

The long-range coupling of e-h pairs on spatially separated donor and acceptor molecules is observed for the first time.
Keywords: Organic semiconductor, donor and acceptor molecules, electron-hole pairs
Abstract
Understanding exciton behavior in organic semiconductor molecules is crucial for the development of organic semiconductor-based excitonic devices such as organic light-emitting diodes and organic solar cells, and the tightly bound electron-hole pair forming an exciton is normally assumed to be localized on an organic semiconducting molecule. We report the observation of long-range coupling of electron-hole pairs in spatially separated electron-donating and electron-accepting molecules across a 10-nanometers-thick spacer layer. We found that the exciton energy can be tuned over 100 megaelectron volts and the fraction of delayed fluorescence can be increased by adjusting the spacer-layer thickness. Furthermore, increasing the spacer-layer thickness produced an organic light-emitting diode with an electroluminescence efficiency nearly eight times higher than that of a device without a spacer layer. Our results demonstrate the first example of a long-range coupled charge-transfer state between electron-donating and electron-accepting molecules in a working device.
INTRODUCTION
When a positive charge carrier and a negative charge carrier are drawn together by coulombic attraction, the carriers in some inorganic semiconductors are transformed into a bound pair, that is, an exciton, through electron-hole coupling. Similarly, when organic semiconducting molecules are excited through carrier recombination of electrons on a lowest unoccupied molecular orbital (LUMO) level and holes on a highest occupied molecular orbital (HOMO) level, they form localized excitons on a molecule, as shown in Fig. 1A for the example of 4,4′,4″-tris(N-3-methylphenyl-N-phenylamino)triphenylamine (m-MTDATA). The excitons are very stable because of their rather large exciton binding energies (Eb) of 0.5 to 1.0 eV (1, 2), so they are usually classified as Frenkel excitons (3, 4).
Fig. 1. Photoluminescence characteristics of the exciplex system.
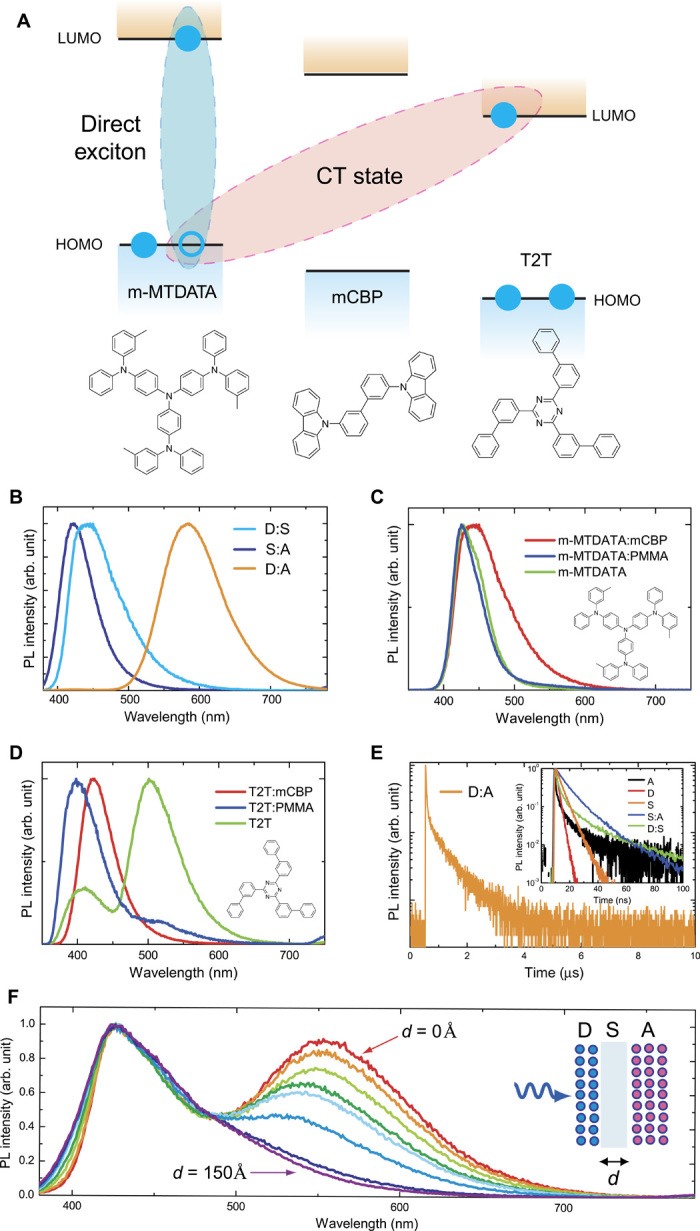
(A) Chemical structures of electron-donating (D), spacer (S), and electron-accepting (A) molecules. (B) Steady-state fluorescence spectra of D:S, A:S, and D:A co-deposited films. (C and D) Steady-state fluorescence spectra of neat and doped films. The broad emission band with a peak at around 500 nm in the T2T neat film is assigned to excimer emission [that is, 1(AA)*]. (E) Room-temperature time-resolved fluorescence decay curve of a D:A film. Inset: Room-temperature time-resolved fluorescence decay curves of D:S and S:A films. (F) Steady-state fluorescence spectra of multilayer thin films [10-nm-thick D/x-nm-thick S (x = 0, 10, 30, 50, 70, 90, 120, and 150 Å)/20-nm-thick A]. Inset: Schematic illustration of a multilayer D (blue)/S/A (red) film. The excitation light (λex = 300 nm) was incident from the side of the D layer.
An important issue in organic electronics is controlled modulation of exciton behavior in organic semiconducting thin films to better understand exciton behavior and develop efficient organic-based excitonic devices such as organic light-emitting diodes (OLEDs) (5, 6) and organic solar cells (OSCs) (7, 8). In particular, methods to control the strength of Eb would be of great use for various device applications such as excitonic transistors (9, 10). However, most organic molecules have rather large Eb, resulting in the formation of Frenkel excitons, that is, excitons strongly localized on individual molecules. Thus, controlling the spatial separation of electron-hole coupling is rather difficult.
In contrast, photon radiation from a charge-transfer (CT) state formed by electron-hole coupling between two distinct molecules, that is, electron-donating (D) and electron-accepting (A) molecules, to form an exciplex is well known, as illustrated in Fig. 1A for the combination of m-MTDATA and 2,4,6-tris(biphenyl-3-yl)-1,3,5-triazine (T2T) (11–17). Because the photon radiation from an exciplex occurs as a result of an electron transition from the LUMO of an acceptor to the HOMO of a donor, a well-defined electron-hole spacer between them will influence the exciton decay characteristics. For efficient electron transfer from an excited donor molecule to the ground state of an acceptor molecule, it is believed that the two molecules should be physically adjacent. The dynamics of exciplex formation through long-range electron-hole coupling of spatially separated D-A pairs in organic thin films have yet to be reported. In addition, because the recombination process of a CT state limits the charge separation process in OSCs (18, 19), understanding the interfacial CT states of D-A heterointerfaces is important to further improve charge separation and photocurrent generation.
Herein, we observe long-range electron-hole coupling for spatially separated organic semiconducting D-A heterointerfaces, and we successfully manipulate the characteristics of CT excitons by finely controlling the D-A separation distance using an organic thin film structure of “D–spacer (S)–A.” To investigate the excited states formed at the thin film interfaces between donor, spacer, and acceptor molecules, we prepared films with m-MTDATA (20) as the electron-donating molecule, 3,3-di(9H-carbazol-9-yl)biphenyl (mCBP) (21) as the spacer molecule, and T2T (22) as the electron-accepting molecule. The chemical structures of these materials are shown in Fig. 1A.
RESULTS
First, the interactions between the excited states of the donor, spacer, and acceptor in codeposited thin films were examined by steady-state photoluminescence (PL) measurements. Figure 1B shows the PL spectra of co-deposited films of m-MTDATA:T2T, m-MTDATA:mCBP, and mCBP:T2T each co-deposited with a 1:1 molar ratio. The PL spectrum of the co-deposited D:A film was greatly red-shifted relative to that of the excited donor [denoted 1(D)*] and the excited acceptor [denoted 1(A)*] (see Fig. 1, C and D). As a result, the film showed orange emission with a peak at 585 nm, which corresponds to an exciton energy of 2.12 eV and a PL quantum efficiency (ΦPL) of 5%. In addition, these co-deposited thin films showed no additional absorption bands in the ground state absorption spectra as shown in fig. S1, indicating that no ground state interactions exist between these molecules. The change in emission can be explained by the formation of a bimolecular excited state, that is, an exciplex, between D and A: a1(1(D)*A) + a2(D1(A)*) → b1(Dδ+Aδ−)* + b2(Dδ−Aδ+)* → hvex + D + A, where a1, a2, b1, and b2 are proportionality constants for the D:A co-deposited film. Although the excitons in both the D:S and S:A systems show short transient lifetimes of less than 100 ns (Fig. 1E, inset), the time-resolved fluorescence of a D:A co-deposited film measured at 300 K revealed the presence of not only a prompt component with a transient decay time (τp) of ~33 ns but also a long-lived delayed component with a decay time (τd) of ~0.7 μs (Fig. 1E). In addition, the emission spectrum of the delayed component nearly coincides with that of the prompt component (fig. S2). Because the energy levels of 3(D)* (~2.61 eV) and 3(A)* (~2.80 eV) are sufficiently higher than the exciton energy level of 1(Dδ+Aδ−)*, these features confine the exciplex triplet excitons well, resulting in the occurrence of thermally activated delayed fluorescence (TADF) (17, 23–25).
Further considering the D/S and S/A interactions, blue emission peaks around 425 and 445 nm were observed for the D:S and S:A co-deposited films, respectively. The emission peaks of these co-deposited films exhibited a slight red shift (~0.2 eV) compared with those of the neat films of the donor and acceptor molecules (Fig. 1, B to D). Although a single-exponential decay with a lifetime of ~2 ns was observed in an m-MTDATA neat film, a multiexponential decay with the components having lifetimes of ~2.5 and ~68 ns was observed in the m-MTDATA:mCBP co-deposited thin film. In addition, the lifetimes of the radiative decay components of the T2T:mCBP co-deposited film (~6 and ~15 ns) were longer than those of the T2T neat film. These PL decay characteristics indicate the presence of weak interactions between 1(D)* and 1(A)* and the ground state of the S molecules. However, these radiative decay lifetimes are quite short compared to that of the D:A system. These results imply that mCBP has bipolar characteristics and acts as a weak electron acceptor for m-MTDATA and a weak electron donor for T2T. Also, we note that whereas broad excimer emission was observed around 500 nm for a neat film of T2T (see Fig. 1D), we ignored the excimer in our discussion of S/A and D/A interactions because the proportion of excimers should be small.
When m-MTDATA molecules were directly excited by photoabsorption, multilayered thin films with the structure of 10-nm-thick D/x-nm-thick S (x = 0, 1, 3, 5, 7, 9, 12, and 15 nm)/20-nm-thick A exhibited clear exciplex emission on the basis of the D/A interaction in addition to the 1(D)* emission (Fig. 1F). These results strongly imply that an electron moves from the excited m-MTDATA layer to the T2T layer through the mCBP spacer layer, involving rather long-range electron transfer through the spacer layer, before formation of the exciplex state 1(Dδ+Aδ−)*. The exciplex formation and emission mechanisms of the multilayer films under optical excitation can be described by Eq. 1
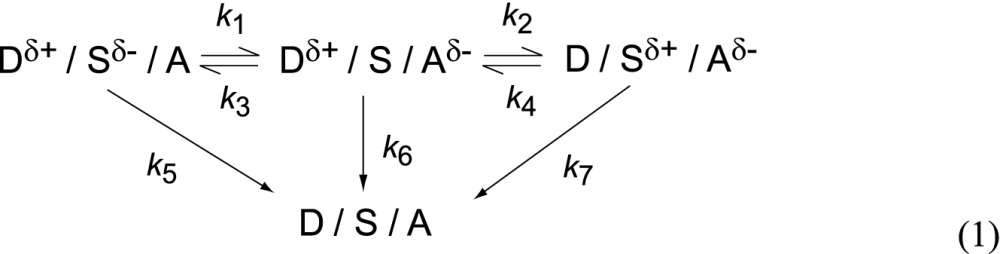 |
Under optical excitation, because the exciplex energy of D:A is smaller than that of D:S, the transfer rate (k1) should be larger than the competitive rate (k3), that is, k3 > k1. In a similar manner, it is expected that k4 > k2. After photoexcitation of either m-MTDATA or T2T, the excitons are transferred to form exciplexes (Dδ+/Sδ−/A) or (D/Sδ+/Aδ−), respectively, and some of these exciplexes then form (Dδ+/S/Aδ−), which is the lowest energy state of the three, through cascade electron transfer between the three molecules. These intermolecular CT states can radiatively decay from the lowest singlet excited state (S1) to the ground state with rate constants of k5, k6, and k7. When the thickness of the spacer layer (d) was increased, a slight broadening of the blue emission band was observed (Fig. 1F), suggesting that the contribution of the radiative decay paths corresponding to k5 and k7 increases because of a decrease in the contribution of that of k6. Although the intensity of exciplex emission gradually decreased with increasing d, resulting in no exciplex emission from the thin films with d = 12 and 15 nm, the most important aspect of these spectra is the presence of a 1(Dδ+Aδ−)* state in spatially separated D-A pairs, even with a 9-nm-thick spacer layer.
The clear interaction between holes on m-MTDATA and electrons on T2T was confirmed through the direct generation of radical cations of m-MTDATA and radical anions of T2T. To demonstrate the long-range coupling of electron-hole pairs in the spatially separated D-S-A films under electrical excitation, we studied the performance of OLEDs with a hole transporting layer (HTL) of m-MTDATA and an electron transporting layer (ETL) of T2T separated by an mCBP spacer layer, the energy diagram of which is depicted in Fig. 2A. In this device architecture, holes are injected from a dipyrazino[2,3-f:20,30-h]quinoxaline-2,3,6,7,10,11-hexacarbonitrile (HAT-CN) layer into the HOMO level of the m-MTDATA layer (−5.1 eV), and electrons are injected from an aluminum electrode into the LUMO level of the T2T layer (−2.8 eV) via the LUMO level of a tris(8-hydroxyquinolinato)aluminum (Alq3) layer. The carriers mainly accumulate at the interface at each side of the mCBP layer because of the rather high energy barriers for carrier injection into mCBP of 0.9 eV for hole injection and 0.4 eV for electron injection.
Fig. 2. Electroluminescence characteristics of the exciplex system.
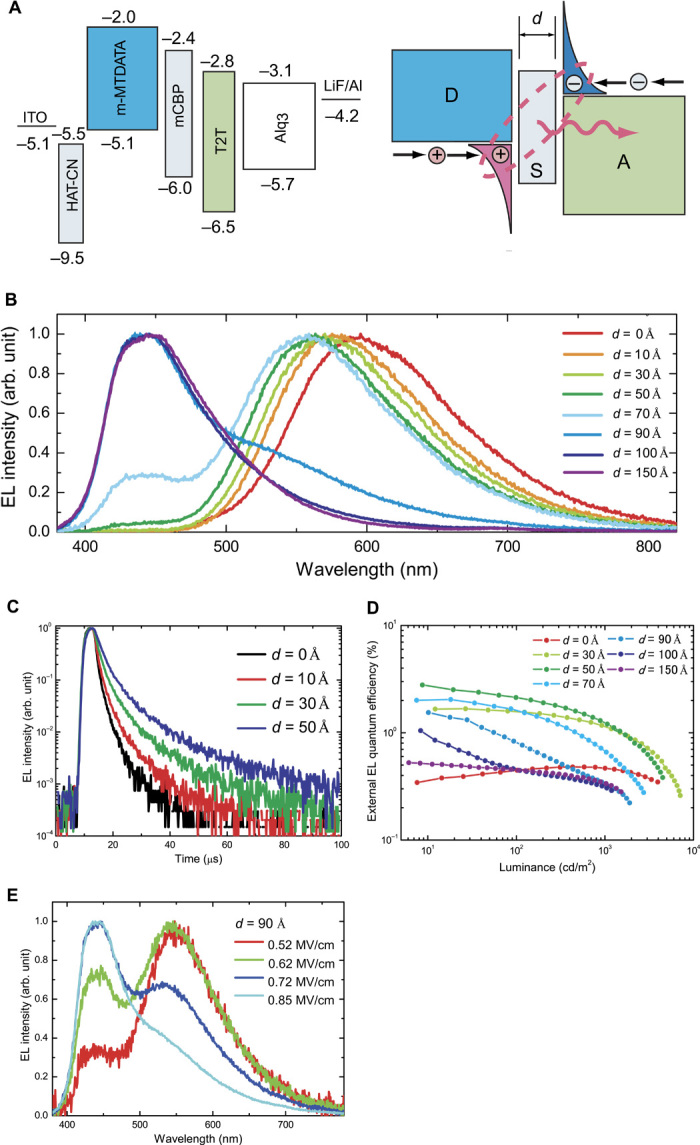
(A) Structures and energy diagram of OLEDs. Energy gaps were estimated from absorption edges of films. The anode and cathode were 100-nm-thick indium tin oxide and 5-nm-thick lithium fluoride/100-nm-thick aluminum, respectively. (B) EL spectra of devices with various spacer-layer thickness (d) at a luminance of 1000 cd/m2. (C) Room-temperature time-resolved EL decay curves for devices with d = 0, 10, 30, and 50 Å. OLEDs were exposed to a pulse voltage with a duration of 5 μs. (D) External EL quantum efficiency against luminance for devices with various d. (E) Dependence of EL spectra on applied electrical field for devices with a spacer-layer thickness of d = 90 Å.
Observation of a spectral response that is dependent on d from 1(Dδ+Aδ−)* states in devices provides initial evidence for long-range coupling of electron-hole pairs under electrical excitation. Figure 2B shows the electroluminescence (EL) spectra for devices with various d at a luminance of 1000 cd/m2. In the device with d = 0 nm, only orange emission with a peak at 593 nm (~2.09 eV) was observed, indicating the formation of a 1(Dδ+Aδ−)* state at the D-A interface. This phenomenon is very similar to the PL process. However, because electrons (radical anions) and holes (radical cations) directly accumulated at the interfaces of mCBP/T2T and m-MTDATA/mCBP, respectively, the exciplex formation process is simply described by the reaction
| (2) |
Thus, EL components originating from 1(m-MTDATA)* and 1(T2T)* are not observed.
Although a gradual blue shift of the EL spectra corresponding to a shift in exciton energy from 2.09 to 2.21 eV was observed as d increased from 0 to 7 nm, the devices still showed broad EL originating mainly from 1(Dδ+Aδ−)* with the gradual appearance of additional EL components in the blue region originating from 1(D)* or 1(A)*, indicating that the electrons and holes were still interacting through a rather thick spacer layer. The blue shift of EL from 1(Dδ+Aδ−)* could be explained by a decrease in the electron-hole coulombic attraction energy [EC = e2/4πεε0r, where e is elementary charge, ε is the relative permittivity of organic materials (~3.5), ε0 is the permittivity of free space, and r is the distance between the charges, respectively] contributing to the hvex of 1(Dδ+Aδ−)* according to the equation hvex ≈ ID (ionization potential of the donor) − AA (electron affinity of the acceptor) − EC, as shown in fig. S3. Here, the Eb of 1(Dδ+Aδ−)* is calculated to be ~0.21 eV for the device with d = 0 nm from the difference between the exciton energy (~2.09 eV) and the energy gap between ID and AA (~2.3 eV). Eb slightly decreases with an increase in the spacer-layer thickness, resulting in Eb = ~0.09 eV for the device with d = 7 nm. For devices with d > 7 nm, the intensity of the emission band around 450 nm corresponding to 1(Dδ+Sδ−)* gradually increased with a decrease in the 1(Dδ+Aδ−)* emission intensity, indicating that the electrons accumulate at the D/S interface because of electron injection into the LUMO level of mCBP at high forward electrical fields of more than 0.8 MV/cm. Furthermore, whereas broad EL originating from 1(Dδ+Aδ−)* was still observed in the devices with d = 9 and 10 nm at low current density (see fig. S4), the device with d = 15 nm showed only 1(D)* emission even at low current density, indicating the limitation of the distance for electron coupling in this D-S-A combination.
The presence of a long-range coupled 1(Dδ+Aδ−)* state was further confirmed by time-resolved EL measurements of the devices under electrical excitation using short voltage pulses. Figure 2C shows the transient decay curves obtained for the devices. Clear delayed EL components after pulse excitation were observed from the devices with d = 0 to 5 nm for the emission band at >500 nm, suggesting the contribution of the triplet states of the (Dδ+Aδ−) complex, that is, upconversion of 3(Dδ+Aδ−)* to 1(Dδ+Aδ−)* via TADF (17). For the device with d = 0 nm, the EL intensity decayed rapidly, and weak delayed EL with a transient decay time (τd-EL) of ~4.3 μs was observed. We found that τd-EL increased with d, with τd-EL of ~8.1 and ~13.2 μs for devices with d = 3 and 5 nm, respectively. One possible reason for the increase in τd-EL is the reduction in nonradiative decay from the 3(Dδ+Aδ−)* state to the ground state (25). The 10 mol % D:10 mol % A:80 mol % S co-deposited films had a rather high ΦPL of 25%, which is about five times higher than that of 50 mol % D:50 mol % A co-deposited films, indicating that increasing the separation distance between D and A molecules improves the overall probability for radiative decay. From this result, a reduction in the nonradiative decay constant for the 3(Dδ+Aδ−)* state can be expected, which would thereby increase the triplet lifetime and τd-EL. Although a large blue shift of EL was observed for the device with d = 10 nm, weak delayed EL was still detected (fig. S5), confirming the formation of the 1(Dδ+Aδ−)* exciplex at the interfaces even through a 10-nm-thick spacer layer.
The contribution of the delayed EL to the total EL intensity increased with d, as shown in Fig. 2C, suggesting that the 3(Dδ+Aδ−)* state was effectively converted to the 1(Dδ+Aδ−)* state through reverse intersystem crossing (RISC) to obtain efficient radiative decay from the 1(Dδ+Aδ−)* state to the ground state. This can be caused by the increased separation of electron wave functions of the HOMO (ΨH) and LUMO (ΨL) in the 1(Dδ+Aδ−)* state with increasing d, narrowing the energy gap between the singlet and triplet states (ΔEST) (25). To measure the ΔEST of the D-S-A films, the phosphorescent radiative decay from the 3(Dδ+Aδ−)* state to the ground state in 50 mol % D:50 mol % A and 10 mol % D:10 mol % A:80 mol % S co-deposited films was measured at 5 K (see fig. S2). The D:A co-deposited film exhibited weak phosphorescence, and the estimated onset of the phosphorescence spectrum, that is, lowest triplet state (T1), was 2.36 eV. Because the S1 of this film was 2.45 eV, the ΔEST between the energy levels of the 1(Dδ+Aδ−)* and 3(Dδ+Aδ−)* states of the D:A co-deposited film was calculated to be 0.09 eV. Similarly, the 10 mol % D:10 mol % A:80 mol % S co-deposited film showed a rather small ΔEST of 0.04 eV with S1 = 2.56 eV and T1 = 2.52 eV, indicating that artificial control of ΔEST in the same molecular system is possible by spatially separating the D and A molecules.
Because of the large proportion of the delayed EL, the maximum external EL quantum efficiency (ηEQE) markedly increased with d from 0 to 5 nm, resulting in a rather high ηEQE of 2.8% for the device with d = 5 nm. This value is about eight times higher than that of the device without a spacer layer, as shown in Fig. 2D. We note that the ηEQE of 2.8% is also about three times higher than that of a device with 50 mol % m-MTDATA:50 mol % T2T as an emissive layer (see fig. S6). Conversely, when m-MTDATA and T2T layers are separated by >5 nm, ηEQE decreases with increasing d. In addition, for the device with d = 9 nm, although efficiency enhancement by a factor of more than 2 was achieved at low current densities, ηEQE decreased rapidly with increasing luminance, corresponding to the disappearance of the emission band originating from the 1(Dδ+Aδ−)* state, as shown in Fig. 2E and fig. S4.
Here, we discuss the dependence of EL characteristics on the spacer-layer material. When 9,10-di(2-naphthyl)anthracene (ADN) was used instead of mCBP as a spacer layer, although broad orange emission originating from the 1(Dδ+Aδ−)* state was observed from a device with a 5-nm-thick ADN spacer layer, the ηEQE of this device was <0.5%. Because the T1 level of ADN (1.69 eV) (26) is much lower than that of the 3(Dδ+Aδ−)* state of the m-MTDATA–T2T system (2.36 eV), the triplet excitons are quenched by the lower triplet level of the spacer layer. No delayed EL component after pulse voltage excitation was observed for this device (see fig. S7), indicating that there is no contribution from triplet excitons. On the other hand, because mCBP molecules have a high triplet energy level (~2.90 eV), the energy of the 3(Dδ+Aδ−)* state of the m-MTDATA–T2T system can be confined to the exciplex in the D/S/A heterojunction. These results clearly reveal the contribution of the 3(Dδ+Aδ−)* state of m-MTDATA:T2T to the total EL intensity of devices with an mCBP spacer layer.
Finally, we explore the possibility of energy transfer from exciplexes to fluorescent dopants with the goal of enhancing ηEQE. Tuning the exciton energy of exciplexes by spatially separating the D-A pairs enables us to transfer energy to emissive donor molecules. To transfer energy from long-range coupled exciplexes to emissive donor molecules, tetraphenyldibenzoperiflanthene (DBP) was doped as an additional energy acceptor molecule into the spacer layer [that is, mCBP (1.5 nm)/2 wt % DBP:mCBP (2 nm)/mCBP (1.5 nm)], as shown in the inset of Fig. 3A. Figure 3A presents the EL spectrum of this device. Under electrical excitation, additional emission bands with peaks at λ = 610, 661, and 725 nm are clearly observed, corresponding to the emission peaks of DBP in an mCBP host matrix under optical excitation (dashed line).
Fig. 3. Resonant transfer of the exciplex energy to energy acceptor molecules.
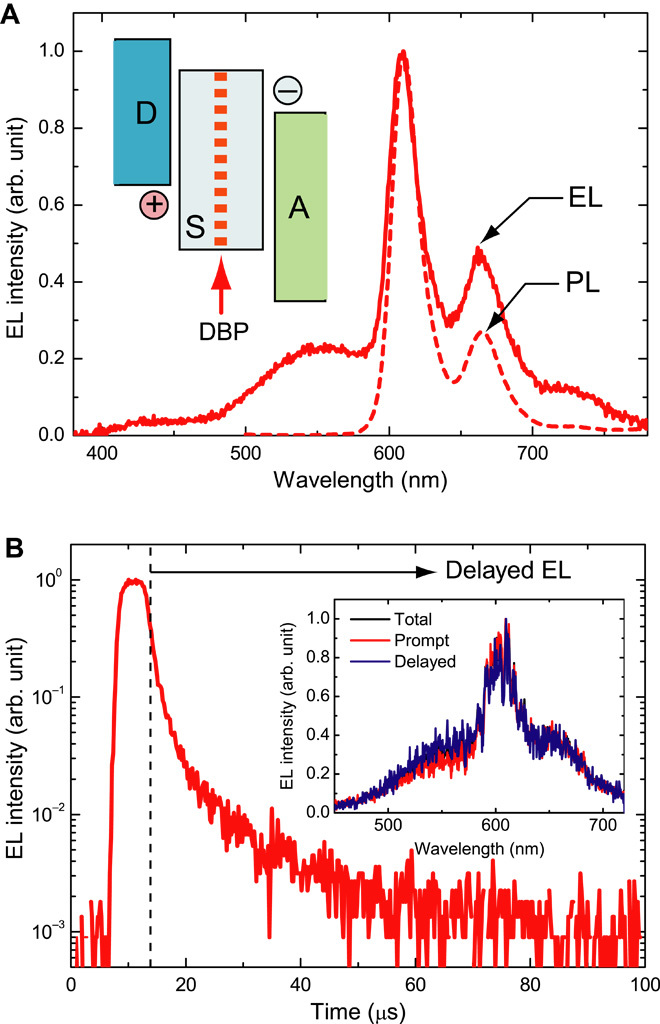
(A) EL spectrum of an energy transfer–type device at a luminance of 200 cd/m2. The dashed line shows the PL spectrum of a 2 wt % DBP:mCBP co-deposited film. Inset: Schematic illustration of the emission layer. (B) Time-resolved EL decay curves measured for a device with d = 50 Å at room temperature. EL was accumulated after exposure of each OLED to a pulse voltage with a duration of 5 μs. Inset: Accumulated emission spectra for total (black), prompt (red), and delayed (blue) components.
Figure 3B shows the transient EL decay curves obtained for this device. A clear delayed EL component was observed after the excitation pulse for the emission band at 610 nm, and the emission spectrum of the delayed component matched that of the prompt component well, indicating that the triplet energy of the exciplex was resonantly transferred to the S1 state of DBP after RISC in the exciplex. Although singlet and triplet excitons generated in the D-S-A system can both be transferred to the DBP molecules, the device showed a limited maximum ηEQE of 2.4%, which is comparable to that of the device with d of 5 nm. The low ηEQE may result from the in-plane distribution of DBP decreasing the Förster energy transfer rate between the exciplex and DBP because the direction of the dipoles in the exciplexes should be perpendicular to the film thickness.
DISCUSSION
We can draw three important conclusions from our D-S-A system. First, an electron on an acceptor molecule and a hole on a donor molecule can interact across a 10-nm-thick spacer layer, and we confirmed the presence of long-range coupled CT states formed in such spatially separated D-A pairs. Second, exciplex characteristics, that is, exciton energy, radiative decay lifetime, and exciton production efficiency, under optical and electrical excitation can be modulated by adjusting the distance of electron-hole coupling. Third, the energy of the spatially delocalized exciplex that forms between a spatially separated D-A pair can be harvested by additional energy acceptor molecules. In this device architecture, because no direct carrier injection into emissive molecules (that is, DBP) is required to obtain DBP emission, the carrier recombination and photon-radiation zones can be spatially separated while a high electrical field is applied to the thin spacer layer. Because numerous organic semiconducting D-A systems for light emission (15) and charge separation (27) have been developed, we expect that our observation of long-range coupling of electron-hole pairs in organic D-S-A multilayers reveals new possibilities for developing organic excitonic devices and molecular electronics (28–30).
MATERIALS AND METHODS
Materials
HAT-CN, m-MTDATA, and mCBP were purchased from NARD Institute Ltd. Alq3 was used as received from Nippon Steel Chemical Co. Ltd. T2T was synthesized according to a reported procedure (22).
Optical characterization of organic thin films
Steady-state PL spectra of organic films were recorded by a spectrophotometer (FP-6500-A-51, JASCO). PL quantum efficiency was measured by an absolute PL quantum yield measurement system (C11347-01, Hamamatsu Photonics) under a flow of nitrogen gas with an excitation wavelength of 337 nm. Low-temperature PL intensity and emission lifetime were measured using a streak camera (C4334, Hamamatsu Photonics) and cryostat (Iwatani Industrial Gases Co.) with a nitrogen gas laser (MNL200, Laser Technik) as an excitation light source under a pressure of about 3 Pa. HOMO levels were measured by photoelectron spectroscopy (Riken Keiki, AC-3).
Fabrication of OLEDs
Glass substrates with prepatterned 100 ohm/sq tin-doped indium oxide (ITO) were used as anodes. Substrates were washed by sequential ultrasonication in neutral detergent, distilled water, acetone, and isopropanol, and then exposed to ultraviolet (UV)–ozone (NL-UV253, Nippon Laser & Electronics Lab) to remove adsorbed organic species. After precleaning, effective device areas of 1 or 4 mm2 were defined on the glass substrates with a 100-nm-thick ITO coating by a polyimide insulation layer using a conventional photolithography technique. Substrates were treated with UV-ozone for 25 min and then immediately transferred into an evaporation chamber.
Organic layers were formed by thermal evaporation. Doped layers were deposited by coevaporation. Deposition was performed under vacuum at a pressure of <5 × 10−5 Pa. After fabrication, devices were immediately encapsulated with glass lids using epoxy glue in a nitrogen-filled glove box (O2 <0.1 ppm, H2O <0.1 ppm). Commercial calcium oxide desiccant (Dynic Co.) was included in each encapsulated package.
Characterization of OLEDs
The current density–voltage–luminance characteristics of the OLEDs were evaluated using a source meter (Keithley 2400, Keithley Instruments Inc.) and an absolute external quantum efficiency measurement system (C9920-12, Hamamatsu Photonics). Each EL spectrum was collected by an optical fiber connected to a spectrometer (PMA-12, Hamamatsu Photonics). Time-resolved EL decay curves were obtained using a streak camera (C4334, Hamamatsu Photonics) with a pulse generator (81101A, Agilent) as an electrical excitation source. We used a short pulse with a width of 5 μs and a fixed voltage of 7 V. All measurements were performed in ambient atmosphere at room temperature.
Supplementary Material
Acknowledgments
We thank W. J. Potscavage Jr. for his assistance with the preparation of the manuscript. Funding: This work was supported by the Japan Science and Technology Agency (JST), ERATO, Adachi Molecular Exciton Engineering Project. Author contributions: H.N. developed the basic concepts. The experiments were conceived, designed, and carried out by H.N., T.F., and K.M. H.N. and C.A. wrote the manuscript. The project was supervised by C.A. All the authors discussed the results and contributed to the article. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501470/DC1
Fig. S1. UV-vis absorption spectra.
Fig. S2. Time-resolved PL spectra of D:A co-deposited films.
Fig. S3. The dependence of exciton energy on distance between D and A.
Fig. S4. Dependence of EL spectra on luminance.
Fig. S5. Time-resolved EL spectra for a device with spacing layer thickness d = 100 Å.
Fig. S6. External EL quantum efficiency for a device with spacing layer and for a device with a D:A co-deposited film as an emissive layer.
Fig. S7. Performance of an OLED with an ADN spacer layer.
REFERENCES AND NOTES
- 1.Hill I. G., Kahn A., Soos Z. G., Pascal R. A. Jr, Charge-separation energy in films of π-conjugated organic molecules. Chem. Phys. Lett. 327, 181–188 (2000). [Google Scholar]
- 2.Nayak P. K., Periasamy N., Calculation of electron affinity, ionization potential, transport gap, optical band gap and exciton binding energy of organic solids using “solvation” model and DFT. Org. Electron. 10, 1396–1400 (2009). [Google Scholar]
- 3.N. S. Sariciftci, Ed., Primary Photoexcitations, in Conjugated Polymers: Molecular Exciton Versus Semiconductor Band Model (World Scientific, Singapore, 1997). [Google Scholar]
- 4.M. Pope, C. E. Swenberg, Electronic Processes in Organic Crystals and Polymers (Oxford Univ. Press, New York, ed. 2, 1999) 1328 pp. [Google Scholar]
- 5.Tang C. W., VanSlyke S. A., Organic electroluminescent diodes. Appl. Phys. Lett. 51, 913–915 (1987). [Google Scholar]
- 6.Burroughes J. H., Bradley D. D. C., Brown A. R., Marks R. N., Mackay K., Friend R. H., Bums P. L., Holmes A. B., Light-emitting diodes based on conjugated polymers. Nature 347, 539–541 (1990). [Google Scholar]
- 7.Tang C. W., Two-layer organic photovoltaic cell. Appl. Phys. Lett. 48, 183–185 (1986). [Google Scholar]
- 8.Yu G., Gao J., Hummelen J. C., Wudl F., Heeger A. J., Polymer photovoltaic cells: Enhanced efficiencies via a network of internal donor-acceptor heterojunctions. Science 270, 1789–1791 (1995). [Google Scholar]
- 9.High A. A., Novitskaya E. E., Butov L. V., Hanson M., Gossard A. C., Control of exciton fluxes in an excitonic integrated circuit. Science 321, 229–231 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Grosso G., Graves J., Hammack A. T., High A. A., Butov L. V., Hanson M., Gossard A. C., Excitonic switches operating at around 100 K. Nat. Photon. 3, 577–580 (2009). [Google Scholar]
- 11.Foster R., Electron donor-acceptor complexes. J. Phys. Chem. 84, 2135–2141 (1980). [Google Scholar]
- 12.Mattes S. L., Farid S., Exciplexes and electron transfer reactions. Science 226, 917–921 (1984). [DOI] [PubMed] [Google Scholar]
- 13.Jenekhe S. A., Osaheni J. A., Excimers and exciplexes of conjugated polymers. Science 265, 765–768 (1994). [DOI] [PubMed] [Google Scholar]
- 14.Jenekhe S. A., Excited-state complexes of conjugated polymers. Adv. Mater. 7, 309–311 (1995). [Google Scholar]
- 15.Wang J.-F., Kawabe Y., Shaheen S. E., Morrell M. M., Jabbour G. E., Lee P. A., Anderson J., Armstrong N. R., Kippelen B., Mash E. A., Peyghambarian N., Exciplex electroluminescence from organic bilayer devices composed of triphenyldiamine and quinoxaline derivatives. Adv. Mater. 10, 230–233 (1998). [Google Scholar]
- 16.Noda T., Ogawa H., Shirota Y., A blue-emitting organic electroluminescent device using a novel emitting amorphous molecular material, 5,5′-bis(dimesitylboryl)-2,2′-bithiophene. Adv. Mater. 11, 283–285 (1999). [Google Scholar]
- 17.Goushi K., Yoshida K., Sato K., Adachi C., Organic light-emitting diodes employing efficient reverse intersystem crossing for triplet-to-singlet state conversion. Nat. Photon. 6, 253–258 (2012). [Google Scholar]
- 18.Brédas J.-L., Norton J. E., Cornil J., Coropceanu V., Molecular understanding of organic solar cells: The challenges. Acc. Chem. Res. 42, 1691–1699 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Clarke T. M., Durrant J. R., Charge photogeneration in organic solar cells. Chem. Rev. 110, 6736–6767 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Shirota Y., Kuwabara Y., Inada H., Wakimoto T., Nakada H., Yonemoto Y., Kawami S., Imai K., Multilayered organic electroluminescent device using a novel starburst molecule, 4,4′,4′-tris(3-methylphenylphenylamino)triphenylamine, as a hole transport material. Appl. Phys. Lett. 65, 807–809 (1994). [Google Scholar]
- 21.Schrögel P., Langer N., Schildknecht C., Wagenblast G., Lennartz C., Strohriegl P., Meta-linked CBP-derivatives as host materials for a blue iridium carbene complex. Org. Electron. 12, 2047–2055 (2011). [Google Scholar]
- 22.Chen H.-F., Yang S.-J., Tsai Z.-H., Hung W.-Y., Wang T.-C., Wong K.-T., 1,3,5-Triazine derivatives as new electron transport–type host materials for highly efficient green phosphorescent OLEDs. J. Mater. Chem. 19, 8112–8118 (2009). [Google Scholar]
- 23.Endo A., Ogasawara M., Takahashi A., Yokoyama D., Kato Y., Adachi C., Thermally activated delayed fluorescence from Sn4+–porphyrin complexes and their application to organic light emitting diodes—A novel mechanism for electroluminescence. Adv. Mater. 21, 4802–4806 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Q., Li J., Shizu K., Huang S., Hirata S., Miyazaki H., Adachi C., Design of efficient thermally activated delayed fluorescence materials for pure blue organic light emitting diodes. J. Am. Chem. Soc. 134, 14706–14709 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Uoyama H., Goushi K., Shizu K., Nomura H., Adachi C., Highly efficient organic light-emitting diodes from delayed fluorescence. Nature 492, 234–238 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Zhang Y., Forrest S. R., Existence of continuous-wave threshold for organic semiconductor lasers. Phys. Rev. B. 84, 241301 (2011). [Google Scholar]
- 27.Congreve D. N., Lee J., Thompson N. J., Hontz E., Yost S. R., Reusswig P. D., Bahlke M. E., Reineke S., Voorhis T. V., Baldo M. A., External quantum efficiency above 100% in a singlet-exciton-fission–based organic photovoltaic cell. Science 340, 334–337 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Albinsson B., Mårtensson J., Long-range electron and excitation energy transfer in donor–bridge–acceptor systems. J. Photochem. Photobiol. C 9, 138–155 (2008). [DOI] [PubMed] [Google Scholar]
- 29.Helms A., Heiler D., McLendon G., Electron transfer in bis-porphyrin donor-acceptor compounds with polyphenylene spacers shows a weak distance dependence. J. Am. Chem. Soc. 114, 6227–6238 (1992). [Google Scholar]
- 30.Davis W. B., Svec W. A., Ratner M. A., Wasielewski M. R., Molecular-wire behaviour in p-phenylenevinylene oligomers. Nature 396, 60–63 (1998). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/2/e1501470/DC1
Fig. S1. UV-vis absorption spectra.
Fig. S2. Time-resolved PL spectra of D:A co-deposited films.
Fig. S3. The dependence of exciton energy on distance between D and A.
Fig. S4. Dependence of EL spectra on luminance.
Fig. S5. Time-resolved EL spectra for a device with spacing layer thickness d = 100 Å.
Fig. S6. External EL quantum efficiency for a device with spacing layer and for a device with a D:A co-deposited film as an emissive layer.
Fig. S7. Performance of an OLED with an ADN spacer layer.