

Abstract
Atherosclerosis is mediated by local and systematic inflammation. The multi-ligand receptor for advanced glycation end products (RAGE) has been studied in animals and humans, and is an important mediator of inflammation and atherosclerosis. This review focuses on S100/calgranulin proteins (S100A8, S100A9, and S100A12) and their receptor RAGE in mediating vascular inflammation. Mice lack the gene for S100A12, which in humans is located on chromosome 3 between S100A8 and S100A9. Transgenic mice with smooth muscle cell targeted expression of S100A12 demonstrate increased coronary and aortic calcification as well as increased plaque vulnerability. Serum S100A12 has recently been shown to predict future cardiovascular events in a longitudinal population study, underscoring a role for S100A12 as a potential biomarker for coronary artery disease. Genetic ablation of S100A9 or RAGE in atherosclerosis susceptible Apolipoprotein E (ApoE) null mice results in reduced atherosclerosis. Importantly, S100A12 and the RAGE axis can be modified pharmacologically. For example, soluble RAGE reduces murine atherosclerosis and vascular inflammation. Additionally, a class of compounds currently in phase III clinical trials for multiple sclerosis and rheumatologic conditions, the Quinoline-3-carboxamides, reduce atherosclerotic plaque burden and complexity in transgenic S100A12 ApoE null mice, but have not been tested with regards to human atherosclerosis. The RAGE axis is an important mediator for inflammation-induced atherosclerosis and S100A12 has emerged as biomarker for human atherosclerosis. Decreasing inflammation by inhibiting S100/calgranulin-mediated activation of RAGE attenuates murine atherosclerosis, and future studies in patients with coronary artery disease are warranted to confirm S100/RAGE as therapeutic target for atherosclerosis.
Keywords: Atherosclerosis, Inflammation, S100/Calgranulins, S100A12
Introduction
With an estimated 17.5 million deaths yearly, cardiovascular diseases remain the leading cause of death in the world.[1] Atherosclerosis is a chronic inflammatory process of the vascular wall mediated by activated macrophages, T lymphocytes, B lymphocytes, and smooth muscle cells (SMC).[2,3] Accordingly, multiple inflammatory serum markers have been investigated for their association with atherosclerosis.[4–7] Although there is evidence that statins reduce high sensitivity C-reactive protein (hs-CRP), there have been only few, and mostly small randomized controlled trials of immune modulating drugs with respect to atherosclerosis.[8, 9] Therefore, the results of the two large ongoing trials, the Cardiovascular Inflammation Reduction Trial (CIRT), testing low dose methotrexate in patients with prior myocardial infarction (MI) and either diabetes mellitus (DM) or metabolic syndrome and the CANTOS trial, testing the monoclonal antibody Canakinumab [10,11] are eagerly awaited.
The multi-ligand receptor for advanced glycation end products (RAGE) is a strong candidate for a common pathway mediating arterial inflammation.[12] RAGE transmits signals released or up-regulated in the inflammatory response. The ligands of RAGE include the S100/calgranulin family, Advanced Glycation End products (AGE), Mac-1, high mobility group box-1, amyloid-β peptide, β-sheet fibrils, and lysophosphatidic acid.[12–14] S100A12 is a member of the S100 family of proteins. Serum concentration of S100A12 has been associated with more extensive coronary atherosclerosis in patients with coronary artery disease (CAD), DM, and chronic kidney disease (CKD).[15–20] Furthermore, serum S100A12 has also been associated with disease activity in rheumatoid arthritis, psoriatic arthritis, and with cardiovascular complications in lupus. Importantly, the anti-inflammatory effects of methotrexate in patients with inflammatory arthritis were associated with reduction in serum S100A12, raising the hypothesis that S100A12 itself could be a therapeutic target.[21, 22] Although S100A12 is expressed locally in various cells within the inflamed tissue including epithelial cells, SMCs, and myeloid cells, the exact mechanisms leading to increased serum concentrations of S100/calgranulins are lesser known. However, it is likely that elevated S100A12 concentration in the serum or plasma stems, at least in part, from circulating myeloid cells, since a strong correlation exists between serum S100A12 protein and relative quantities of S100A12 mRNA in peripheral blood mononuclear cells.[23]
The S100 Proteins and Inflammation
There are at least 24 different members of the low molecular weight (10–14 kilo-dalton) S100 protein family, all of which bind calcium and have a conserved calcium-binding motif called the elongation factor (EF) hand. Despite the structural and functional differences among the different S100 proteins, the shared characteristic of being soluble in 100%-saturated ammonium sulphate at neutral ph, lead to the name of “S100 proteins”. Several names are in use for the S100/calgranulins: S100A8 is also known as calgranulin A or myeloid-related Protein (MRP) 8, S100A9 is also known as calgranulin B or MRP 14, and the heterodimer is termed S100A8/A9, MRP 8/14 or calprotectin. Other names for S100A12 includes calgranulin C, or extracellular newly identified RAGE binding protein (EN-RAGE), and calcium-binding protein in amniotic fluid fluid-1 (CAAF1). S100 proteins are found both within cells and extra-cellularly. They act on a variety of target proteins and receptors and are involved in diverse functions, including cellular proliferation, differentiation, apoptosis, calcium homeostasis, energy metabolism, regulation of the cell cytoskeleton and inflammation. When calcium binds to the EF-hand, the protein undergoes changes in the spatial conformation leading to interact with various target proteins. Some of the S100 protein-mediated events, such as signaling in the extracellular space, where levels of calcium are already high, are unlikely to be calcium regulated, and the biological activity is therefore more likely regulated by Zinc and Copper. S100A8, S100A9, and S100A12 share structural and functional homologies, and are often named S100/calgranulins.[24, 25]
The S100/calgranulins are expressed in myeloid cells including neutrophils, monocytes, and dendritic cells. S100A8/9 comprise 40% of the cytosolic protein in neutrophils, and S100A12 comprises 5%.[26,27] At the site of inflammation, myeloid cells release S100/calgranulins from the cytoplasm upon cell activation, and for example, S100A8/9 are abundantly enriched in Neutrophil Extracellular Traps, and found to be critically important for the clearance of fungal or bacterial infections.[28,29] Other effects of S100/calgranulin includes cytokine-like effects with activation of cell surface receptors, including RAGE and toll-like receptor-4(TLR-4).[30–32] Therefore, the S100/calgranulins are part of the pro-inflammatory molecules referred to as Damage-Associated Molecular Pattern proteins that activate innate immune pathways. Although the S100/calgranulins are considered pro-inflammatory, there are significant differences between S100A8, S100A9, and S100A12. For example, oxidation of methionines 63 and 83 on S100A9 and of cysteine 42 on S100A8 inhibits both the chemotactic and repellent effects of these proteins on neutrophils, whereas S100A12 is more resistant to oxidation and remains biological active in conditions of increased oxidative stress.[33] Interestingly, S100A8 and S100A9 are more sensitive to oxidation compared to low density lipoproteins and albumin, and it is proposed that the high amounts of S100A8 and 9 present in atherosclerotic plaque might contribute to oxidant scavenging and protect other tissues components from oxidative damage during inflammation.[34]
S100/calgranulins bind to other receptors in addition to RAGE. For example, S100A12 has been shown to activate mast cells by releasing histamine and other cytokines including IL-6 and monocyte chemotactic protein-1, even in the absence of RAGE expression on mast cells.[35] Furthermore, S100A12 has chemotactic activity for mast cells and monocytes, which appears to be mediated by a G-protein coupled receptor and independent of RAGE.[36] S100A8 binds TLR-4 and induces MyD88 translocation and IRAK-1 phosphorylation.[32] These studies collectively demonstrate that S100/calgranulin are potent stimulator for acute inflammatory responses even in the absence of RAGE. Other studies, including from our laboratory, showed enhanced transcription of genes critically involved in the regulation of inflammation in white blood cells in S100/calgranulin transgenic mice, and this transcription profile was abolished in mice with global RAGE-deficiency [31], demonstrating a critical role of RAGE in mediating activation of white blood cells.
RAGE activates multiple signaling pathways and its activation is important in endothelial dysfunction and inflammation as demonstrated by attenuated atherosclerosis in ApoE null mice lacking RAGE.[37] RAGE was identified in 1992 from bovine lung as a protein that bound AGEs.[38] AGEs are formed through non-enzymatic glycation or glycoxidations of proteins, lipids, and nucleic acids and this process is accelerated during oxidative stress and inflammation, although AGE also accumulate in tissues during normal aging.
RAGE contains a V-type domain, two C-type immunoglobulin (Ig)-like domains (C1 and C2), a trans-membrane spanning helix, and a cytosolic domain for signaling. S100A12 binds to the V-C1 domain, while the S100A8 and S100A9 binding sites are not known. It has been demonstrated that increasing concentrations of heparin sulfate minimally impaired the binding of S100A12 to RAGE, but competes with binding of S100A9 or S100A8/9 to RAGE.[39] Also, glycan-enrichment of RAGE led to a 30-fold higher binding capacity of S100A12 to RAGE, which was specific for S100A12.[40] This suggests that S100A12 may have the strongest potential to activate RAGE. When S100A12 binds to RAGE a pro-inflammatory response has been demonstrated in vivo and in vitro with the activation of nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) and increased secretion of Interleukin (IL)-6, IL-1β, and tumor necrosis factor (TNF) α.(figure 1)[3,30]. Blocking RAGE using anti-RAGE IgG or soluble RAGE, a decoy form of the receptor that limits access of ligands to RAGE, reduced inflammation and atherosclerosis.[41–43]
Figure 1. Ligand-mediated activation of the receptor for advanced glycation endproducts (RAGE) mediates inflammation.
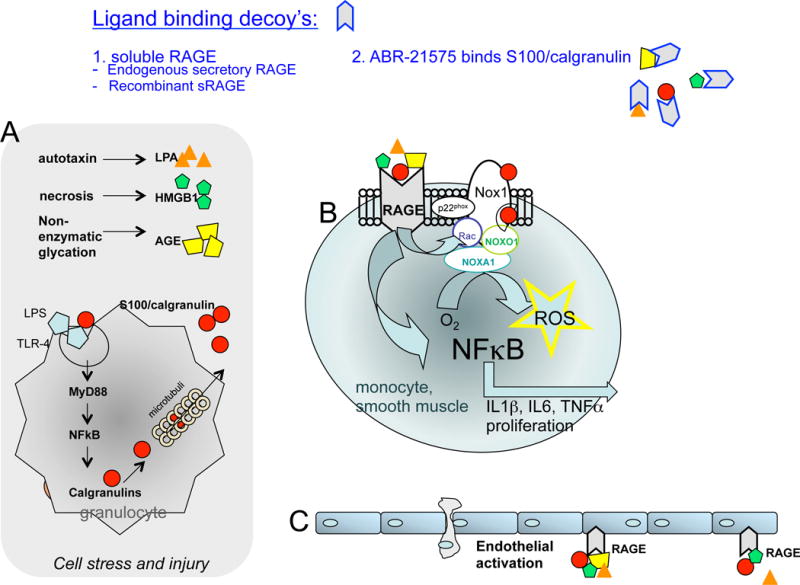
A) Ligands of RAGE, including advanced glycation endproducts (AGEs), high mobility group protein box 1 (HMGB1), S100/calgranulins, and lysophosphatidic acid (LPA) are released during cell stress and bind to RAGE on monocytes and smooth muscle cells (B) and endothelial cells (C). This activates pathways, leading to the translocation of transcription factor kappa B (NFkB) and increased reactive oxygen species (ROS), which results in a proinflammatory cell phenotype. Endogenous and pharmacological compounds with the ability to bind RAGE-ligands are suggested as possible therapeutic interventions to block activation of RAGE and other targets [modified from reference 3].
S100A12/RAGE in Animal Models of Vascular Disease
Both RAGE and TLR-4 play important roles in mediating acute and chronic inflammation and are relevant for atherosclerosis, as demonstrated by decreased atherosclerosis in apolipoprotein (Apo) E null mice lacking either RAGE or TLR-4 signaling.[37,44] However, RAGE may play a lesser role in normal physiologic development and in healthy homeostasis, as mice lacking RAGE develop normally and have normal phenotypes, except mildly increased lung fibrosis in mice ages > 15 months. In contrast, lack of TLR-4 signaling contributes to the growth of tumors in various organs.[45] This potentially makes RAGE or RAGE-mediated signaling an ideal therapeutic target.
Our laboratory took advantage of the fact that S100A12 is not present in mice and developed transgenic mice expressing human S100A12 in SMCs driven by the SM22α promoter. After backcrossing into hyperlipidemic ApoE deficient mice we demonstrated that S100A12 transgenic mice had a 1.4 fold increase in atherosclerotic plaque size and a large increase in necrotic core, calcified plaque area, and reduced extracellular matrix compared to ApoE −/− wild type (WT) mice (figure 2 a–f).[46] Importantly, these are histological criteria characteristic for vulnerable plaques and are often associated with plaque rupture and MI in patients with CAD.[47] Mechanisms underlying the accelerated pathological vascular remodeling observed in the transgenic mice with expression of human S100A12 in SMCs are mediated, at least in part, by enhanced oxidative stress. Oxidative stress can activate Runx-2 in SMCs and thereby regulate the expression of many osteoblastic genes promoting vascular calcification.[48] Indeed, aortic tissue or cultured primary aortic SMCs from young S100A12 transgenic mice demonstrated increased ROS and up-regulation of many bone-regulating genes even before onset of overt vascular calcification.[46] Other potential mechanisms of S100/calgranulins promoting vascular calcification might be mediated by binding of S100 proteins to annexins, a dominant group of proteins in matrix vesicles.[49] Matrix vesicles are released from SMCs, macrophages, bone- and other cells and provide a nidus for mineral formation once they accumulate in the extracellular matrix.[50–52]
Figure 2.
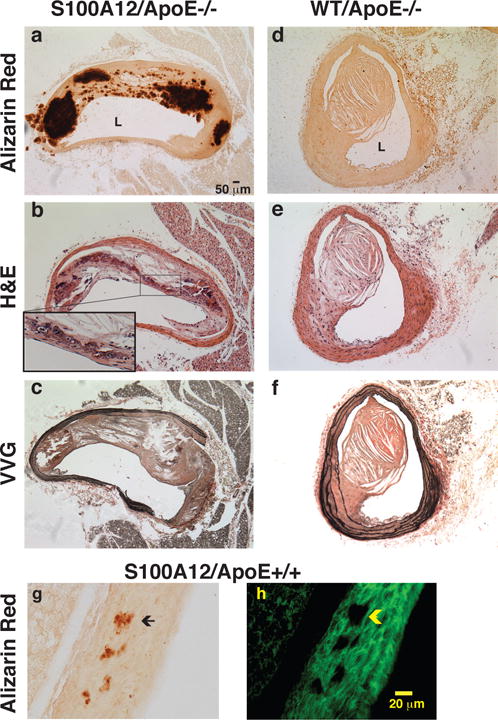
Transgenic expression of human S100A12 targeted to smooth muscle accelerates atherosclerotic remodeling with calcification, necrotic core, and elastic fiber degradation in Apolipoprotein (Apo)E null mice (a–c) compared to wild type (WT/ApoE) littermate mice (d–f) and promotes medial calcification in normolipidemic C57BL6/J mice (g,h). Alizarin Red stain for calcium (a,d,g), hematoxylin and eosin stain (b,e) and Verhoeff van Giessen stain for elastic fibers (c,f). L=lumen. Insert in b magnifies osteoblast like cell. [modified from reference 46]
Transgenic S100A12 mice with normal lipid profiles (ApoE +/+, C57BL6J) develop scant vascular calcification with aging (figure 2 g–h)[46], and other risk factors, aside from hyperlipidemia, accelerate medial aortic calcification as demonstrated in S100A12 transgenic mice with surgically induced CKD.[53] In contrast, WT mice lacking S100A12 did not demonstrate medial aortic calcification when subjected to the same model of surgically induced CKD. Calcification of the tunica media usually occurs in the absence of macrophages and hyperlipidemia and is thought to involve SMCs that undergo a phenotypic switch to osteoblast-like cells with a gain of a pro-calcific phenotype.[54] Indeed, our data demonstrate that S100A12 increases the susceptibility of SMCs to gain a transcriptional profile typical of osteoblast-like cells. Mechanistically, pre-treatment of cultured aortic SMCs with soluble RAGE, a binding domain of RAGE that limits the access of S100A12 and other ligands to RAGE, as well as knockdown of Nox1, a critical component of NADPH oxidase in SMCs, both attenuated calcification of aortic SMCs.[53] These data indicate that S100A12-mediated increase in reactive oxygen species in vascular smooth muscle activates RAGE, Nox1, or a RAGE-Nox1 signaling complex, which initiates an osteogenic phenotype in an appropriate cellular environment. We speculate that S100A12-mediated arterial calcification as seen in mice with hyperlipidemia [46], or in chronic uremia induced by ureter ligation [53], is a possible mechanistic explanation for the strong positive association between elevated serum S100A12 in patients with DM or CKD with accelerated vascular calcification and increased adverse cardiovascular events.
Furthermore, transgenic expression of S100A12 in SMCs mediates other pathologic vascular features even without CKD or hyperlipidemia. S100A12 modulates the phenotype of SMCs and promotes a switch from a contractile state to a synthetic state with increased oxidant stress and increased production of matrix metalloproteinases 2/9, IL-6, and Smad2, which leads over time to the formation of thoracic aortic aneurysms in S100A12 transgenic mice.[55] The aortic aneurysms in S100A12 mice have similar histological features of aortic remodeling as seen in mice with a fibrillin-1mutation, suggesting that different signaling pathways converging in aneurysmal disease share similar histo-pathologic features with loss of SMCs, disruption of elastic fibers and increased extracellular matrix. While S100A12 is not expressed in healthy human vascular SMCs, there is growing experimental evidence that S100A12 and other S100 proteins are up-regulated in SMCs in response to injury, including lipopolysaccharide or endothelial wire injury, and importantly, S100A12 is highly expressed in SMCs in ruptured coronary artery plaques associated with sudden death [56], and in SMCs in dissecting human aortic aneurysms.[57] Although acute MI and aortic dissections are clinically distinctly different vascular diseases, increased apoptosis or other forms of cell death leading to an acute loss of SMCs may possibly be a shared mechanism underlying both vascular diseases characterized by a sudden instability of the vasculature. Indeed, treatment of various cell lines with recombinant S100A8/9 protein was previously shown to induce apoptosis [58], and our laboratory showed a significant reduction of apoptosis and inflammation in primary aortic SMCs harvested from patients with thoracic aortic aneurysms upon silencing of S100A12.[57] This suggests that up regulation of S100A12 in SMCs in response to cellular injury may initiate pathways of cell death and thereby potentially renders a previously stable vascular lesion unstable. This view is supported by recent findings of increased incidence of acute type A thoracic aortic dissection to coincidence with influenza activity [59] and by the association of S100A12 with acute myocardial infarction.
There is growing evidence that implicates enhanced myelopoiesis as a potential mechanism for vascular inflammation relevant to atherosclerosis and myocardial infarction, with some of the effects mediated by the RAGE axis.[60,61] Diabetic mice with either chemically (streptozotocin) or genetically (C57BL/6-Ins2Akita) induced islet cell destruction had increased number of circulating Ly6-Chi proinflammatory monocytes and neutrophils, and this leukocytosis was due to enhanced bone marrow myelopoiesis with increased production of granulocyte macrophage progenitors and common myeloid progenitors. The myelopoiesis induced by hyperglycemia was driven by S100A8/9 and RAGE as demonstrated by increased proliferation of wild type and myd88−/− bone marrow in response to S100A8/9, whereas RAGE−/− cells did not proliferate in excess. Mechanistically, S100A8/9 dramatically elevated macrophage colony-stimulating factor (M-CSF), granulocyte-macrophage colony stimulating factor (GM-CSF), and granulocyte colony stimulating factor (G-CSF) in bone marrow cells via NFkB, and this was abolished in cells lacking RAGE. [60] Interestingly, anti-diabetic therapy achieving normoglycemia corrected enhanced myelopoiesis, reduced circulating Ly6-Chi cells and their entry into atherosclerotic lesions and thereby regressed atherosclerotic lesions in this diabetic LDLR−/− mouse model of atherosclerosis. [60] Collectively, these findings suggest that hyperglycemia per se promotes monocyte production as a contributing factor for atherosclerosis, and provides a rational to develop strategies that aim at therapeutic targets in the signaling pathways linking hyperglycemia to S100A8/9/RAGE-mediated myelopoiesis and atherosclerosis. Dutta et al. demonstrated enhanced myelopoiesis after experimental myocardial infarction as a causative factor for further progression of atherosclerosis in ApoE null mice and suggests this pathway as a potential therapeutic target since inhibition of extramedullary hematopoiesis by either splenectomy or b3-adrenorecptor blockade attenuated progression of atherosclerosis after myocardial infarction.[61]
As a result of their association with chronic inflammation, the S100/calgranulins have been looked at as potential therapeutic targets. Quinoline-3-carboxamides (Q-compounds) have been around for over 30 years and decrease autoimmune and inflammatory disease in multiple animal models, with relative preservation of innate immunity.[62–65] Until recently, their exact mechanism of action was unknown. It was demonstrated that 6 different Q-compounds, including ABR-215757 (Paquinimod, currently in development for the treatment of lupus) bind S100A9 in peripheral blood mononuclear cells with high affinity and thereby decrease the binding of S100A9 to TLR4 and RAGE.[64] Mice treated with ABR-215757 and injected with lipopolysaccharide demonstrated the same degree of inhibition of TNFα production as it was seen when mice were treated with monoclonal antibodies to S100A9, and this identified S100A9 as one molecular target of the Q-compounds.[66]
We demonstrated that ABR-215757 also binds S100A12 in vitro with a slightly different kinetic profile then S100A9.[39] Moreover, when S100A12 transgenic ApoE−/− mice with established atherosclerotic disease were treated with ABR-215757, we found a 20% reduction in atherosclerotic lesion size in the innominate artery and aortic root, compared to placebo treated mice. Importantly, they also had smaller necrotic core size, decreased intima and media calcification, minimal elastic fiber disruption, and more plaque area covered with SMCs (figure 3). Additionally, the expression of leukocyte markers CD68, CD4, and CD11c was reduced by 55–60% in the S100A12 mice treated with ABR-215757, indicating a reduction in inflammation. ApoE−/− WT mice (without S100A12) exhibited less of an effect from ABR-215757, showing no effect on atherosclerosis in the aortic root, but a reduction in atherosclerotic lesion size and cellular complexity in the innominate artery, likely related to ABR-215757 targeting S100A9 in WT mice. There was also a significant reduction in T cell accumulation in the atherosclerotic lesions, but no change in macrophage accumulation, indicating that the effect of ABR-215757 may be primarily related to T cell lymphocytes. These findings suggest that ABR-215757 treatment may attenuate or even halt S100/calgranulin-mediated acceleration of atherosclerosis.
Figure 3. Treatment with ABR-21757 improves features of atherosclerotic plaque morphology (A–J) and reduces vascular inflammation (K–O).
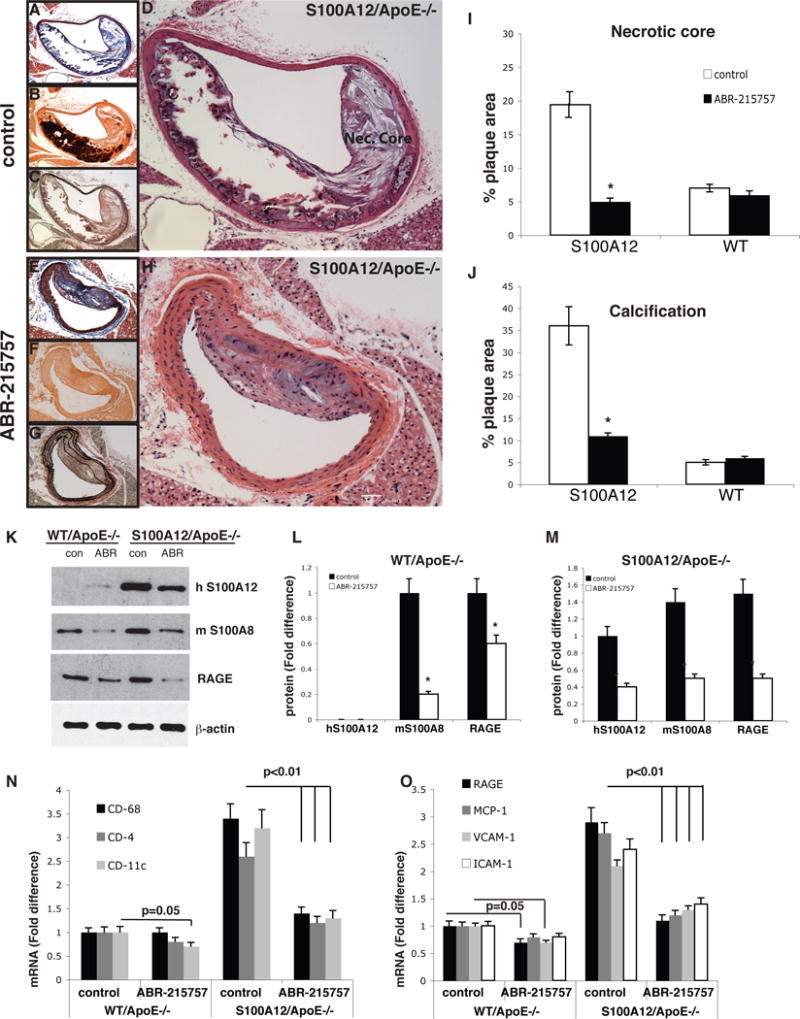
Innominate artery lesions from control S100A12/Apolipoprotein (Apo)E−/− (A–D) and in mice receiving treatment with ABR-215757 (E–H) stained with Masson trichrome (A,E), Alizarin Red S (calcium phosphate in red, B,F), Verhoeff-Van Gieson (elastic fibers in black, C,G), and Hematoxin & Eosin (D,H). Original magnification, 10×, scale bare, 10 μm. Quantification of lesion characteristics for necrotic area (I) and Alizarin Red stained plaque area (J). K–M: Protein level for S100/calgranulin and RAGE in aortic tissue lysates and mRNA (N–O) in aortic tissue in WT/ApoE−/− and S100A12/ApoE−/− mice after 5 weeks of ABR-215757 or vehicle treatment. [modified from reference 39.]
Association of S100/calgranulins with human vascular disease
Elevated serum concentrations of the S100/calgranulins have been correlated with disease activity in chronic inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, asthma, and Kawasaki vasculitis, and collectively, levels of S100/calgranulins are considered biomarkers of inflammation.[25] The S100/calgranulins and sRAGE have also been associated with traditional risk factors for vascular disease including hyperglycemia, insulin resistance, and with the presence of vascular disease itself. A study of S100A12 levels and soluble RAGE levels in subjects with and without DM demonstrated that S100A12 levels were increased in diabetics and inversely related to soluble RAGE levels.[17] High S100A12 levels and low soluble RAGE levels were also associated with increased risk of cardiovascular disease as determined by the Framingham score [17], and other cross sectional studies confirmed serum S100A12 as an independent predictor of increased glycosylated hemoglobin levels [67]. Serum S100A12 levels have been found elevated in patients with CAD in many studies as listed in table 1. Importantly, intervention with a moderate intensity exercise program in diabetics showed a significant improvement of glucose control, lipid profiles and increases in soluble RAGE levels compared to baseline. [68] Similarly, S100A12 mRNA in peripheral blood cells was significantly reduced (among 142 other genes that were differentially expressed from baseline) after 52 weeks of a rigorous cardiovascular disease risk reduction program with comprehensive life style changes in 63 participants with CAD or ≥2 CAD risk factors [69]. This suggests that S100/calgranulins play an important role within the inflammatory networks, acting as inflammation-responsive proteins but yet amplifying inflammatory signals, ultimately contributing to a positive feedback cycle conducive to unchecked inflammation that subsequently may promote development and progression of atherosclerosis.
Table 1.
Studies linking S100A12 to human cardiovascular disease
| Year | Study type | n | Patient Population | Outcome | Statistical Significance |
|---|---|---|---|---|---|
| 200456 | Autopsy | 112 | Diabetic and non-diabetics with Sudden cardiac Death | S100A12 expression in SMCs and macrophages | Not quantified |
| 200894 | Retrospective | 41, 215, 86# | Angiographically significant CAD | S100A12 gene expression in monocytes | p = 0.00001 |
| 200920 | Retrospective | 72 | Patients on HD | Plasma S100A12, carotid intimal medial thickness | p = 0.014 |
| 200915 | Cross-sectional | 67 | Symptomatic angiographically significant CAD | Serum S100A12 in patients with and without CAD | (775 ± 87.6 ng/ml) vs (282.1 ±22.9 ng/ml), p = 0.0002 |
| 201018 | Retrospective | 184 | Patients on HD | Serum S100A12, cardiovascular mortality | HR 3.23, CI 1.48–7.01, p = 0.003 |
| 201078 | Prospective | 526 | Non-diabetic patients referred for angiography | Panel of genes (including S100A12), obstructive CAD | ROC AUC 0.70 ± 0.02, p < 0.001 - for the entire panel |
| 201119 | Cross-sectional | 550 | Patients on HD | Plasma S100A12, presence of cardiovascular disease | OR 1.28, CI 1.13–1.44, p < 0.001 |
| 201279 | Prospective | 652 | PCI for stable CAD | Plasma S100A12, MACE | HR 1.64, CI 1.06–2.53, p = 0.025 |
| 201316 | Prospective | 188 | Diabetics referred for angiography | Serum S100A12, CAD | OR 1.018, CI 1.009–1.016, p <0.001 |
| 201322 | Prospective | 237 | Patients with inactive lupus | Serum S100A12, history of cardiovascular disease | 305 ng/ml vs 177 ng/ml, p = 0.03 |
| 201480 | Prospective | 240 | Patients with CAD referred for angiography | Serum S100A12, lesion complexity on angiography | OR 1.02, CI 1.01–1.04, p <0.01 |
| 201583 | Prospective | 839 | Healthy people without CAD | Serum S100A12 level, incident CAD | HR 1.30, CI 1.06–1.59 |
The association was studied in multiple cohorts in this study.
SCD = sudden cardiac death, CAD = coronary artery disease, HD = hemodialysis, PCI = percutaneous intervention, SMC = smooth muscle cell, MACE = major adverse cardiac event, ng = nanogram, ml = milliliter, HR = hazard ratio, CI = 95% confidence interval, ROC = receiver operator curve, AUC = area under the curve, OR = odds ratio
The S100/calgranulins are a biomarker of obesity and S100A8/9 serum level are associated with body mass index even after adjustment for hs-CRP, TNF-α, and neutrophil granulocyte counts in non-diabetic obese subjects.[70] Serum S100A8/9 levels decrease to normal levels in obese subjects who lose significant weight.[71] There is genetic evidence that supports a possible role for S100/calgranulins as mediator of obesity beyond serving as a biomarker. Changes in fat free body mass after a 20 week endurance exercise program were strongest linked to a polymorphism in the S100 gene cluster on chromosome 1 in the Heritage family study of 364 sibling pairs from 99 Caucasian families.[72] A cardiovascular genome study including 1252 men identified that obese subjects homozygous for the S/S allele in the RAGE G82S polymorphisms have significantly increased hs-CRP compared to obese subjects with the common G/G or G/S variant of the RAGE G82S.[73] Our laboratory was the first to identify the G82S variant as a variant within the ligand binding domain of RAGE that promotes enhanced binding of S100A12 with subsequently increased NFκB, IL-6, and TNF-α signaling in peripheral blood mononuclear cells in response to S100A12.[74] Taken together, this supports a role for S100/RAGE activation as a highly relevant pathway to promote inflammation and obesity in human subjects. This view is further supported by experimental data demonstrating reduced weight gain upon feeding of a high fat diet in mice with global deficiency in RAGE [75].
Regarding cardiovascular disease, a nested case-study among patients enrolled in the TIMI 22 study identified the serum concentration of S100A8/9 and hs-CRP obtained 30 days after acute coronary syndrome as predictors of recurrent cardiovascular events over the ensuing 18–36 months.[76] A similar predictive power was shown for serum S100A12 concentration in two cross-sectional studies of patients undergoing chronic hemodialysis where serum S100A12 levels predicted both the presence of cardiovascular disease and cardiovascular mortality.[18,19] In those studies, the level of S100A12 remained a predictor of adverse cardiovascular outcomes even after stratification for other inflammatory markers, like hs-CRP and IL-6 levels. An autopsy study of victims of sudden cardiac death found enhanced expression of S100A12 and RAGE in macrophages and SMCs in the ruptured coronary artery plaque [56], suggesting a potential role of locally expressed S100A12 within the atherosclerotic lesion for increased plaque instability. Additional support linking S100/calgranulin to acute myocardial infarction stems from a gene expression study on platelet-derived mRNA which demonstrated increased gene expression of only two genes, CD69 and S100A9, in patients with ST elevation MI, compared with patients with stable CAD.[77] Since the mRNA in platelets is a remnant from the megakaryocytes prior to their release from the bone marrow, the gene expression levels found in platelets are likely reflective of conditions prior to the onset of ST elevation MI, suggesting that increased levels of CD69 and S100A9 in platelets could be causal for acute MI. Other studies found that gene expression of S100A12 in peripheral blood mononuclear cells was an independent predictor of angiographically confirmed obstructive CAD in the PREDICT trial, a multicenter trial of 526 non-diabetic patients.[78] Another study looked at 652 patients with stable CAD who underwent percutaneous intervention and found that plasma S100A12 was a significant independent predictor for major adverse cardiovascular events (MACE), where as hs-CRP was not.[79] A recent Chinese study demonstrated that S100A12 was increased in patients with acute coronary syndrome and lesion complexity angiographically, demonstrating that S100A12 may be a marker for plaque vulnerability in humans.[80] Moreover, in patients with end stage renal disease S100A12 plasma levels were associated with increased carotid intimal medial thickness[81], and with peripheral arterial disease.[82]
While these mostly small case control studies mentioned above and shown in table 1 demonstrate an association between S100A12 and CAD in specifically selected populations, it has only recently been studied longitudinally. The Rotterdam study, a population based cohort study of 839 participants without coronary heart disease followed for 10.6 years, identified S100A12 as the only biomarker among 16 other measured biomarkers of inflammation that significantly associates with CAD when adjusted for age and sex.[83] Participants in the highest tertile of S100A12 levels had a 2.6 fold higher risk of developing CAD compared with participants in the lowest tertile (Hazard ratio, 2.59; 95% confidence interval, 1.52–4.40), (figure 4). Further adjustments for other inflammatory markers and traditional risk factors (DM, hypertension, CKD, smoking, body mass index, hyperlipedemia) did not change the association. Given that S100A12 was associated with CAD even after correction for hs-CRP, CD40, IL-8, IL-18, and other inflammatory markers, this suggests that S100A12 represents a distinct inflammatory pathway. There was also a stronger association for S100A12 with future MI and CAD mortality compared with revascularization, which suggests that, as confirmed in animal studies and suggested in cross-sectional data, S100A12 may be a determinant of plaque instability. These results suggest that S100A12 predicts future cardiovascular events, and may have a role in primary prevention and intensive risk factor modifications.
Figure 4.
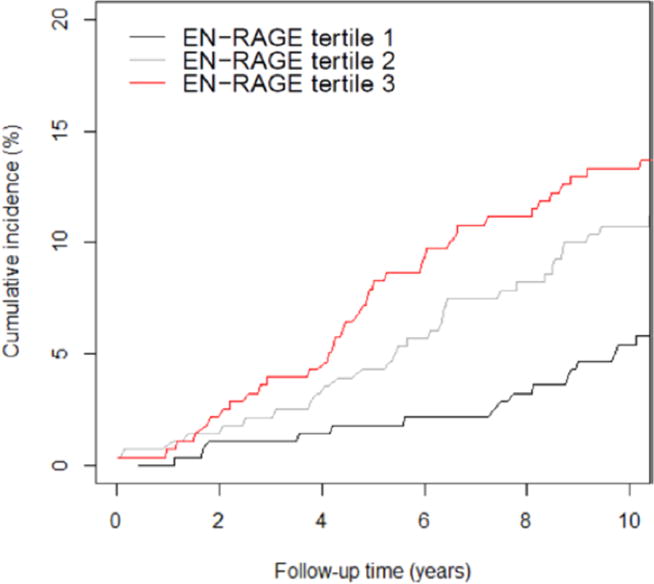
Cumulative incidence curves for first, second, and third tertile of serum S100A12 (EN-RAGE) in relation to incidence of coronary artery disease adjusting for competing noncoronary heart disease death ≤10 years of follow up. EN-RAGE = extra-cellular newly identified receptor for advanced glycation endprodcuts binding protein. [Reproduced with permission from 83]
Therapeutic Potential
Genetic ablation of S100A9 [84] or RAGE [37] in atherosclerosis susceptible ApoE null mice results in reduced atherosclerosis. Importantly, S100A12 and the RAGE axis can also be modified pharmacologically. Q-compound ABR-215757 binds with S100A12 in vitro and reduces atherosclerosis in hyperlipidemic mice with transgenic S100A12 expression, demonstrating that S100A12 can serve as a pharmacological target. [39]
The current Q-compounds undergoing clinical study include ABR-215757 (Paquinimod), Laquinimod, and Tasquinimod. It should be noted that a previous generation Q-compound, Linomide, was in phase III clinical trials for multiple sclerosis, but those trials were terminated due to increased cardiovascular events – primarily MI.[85] The molecular mechanisms of Linomide leading to MI remains unclear, however there were pre-clinical data on Linomide demonstrating pro-inflammatory arteritis in medium sized arteries, hemorrhage, necrosis, and fibrosis of the myocardium in beagle dogs that were not seen in mice studies.[86] The newer Q-compounds are derivatives of the original compound Linomide and the propensity for pro-inflammatory response of different derivative Q-compounds were studied in beagle dogs.[87] Based on these data, it was suggested that the pro-inflammatory response could be minimized by replacing a methyl group with an ethyl group in the N-carboxamide moiety of the compound.[87] Laquinimod and Paquinimod both share this substitution and thus far have not demonstrated the unexpected pro-inflammatory response in dogs that was seen with Linomide.[87,88] Paquinimod is currently approved in the United States as an orphan drug for systemic sclerosis and has been demonstrated to be safe in patients with lupus.[89] In phase III trials, Laquinimod has slowed the progression of disability and reduced the rate of relapse in patients with multiple sclerosis.[90,91] Tasquinimod was tested for metastatic castrate-resistance prostate cancer in a phase clinical III trial.[92] Besides the Q compounds, three distinct anti-allergic drugs, amlexanox, cromolyn and tranilast, bind S100A12, although there effect on atherosclerosis has not been examined in murine or human disease. [93].
One anti-inflammatory drug that is currently tested in a prospective randomized clinical trial to examine whether it improves clinical endpoints of CAD is methotrexate (target dose of 20 mg/week) in the ongoing CIRT study. Although the anti-inflammatory effects of methotrexate are multifactorial, it is possible that reducing circulating levels of S100A12 as is was previously demonstrated in patients with systemic arthritis [21], might be a potential underlying mechanism of any potential beneficial effects of methotrexate on vascular inflammation. Therefore, it will be of interest to examine the S100/calgranulins as potential biomarker to predict response to methotrexate therapy in CIRT.
In summary, S100/calgranulins and the RAGE axis are exciting emerging pathways linking inflammation to atherosclerosis and to vascular calcification. S100A12 may have a new role as a marker of future cardiovascular disease, and may be able to be incorporated into primary prevention and risk factor modification. However, there is no definitive evidence that S100A12 is a therapeutic target in human atherosclerosis since it is associated with other mechanisms of inflammation and has not been specifically targeted in prospective clinical trials. Further prospective studies are required. With this caveat, S100/calgranulins have emerged as possible therapeutic targets of the Q-compounds, and with further drug development, modification of the RAGE axis may reduce cardiovascular disease burden and mortality.
Significance.
While significant advances have been made in the treatment of atherosclerosis, it remains the leading cause of death worldwide and further treatment avenues are needed. The proinflammatory S100/calgranulins, in particular S100A8, S100A9, and S100A12, and their interaction with RAGE are important mediators of inflammation and atherosclerosis as it has been demonstrated in both mouse and human models. S100A12 is a biomarker for coronary artery disease in a recent longitudinal population-based study. S100A9 and S100A12 are therapeutic targets for a new class of immune modulators, the Quinoline-3-carboxamides, and one of them is undergoing a phase III clinical trial for multiple sclerosis. It is possible that these compounds have potential to improve outcomes in atherosclerosis and further studies may yield important therapeutic potential.
Acknowledgments
None
Sources of Funding: R01HL114821
Abbreviations
- SMC
Smooth muscle cell
- hs-CRP
High sensitivity C-reactive Protein
- MI
Myocardial infarction
- DM
Diabetes Mellitus
- RAGE
receptor for advanced glycation end products
- AGE
Advanced Glycation Endproducts
- CAD
Coronary artery disease
- CKD
Chronic kidney disease
- EF
Elongation factor
- TLR-4
Toll like receptor 4
- MRP
Myeloid-related Protein
- Ig
Immunoglobulin
- NFκB
nuclear factor kappa-light-chain-enhancer of activated B cells
- IL
Interleukin
- TNF
Tumor necrosis factor
- Apo
Apolipoprotein
- WT
Wild type
- Q-compounds
Quinoline-3-carboxamides
- CD
Cluster of differentiation
- ACS
Acute coronary syndrome
- MACE
Major adverse cardiac events
Footnotes
Disclosures: None
References
- 1.Mendis S, Davis S, Norrving B. Organizational update: the world health organization global status Report on noncommunicable Diseases 2014; one more landmark step in the combat against stroke and vascular disease. Stroke. 2015;46(5):121–122. doi: 10.1161/STROKEAHA.115.008097. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Inflammation in atherosclerosis. Arterioscler Thromb Vasc Biol. 2012;32:2045–2051. doi: 10.1161/ATVBAHA.108.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hofmann MA, Schmidt AM. The Next Generation of RAGE Modulators: Implications for Soluble RAGE Therapies in Vascular Inflammation. J Mol Med. 2013;91:1329–1331. doi: 10.1007/s00109-013-1097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, Lowe GD, Pepys MB, Gudnason V. C-Reactive Protein and Other Circulating Markers of Inflammation in the Prediction of Coronary Heart Disease. N Engl J Med. 2004;350:1387–1397. doi: 10.1056/NEJMoa032804. [DOI] [PubMed] [Google Scholar]
- 5.Sarwar N, Butterworth AS, Freitag DF, et al. Interleukin-6 Receptor Pathways in Coronary Heart Disease: A Collaborative Meta-Analysis of 82 Studies. Lancet. 2012;379:1205–1213. doi: 10.1016/S0140-6736(11)61931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jefferis BJ, Papacosta O, Owen CG, Wannamethee SG, Humphries SE, Woodward M, Lennon LT, Thompson A, Welsh P, Rumley A, Lowe GD, Whincup PH. Interleukin 18 and Coronary Heart Disease: Prospective Study and Systemic Review. Atherosclerosis. 2011;217:227–233. doi: 10.1016/j.atherosclerosis.2011.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kavousi M, Elias-Smale S, Rutten JH, et al. Evaluation of Newer Risk Markers for Coronary Heart Disease Risk Classification. Ann Intern Med. 2012;156:438–444. doi: 10.7326/0003-4819-156-6-201203200-00006. [DOI] [PubMed] [Google Scholar]
- 8.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. N Engl J Med. 2008;359:2195–2007. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 9.Nidorf SM, Eikelboom JW, Budgeon A, Thompson PL. Low-dose Colchicine for Secondary Prevention of Cardiovascular Disease. J Am Coll Cardiol. 2013;61:404–410. doi: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 10.Everett BM, Pradhan AD, Solomon DH, et al. Rationale and Design of the Cardiovascular Inflammation Reduction Trial: A Test of the Inflammatory Hypothesis of Atherothrombosis. Am Heart J. 2013;166:199–207. doi: 10.1016/j.ahj.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ridker PM, Thuren T, Zalewski A, Libby P. Interleukin-1β Inhibition and the Prevention of Cardiovascular Events: Rationale and Design of the Canakinumab Anti-inflammatory Thrombosis Outcomes Study. Am Heart J. 2011;162:597–605. doi: 10.1016/j.ahj.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Yan S, Ramasamy R, Schmidt AM. The RAGE Axis: A Fundamental Mechanism Signaling Danger to the Vulnerable Vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schmidt AM, Sahagan B, Nelson RB, Selmer J, Rothlein R, Bell JM. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr Opin Investig Drugs. 2009;10:672–680. [PubMed] [Google Scholar]
- 14.Rai V, Toure F, Chitayat S, Pei R, Song F, Li Q, Zhang J, Rosario R, Ramasamy R, Chazin WJ, Schmidt AM. Lysophosphatidic Acid Targets Vascular and Oncogenic Pathways via RAGE Signaling. J Exp Med. 2012;209:2339–2350. doi: 10.1084/jem.20120873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goyette J, Yan WX, Yamen E, et al. Pleiotropic Roles of S100A12 in Coronary Atherosclerotic Plaque Formation and Rupture. J Immunol. 2009;183:593–603. doi: 10.4049/jimmunol.0900373. [DOI] [PubMed] [Google Scholar]
- 16.Zhao P, Wu M, Yu H, Huang Y, Wang Y, Wang W, Yin W. Serum S100A12 Levels are Correlated with the Presence and Severity of Coronary Artery Disease in Patients with Type 2 Diabetes Mellitus. J Investig Med. 2013;61:861–866. doi: 10.2310/JIM.0b013e318292fb1e. [DOI] [PubMed] [Google Scholar]
- 17.Basta G, Sironi AM, Lazzerini G, Del Turco S, Buzzigoli E, Casolaro A, Natali A, Ferrannini E, Gastaldelli A. Circulating Soluble Receptor for Advanced Glycation End Products is Inversely Associated with Glycemic Control and S100A12 Protein. J Clin Endocrinol Metab. 2006;91:4628–4634. doi: 10.1210/jc.2005-2559. [DOI] [PubMed] [Google Scholar]
- 18.Nakashima A, Carrero JJ, Qureshi AR, Miyamoto T, Anderstam B, Baraby P, Heimbuerger O, Stenvinkel P, Lindjolm B. Effect of Circulating Soluble Receptor for Advanced Glycation End Products (sRAGE) and the Proinflammatory RAGE Ligand (EN-RAGE, S100A12) on Mortality in Hemodialysis Patients. Clin J Am Soc Nephrol. 2010;5:2213–2219. doi: 10.2215/CJN.03360410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shiotsu Y, Mori Y, Nishimura M, et al. Plasma S100A12 Level is Associated with Cardiovascular Disease in Hemodialysis Patients. Clin J Am Soc Nephrol. 2011;6:718–723. doi: 10.2215/CJN.08310910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mori Y, Kosaki A, Kishimoto N, et al. Increased Plasma S100A12 (EN-RAGE) Levels in Hemodialysis Patients with Atherosclerosis. Am J Nephrol. 2009;29:18–24. doi: 10.1159/000148646. [DOI] [PubMed] [Google Scholar]
- 21.Foell D, Kane D, Bresnihan B, Vogel T, Nacken W, Sorg C, Fitzgerald O, Roth J. Expression of the Pro-Inflammatory Protein S100A12 (EN-RAGE) in Rheumatoid and Psoriatic Arthritis. Rheumatology. 2003;42:1–7. doi: 10.1093/rheumatology/keg385. [DOI] [PubMed] [Google Scholar]
- 22.Tyden J, Lood C, Gullstrand B, Joensen A, Nived O, Sturfelt G, Truedsson L, Ivars F, Leanderson T, Bengtsson AA. Increased Serum Levels of S100A8/A9 and S100A12 are Associated with Cardiovascular Disease in Patients with Inactive Systemic Lupus Erythematosus. Rheumatology. 2013;52:2048–2055. doi: 10.1093/rheumatology/ket263. [DOI] [PubMed] [Google Scholar]
- 23.Hara M, Ando M, Morito T, Nokiba H, Iwasa Y, Tsuchiya K, Nitta K. S100A12 gene expression is increased in peripheral leukocytes in chronic kidney disease stage 4–5 patients with cardiovascular disease. Nephron Clin Prac. 2013;123:202–208. doi: 10.1159/000353808. [DOI] [PubMed] [Google Scholar]
- 24.Donato R, Cannon BR, Sorci G, Riuzzi F, Hsu K, Weber DJ, Geczy CL. Functions of S100 Proteins. Curr Mol Med. 2013;13(1):24–57. [PMC free article] [PubMed] [Google Scholar]
- 25.Hofmann MA, Schmidt AM. S100/calgranulins EN-RAGEing the Blood Vessels: Implications for Inflammatory Responses and Atherosclerosis. Am J Cardiovasc Dis. 2011;1(1):92–100. [PMC free article] [PubMed] [Google Scholar]
- 26.Vogl T, Pröpper C, Hartmann M, Strey A, Strupat K, van den Bos C, Sorg C, Roth J. S100A12 is expressed exclusively by granulocytes and acts independently from MRP8 and MRP14. J BiolChem. 1999;274:25291–25296. doi: 10.1074/jbc.274.36.25291. [DOI] [PubMed] [Google Scholar]
- 27.Averill MM, Kerkhoff C, Bornfeldt KE. S100A8 and S100A9 in cardiovascular biology and disease. Arterioscler Thromb Vasc Biol. 2012;32:223–229. doi: 10.1161/ATVBAHA.111.236927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, Brinkman V, Jungblut PR, Zychlinsky A. Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. Plos Pathog. 2009;5:e1000639. doi: 10.1371/journal.ppat.1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Filippo K, Neill DR, Mathies M, Bangert M, McNeil E, Kadioglu A, Hogg N. A new protective role for S100A9 in regulation of neutrophil recruitment during invasive pneumococcal pneumonia. FASEB J. 2014;28:3600–3608. doi: 10.1096/fj.13-247460. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann MA, Drury S, Fu C, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 31.Yan L, Mathew L, Chellan B, Gardner B, Earley J, Puri TS, Hofmann Bowman MA. S100/Calgranulin-mediated Inflammation accelerates left ventricular hypertrophy and aortic valve sclerosis in chronic kidney disease in a receptor for advanced glycation end products-dependent manner. Arterioscler Thromb Vasc Biol. 2014;34(7):1399–1411. doi: 10.1161/ATVBAHA.114.303508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vogl T, Tenbrock K, Ludwig S, Leukert N, Ehrhardt C, van Zoelen MA, Nacken W, Foell D, van der Poll T, Sorg C, Roth J. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13:1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 33.Sroussi HY, Berline J, Palefsky JM. Oxidation of methionine 63 and 83 regulates the effect of S100A9 on the migration of neutrophils in vitro. Journal of Leukocyte Biology. 2007;81:818–824. doi: 10.1189/jlb.0706433. [DOI] [PubMed] [Google Scholar]
- 34.Lim SY, Raftery MJ, Goyette J, Hsu K, Geczy CL. Oxidative Modifications of S100 Proteins: Functional Regulation by Redox. J Leukoc Biol. 2009;86:577–587. doi: 10.1189/jlb.1008608. [DOI] [PubMed] [Google Scholar]
- 35.Yang Z, Yan WX, Cai H, et al. S100A12 Provokes Mast Cell Activation: A Potential Amplification Pathway in Asthma and Innate Immunity. J Allergy Clin Immunol. 2007;119:106–114. doi: 10.1016/j.jaci.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 36.Yan WX, Armishaw C, Goyette J, Yang Z, Cai H, Alewood P, Geczy CL. Mast Cell and Monocyte Recruitment by S100A12 and its Hinge Domain. J Biol Chem. 2008;283:13035–13043. doi: 10.1074/jbc.M710388200. [DOI] [PubMed] [Google Scholar]
- 37.Harja E, Bu DX, Hudson BI, et al. Vascular and Inflammatory Stresses mediate Atherosclerosis via RAGE and its Ligands in apo E−/− mice. J Clin Invest. 2008;118:183–194. doi: 10.1172/JCI32703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, Elliston K, Stern D, Shaw A. Cloning and Expression of a Cell Surface Receptor for Advanced Glycosylation End Products of Proteins. J Biol Chem. 1992;267:14998–15004. [PubMed] [Google Scholar]
- 39.Yan L, Bjork P, Butuc R, Gawdzik J, Early J, Kim G, Hofmann Bowman MA. Beneficial effects of quinolone-3-carboxamide (ABR-215757) on atherosclerotic plaque morphology in S100A12 transgenic ApoE null mice. Atherosclerosis. 2013;228:69–79. doi: 10.1016/j.atherosclerosis.2013.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Srikrishna G, Nayak J, Weigle B, Temme A, Foell D, Hazelwood L, Olsson A, Volkman N, Nanein D, Freeze HH. Carboxylated N-Glycans on RAGE Promote S100A12 Binding and Signaling. J Cell Biochem. 2010;110:645–659. doi: 10.1002/jcb.22575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Jr, Chow WS, Stern D, Schmidt AM. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med. 1998;4:1025–1031. doi: 10.1038/2012. [DOI] [PubMed] [Google Scholar]
- 42.Sakaguichi T, Yan SF, Yan SD, et al. Central Role of RAGE-Dependent Neointimal Expansion in Arterial Restenosis. J Clin Invest. 2003;111:959–972. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tae HJ, Kim JM, Park S, et al. The N-glycoform of sRAGE is the key determinant for its therapeutic efficacy to attenuate injury-elicited arterial inflammation and neointimal growth. J Mol Med. 2013;91(12):1369–1381. doi: 10.1007/s00109-013-1091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bjorkbacka H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, Lee MA, Means T, Halmen K, Luster AD, Golenbock DT, Freeman MW. Reduced Atherosclerosis in MyD88-null Mice links Elevated Serum Cholesterol Levels to Activation of Innate Immunity Signaling Pathways. Nat Med. 2004;10:416–421. doi: 10.1038/nm1008. [DOI] [PubMed] [Google Scholar]
- 45.Rakoff-Nahoum S, Medzhitov R. Toll-like receptors and cancer. Nat Rev Cancer. 2009;9:57–63. doi: 10.1038/nrc2541. [DOI] [PubMed] [Google Scholar]
- 46.Hofmann Bowman MA, Gawdzik J, Bukhari U, Husain AN, Toth PT, Kim G, Earley J, McNally EM. S100A12 in Vascular Smooth Muscle Accelerates Vascular Calcification in Apolipoprotein E-Null Mice by Activating an Osteogenic Gene Regulatory Program. Arterioscler Thromb Vasc Biol. 2011;31:337–344. doi: 10.1161/ATVBAHA.110.217745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Virmani R, Kolodgie FD, Burke AP, Farb A, Schwartz SM. Lessons from Sudden Coronary Death: A Comprehensive Morphologic Classification Scheme for Atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–1275. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 48.Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, McDonald JM, Chen Y. Oxidative Stress Induces Vascular Calcification through Modulation of the Osteogenic Transcription Factor Runx2 By AKT Signaling. J Biol Chem. 2008;283:15319–15327. doi: 10.1074/jbc.M800021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cmoch A, Strzelecka-Kiliszek A, Palczewska M, Groves P, Pikula S. Matrix Vesicles Isolated from Mineralization-Competent Saos-2 cells are Selectively Enriched with Annexins and S100 Proteins. Biochem and Biophys Res Comm. 2011;412:683–687. doi: 10.1016/j.bbrc.2011.08.025. [DOI] [PubMed] [Google Scholar]
- 50.Hofmann Bowman MA, McNally EM. Genetic pathways of vascular calcification. Trends Cardiovasc Med. 2012;22:93–98. doi: 10.1016/j.tcm.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapustin AN, Chatrou ML, Drozdov I, et al. Circ Res. 2015;116(8):1312–23. doi: 10.1161/CIRCRESAHA.116.305012. [DOI] [PubMed] [Google Scholar]
- 52.New SE, Aikawa E. Role of extracellular vesciles in de novo mineralization:and additional novel mechanisms of cardiovascular calcification. Arterioscler Thromb Vasc Biol. 2013;33(8):1753–1758. doi: 10.1161/ATVBAHA.112.300128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gawdzik J, Mathew L, Kim G, Puri TS, Hofmann Bowman MA. Vascular Remodeling and Arterial Calcification Are Directly Mediated by S100A12 (EN-RAGE) in Chronic Kidney Disease. Am J Nephrol. 2011;33:250–259. doi: 10.1159/000324693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Burton DG, Matsubara H, Ikeda K. Pathophysiology of Vascular Calcification: Pivotal Role of Cellular Senescence in Vascular Smooth Muscle Cells. Exp Gerontol. 2010;45:819–824. doi: 10.1016/j.exger.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 55.Hofmann Bowman M, Wilk J, Heydemann A, Kim G, Rehman J, Lodato J, Raman J, McNally EM. S100A12 Mediates Aortic Wall Remodeling and Aortic Aneurysm. Circ Res. 2010;106:145–154. doi: 10.1161/CIRCRESAHA.109.209486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burke AP, Kolodgie FD, Zieske A, Fowler DR, Weber DK, Varghese PJ, Farb A, Virmani R. Morphologic Findings of Coronary Atherosclerotic Plaques in Diabetics: A Postmortem Study. Arterioscler Thromb Vasc Biol. 2004;24:1266–1271. doi: 10.1161/01.ATV.0000131783.74034.97. [DOI] [PubMed] [Google Scholar]
- 57.Das D, Gawdzik J, Dellefave-Castillo L, McNally EM, Husain A, Raman J, Hofmann Bowman MA. S100A12 expression in thoracic aortic aneurysm is associated with increased risk of dissection and perioperative complications. JACC. 2012;60:775–785. doi: 10.1016/j.jacc.2012.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yui S, Nakatani Y, Mikami M. Calprotectin (S100A8/S100A9), an inflammatory protein complex from neutrophils with a broad apoptosis-inducing activity. Biol Pharm Bull. 2003;26:753–760. doi: 10.1248/bpb.26.753. [DOI] [PubMed] [Google Scholar]
- 59.Sandhu HK, Charlton-Ouw KM, Leake SS, Azizzadeh A, Nguyen TC, Estrera AL, Safi HJ, Miller CC. Is the Seasonal Variation in Acute Aortic Dissection Occurrence Associated with Flu Activity? Circulation. 2014;130:A19298. [Google Scholar]
- 60.Nagareddy PR, Murphy AJ, Stirzaker RA, Hu Y, Yu S, Miller RG, Ramkhelawon B, Distel E, Westerterp M, Huang LS, Schmidt AM, Orchard TJ, Fisher EA, Tall AR, Goldberg IJ. Hyperglycemia Promotes Myelopoiesis and Impairs the Resolution of Atherosclerosis. Cell Metabolism. 2013;17:695–708. doi: 10.1016/j.cmet.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dutta P, Courties G, Wei Y, et al. Myocardial Infarction Accelerates Atherosclerosis. Nature. 2012;487:325–329. doi: 10.1038/nature11260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Karussis DM, Lehmann D, Slavin S, Vourka-Karussis U, Mizrachi-Koll R, Ovadia H, Kalland T, Abamsky O. Treatment of Chronic-Relapsing Experimental Autoimmune Encephalomyelitis with the Synthetic Immunomodulator Linomide (Quinoline-3-Carboxamide) Proc Natl Acad Sci USA. 1993;90:6400–6404. doi: 10.1073/pnas.90.14.6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tarkowski A, Gunnarsson K, Stalhandske T. Effects of LS-2616 Administration upon the Autoimmune Disease of (NZB × NZW) F1 Hybrid Mice. Immunology. 1986;59:589–594. [PMC free article] [PubMed] [Google Scholar]
- 64.Bjork J, Kleinau S. Paradoxical Effects of LS-2616 (Linomide) Treatment in the Type II Collagen Arthritis Model in Mice. Agents Actions. 1989;27:319–321. doi: 10.1007/BF01972810. [DOI] [PubMed] [Google Scholar]
- 65.Brunmark C, Runstrom A, Ohlsson L, Sparre B, Brodin T, Astrom M, Hedlung G. The New Orally Active Immunoregulator Laquinimod (ABR-215062) Effectively Inhibits Development and Relapses of Experimental Autoimmune Encephalomyelitis. J Neuroimmunol. 2002;130:163–172. doi: 10.1016/s0165-5728(02)00225-4. [DOI] [PubMed] [Google Scholar]
- 66.Bjork P, Bjork A, Vogl T, Stenstroem M, Liberg D, Olsson A, Roth J, Ivars F, Leanderson T. Identification of human S100A9 as a novel target for treatment of autoimmune disease via binding to quinoline-3- carboxamides. PLoS Biol. 2009;7:e97. doi: 10.1371/journal.pbio.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kosaki A, Hasegawa T, Kimura T, Iida K, Hitomi J, Matsubara H, Mori Y, Okigaki M, Toyoda N, Masaki H, Inoue-Shibata M, Nishikawa M, Iwasaka T. Increased Plasma S100A12 (EN-RAGE) Levels in Patients with Type 2 Diabetes. J Clin Endocrinol Metab. 2004;89:5423–5428. doi: 10.1210/jc.2003-032223. [DOI] [PubMed] [Google Scholar]
- 68.Choi KM, Han KA, Ahn HE, Hwang SY, Hong HC, Choi HY, Yang SJ, Yoo HJ, Baik SH, Choi DS, Min KW. Effects of Exercise on sRAGE Levels and Cardiometabolic Risk Factors in Patients with Type 2 Diabetes: A Randomized Controlled Trial. J Clin Endocrinol Metab. 2012;97:3751–3758. doi: 10.1210/jc.2012-1951. [DOI] [PubMed] [Google Scholar]
- 69.Ellsworth DL, Croft DT, Weyandt J, et al. Intensive Cardiovascular Risk Reduction Induces Sustainable Changes in Expression of Genes and Pathways Important to Vascular Function. Circ Cardiovasc Genet. 2014;7:151–160. doi: 10.1161/CIRCGENETICS.113.000121. [DOI] [PubMed] [Google Scholar]
- 70.Mortenson OH, Nielsen AR, Erikstrup C, Plomgaard P, Fischer CP, Krogh-Madsen R, Lindegaard B, Petersen AM, Taudorf S, Pedersen BK. Calprotectin—a Novel Marker of Obesity. PLoS One. 2009;4:e7419. doi: 10.1371/journal.pone.0007419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Rotellar F, Valenti V, Silva C, Gil MJ, Fernandez-Real JM, Salvador J, Fruhbeck G. Increased Levels of Calprotectin in Obesity are Related to Macrophage Content: Impact on Inflammation and Effect of Weight loss. Mol Med. 2011;17:1157–1167. doi: 10.2119/molmed.2011.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chagnon YC, Rice T, Perusse L, Borecki IB, Ho-Kim MA, Lacaille M, Pare C, Bouchard L, Gagnon J, Leon AS, Skinner Js, Wilmore JH, Rao DC, Bouchard C. Genomic Scan for Genes Affecting Body Composition Before and After Training in Caucasians from HERITAGE. J Appl Physiol. 2001;90:1777–1787. doi: 10.1152/jappl.2001.90.5.1777. [DOI] [PubMed] [Google Scholar]
- 73.Kim OY, Jo SH, Jang Y, Chae JS, Kim JY, Hyun YJ, Lee JH. G Allele at RAGE SNP82 is Associated with Proinflammatory Markers in Obese Subjects. Nutr Res. 2009;29:106–113. doi: 10.1016/j.nutres.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 74.Hofmann MA, Drury S, Hudson BI, et al. RAGE and Arthritis: the G82S Polymorphism Amplifies the Inflammatory Response. Genes Immun. 2002;3:123–135. doi: 10.1038/sj.gene.6363861. [DOI] [PubMed] [Google Scholar]
- 75.Song F, del Pozo CH, Rosario R, et al. RAGE Regulates the Metabolic and Inflammatory Response to High Fat Feeding in Mice. Diabetes. 2014;63(3):1948–1965. doi: 10.2337/db13-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Morrow DA, Wang Y, Croce K, et al. Myeloid-Related Protein 8/14 and the Risk of Cardiovascular Death or Myocardial Infarction after an Acute Coronary Syndrome in the Pravastatin or Atorvastatin Evaluation and Infection Therapy: Thrombolysis in Myocardial Infarction (PROVE IT-TIMI 22) Trial. Am Heart J. 2008;155:49–55. doi: 10.1016/j.ahj.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Healy AM, Pickard MD, Pradhan AD, et al. Platelet Expression Profiling and Clinical Validation of Myeloid-Related Protein-14 as a Novel Determinant of Cardiovascular Events. Circulation. 2006;113:2278–2284. doi: 10.1161/CIRCULATIONAHA.105.607333. [DOI] [PubMed] [Google Scholar]
- 78.Rosenberg S, Elashoff MR, Beineke P, et al. Multicenter Validation of the Diagnostic Accuracy of a Blood-Based Gene Expression Test for Assessing Obstructive Coronary Artery Disease in Nondiabetic Patients. Ann Intern Med. 2010;153:425–434. doi: 10.7326/0003-4819-153-7-201010050-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Saito T, Hojo Y, Ogoyama Y, Hirose M, Ikemoto T, Katsiuki T, Shimada K, Kario K. S100A12 as a marker to Predict Cardiovascular Events in Patients with Chronic Coronary Artery Disease. Circ J. 2012;76:2647–2652. doi: 10.1253/circj.cj-12-0093. [DOI] [PubMed] [Google Scholar]
- 80.Liu J, Ren Y, Zhang L, Tong Y, Kang L. Serum S100A12 Concentrations are correlated with Angiographic Coronary Lesion Complexity in Patients with Coronary Artery Disease. Scand J Clin Lab Invest. 2014;74:149–154. doi: 10.3109/00365513.2013.864786. [DOI] [PubMed] [Google Scholar]
- 81.Kim JK, Park S, Lee MJ, Song YR, Han SH, Kim SG, Kang SW, Choi KH, Kim HJ, Yoo TH. Plasma Levels of Soluble Receptor for Advanced Glycation End Products (sRAGE) and Proinflammatory Ligand for RAGE (EN-RAGE) are Associated with Carotid Atherosclerosis in Patients with Peritoneal Dialysis. Atherosclerosis. 2012;220:208–214. doi: 10.1016/j.atherosclerosis.2011.07.115. [DOI] [PubMed] [Google Scholar]
- 82.Shiotsu Y, Mori Y, Hatta T, et al. Plasma S100A12 Levels and Peripheral Arterial Disease in End-Stage Renal Disease. Nephron Extra. 2011;1:242–250. doi: 10.1159/000335198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ligthart S, Sedaghat S, Ikram A, Hofman A, Franco OH, Dehghan A. EN-RAGE: A Novel Inflammatory Marker for Incident Coronary Heart Disease. Arterioscler Thromb Vasc Biol. 2014;34:2695–2699. doi: 10.1161/ATVBAHA.114.304306. [DOI] [PubMed] [Google Scholar]
- 84.Croke K, Gao H, Wang Y, Mooroka T, Sakuma M, Shi C, Sukhova GK, Packard RR, Hogg N, Libby P, Simon DI. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation. 2009;120:427–36. doi: 10.1161/CIRCULATIONAHA.108.814582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tan Il, Nijeholt G, Polman CH, Ader HJ, Barkhof F. Linomide in the Treatment of Multiple Sclerosis: MRI Results from Prematurely Terminated Phase III Trials. Multiple Sclerosis. 2000;6:99–104. doi: 10.1177/135245850000600208. [DOI] [PubMed] [Google Scholar]
- 86.Noseworthy JH, Wolinsky JS, Lublin FD, Whitaker JN, Linde A, Gjorstrup P, Sullivan HC. Linomide in Relapsing and Secondary Progressive MS: Part 1: Trial Design and Clinical Results. Neurology. 2000;54:1726–1733. doi: 10.1212/wnl.54.9.1726. [DOI] [PubMed] [Google Scholar]
- 87.Jonsson S, Andersson G, Fex T, Fristedt T, Hedlund G, Jansson K, Abramo L, Fritzson I, Pekarski O, Runstrom A, Sandin H, Thuvesson I, Bjork A. Synthesis and Biological Evaluation of New 1,2-Dihydro-4-hydrox-2-oxo-3-quinolinecarboxamides for Treatment of Autoimmune Disorders: Structure-Activity Relationship. J Med Chem. 2004;47:2075–2088. doi: 10.1021/jm031044w. [DOI] [PubMed] [Google Scholar]
- 88.Carlsten H, Jonsson C, Bokarewa M, Svensson L, Tarkowski A. The Impact of a new Immunomodulator Oxo-quinoline-3-carboxamide on the Progression of Experimental Lupus. International Immunopharmacology. 2004;4:1515–1523. doi: 10.1016/j.intimp.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 89.Bengtsson AA, Sturfelt G, Lood C, Roenningblom L, van Vollenhoven RF, Axelsson B, Sparre B, Tuvesson H, Ohman MW, Leanderson T. Pharmacokinetic, Tolerability, and Preliminary Efficacy of Paquinimod (ABR-215757), A New Quinoline-3-Carboxamide Derivative. Arthritis and Rheum. 2012;64:1579–1588. doi: 10.1002/art.33493. [DOI] [PubMed] [Google Scholar]
- 90.Comi G, Jeffery D, Kappos L, Montalban X, Boyko A, Rocca MA, Filippi M. Placebo-Controlled Trial of Oral Laquinimod for Multiple Sclerosis. N Engl J Med. 2012;366:1000–1009. doi: 10.1056/NEJMoa1104318. [DOI] [PubMed] [Google Scholar]
- 91.Vollmer TL, Sorensen PS, Selmaj K, Zipp F, Havrodova E, Cohen JA, Sasson N, Gilgun-Sherki Y, Arnold DL. A Randomized Placebo-Controlled Phase III Trial of Oral Laquinimod for Multiple Sclerosis. J Neurol. 2014;261:773–783. doi: 10.1007/s00415-014-7264-4. [DOI] [PubMed] [Google Scholar]
- 92.Pili R, Haggman M, Stadler WM, Gingrich JR, Assikis VJ, Bjoerk A, Nordle O, Forsberg G, Carducci MA, Armstrong AJ. Phase II Randomized, Double-Blind, Placebo-Controlled Study of Tasquinimod in Men with Minimallly Symptomatic Metastatic Castrate-Resistant Prostate Cancer. J Clin Oncol. 2011;29:4022–4028. doi: 10.1200/JCO.2011.35.6295. [DOI] [PubMed] [Google Scholar]
- 93.Shishibori T, Oyama Y, Matsushita O, Yamashita K, Furuichi H, Okabe A, Maeta H, Hata Y, Kobayashi R. Three distinct anti-allergic drugs, amlexanox, cromolyn and tranilast, bind to S100A12 and S100A13 of the S100 protein family. Biochem J. 1999;338:583–589. [PMC free article] [PubMed] [Google Scholar]
- 94.Wingrove JA, Daniels SE, Sehnert AJ, et al. Correlation of Peripheral-Blood Gene Expression with Extent of Coronary Artery Stenosis. Circ Cardiovasc Genet. 2008;1:31–38. doi: 10.1161/CIRCGENETICS.108.782730. [DOI] [PubMed] [Google Scholar]