors (TLRs) or IL-1R (Interleukine-1 Receptor); and M2c, induced by IL-10 and glucocorticoid hormones. M2a macrophages display the alternatively activated phenotype typically attributed to M2 cells, i.e. stimulation by IL-4 and IL-13 leads to,STAT6 activation and translocation, and activation of other proteins, such as c-Myc, IRF-4, transglutaminase 2 (TGM2), mannose receptor C type 1 (MRC1), cholesterol 25-hydroxylase (CH25H) and the prostaglandin-endoperoxide, synthase 1 (PTGS1), Krüppel-like factor 4 (KLF4) and the signaling modulators CISH (cytokine inducible SH2-containing protein) and SOCS1 (suppressor of cytokine signaling 1) [51]. M2b macrophages exhibit a high production and secretion of IL-10, and their activation turns off IL-12. They express both pro- and anti-inflammatory cytokines (TNF-α, IL-1, IL-6, IL-10high and IL-12low). Moreover they up-regulate antigen presentation and recruit TH2 (T helper cells type 2) responses [52,53]. Finally, M2c macrophages are responsible for the production of IL-10, TGF-β and extracellular matrix components [44]. In general, the M2 population is characterized by IL-10high and IL-12low, and low production of pro-inflammatory cytokines (IL-1, TNF and IL-6), except for M2b, which expresses high levels of inflammatory cytokine production in contact with immune complexes and LPS. On their cell surface, they exhibit non-opsonic receptors such as mannose receptor. Opposite to the classical M1 macrophages, the alternative M2 macrophages use oxidative metabolism. They express a high level of fatty acid β-oxidation (FAO) and oxidative phosphorylation (OXPHOS) via the IL-4 signaling pathway. Usually, mannose receptor (MRC1 or CD206), TMG2 and the chitinase-like secretory protein YM-1 have been used to identify them. The roles associated to alternative M2 macrophages are sometimes confusing. Song and collaborators [54] demonstrated in vitro that these cells produced pro-fibrogenic factors such as platelet-derived growth factor BB (PDGFBB) and TGF-β. Consequently, they induced proliferation of myofibroblasts (for example, activated HSCs), facilitated collagen production by differentiated HSCs and encouraged fibrogenesis, tissue remodeling and angiogenesis. In contrast, results from other studies seemed to associate alternative M2 macrophages to the resolution of fibrosis by phagocytizing apoptotic cells and matrix components via mannose and scavenger receptors [45]. In some studies, data showed that activated macrophages participated in the suppression or promotion of cancer. Two populations of macrophages distinguished by their metabolism were identified in cancer: M1 tumor-associated macrophages (TAMs) and M2 TAMs. M1 TAMs were able to suppress tumor growth, whereas M2 TAMs played a role in cancer progression. M1 TAM metabolism is especially based on iNOS, which converts arginine to NO, which is toxic for cancer cells. However, M2 TAMs express the enzyme arginase I, which metabolizes arginine to ornithine and also promotes M2 polarization and impairs the anti-tumor activity of T cells [30].
Kupffer Cells in Physiological Condition
Tolerogenic function of Kupffer cells
The liver is located at a strategic position that allows it to carry out its metabolic functions in lipid, carbohydrate and protein generation and in the degradation of toxic and waste products. Kupffer cells, the largest population of tissue-resident macrophages, are found in the sinusoidal lumen and display an important tolerogenic function to avoid the induction of immunity against innocuous antigens, such as gut-derived nutrients and antigens from aged or dead cells that have been cleared from the bloodstream. Along with dendritic cells (DCs) and liver sinusoidal endothelial cells (LSECs), KCs constitute the reticulo-endothelial system, whose functions are to clear antigens and pathogen- associated molecular patterns (PAMPs) and to degrade products and toxins from sinusoidal blood. Because Kupffer cells are positioned in the liver, they have the ability to encounter T cells, NK (Natural Killer) cells and NKT (Natural Killer T) cells [55,56]. Under normal conditions, KCs express low levels of MHC (Major Histocompatibility Complex) class II and co-stimulatory molecules and can inhibit DC-induced antigen-specific T cell activation via the production of prostaglandin E (PGE) and 15-deoxy-delta12, 14-PGJ2 [57]. Moreover, KCs can induce the suppressive activity of Treg cells by interacting with the Treg cells and stimulating the production of IL-10, which is crucial for the induction of tolerance to hepatocyte- expressed antigens [58]. To accomplish their tolerogenic function, KCs have the ability to express and secrete IL-10 [59] and TGF-β [60] and then suppress T cells. This tolerogenic property of KCs is essential to prevent undesired immune responses under the physiological conditions.
Kupffer cells and clearance function
The complement system exhibits an important function in the clearance of pathogens, immune complexes and apoptotic cells present in the bloodstream. The complement component C3 is an important protein complex involved in complement activation; C3 is able to bind to bacterial surfaces via a thioester bond after cleavage to C3b, a subunit of active C3 convertase. Active C3 products present on the surface of pathogens are then recognized by complement receptors expressed on phagocytic cells [61]. Complement C3 contains four fragment receptors: CR1, CR2, CR3 and CR4. In 2005, Helmy and co-workers identified a new receptor, CRig, belonging to complement C3, the majority of which is expressed on the KC surface [62]. CRIg binds to C3b and iC3b, cleavage products of C3, and induces the opsonisation of pathogens by KCs.
Other complement receptors are found on the KC surface, such as anaphylatoxin C3a receptor, C5a receptor [63,64] and complement receptors 1 and 4 [65]. These properties give KCs a key role in the clearance of pathogens and dead or dying erythrocytes from the blood circulation by their phagocytosis activity [14]. Moreover, KCs express the enzyme heme oxygenase 1 (HO-1) in their endoplasmic reticulum and peri-nuclear envelope. This enzyme functions in oxidative degradation of the heme molecules contained inside senescent erythrocytes and controls heme metabolism by generating carbon monoxide and bile pigments.
Kupffer cells and hepatocyte metabolism
Chawla’s group [66] identified peroxisome proliferator activated receptor δ and γ (PPARδ and PPARγ) expression on the cell membrane of KCs. Activation of PPARγ signaling regulates metabolic programs in M2-like activated KCs, and that induces the expression of arginase I, an essential enzyme that activates KCs into alternative M2 macrophages. Moreover, PPARδ is required for the full expression of the immune phenotype of activated KCs. This activation consists of the expression of recognition receptors, such as Mrc1 and Clec7a, but also of co-stimulatory molecules, such as Pdcd1lg2, and of suppression of macrophage-inflammatory responses. The investigators observed first that chimeric PPARδ−/−-depleted mice exhibited a reduced expression of β-oxidation and OXPHOS genes in liver, suggesting that PPARδ−/− KCs could modulate hepatocyte metabolism. To confirm this hypothesis, they performed an in vitro experiment that consisted of co-culturing wild-type hepatocytes in the presence of PPARδ−/− macrophages or wild-type macrophages. They observed that hepatocytes co- cultured with PPARδ−/−-depleted macrophages exhibited an approximately 25% decrease in the rate of fatty acid oxidation compared to controls. Moreover, histological analysis revealed that mouse livers with PPARδ−/− KCs showed the presence of hepatic steatosis and an approximately 50% increase in extractable liver triglycerides. These data suggest that PPARδ−/− KCs synthetized and secreted factors that could directly modulate oxidative metabolism in parenchymal cells.
Kupffer Cells in Pathological Conditions
Kupffer cells and anti-bacterial defense
KCs, as macrophages, have an important function in the innate immune response in liver. Liver expresses in normal conditions low levels of mRNAs encoding TLRs and their downstream signaling pathways, such as myeloid differentiation primary response gene-88 (MyD88) [67,68]. KCs exhibit TLR2, TLR3, TLR9 and TLR4, which are responsive to LPS, the Gram-negative bacteria cell wall component [69,70]. Recognition of LPS from intestinal microbiota by TLRs present on the KC surface induces both immune activation and tolerance under specific conditions. For example, increased exposure of TLR4 to LPS and/or increased expression or sensibility of TLR4 could remove the tolerogenic phenotype of KCs. The binding of LPS on their TLR4 initiates specific signaling pathways that activate transcription factors such as nuclear factor NF-κB, activator protein 1 (AP-1) and IRFs. These activated transcription factors induce the transcription of specific genes involved in pro- inflammatory, anti-viral and anti-bacterial responses, and genes involved in the control of cell survival and apoptosis [71]. Data have shown that mice depleted of KCs died with a sublethal dose of Listeria monocytogenes [72]. The absence of KCs facilitated bacterial growth in the liver, especially in apoptotic hepatocytes and in the spleen. Moreover, Brenner’s group observed that in KC-depleted mice, there was less neutrophil accumulation and infiltration into the liver. KCs seemed to participate in the recruitment of neutrophils to protect hepatocytes from bacterial infection. Activation of KCs during the bacterial infection involves the recognition between bacterial surface sugars and lectins and receptors expressed on the KC surface [10]. These ligand-receptor interactions activated the synthesis of inflammatory mediators such as IL-6, IL-12, IL-1β, TNF-α and NO. Thus, these chemokines inhibited the proliferation of microorganisms [73,74]. NO produced by KCs in hepatic injury had a double role [10]; it can protect hepatocytes via inhibition of caspases and apoptosis in endotoxemia or CCl4-induced damage, but in other conditions such as ischemia/ reperfusion injury, shock or galactosamine-induced liver injury, NO can increase oxidative stress or the expression of inflammatory mediators [75]. Furthermore, KCs produced cytokines such as MIP-1α, MIP-1β, MCP-1 and MCP-2 to recruit monocytes and neutrophils into the liver to control bacterial infection [76].
Kupffer cells and non-alcoholic liver diseases
Non-alcoholic fatty liver diseases (NAFLDs) are a spectrum of disorders that include non-alcoholic fatty liver (NAFL), steatosis with inflammation, non-alcoholic steatohepatitis (NASH) and NASH with fibrosis. 10% to 29% of patients with NASH will progress to cirrhosis within 10 years, and of these, 4% to 27% are expected to develop hepatocellular carcinomas [77]. The shift from NFL to NASH is characterized first by various metabolic syndromes and insulin resistance, which induce accumulation of free fatty acids and lipids in peripheral blood and hepatocytes, and secondly, by a series of innate immune responses that result from the stimulation of lipotoxins and LPS [78]. KCs, liver-resident macrophages, display a critical mediator in the development of NAFLD, and specifically in the second step of this disease progression [79]. Results obtained by different research groups showed that: 1) chemical depletion of KCs prevents the release of pro-inflammatory cytokines and alleviates hepatocellular damage [80] and 2) ablation of KCs protects against the development of hepatic insulin resistance in response to high-fat diets [79] and hepatic steatosis after longer feeding of high-fat diets [81]. One of the signaling pathways that appear to be involved in this disorder is LPS/TLR4, and TLR4 is widely present on the KC surface. Yang, et al [82] demonstrated that genetically obese ob/ob (leptin-deficient) mice were more sensitive to low-dose LPS compared to wild-type mice. Rivera and co-workers showed that TLR4−/− mice fed with a methionine/choline-deficient diet, a model system for NASH, exhibited less severe hepatic injury and less accumulation of intrahepatic lipids compared to TLR4+/+ mice [83]. More recently, it was shown that over-expression of CD14, a co-receptor of TLR4, in KCs of mice with high-fat diet (HFD)-induced steatosis increased the hypersensitivity against low-dose LPS [84]. Together these data support the implication of TLR4 signaling pathway in NAFLD.
High-glucose and high-fat diets increase the gut permeability and trigger the accumulation of LPS. Binding of LPS to their receptors on the KC surface promotes the production and secretion of pro-inflammatory cytokines that recruit T and B lymphocytes and other leukocytes [85,86]. The aggregation of innate immune system cells in liver encourages steatohepatitis and inflammatory necrosis in hepatocytes, followed by NASH progression [87]. Activation of the TLR4 signaling pathway induces MYD88- dependent and MYD88-independent pathway responses. The first one involved the participation of interleukin receptor associative kinases (IRAKs) and tumor necrosis receptor associated factor 6 (TRAF-6) [88,89]. This MYD88-dependent pathway activates first the complexes JNK/MAPK and IKK that stimulate the synthesis and secretion of cytokines such as TNF-α, IL-6 and IL-10. On the other hand, LPS/TLR4 can also activate MYD88-independent pathway, which is mediated by the Toll/IL-1 receptor, and promotes the expression of IFN-1β and IL-2 [90]. Thus, activated KCs secrete many cytokines that encourage the infiltration of neutrophils, NK cells, NK T cells, T cells, and monocytes. Experiments conducted by Ma et al showed the presence of KCs containing a significant accumulation of intracellular toxic lipids in a NAFLD mouse model [91]. This high accumulation of lipids in KCs may affect the function of mitochondria and induced oxidative and endoplasmic reticulum (ER) stress [92]. Oxidative stress resulted from insufficient free fatty acid (FFA) β-oxidation and dysfunction of mitochondria and leads to the activation of the NF-κB/JNK pathway, high mobility group box 1 (HMGB1)/TLRs, cytokines and chemokines [91,93,94]. ER stress in KCs promotes activation of the JNK/NF-κB/(C/EBP) pathway, which results in insulin resistance and apoptosis.
Kupffer cells and alcoholic liver diseases
Alcoholic liver diseases (ALD) and NAFLDs are the major liver- associated causes of morbidity and mortality in Western countries [95,96]. In the United States of America, 18 million people are affected by alcohol abuse [97], and fatty liver was observed in up to 90% of alcoholics [98]. Alcohol consumption encourages intestinal permeability and increases plasma and liver endotoxin and LPS levels [99]. The presence of a high level of LPS in the alcoholic liver activates KCs, and they release active mediators such as pro- inflammatory cytokines, eicosanoids and ROS. It has been shown that ALD affect more women than men [100], even though the underlying mechanisms remain unclear. Iimuro et al [101] observed that after exposure to ethanol, female rats exhibited higher plasma endotoxin levels than males; this could be due to the presence of estriol in female rats, which increases intestinal permeability and endotoxin accumulation in the portal vein [102]. Indeed, KCs isolated from rats treated with estriol and injected with a sublethal dose of LPS exhibited higher expression of TNF-α, and CD14 compared to control rats [103]. Activation of TLR4 signaling pathway was widely described in the previous paragraph. Here, we will focus on the function of TNF-α and ROS produced by KCs in ALD.
TNF-α was known to be strongly involved in ALD and can induce hepatocellular damage via the generation of superoxide anions by hepatocytes and can increase the synthesis and secretion of IL-8, which recruits neutrophils [104]. Mice deleted of TNF-α receptors are more resistant to ethanol-induced liver damage [105]. Activated KCs are the major source of TNF-α production and secretion after activation of the LPS/TLR4 signaling pathway. Stabilization of TNF-α mRNA in KCs seems to play a crucial role in the high TNF-α level in ALD. Data obtained by Saklatava et al showed that after chronic ethanol exposure, p38 mitogen-activated protein, an important regulator of TNF-α mRNA stability in macrophages, exhibited a higher phosphorylation level [106]. McMullen and co-workers observed that rats chronically exposed to ethanol translocated HuR (Human antigen R), a TNF-α mRNA- binding protein, from the nucleus to the cytoplasm and showed a higher binding capacity of HuR on TNF-α mRNA in KCs, which allowed stabilization of TNF-α mRNA [107]. Thus, stabilization of TNF-α mRNA in KCs could be an important mechanism for ALD progression. Oxidative stress was observed during chronic alcohol consumption, suggesting its involvement in ALD. ROS can be generated by various enzymes in liver: CYP2E1, NADH/NADPH oxidase, xanthine oxidase and arachidonic pathway enzymes such as lipoxygenase (LOX) and cyclooxygenase (COX). KCs express superoxide dismutase (SOD), which uses superoxide anion to produce hydrogen peroxide. Superoxide and hydrogen peroxide can interact and achieve more cytotoxic radicals such as hydroxyl radical [108]. Under normal conditions, hydrogen peroxide is very quickly metabolized by glutathione peroxidase to produce H2O and O2. Under alcohol abuse conditions, enhanced ROS release [109] and reduced glutathione [110] were observed in KCs, as well as lipid peroxidation and mitochondrial dysfunction [111,112].
Kupffer cells and hepatitis
Hepatitis B virus (HBV) and Hepatitis C virus (HCV) have infected more than 500 million people worldwide. These viruses cause liver inflammation, fibrosis, cirrhosis and hepatocellular carcinoma. The modes of transmission of HBV and HCV are percutaneous and sexual exposure, albeit perinatal exposure is often observed for HBV [113,114]. HBV is a 3.2 kb partially double-strand DNA- envelope virus that replicates via RNA intermediates. HBV is composed of two particles: HBcAg (Hepatitis B core protein)-encapsulated viral DNA and HBsAg (Hepatitis B surface antigen). HBsAg and a truncated form of HBcAg, HBeAg (Hepatitis B extracellular form of HBcAg), are secreted by infected hepatocytes and can be detected in HBV patient sera [115,116].
HCV contains a 9.6 kb positive-strand RNA genome that translates into the structural E1 core protein and E2 envelope protein, and the non-structural proteins NS1-NS5 [117]. HBV and HCV only infect and replicate in humans and non-human primates, and immunocompetent small animal models for viral hepatitis are not yet available [118]. Several mouse models infected with lymphocytic choriomeningitis virus (LCMV), murine cytomegalovirus (MCMV), mouse hepatitis virus (MHV) and adenovirus were used to understand the function of KCs in viral infection. However, unlike HBV and HCV, which infect only hepatocytes, these viruses infect not only hepatocytes but also other cells and organs.
Little is known about the interaction between KCs and HBV/ HCV, and how HBV/HCV can activate KC responses. Studies using THP-1 monocytic cells suggested that HBcAg could bind to TLR2 and HSPG (heparan sulfate proteoglycan) present on the THP-1 cell surface. These ligand/receptor interactions activate THP-1, which in turn produced IL-6, IL-12p40 and TNF [119]. Other studies with different systems showed that HBV can bind to receptors expressed on the KC surface, such as HSPG, CD14, and mannose receptor, and induce production and secretion of IL-1β, IL-6, TNF, TGF-β, CXCL8, PD-L2, galectin-9, TRAIL, FasL, granzyme B, perforin, and ROS [120,121]. More is known concerning the interaction of HCV on hepatocyte receptors. Some of these receptors are present on the KC surface, such as HSPG, SR-B1, LDL-receptor, DC-SIGN, TLR2, TLR4, CD40, CD80 and MHC class II, and activated KCs can then express IL-1β, TNF, IL-10, PD-L1, galectin-9, TRAIL, granzyme B and perforin [121].
The activation of KCs and resulting secretion of pro-inflammatory cytokines positively promotes the NF-κB pathway to inhibit HBV replication in primary hepatocytes [122]. During HCV infection, the number of KCs increases in liver and produce TNF, which encourages the further susceptibility of hepatocytes to HCV infection [123]. On the other hand, HCV can promote production and secretion of IL-6, IL-1β, and IFN-β, which may inhibit HCV replication [124–126]. Cytokines secreted by activated KCs encourage differentiation of neighboring cells, such as HSC to promote fibrogenesis, and recruit and activate other immune cells from bone marrow, which will further increase the anti-viral and inflammatory response.
Kupffer cells and liver fibrosis
Liver fibrosis is caused by chronic damage of the liver and is one of the resulting syndromes of chronic HCV infection, alcohol abuse and NAFLDs. In healthy liver, HSCs are quiescent, and initiation of fibrogenesis occurs with activation and differentiation of the HSCs. These activated HSCs synthesize and secrete different types of collagens into the extracellular matrix (ECM). Some studies suggest that KCs have a significant role in initiation of fibrogenesis whereas other studies suggest a more minimal role [127]. The group of D. Brenner [72] identified the importance of the CCR2 receptor, which is expressed on the KC surface in liver fibrosis. They performed bile duct ligation (BDL) on wild-type and CCR2−/− mice or treated the mice with 12 injections of CCl4. They observed that CCR2−/− mice presented significantly less hepatic fibrosis, reduced collagen deposition and HSC activation. At the same time, other groups showed the involvement of KCs in liver fibrosis by inducing the decreased synthesis of metalloproteinases (MMPs) and the increased production of specific tissue inhibitors of metalloproteinases (TIMPs) or non-specific metalloproteinase inhibitors [128,129]. KCs appeared to synthesize and secrete different molecules that facilitate HCS proliferation and activation, such as TGF-β1 and growth factors [130]. Moreover, KCs induced the expression of platelet-derived growth factor (PDGF) receptors on activated HSCs, thus enhancing HSC proliferation [131], and/or they synthesized and secreted TNF-α, IL-1 and MCP-1, which are mitogenic and chemoattractant molecules for HSCs [132,133]. In addition, activated HSCs initiated production of specific collagens and proteoglycans. Furthermore, Benyon and collaborators demonstrated that gelatinases produced by KCs could induce the phenotypic change of HSCs, since these enzymes could degrade collagen type IV, essential for the maintenance of normal function of quiescent HSCs, and facilitate the synthesis of collagen type I, which triggers the phenotypic change of HSCs [134]. Other data showed that activated KCs in NASH expressed chitotriosidase enzyme, which can influence HSC activation [135]. Activated and differentiated HSCs are not the only source of myofibroblasts, which are involved in liver fibrosis. Other cells come from bone marrow, and portal fibroblasts could be differentiated into myofibroblasts [136,137]. In 1994, Bucala and colleagues [138] discovered the role of bone marrow-derived fibrocytes in fibrosis. They are CD45+ and express collagen-α1, α-SMA and other cytoskeletal proteins. Under normal conditions, fibrocytes exhibit both fibroblast markers (fibronectin, vimentin, collagen type I) [139] and hematopoietic markers (CD45, CD34, MHCII, CD11b, Gr1, Ly6C, and CD54) [140]. In response to liver injury, and specifically to TGF-β produced and secreted by activated KCs, these bone marrow-derived fibrocytes are recruited to liver and initiate their activation and differentiation to fibroblasts by reducing the expression of hematopoietic markers and by activating the expression of α-SMA, and collagen type 1 [141]. A higher number of CD68+ cells was found in fibrotic livers after CCl4-induced liver damage, and these macrophages are concentrated in scars during advanced fibrosis [45]. Moreover, the position of these CD68+ cells in fibrotic liver was overlaid with the expression of YM-1 protein (chitinase). During fibrotic liver resolution, the number of macrophages expressing CD68 surface marker was significantly decreased, and the expression of YM-1, a marker of M2-like macrophages, almost completely disappeared, whereas the amount of M1-like macrophages did not change. These data suggest that alternative M2 macrophages played a pro-fibrogenesis role and were resorbed as soon as the liver fibrosis resolution started.
Kupffer cells and liver cancer
It was shown previously that KCs could be activated directly or indirectly by various components. The results of this activation involve production and secretion of multiple inflammatory cytokines, ROS and growth control mediators. More recently, some experiments demonstrated the implication of activated KCs in the process of hepatic carcinogenesis. More specifically, they are involved in the enhancement of clonal expansion of preneoplastic cells leading to neoplasia. Neoplasia induction requires two processes: DNA damage and alteration in cell growth control. Genotoxic agents or non-genotoxic agents are known to modulate cell growth and cell death, with changes of gene expression, by increasing DNA replication with accumulation of DNA damage. Also, they encourage the clonal expansion of preneoplastic hepatocytes [142].
Injured liver, and specifically apoptotic hepatocytes, induces activation of KCs and production of cytokines, in particular TNF-α [143], interleukines and ROS, which modulate the hepatocellular growth [144]. Indeed, inactivation, but not depletion, of KCs by injecting glycine or methyl palmitate in mice, has been shown to decrease hepatocellular growth after treatment with genotoxic and non-genotoxic agents [145,146]. Klaunig’s group utilized clodronate-encapsulated liposomes to delete KCs [147] in male B6C3F1 mice, and they injected LPS into control and KC-depleted mice. They observed that LPS induced increased DNA synthesis in control mice, whereas in KC-depleted mice, DNA synthesis was decreased 80% compared to control. These data demonstrated the implication of KCs in the induction of cell growth. They also wondered whether KCs might play a role in the modulation of preneoplastic lesion growth. To answer this question, control or KC-depleted B6C3F1 mice pretreated with DEN, a DNA-damaging compound that promotes preneoplastic foci, were treated with LPS. In normal mice, LPS increased the relative volume of hepatic focal lesions 4 fold compared to KC-depleted mice. Moreover, LPS induced enhanced DNA synthesis (3 fold) within focal lesions in normal mice, while DNA synthesis was decreased in KC-depleted mice [148]. To better understand the mechanisms implicating KCs in hepatocyte proliferation and tumor modulation, experiments with peroxisome proliferators, a non-genotoxic agent, were performed. Peroxisome proliferators, a class of rodent liver carcinogens, display an important role in activation of the PPAR-α signaling pathway [149]. Activation of PPAR-α is essential for liver carcinogenesis induction in animals fed with peroxisome proliferators. Peroxisome proliferators are known to increase proliferation of rodent hepatocytes both in vitro and in vivo. Interestingly, proliferation of hepatocytes induced by peroxisome proliferators in vivo is increased 8–10 fold, while it was increased only 2 fold in vitro. Moreover, purified primary hepatocytes cultured in the presence of peroxisome proliferators failed to increase the level of DNA synthesis [148]. After treatment with peroxisome proliferators, TNF-α mRNA and protein levels were increased in whole liver and in serum. Neutralizing antibody of TNF-α prevented liver cell proliferation in rats [150], and inactivation of KCs with glycine or methyl palmitate prevented enhanced TNF-α mRNA and protein levels, as well as cell proliferation [145] after peroxisome proliferator treatment. These data suggested that KCs are crucial to induce hepatocyte proliferation. Peroxisome proliferators act directly on KCs and induce activation of NADPH, and consequently increase superoxide anion [151].
Studies performed in our laboratory demonstrated that DEN- induced liver injury increases the number of mouse necrotic hepatocytes. Hepatocytes undergoing cell death released high-mobility group protein B1 (HMGB1) proteins, which activated KCs by binding to the triggering receptor expressed on myeloid cells 1 (TREM-1). Activation of KCs showed increased transcriptional and translational expression of TREM-1 and induced inflammatory responses that drive hepatocarcinogenesis [152]. Furthermore, activated KCs produced high levels of chemokines, such as CCL2 and CXCL10, which are important for the regulation of inflammatory and immune cell migration, differentiation and function. Another study showed that KCs are attracted to liver tumor cells and have the ability to phagocytize them [153,154]. Moreover, activated KCs can produce NO, which is an effective weapon of the KC machinery against tumor cells. Another indirect mechanism of KC action against tumor cells is the secretion of IL-12, which recruits and induces NK cell cytotoxicity [155].
Kupffer cells in liver metastases
KCs play an essential function in the host tumoral surveillance system. Their strategic position in liver allows to them discriminate and remove neoplastic cells that rich to liver. During metastasis, metastatic cells migrate via the bloodstream to colonize other organs. Liver is the main site of metastatic disease for many gastrointestinal and extra-gastrointestinal cancers, as melanoma, breast, pancreatic and renal cancer [156]. Four different stages of liver metastasis have been identified: 1) the microvascular phase, which implicates tumor cell arrest in the sinusoidal vessels, tumor cell death or extravasation; 2) the extravascular, preangiogenic phase, during which host stromal cells are recruited into avascular micrometastases; 3) the angiogenic phase, the stage which recruits endothelial cells and tumors become vascularized; and 4) the growth phase, which leads to establishment of clinical metastases [157]. Obstruction of sinusoidal vessels by tumor cells promote local release of NO and ROS by KCs and by LSECs [158]. In addition, KCs may recruit inflammatory cells, and together they may arrest metastatic cells by inhibiting their growth and eliminating them [151]. Experiments performed on rat showed that in early states of colorectal cancer liver metastasis, KCs display tumoricidal activity in cooperation with NK cells [56]. NK cells were recruited by activated KCs and, in turn, they secreted pro-inflammatory cytokines such as GM-CSF and IFN-γ, that activate KCs, enhance KC phagocytosis capacity or sensitize tumor cells to cytotoxic effects [159]. To defend against innate immune cells, tumors cells can produce and secrete HMGB1, which triggers macrophage and monocyte apoptosis; when the HMGB1 level increases, the KC number decreases, and HMGB1 promotes liver metastases [160].
Recently, interesting studies performed by Wen and collaborators showed a dual role of Kupffer cells during colorectal cancer liver metastasis [161]. Their results showed that KC depletion by gadolinium chloride (GdCl3) before tumor induction and during the early time points (day 0, 10 and 14) of tumor induction increased tumor growth. Whereas late KC depletion (after day 14) has a completely inverse effect: it decreased tumor growth. These data suggest that KCs exhibit an anti-tumor function during the early stages of tumor progression and they display a pro-tumor effect during the later stages. To promote liver metastasis, tumor cells can initiate by distance a pre-metastic niche formation via activation of KCs by exosomes secretion in the bloodstream [162]. Costa-Silva et al used a model of pancreatic ductal adenocarcinoma (PDAC) to understand how pancreatic tumor cell can colonize liver and induce metastasis. Pancreatic cancers are one of the most lethal cancers with five years survival rate of about 6% and PDAC represents 90% of cases [163]. In their study, they showed that these PDAC primary tumor cells secrete exosomes, which migrate to liver via the bloodstream. Mass spectrometry analysis reveals that these PDAC-derived exosomes highly contain migration inhibitory factor (MIF). Furthermore, they demonstrated that PDAC-derived exosomes have been taken up by KCs in liver leading to KC activation. Activated KC produce MIF-dependent cytokines (TGF-β), which activate HSCs to produce fibronectin to create a fibrotic microenvironment leading to recruit bone marrow-derived cells, as macrophages and neutrophils. The pre-metastatic niche formation is essential for the tumor cells to establish in the liver and induce liver metastasis. Additionally, KCs produce many growth factors such as HGF, which encourages tumor cell proliferation [5], and MMPs, especially MMP-9 and MMP-14, which facilitate angiogenesis and tumor invasion, via ECM alterations [129].
Summary
Kupffer cells assume various functions under physiological conditions and controversial functions in liver injury and repair. Under normal conditions, KCs are the major immune cells that are permanently present in the liver. Their strategic position gives them the key role in dead or dying erythrocyte clearance and in the fight against bacterial infections. At the same time, they interact with other hepatic cells, and parenchymal or non-parenchymal cells, to maintain their metabolism homeostasis. Under pathological conditions (such as ALD/NAFLD) chemical reagent-induced injury (such as DEN, CCl4, or acetaminophen), exposure to LPS from bacterial degradation, and/or the tumor process, necrotic hepatocytes release many signaling molecules that activate KCs. Activation of KCs results in differentiation of these cells into classical M1 or alternative M2 macrophages. Macrophage differentiation induces metabolism modifications and specialized gene expression patterns.
Activated KCs synthesize and secrete many inflammatory chemokines and cytokines in order to recruit innate immune cells to the injured site and induce their differentiation. At the same time, they are able to induce HSCs from a quiescent state to an activated state and recruit bone marrow-derived fibrocytes/fibroblasts or portal fibroblasts, although the signaling molecules and pathways involved in this process remain to be elucidated. Activation of HSCs, bone marrow-derived fibroblast and portal fibroblasts to myofibroblasts allows the production of different collagens and initiation of liver fibrogenesis.
Some studies reported that Kupffer cells play a role in tumor cell phagocytosis in hepatic carcinomas. However, they also produce, at the same time, some cytokines and chemokines that promote hepatocyte proliferation (Figure 1). Still, functions of KCs during fibrogenesis and its resolution, as well as its role in hepatic carcinogenesis remain elusive. Some studies reported that classical M1 macrophages are anti-inflammatory, whereas alternative M2 macrophages are pro-inflammatory and pro-fibrogenic. However, little is known about the signaling pathways and molecules required for KC differentiation and about themechanisms that modulate and regulate these processes. As we know, activation of KCs involves PPAR-γ signaling pathway, and inhibition of this pathway may modulate KC functions. Thalidomide (α-N- phthalimidoglutarimide) and pioglitazone are ligands for PPAR-γ. Thalidomide was recognized tosuppress TNF-α production by macrophages and other cell types, such as activated T cells [164]. Furthermore, thalidomide prevents the LPS-induced increase in CD14 expression [165]. Unfortunately, thalidomide also presents teratogenic effects. The possibility to synthetize thalidomide analogs lacking these teratogenic effects could be a next step to modulate KC functions in pathological conditions. Further analyses of the regulatory mechanisms in KC differentiation and function should allow the development of a new range of therapeutics.
Figure 1. Implication of Kupffer cells in liver injury.
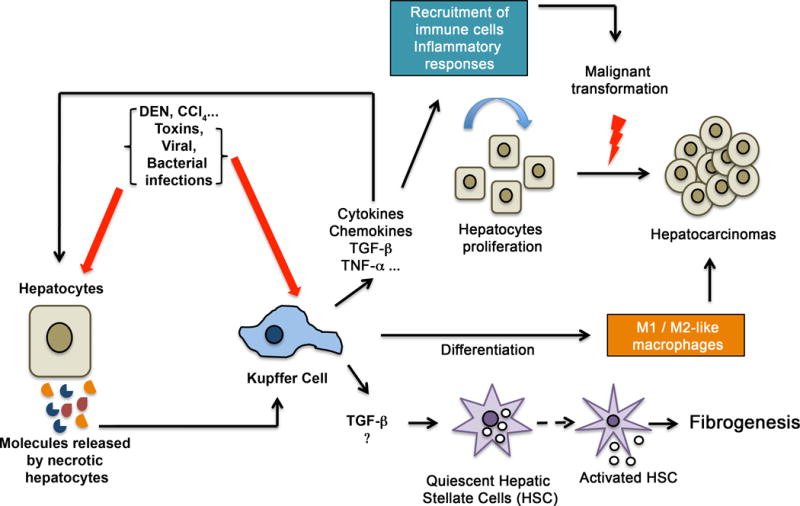
Toxic components, bacterial/viral infections and other injuries target hepatocytes. Necrotic hepatocytes release various molecules that activate Kupffer cells. These latter synthesize and secrete important cytokines, chemokines, TNF-α, and TGF-β, allowing the recruitment of inflammatory responses into the injured liver and the activation of HSCs. The inflammatory responses induce chronic inflammation, which induces hepatocyte proliferation and transformation to generate hepatocellular carcinoma. Activation of HSCs drives the synthesis of collagens and specific proteins facilitating fibrogenesis. Activated KCs differentiate into classical M1 or alternative M2 macrophages, which participate in fibrosis or tumor progression or fibrosis resolution and elimination of tumor cells.
Acknowledgments
We thank Drs. Rhea-Beth Markowitz and Vera Portik-Dobos for critical reading of the manuscript. Research in the author’s laboratory is supported by a grant from the National Institutes of Health NIH CA172230 (A. Horuzsko).
Abbreviations
- ALD
alcoholic liver diseases
- AP-1
activator protein 1
- ATP
adenosine triphosphate
- BDL
bile duct ligation
- CCl4
carbon tetrachloride
- CH25H
cholesterol 25-hydroxylase
- CISH
cytokine inducible SH2-containing protein
- COX
cyclooxygenase
- DCs
dendritic Cells
- DEN
diethylnitrosamine
- ECM
extracellular matrix
- FAO
fatty acid β-oxidation
- G-CSF
granulocyte colony-stimulating factor
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HMGB1
high mobility group box 1
- HO-1
heme oxygenase
- HSCs
hepatic stellate cells
- INF-γ
interferon gamma
- IL
interleukin
- iNOS
induced nitric oxide synthase
- IRF
interferon regulatory factor
- KCs
Kupffer Cells
- KLF
Krüppel-like factor
- LCMV
lymphocytic choriomeningitis virus
- LOX
lipoxygenase
- LPS
lipopolysaccharide
- LSECs
liver sinusoidal endothelial cells
- MCMV
murine cytomegalovirus
- MHV
mouse hepatitis virus
- MHC
major histocompatibility complex
- MMPs
matrix metalloproteinases
- MRC
mannose receptor C
- MyD88
myeloid differentiation primary response gene-88
- NAFLD
non-alcoholic fatty liver diseases
- NAFL
non-alcoholic fatty liver
- NASH
non-alcoholic steatohepatitis
- NF-κB
nuclear factor kappa B
- NK
natural killer
- NKT
natural killer T cells
- NO
nitric oxide
- NOS
nitric oxide synthase
- OXPHOS
oxidative phosphorylation
- PPAR
peroxisome proliferator activated receptor
- PFKFB
6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase
- PGE
prostaglandin E
- PTGS1
prostaglandin-endoperoxide synthase 1
- ROS
reactive oxygen species
- SOCS
suppressor of cytokine signaling
- SOD
superoxide dismutase
- STAT
signal transducers and activators of transcription
- TAMs
tumor-associated macrophages
- TGF-β
transforming growth factor beta
- TGM
transglutaminase
- TH1
T helper cells type 1
- TH2
T helper cells type 2
- TIMPs
tissue inhibitors of metalloproteinases
- TLRs
Toll-like receptors
- TNF
tumor necrosis factor
- TREM-1
triggering receptor expressed on myeloid cells 1
Footnotes
Competing Interests Statement
The authors declare no competing financial interests.
References
- 1.Williams GM, Iatropoulos MJ. Alteration of liver cell function and proliferation: differentiation between adaptation and toxicity. Toxicol Pathol. 2002;30(1):41–53. doi: 10.1080/01926230252824699. [DOI] [PubMed] [Google Scholar]
- 2.Kupffer C. Ueber Sternzellen der Leber. Archiv für mikroskopische Anatomie. 1876;12:353–358. [Google Scholar]
- 3.Sródka A, Gryglewski RW, Szczepariski W. Browicz or Kupffer cells? Pol J Pathol. 2006;57(4):183–5. [PubMed] [Google Scholar]
- 4.Fox ES, Thomas P, Broitman SA. Comparative studies of endotoxin uptake by isolated rat Kupffer and peritoneal cells. Infect Immun. 1987;55(12):2962–6. doi: 10.1128/iai.55.12.2962-2966.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bilzer M, Roggel F, Gerbes AL. Role of Kupffer cells in host defense andliver disease. Liver Int. 2006;26(10):1175–86. doi: 10.1111/j.1478-3231.2006.01342.x. [DOI] [PubMed] [Google Scholar]
- 6.Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 2015;42(1):145–58. doi: 10.1016/j.immuni.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 7.Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol. 2009;27:147–63. doi: 10.1146/annurev.immunol.021908.132629. [DOI] [PubMed] [Google Scholar]
- 8.Parker GA, Picut CA. Liver immunobiology. Toxicol Pathol. 2005;33(1):52–62. doi: 10.1080/01926230590522365. [DOI] [PubMed] [Google Scholar]
- 9.Karimi MH, Geramizadeh B, Malek-Hosseini SA. Tolerance Induction in Liver. Int J Organ Transplant Med. 2015;6(2):45–54. [PMC free article] [PubMed] [Google Scholar]
- 10.Kolios G, Valatas V, Kouroumalis E. Role of Kupffer cells in the pathogenesis of liver disease. World J Gastroenterol. 2006;12(46):7413–20. doi: 10.3748/wjg.v12.i46.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi K, Yamamura F, Naito M. Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: a light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. J Leukoc Biol. 1989;45(2):87–96. doi: 10.1002/jlb.45.2.87. [DOI] [PubMed] [Google Scholar]
- 12.Naito M, Yamamura F, Nishikawa S, Takahashi K. Development, differentiation, and maturation of fetal mouse yolk sac macrophages in cultures. J Leukoc Biol. 1989;46(1):1–10. doi: 10.1002/jlb.46.1.1. [DOI] [PubMed] [Google Scholar]
- 13.Naito M, Hasegawa G, Takahashi K. Development, differentiation, and maturation of Kupffer cells. Microsc Res Tech. 1997;39(4):350–64. doi: 10.1002/(SICI)1097-0029(19971115)39:4<350::AID-JEMT5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 14.Naito M, Hasegawa G, Ebe Y, Yamamoto T. Differentiation and function of Kupffer cells. Med Electron Microsc. 2004;37(1):16–28. doi: 10.1007/s00795-003-0228-x. [DOI] [PubMed] [Google Scholar]
- 15.Bouwens L, Knook DL, Wisse E. Local proliferation and extrahepatic recruitment of liver macrophages (Kupffer cells) in partial-body irradiated rats. J Leukoc Biol. 1986;39(6):687–97. doi: 10.1002/jlb.39.6.687. [DOI] [PubMed] [Google Scholar]
- 16.Steinhoff G, B M, Sorg C, Wonigeit K, Pichlmayr R. Sequential analysis of macrophage tissue differentiation and Kupffer cell exchange after human liver transplantation. Cells of the hepatic sinusoid. 1989;2:406–409. [Google Scholar]
- 17.van Furth R. Monocyte origin of Kupffer cells. Blood Cells. 1980;6(1):87–92. [PubMed] [Google Scholar]
- 18.Diesselhoff-den Dulk MM, Crofton RW, van Furth R. Origin and kinetics of Kupffer cells during an acute inflammatory response. Immunology. 1979;37(1):7–14. [PMC free article] [PubMed] [Google Scholar]
- 19.Yamamoto T, Naito M, Moriyama H, Umezu H, Matsuo H, Kiwada H, et al. Repopulation of murine Kupffer cells after intravenous administration of liposome-encapsulated dichloromethylene diphosphonate. Am J Pathol. 1996;149(4):1271–86. [PMC free article] [PubMed] [Google Scholar]
- 20.Shepard JL, Zon LI. Developmental derivation of embryonic and adult macrophages. Curr Opin Hematol. 2000;7(1):3–8. doi: 10.1097/00062752-200001000-00002. [DOI] [PubMed] [Google Scholar]; Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110(12):4077–85. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zigmond E, Samia-Grinberg S, Pasmanik-Chor M, Brazowski E, Shibolet O, Halpern Z, et al. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J Immunol. 2014;193(1):344–53. doi: 10.4049/jimmunol.1400574. [DOI] [PubMed] [Google Scholar]
- 22.Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ, et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110(12):4077–85. doi: 10.1182/blood-2007-02-073841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science. 2012;336(6077):86–90. doi: 10.1126/science.1219179. [DOI] [PubMed] [Google Scholar]
- 24.Ebe Y, Hasegawa G, Takatsuka H, Umezu H, Mitsuyama M, Arakawa M, et al. The role of Kupffer cells and regulation of 894 neutrophil migration into the liver by macrophage inflammatory protein-2 in primary listeriosis in mice. Pathol Int. 1999;49(6):519–32. doi: 10.1046/j.1440-1827.1999.00910.x. [DOI] [PubMed] [Google Scholar]
- 25.Hardonk MJ, Dijkhuis FW, Grond J, Koudstaal J, Poppema S. Evidence for a migratory capability of rat Kupffer cells to portal tracts and hepatic lymph nodes. Virchows Arch B Cell Pathol Incl Mol Pathol. 1986;51(5):429–42. doi: 10.1007/BF02899050. [DOI] [PubMed] [Google Scholar]
- 26.Sato A, Nakashima H, Nakashima M, Ikarashi M, Nishiyama K, Kinoshita M, et al. Involvement of the TNF and FasL produced by CD11b Kupffer cells/macrophages in CCl4-induced acute hepatic injury. PLoS One. 2014;9(3):e92515. doi: 10.1371/journal.pone.0092515. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Austyn JM, Gordon S. F4/80, a monoclonal antibody directed specifically against the mouse macrophage. Eur J Immunol. 1981;11(10):805–15. doi: 10.1002/eji.1830111013. [DOI] [PubMed] [Google Scholar]
- 28.Hume DA, Robinson AP, MacPherson GG, Gordon S. The mononuclear phagocyte system of the mouse defined byimmunohistochemical localization of antigen F4/80. Relationship between macrophages, Langerhans cells, reticular cells, and dendritic cells in lymphoid and hematopoietic organs. J Exp Med. 1983;158(5):1522–36. doi: 10.1084/jem.158.5.1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanchez-Madrid F, Simon P, Thompson S, Springer TA. Mapping of antigenic and functional epitopes on the alpha- and beta-subunits of two related mouse glycoproteins involved in cell interactions, LFA-1 and Mac-1. J Exp Med. 1983;158(2):586–602. doi: 10.1084/jem.158.2.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith MJ1, Koch GL. Differential expression of murine macrophage surface glycoprotein antigens in intracellular membranes. J Cell Sci. 1987;87(Pt 1):113–9. doi: 10.1242/jcs.87.1.113. [DOI] [PubMed] [Google Scholar]
- 31.Kinoshita M, Uchida T, Sato A, Nakashima M, Nakashima H, Shono S, et al. Characterization of two F4/80-positive Kupffer cell subsets by their function and phenotype in mice. J Hepatol. 2010;53(5):903–10. doi: 10.1016/j.jhep.2010.04.037. [DOI] [PubMed] [Google Scholar]
- 32.Movita D, Kreefft K, Biesta P, van Oudenaren A, Leenen PJ, Janssen HL, et al. Kupffer cells express a unique combination of phenotypic and functional characteristics compared with splenic and peritoneal macrophages. J Leukoc Biol. 2012;92(4):723–33. doi: 10.1189/jlb.1111566. [DOI] [PubMed] [Google Scholar]
- 33.Winwood PJ, Arthur MJ. Kupffer cells: their activation and role in animal models of liver injury and human liver disease. Semin Liver Dis. 1993;13(1):50–9. doi: 10.1055/s-2007-1007337. [DOI] [PubMed] [Google Scholar]
- 34.Su GL. Lipopolysaccharides in liver injury: molecular mechanisms of Kupffer cell activation. Am J Physiol Gastrointest Liver Physiol. 2002;283(2):G256–65. doi: 10.1152/ajpgi.00550.2001. [DOI] [PubMed] [Google Scholar]
- 35.Luckey SW, Petersen DR. Activation of Kupffer cells during the course of carbon tetrachloride-induced liver injury and fibrosis in rats. Exp Mol Pathol. 2001;71(3):226–40. doi: 10.1006/exmp.2001.2399. [DOI] [PubMed] [Google Scholar]
- 36.Arthur MJ, Bentley IS, Tanner AR, Saunders PK, Millward-Sadler GH, Wright R. Oxygen-derived free radicals promote hepatic injury in the rat. Gastroenterology. 1985;89(5):1114–22. doi: 10.1016/0016-5085(85)90218-5. [DOI] [PubMed] [Google Scholar]
- 37.Shiratori Y, Takikawa H, Kawase T, Sugimoto T. Superoxide anion generating capacity and lysosomal enzyme activities of Kupffer cells in galactosamine induced hepatitis. Gastroenterol Jpn. 1986;21(2):135–44. doi: 10.1007/BF02774831. [DOI] [PubMed] [Google Scholar]
- 38.Laskin DL. Nonparenchymal cells and hepatotoxicity. Semin Liver Dis. 1990;10(4):293–304. doi: 10.1055/s-2008-1040485. [DOI] [PubMed] [Google Scholar]
- 39.Herrold KM. Effect of Route of Administration on the Carcinogenic Action of Diethylnitrosamine (N-Nitrosodiethylamine) Br J Cancer. 1964;18:763–7. doi: 10.1038/bjc.1964.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–95. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mackaness GB. Cellular resistance to infection. J Exp Med. 1962;116:381–406. [PubMed] [Google Scholar]
- 42.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176(1):287–92. doi: 10.1084/jem.176.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Doyle AG, Herbein G, Montaner LJ, Minty AJ, Caput D, Ferrara P, et al. Interleukin-13 alters the activation state of murine macrophages in vitro: comparison with interleukin-4 and interferon-gamma. Eur J Immunol. 1994;24(6):1441–5. doi: 10.1002/eji.1830240630. [DOI] [PubMed] [Google Scholar]
- 44.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677–86. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 45.Beljaars L, Schippers M, Reker-Smit C, Martinez FO, Helming L, Poelstra K, et al. Hepatic Localization of Macrophage Phenotypes during Fibrogenesis and Resolution of Fibrosis in Mice and Humans. Front Immunol. 2014;5:430. doi: 10.3389/fimmu.2014.00430. eCollection 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon gamma: implications for immune responses and autoimmune diseases. Immunity. 2009;31(4):539–50. doi: 10.1016/j.immuni.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dinarello CA. Interleukin-1 and interleukin-1 antagonism. Blood. 1991;77(8):1627–52. [PubMed] [Google Scholar]
- 48.Mills CD. Macrophage arginine metabolism to ornithine/urea or nitric oxide/citrulline: a life or death issue. Crit Rev Immunol. 2001;21(5):399–425. [PubMed] [Google Scholar]
- 49.Edin S, Wikberg ML, Dahlin AM, Rutegård J, Öberg Å, Oldenborg PA, et al. The distribution of macrophages with a M1 or M2 phenotype in relation to prognosis and the molecular characteristics of colorectal cancer. PLoS One. 2012;7(10):e47045. doi: 10.1371/journal.pone.0047045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–73. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 51.Martinez FO, Helming L, Milde R, Varin A, Melgert BN, Draijer C, et al. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. 2013;121(9):e57–69. doi: 10.1182/blood-2012-06-436212. [DOI] [PubMed] [Google Scholar]
- 52.Edwards JP, Zhang X, Frauwirth KA, Mosser DM. Biochemical and functional characterization of three activated macrophage populations. J Leukoc Biol. 2006;80(6):1298–307. doi: 10.1189/jlb.0406249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anderson CF, Mosser DM. A novel phenotype for an activated macrophage: the type 2 activated macrophage. J Leukoc Biol. 2002;72(1):101–6. [PubMed] [Google Scholar]
- 54.Song E, Ouyang N, Hörbelt M, Antus B, Wang M, Exton MS. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000;204(1):19–28. doi: 10.1006/cimm.2000.1687. [DOI] [PubMed] [Google Scholar]
- 55.Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol. 2010;10(11):753–66. doi: 10.1038/nri2858. [DOI] [PubMed] [Google Scholar]
- 56.Li Y, Cao G, Zheng X, Wang J, Wei H, Tian Z, et al. CRACC-CRACC interaction between Kupffer and NK cells contributes to poly I:C/D-GalN induced hepatitis. PLoS One. 2013;8(9):e76681. doi: 10.1371/journal.pone.0076681. eCollection 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.You Q, Cheng L, Kedl RM, Ju C. Mechanism of T cell tolerance induction by murine hepatic Kupffer cells. Hepatology. 2008;48(3):978–90. doi: 10.1002/hep.22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50(2):612–21. doi: 10.1002/hep.23043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knolle PA, Uhrig A, Protzer U, Trippler M, Duchmann R, Meyer zum Büschenfelde KH, et al. Interleukin-10 expression is autoregulated at the transcriptional level in human and murine Kupffer cells. Hepatology. 1998;27(1):93–9. doi: 10.1002/hep.510270116. [DOI] [PubMed] [Google Scholar]
- 60.Bissell DM, Wang SS, Jarnagin WR, Roll FJ. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest. 1995;96(1):447–55. doi: 10.1172/JCI118055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pangburn MK, Müller-Eberhard HJ. Relation of putative thioester bond in C3 to activation of the alternative pathway and the binding of C3b to biological targets of complement. J Exp Med. 1980;152(4):1102–14. doi: 10.1084/jem.152.4.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Helmy KY, Katschke KJ, Jr, Gorgani NN, Kljavin NM, Elliott JM, Diehl L, et al. CRIg: a macrophage complement receptor required for phagocytosis of circulating pathogens. Cell. 2006;124(5):915–27. doi: 10.1016/j.cell.2005.12.039. [DOI] [PubMed] [Google Scholar]
- 63.Pestel S, Jungermann K, Götze O, Schieferdecker HL. Inhibition by prostaglandin E(2) of anaphylatoxin C5a- but not zymosan-induced prostanoid release from rat Kupffer cells. Lab Invest. 2002;82(4):463–71. doi: 10.1038/labinvest.3780439. [DOI] [PubMed] [Google Scholar]
- 64.Schlaf G, Schmitz M, Rothermel E, Jungermann K, Schieferdecker HL, Götze O. Expression and induction of anaphylatoxin C5a receptors in the rat liver. Histol Histopathol. 2003;18(1):299–308. doi: 10.14670/HH-18.299. [DOI] [PubMed] [Google Scholar]
- 65.Hinglais N, Kazatchkine MD, Mandet C, Appay MD, Bariety J. Human liver Kupffer cells express CR1, CR3, and CR4 complement receptor antigens. An immunohistochemical study. Lab Invest. 1989;61(5):509–14. [PubMed] [Google Scholar]
- 66.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, et al. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008;7(6):496–507. doi: 10.1016/j.cmet.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Creus A, Abe M, Lau AH, Hackstein H, Raimondi G, Thomson AW. Low TLR4 expression by liver dendritic cells correlates with reduced capacity to activate allogeneic T cells in response to endotoxin. J Immunol. 2005;174(4):2037–45. doi: 10.4049/jimmunol.174.4.2037. [DOI] [PubMed] [Google Scholar]
- 68.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002 Jan;168(2):554–61. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 69.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48(1):322–35. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 70.Su GL, Klein RD, Aminlari A, Zhang HY, Steinstraesser L, Alarcon WH, et al. Kupffer cell activation by lipopolysaccharide in rats: role for lipopolysaccharide binding protein and toll-like receptor 4. Hepatology. 2000;31(4):932–6. doi: 10.1053/he.2000.5634. [DOI] [PubMed] [Google Scholar]
- 71.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 72.Seki E, de Minicis S, Inokuchi S, Taura K, Miyai K, van Rooijen N, et al. CCR2 promotes hepatic fibrosis in mice. Hepatology. 2009;50(1):185–97. doi: 10.1002/hep.22952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ehlers S, Mielke ME, Blankenstein T, Hahn H. Kinetic analysis of cytokine gene expression in the livers of naive and immune mice infected with Listeria monocytogenes. The immediate early phase in innate resistance and acquired immunity. J Immunol. 1992;149(9):3016–22. [PubMed] [Google Scholar]
- 74.Ofek I, Sharon N. Lectinophagocytosis: a molecular mechanism of recognition between cell surface sugars and lectins in the phagocytosis of bacteria. Infect Immun. 1988;56(3):539–47. doi: 10.1128/iai.56.3.539-547.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sass G, Koerber K, Bang R, Guehring H, Tiegs G. Inducible nitric oxide synthase is critical for immune-mediated liver injury in mice. J Clin Invest. 2001;107(4):439–47. doi: 10.1172/JCI10613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Barsig J, Flesch IE, Kaufmann SH. Macrophages and hepatocytic cells as chemokine producers in murine listeriosis. Immunobiology. 1998;199(1):87–104. doi: 10.1016/S0171-2985(98)80066-1. [DOI] [PubMed] [Google Scholar]
- 77.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332(6037):1519–23. doi: 10.1126/science.1204265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fujimoto M, Uemura M, Nakatani Y, Tsujita S, Hoppo K, Tamagawa T, et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: relation to severity of liver disturbance. Alcohol Clin Exp Res. 2000;24(4 Suppl):48S–54S. [PubMed] [Google Scholar]
- 79.Lanthier N, Molendi-Coste O, Cani PD, van Rooijen N, Horsmans Y, Leclercq IA. Kupffer cell depletion prevents but has no therapeutic effect on metabolic and inflammatory changes induced by a high-fat diet. FASEB J. 2011;25(12):4301–11. doi: 10.1096/fj.11-189472. [DOI] [PubMed] [Google Scholar]
- 80.Parlesak A, Schäfer C, Schütz T, Bode JC, Bode C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J Hepatol. 2000;32(5):742–7. doi: 10.1016/s0168-8278(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 81.Neyrinck AM, Cani PD, Dewulf EM, De Backer F, Bindels LB, Delzenne NM. Critical role of Kupffer cells in the management of dietinduced diabetes and obesity. Biochem Biophys Res Commun. 2009;385(3):351–6. doi: 10.1016/j.bbrc.2009.05.070. [DOI] [PubMed] [Google Scholar]
- 82.Yang SQ, Lin HZ, Lane MD, Clemens M, Diehl AM. Obesity increases sensitivity to endotoxin liver injury: implications for the pathogenesis of steatohepatitis. Proc Natl Acad Sci U S A. 1997;94(6):2557–62. doi: 10.1073/pnas.94.6.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rivera CA, Adegboyega P, van Rooijen N, Tagalicud A, Allman M, Wallace M. Toll-like receptor-4 signaling and Kupffer cells play pivotal roles in the pathogenesis of non-alcoholic steatohepatitis. J Hepatol. 2007;47(4):571–9. doi: 10.1016/j.jhep.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012;16(1):44–54. doi: 10.1016/j.cmet.2012.05.012. [DOI] [PubMed] [Google Scholar]
- 85.Harte AL, da Silva NF, Creely SJ, McGee KC, Billyard T, Youssef-Elabd EM, et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J Inflamm (Lond) 2010;7:15. doi: 10.1186/1476-9255-7-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Duarte N, Coelho IC, Patarrao RS, Almeida JI, Penha-Goncalves C, Macedo MP. How Inflammation Impinges on NAFLD: A Role for Kupffer Cells. Biomed Res Int. 2015;2015:984578. doi: 10.1155/2015/984578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Farrell GC, van Rooyen D, Gan L, Chitturi S. NASH is an Inflammatory Disorder: Pathogenic, Prognostic 1064 and Therapeutic Implications. Gut Liver. 2012;6(2):149–71. doi: 10.5009/gnl.2012.6.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin M, Bradford BU, Wheeler MD, Uesugi T, Froh M, Goyert SM, et al. Reduced early alcohol-induced liver injury in CD14-deficient mice. J Immunol. 2001;166(7):4737–42. doi: 10.4049/jimmunol.166.7.4737. [DOI] [PubMed] [Google Scholar]
- 89.Uesugi T, Froh M, Arteel GE, Bradford BU, Wheeler MD, Gabele E, et al. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J Immunol. 2002;168(6):2963–9. doi: 10.4049/jimmunol.168.6.2963. [DOI] [PubMed] [Google Scholar]
- 90.Uesugi T, Froh M, Arteel GE, Bradford BU, Thurman RG. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology. 2001;34(1):101–8. doi: 10.1053/jhep.2001.25350. [DOI] [PubMed] [Google Scholar]
- 91.Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol. 1999;276(4 Pt 1):G965–74. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- 92.Mari M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4(3):185–98. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 93.Sheth P, Seth A, Thangavel M, Basuroy S, Rao RK. Epidermal growth factor prevents 1075 acetaldehyde-induced paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res. 2004;28(5):797–804. doi: 10.1097/01.alc.0000125358.92335.90. [DOI] [PubMed] [Google Scholar]
- 94.Suzuki T, Seth A, Rao R. Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J Biol Chem. 2008;283(6):3574–83. doi: 10.1074/jbc.M709141200. [DOI] [PubMed] [Google Scholar]
- 95.Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology. 2011;141(5):1572–85. doi: 10.1053/j.gastro.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sakaguchi S, Takahashi S, Sasaki T, Kumagai T, Nagata K. Progression of alcoholic and non-alcoholic steatohepatitis: common metabolic aspects of innate immune system and oxidative stress. Drug Metab Pharmacokinet. 2011;26(1):30–46. doi: 10.2133/dmpk.dmpk-10-rv-087. [DOI] [PubMed] [Google Scholar]
- 97.Mandayam S, Jamal MM, Morgan TR. Epidemiology of alcoholic liver disease. Semin Liver Dis. 2004;24(3):217–32. doi: 10.1055/s-2004-832936. [DOI] [PubMed] [Google Scholar]
- 98.Wilfred de Alwis NM, Day CP. Genetics of alcoholic liver disease and nonalcoholic fatty liver disease. Semin Liver Dis. 2007;27(1):44–54. doi: 10.1055/s-2006-960170. [DOI] [PubMed] [Google Scholar]
- 99.Malaguarnera G, Giordano M, Nunnari G, Bertino G, Malaguarnera M. Gut microbiota in alcoholic liver disease: pathogenetic role and therapeutic perspectives. World J Gastroenterol. 2014;20(44):16639–48. doi: 10.3748/wjg.v20.i44.16639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Becker U, Deis A, Sørensen T, Grønbaek M, Borch-Johnsen K, Müller CF, et al. Prediction of risk of liver disease by alcohol intake, sex, and age: a prospective population study. Hepatology. 1996;23(5):1025–9. doi: 10.1002/hep.510230513. [DOI] [PubMed] [Google Scholar]
- 101.Iimuro Y, Frankenberg MV, Arteel GE, Bradford BU, Wall CA, Thurman RG. Female rats exhibit greater susceptibility to early alcohol induced liver injury than males. Am J Physiol. 1997;272(5 Pt 1):G1186–94. doi: 10.1152/ajpgi.1997.272.5.G1186. [DOI] [PubMed] [Google Scholar]
- 102.Enomoto N, Yamashina S, Schemmer P, Rivera CA, Bradford BU, Enomoto A, et al. Estriol sensitizes rat Kupffer cells via gut-derived endotoxin. Am J Physiol. 1999;277(3 Pt 1):G671–7. doi: 10.1152/ajpgi.1999.277.3.G671. [DOI] [PubMed] [Google Scholar]
- 103.Ikejima K, Enomoto N, Iimuro Y, Ikejima A, Fang D, Xu J, Forman DT, et al. Estrogen increases sensitivity of hepatic Kupffer cells to endotoxin. Am J Physiol. 1998;274(4 Pt 1):G669–76. doi: 10.1152/ajpgi.1998.274.4.G669. [DOI] [PubMed] [Google Scholar]
- 104.Tilg H, Diehl AM. Cytokines in alcoholic and nonalcoholic steatohepatitis. N Engl J Med. 2000;343(20):1467–76. doi: 10.1056/NEJM200011163432007. [DOI] [PubMed] [Google Scholar]
- 105.Thurman RG. II. Alcoholic liver injury involves activation of Kupffer cells by endotoxin. Am J Physiol. 1998;275(4 Pt 1):G605–11. doi: 10.1152/ajpgi.1998.275.4.G605. [DOI] [PubMed] [Google Scholar]
- 106.Saklatvala J, Dean J, Clark A. Control of the expression of inflammatory response genes. Biochem Soc Symp. 2003;70:95–106. doi: 10.1042/bss0700095. [DOI] [PubMed] [Google Scholar]
- 107.McMullen MR, Cocuzzi E, Hatzoglou M, Nagy LE. Chronic ethanol exposure increases the binding of HuR to the TNFalpha 3′-untranslated region in macrophages. J Biol Chem. 2003;278(40):38333–41. doi: 10.1074/jbc.M304566200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Granger DN. Ischemia-reperfusion: mechanisms of microvascular dysfunction and the influence of risk factors for cardiovascular disease. Microcirculation. 1999;6(3):167–78. [PubMed] [Google Scholar]
- 109.Wheeler MD, Kono H, Yin M, Nakagami M, Uesugi T, Arteel GE, et al. The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic Biol Med. 2001;31(12):1544–9. doi: 10.1016/s0891-5849(01)00748-1. [DOI] [PubMed] [Google Scholar]
- 110.Carmiel-Haggai M, Cederbaum AI, Nieto N. Binge ethanol exposure increases liver injury in obese rats. Gastroenterology. 2003;125(6):1818–33. doi: 10.1053/j.gastro.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 111.Masini A, Ceccarelli D, Gallesi D, Giovannini F, Trenti T. Lipid hydroperoxide induced mitochondrial dysfunction following acute ethanol intoxication in rats. The critical role for mitochondrial reduced glutathione. Biochem Pharmacol. 1994;47(2):217–24. doi: 10.1016/0006-2952(94)90009-4. [DOI] [PubMed] [Google Scholar]
- 112.Ceni E, Mello T, Galli A. Pathogenesis of alcoholic liver disease: role of oxidative metabolism. World J Gastroenterol. 2014;20(47):17756–72. doi: 10.3748/wjg.v20.i47.17756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Margolis HS, Alter MJ, Hadler SC. Hepatitis B: evolving epidemiology and implications for control. Semin Liver Dis. 1991;11(2):84–92. doi: 10.1055/s-2008-1040427. [DOI] [PubMed] [Google Scholar]
- 114.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19(7):859–68. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ganem D, Prince AM. Hepatitis B virus infection–natural history and clinical consequences. N Engl J Med. 2004;350(11):1118–29. doi: 10.1056/NEJMra031087. [DOI] [PubMed] [Google Scholar]
- 116.Locarnini S. Molecular virology of hepatitis B virus. Semin Liver Dis. 2004;24(Suppl 1):3–10. doi: 10.1055/s-2004-828672. [DOI] [PubMed] [Google Scholar]
- 117.Ashfaq UA, Javed T, Rehman S, Nawaz Z, Riazuddin S. An overview of HCV molecular biology, replication and immune responses. Virol J. 2011;8:161. doi: 10.1186/1743-422X-8-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Chayama K, Hayes CN, Hiraga N, Abe H, Tsuge M, Imamura M. Animal model for study of human hepatitis viruses. J Gastroenterol Hepatol. 2011;26(1):13–8. doi: 10.1111/j.1440-1746.2010.06470.x. [DOI] [PubMed] [Google Scholar]
- 119.Cooper A, Tal G, Lider O, Shaul Y. Cytokine induction by the hepatitis B virus capsid in macrophages is facilitated by membrane heparan sulfate and involves TLR2. J Immunol. 2005;175(5):3165–76. doi: 10.4049/jimmunol.175.5.3165. [DOI] [PubMed] [Google Scholar]
- 120.Boltjes A, van Montfoort N, Biesta PJ, Op den Brouw ML, Kwekkeboom J, van der Laan LJ, et al. Kupffer cells interact with hepatitis B surface antigen in vivo and in vitro, leading to proinflammatory cytokine production and natural killer cell function. J Infect Dis. 2015;211(8):1268–78. doi: 10.1093/infdis/jiu599. [DOI] [PubMed] [Google Scholar]
- 121.Boltjes A, Movita D, Boonstra A, Woltman AM. The role of Kupffer cells in hepatitis B and hepatitis C virus infections. J Hepatol. 2014;61(3):660–71. doi: 10.1016/j.jhep.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 122.Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50(6):1773–82. doi: 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]
- 123.Fletcher NF, Sutaria R, Jo J, Barnes A, Blahova M, Meredith LW, et al. Activated macrophages promote hepatitis C virus entry in a tumor necrosis factor-dependent manner. Hepatology. 2014;59(4):1320–30. doi: 10.1002/hep.26911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Broering R, Wu J, Meng Z, Hilgard P, Lu M, Trippler M, Szczeponek A, et al. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48(6):914–22. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 125.Zhu H, Shang X, Terada N, Liu C. STAT3 induces anti-hepatitis C viral activity in liver cells. Biochem Biophys Res Commun. 2004;324(2):518–28. doi: 10.1016/j.bbrc.2004.09.081. [DOI] [PubMed] [Google Scholar]
- 126.Srivastava B, Gimson A. Hepatic changes in systemic infection. Best Pract Res Clin Gastroenterol. 2013;27(4):485–95. doi: 10.1016/j.bpg.2013.06.011. [DOI] [PubMed] [Google Scholar]
- 127.Pradere JP, Kluwe J, De Minicis S, Jiao JJ, Gwak GY, Dapito DH, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58(4):1461–73. doi: 10.1002/hep.26429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Arthur MJ. Fibrosis and altered matrix degradation. Digestion. 1998;59(4):376–80. doi: 10.1159/000007492. [DOI] [PubMed] [Google Scholar]
- 129.Knittel T, Mehde M, Kobold D, Saile B, Dinter C, Ramadori G. Expression patterns of matrix metalloproteinases and their inhibitors in parenchymal and non-parenchymal cells of rat liver: regulation by TNF-alpha and TGF-beta1. J Hepatol. 1999;30(1):48–60. doi: 10.1016/s0168-8278(99)80007-5. [DOI] [PubMed] [Google Scholar]
- 130.Meng F, Wang K, Aoyama T, Grivennikov SI, Paik Y, Scholten D, et al. Interleukin-17 signaling in inflammatory, 1163 Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143(3):765–76. e1–3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Friedman SL, Arthur MJ. Activation of cultured rat hepatic lipocytes by Kupffer cell conditioned medium. Direct enhancement of matrix synthesis and stimulation of cell proliferation via induction of platelet- derived growth factor receptors. J Clin Invest. 1989;84(6):1780–5. doi: 10.1172/JCI114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Marra F, Romanelli RG, Giannini C, Failli P, Pastacaldi S, Arrighi MC, et al. Monocyte chemotactic protein-1 as a chemoattractant for human hepatic stellate cells. Hepatology. 1999;29(1):140–8. doi: 10.1002/hep.510290107. [DOI] [PubMed] [Google Scholar]
- 133.Matsuoka M, Pham NT, Tsukamoto H. Differential effects of interleukin-1 alpha, tumor necrosis factor alpha, and transforming growth factor beta 1 on cell proliferation and collagen formation by cultured fat-storing cells. Liver. 1989;9(2):71–8. doi: 10.1111/j.1600-0676.1989.tb00382.x. [DOI] [PubMed] [Google Scholar]
- 134.Benyon RC, Hovell CJ, Da Gaça M, Jones EH, Iredale JP, Arthur MJ. Progelatinase A is produced and activated by rat hepatic stellate cells and promotes their proliferation. Hepatology. 1999;30(4):977–86. doi: 10.1002/hep.510300431. [DOI] [PubMed] [Google Scholar]
- 135.Malaguarnera L, Di Rosa M, Zambito AM, dell’Ombra N, Nicoletti F, Malaguarnera M. Chitotriosidase gene expression in Kupffer cells from patients with non-alcoholic fatty liver disease. Gut. 2006;55(9):1313–20. doi: 10.1136/gut.2005.075697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kisseleva T, Brenner DA. Fibrogenesis of parenchymal organs. Proc Am Thorac Soc. 2008;5(3):338–42. doi: 10.1513/pats.200711-168DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kisseleva T, Brenner DA. Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol. 2011;25(2):305–17. doi: 10.1016/j.bpg.2011.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bucala R. Circulating fibrocytes: cellular basis for NSF. J Am Coll Radiol. 2008;5(1):36–9. doi: 10.1016/j.jacr.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 139.Abe R, Donnelly SC, Peng T, Bucala R, Metz CN. Peripheral blood fibrocytes: differentiation pathway and migration to wound sites. J Immunol. 2001;166(12):7556–62. doi: 10.4049/jimmunol.166.12.7556. [DOI] [PubMed] [Google Scholar]
- 140.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36(4):598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 141.Grieb G, Steffens G, Pallua N, Bernhagen J, Bucala R. Circulating fibrocytes–biology and mechanisms in wound healing and scar formation. Int Rev Cell Mol Biol. 2011;291:1–19. doi: 10.1016/B978-0-12-386035-4.00001-X. [DOI] [PubMed] [Google Scholar]
- 142.Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS, et al. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regul Toxicol Pharmacol. 1998;27(1 Pt 1):47–60. doi: 10.1006/rtph.1997.1163. [DOI] [PubMed] [Google Scholar]
- 143.Hasmall SC, James NH, Macdonald N, Gonzalez FJ, Peters JM, Roberts RA. Suppression of mouse hepatocyte apoptosis by peroxisome proliferators: role of PPARalpha and TNFalpha. Mutat Res. 2000;448(2):193–200. doi: 10.1016/s0027-5107(99)00236-5. [DOI] [PubMed] [Google Scholar]
- 144.Vansaun MN, Mendonsa AM, Lee Gorden D. Hepatocellular proliferation correlates with inflammatory cell and cytokine changes in a murine model of nonalchoholic fatty liver disease. PLoS One. 2013;8(9):e73054. doi: 10.1371/journal.pone.0073054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Rose ML, Germolec D, Arteel GE, Schoonhoven R, Thurman RG. Dietary glycine prevents increases in hepatocyte proliferation caused by the peroxisome proliferator WY-14,643. Chem Res Toxicol. 1997;10(10):1198–204. doi: 10.1021/tx970079u. [DOI] [PubMed] [Google Scholar]
- 146.Watanabe M, Chijiiwa K, Kameoka N, Yamaguchi K, Kuroki S, Tanaka M. Gadolinium pretreatment decreases survival and impairs liver regeneration after partial hepatectomy under ischemia/reperfusion in rats. Surgery. 2000;127(4):456–63. doi: 10.1067/msy.2000.104744. [DOI] [PubMed] [Google Scholar]
- 147.van Rooijen N, Sanders A, van den Berg TK. Apoptosis 1209 of macrophages induced by liposome-mediated intracellular delivery of clodronate and propamidine. J Immunol Methods. 1996;193(1):93–9. doi: 10.1016/0022-1759(96)00056-7. [DOI] [PubMed] [Google Scholar]
- 148.Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci. 2007;96(1):2–15. doi: 10.1093/toxsci/kfl173. [DOI] [PubMed] [Google Scholar]
- 149.Peters JM, Cheung C, Gonzalez FJ. Peroxisome proliferator-activated receptor-alpha and liver cancer: where do we stand? J Mol Med (Berl) 2005;83(10):774–85. doi: 10.1007/s00109-005-0678-9. [DOI] [PubMed] [Google Scholar]
- 150.Bojes HK, Germolec DR, Simeonova P, Bruccoleri A, Schoonhoven R, Luster MI, et al. Antibodies to tumor necrosis factor alpha prevent increases in cell replication in liver due to the potent peroxisome proliferator, WY-14,643. Carcinogenesis. 1997;18(4):669–74. doi: 10.1093/carcin/18.4.669. [DOI] [PubMed] [Google Scholar]
- 151.Rose ML, Rivera CA, Bradford BU, Graves LM, Cattley RC, Schoonhoven R, et al. Kupffer cell oxidant production is central to the mechanism of peroxisome proliferators. Carcinogenesis. 1999;20(1):27–33. doi: 10.1093/carcin/20.1.27. [DOI] [PubMed] [Google Scholar]
- 152.Wu J, Li J, Salcedo R, Mivechi NF, Trinchieri G, Horuzsko A. The proinflammatory myeloid cell receptor TREM-1 controls Kupffer cell activation and development of hepatocellular carcinoma. Cancer Res. 2012;72(16):3977–86. doi: 10.1158/0008-5472.CAN-12-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kan Z, Ivancev K, Lunderquist A, McCuskey PA, McCuskey RS, Wallace S. In vivo microscopy of hepatic metastases: dynamic observation of tumor cell invasion and interaction with Kupffer cells. Hepatology. 1995;21(2):487–94. [PubMed] [Google Scholar]
- 154.Gül N, Babes L, Siegmund K, Korthouwer R, Bögels M, Braster R, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014;124(2):812–23. doi: 10.1172/JCI66776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Rushfeldt C, Sveinbjørnsson B, Seljelid R, Smedsrød B. Early events of hepatic metastasis formation in mice: role of Kupffer and NK-cells in natural and interferon-gamma-stimulated defense. J Surg Res. 1999;82(2):209–15. doi: 10.1006/jsre.1998.5532. [DOI] [PubMed] [Google Scholar]
- 156.Van den Eynden GG, Majeed AW, Illemann M, Vermeulen PB, Bird NC, Høyer-Hansen G, et al. The multifaceted role of the microenvironment in liver metastasis: biology and clinical implications. Cancer Res. 2013;73(7):2031–43. doi: 10.1158/0008-5472.CAN-12-3931. [DOI] [PubMed] [Google Scholar]
- 157.Vidal-Vanaclocha F. The Tumor Microenvironment at Different Stages of Hepatic Metastasis. In: Brodt P, editor. Liver metastasis: biology and clinical management. Springer; 2011. pp. 43–87. [Google Scholar]
- 158.Yanagida H, Kaibori M, Yoshida H, Habara K, Yamada M, Kamiyama Y, et al. Hepatic ischemia/reperfusion upregulates the susceptibility of hepatocytes to confer the induction of inducible nitric oxide synthase gene expression. Shock. 2006;26(2):162–8. doi: 10.1097/01.shk.0000223130.87382.73. [DOI] [PubMed] [Google Scholar]
- 159.Timmers M, Vekemans K, Vermijlen D, Asosingh K, Kuppen P, Bouwens L, et al. Interactions between rat colon carcinoma cells and Kupffer cells during the onset of hepatic metastasis. Int J Cancer. 2004;112(5):793–802. doi: 10.1002/ijc.20481. [DOI] [PubMed] [Google Scholar]
- 160.Luo Y1, Ohmori H, Fujii K, Moriwaka Y, Sasahira T, Kurihara M, et al. HMGB1 attenuates anti-metastatic defence of the liver incolorectal cancer. Eur J Cancer. 2010;46(4):791–9. doi: 10.1016/j.ejca.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 161.Wen SW, Ager EI, Christophi C. Bimodal role of Kupffer cells duringcolorectal cancer liver metastasis. Cancer Biol Ther. 2013;14(7):606–13. doi: 10.4161/cbt.24593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Zhang Y, Wang XF. A niche role for cancer exosomes in metastasis. Nat Cell Biol. 2015;17(6):709–11. doi: 10.1038/ncb3181. [DOI] [PubMed] [Google Scholar]
- 163.Fesinmeyer MD, Austin MA, Li CI, De Roos AJ, Bowen DJ. Differences in survival by histologic type of pancreatic cancer. Cancer Epidemiol Biomarkers Prev. 2005;14(7):1766–73. doi: 10.1158/1055-9965.EPI-05-0120. [DOI] [PubMed] [Google Scholar]
- 164.Sampaio EP, Sarno EN, Galilly R, Cohn ZA, Kaplan G. Thalidomide selectively inhibits tumor 1253 necrosis factor alpha production by stimulated human monocytes. J Exp Med. 1991;173(3):699–703. doi: 10.1084/jem.173.3.699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Parman T, Wiley MJ, Wells PG. Free radical-mediated oxidative DNA damage in the mechanism of thalidomide teratogenicity. Nat Med. 1999;5(5):582–5. doi: 10.1038/8466. [DOI] [PubMed] [Google Scholar]