

Abstract
The cerebrovascular disorder moyamoya disease (MMD) was first described in 1957 in Japan, and is typically considered to be an Asian-specific disease. However, it is globally recognized as one of the major causes of childhood stroke. Although several monogenic diseases are known to be complicated by Moyamoya angiopathy, the ring finger protein 213 gene (RNF213) was identified as a susceptibility gene for MMD. RNF213 is unusual, because (1) it induces MMD with no other recognizable phenotypes, (2) the RNF213 p.R4810K variant is an Asian founder mutation common to Japanese, Korean and Chinese with carrier rates of 0.5–2 % of the general population but a low penetrance, and (3) it encodes a relatively largest proteins with a dual AAA+ ATPase and E3 Ligase activities. In this review, we focus on the genetics and genetic epidemiology of RNF213, the pathology of RNF213 R4810K, and the molecular functions of RNF213, and also address the public health contributions to current unresolved issues of MMD. We also emphasize the importance of a more updated definition for MMD, of qualified cohort studies based on genetic epidemiology and an awareness of the ethical issues associated with genetic testing of carriers.
Electronic supplementary material
The online version of this article (doi:10.1007/s12199-015-0498-7) contains supplementary material, which is available to authorized users.
Keywords: Moyamoya disease, RNF213 R4810K, Asian founder mutation, Angiogenesis, Hypoxia
Introduction
Moyamoya disease (MMD) is a steno-occlusive disease of the cerebral arteries, involving smooth muscle cell proliferation with intima hyperplasia causing arterial stenosis and occlusion around the circle of Willis [1, 2] (Fig. 1). This, in turn, stimulates the compensatory development of collateral vessels, which have a “Puff of Smoke” (Moyamoya in Japanese) appearance in cerebral angiography [3].
Fig. 1.

Moyamoya angiopathy. An identical twin first-born twin sister (II-1), who developed MMD at the age of 36 years, had stenosis in her anterior and middle cerebral arteries bilaterally and underwent surgery. The second-born twin sister (II-2) is represented with a solid quadrant. Magnetic resonance angiography (lower panels) was taken when they were 55 years old. Patient II-2 showed stenosis (arrows). Their mother (I-2) died when she was 71 years old from cerebral infarction. Her niece also developed MMD (II-3). Subjects I-3, II-1, II-2, and II-3 all shared the WT/R4810K genotype. We assumed the carrier status for I-2, I-3, and II-2 in our linkage analysis. Due to the rarity of the disease gene, we assumed that I-2 is a carrier of the MMD-associated gene. This pedigree is simplified from the original pedigree 14 [12]
MMD is currently recognized as one of the major causes of stroke in children worldwide [4, 5]. Natural disease progression leads to cerebral hemorrhage or cerebral infarction, so early diagnosis and intervention before the establishment of a neurological deficit are essential for improved social adaptation of pediatric patients [6]. Nationwide epidemiological surveys are available in Japan and Korea because of the existence of registration programs. The prevalence and annual incidence of MMD in Japan were reported to be 10.5 and 0.94 per 100,000, respectively, while in Korea these figures were 18.1 and 4.3 per 100,000, respectively, in 2013 [7]. An estimated 100–15 % of MMD patients have family histories [8].
Several monogenic genetic diseases are known to lead to the development of MMD as a complication, referred to as moyamoya syndrome (Table 1). In such diseases, MMD is not the major phenotypic presentation, but it appears to develop in some but not all cases with low penetrance. A comprehensive review of the genetics of MMD associated with monogenic gene diseases has recently been published [9]. Impaired biological processes (signal transduction, chromatin remodeling, DNA repair, inflammation, hemostasis, and vascular smooth muscle cell coagulation), attributable to mutations of associated genes, have given insights into the mechanisms by which the mutations elevate the risk of MMD. However, no consolidated pathological process for MMD development has yet been proposed.
Table 1.
Single gene diseases showing co-morbidity with moyamoya angiopathy
| Biological processes | Molecular pathology | Disease | References | Gene | |
|---|---|---|---|---|---|
| Signal transduction | Ras signal pathway | Type I neurofibromatosis | [98–101] | NF1 | [117, 118] |
| Noonan syndrome | [102, 103] | BRAF | [119] | ||
| KRAS | |||||
| PTPNII | |||||
| RAFI | |||||
| SOSII | |||||
| Costello syndrome | [104, 105] | HRAS | [120] | ||
| Notch signal pathway | Alagille syndrome | [106, 107] | JAG1 | [72] | |
| NOTCH2 | |||||
| Wnt signal pathway | Robinow syndrome | [108, 109] | ROR2 | [121] | |
| Chromatin remodeling Cell cycle, DNA repair |
Cell cycle | Schimke immuno-osteo dysplasia | [110, 111] | SMARCAL1 | [122] |
| MOPDII | [112, 113] | PCNT | [112] | ||
| Seckel syndrome | [114] | ATR | [123] | ||
| RBBP8 | |||||
| CENPJ | |||||
| CEP63 | |||||
| NIN | |||||
| DNA repair Angiogenesis |
BRCA1 complex BRISC complex |
X-linked moyamoya syndrome | [88] | BRCC3 | ibid. [88] |
| Inflammation | Inflammation activated thrombosis | Sneddon’s syndrome | [66–69] | CECR1 | [124] |
| Excessive Type I interferon production | Aicardi–Goutieres syndrome | [65] | SAMHD1 | [65] | |
| TRX1 | |||||
| ACP5 | |||||
| Vascular smooth muscle cell dysfunction | eNOS production | Moyamoya and achalasia syndrome | [71] | GUCY1A3 | ibid. [71] |
| Excess proliferation | Thoracic aortic aneurysm and dissection | [70] | ACTA2 | ibid. [70] | |
| Coagulopathy | Thrombosis | Sickel cell disease | [73] | β-globin gene | |
| Protein S | [115, 116] | Protein S | |||
| Protein C | [74, 75] | Protein C | |||
| Thrombotic Thrombocytopeic Purpura | [76] | ADAMTS13 | |||
The ring finger protein 213 gene (RNF213), (mysterin), was recently identified as a susceptibility gene for MMD. RNF213 is unusual among susceptibility genes, because it induces MMD with no other phenotypic traits. The RNF213 variant p.R4810K (c.14429G > A, rs112735431, ss179362673, R4810K hereafter) was first reported by the Kyoto group with a high level of association (odds ratio 63.9 95 %, confidence interval 33.9–120.4) [10] and shown to be associated with MMD at large scales [11, 12]. Both R4859K [11] and R4810K [12] correspond to rs112735431, but while R4859K is based on the computer-predicted open-reading frame in the database [11], R4810K is based on the experimental open-reading frame, which was proven by cDNA cloning [12]. Thus, in this review, we use R4810K. Liu et al. [12] later reported that RNF213 R4810K is a founder variant in East Asian (Japanese, Korean, and Chinese) patients. Indeed, in Japan and Korea, the majority (~80 %) of MMD patients carry at least one allele of RNF213 R4810K [12–17]. A much larger proportion of carriers with RNF213 R4810K is known to develop MMD than that of wild-type (WT) subjects, even though most carriers are unaffected by the disease. This can be explained by the effect of environmental or other genetic factors that elevate the risk of MMD in concert with genetic predisposition. Because the total number of these carriers is estimated to be 15 million in Asian countries, the social impact as a single health issue is extremely significant [18].
RNF213 is composed of 5207 amino acids and has an estimated molecular size of 591 kDa. Its large size initially hampered full-length cDNA cloning, which was first achieved in 2011 [12]. Since then, the biochemical and functional characterization of RNF213 has progressed [12, 19–21], especially through the use of mouse gene ablation technology [20, 22, 23], transgenic mouse models [21], and an induced pluripotent stem cell (iPSC) model established from patients with MMD [24].
This review addresses recent research progress in MMD with regard to effective prevention and intervention programs, enabling public health researchers to identify clear public health goals. In particular, it focuses on RNF213 in terms of the public health aspect of MMD.
Multiple genetic loci on 17q25.3 in Japanese patients with familial MMD
MMD has two phenotypic characteristics. The first is apparent from the pathological investigation of cerebral arteries, and involves smooth muscle cell proliferation and neointimal formation with thrombi at the occlusive lesion [25, 26]. This characteristic forms the basis for the alternative name of MMD; sontaneous occlusion of the circle of Willis [27, 28]. Confirmation of this characteristic requires tissue samples for pathological examination, and so it is not practical. The second characteristic is the appearance of moyamoya vessels [3] in angiography, which has been widely used as the diagnostic criterion because of the ease of access in a clinical setting [29]. Current diagnostic criteria of MMD require bilateral stenosis and moyamoya vessels to be observed, while cases with stenosis around the circle of Willis, but the absence of moyamoya vessels, or unilateral stenosis are excluded. However, MMD disease progression starts with stenotic lesions, then leads to unilateral MMD, and culminates in bilateral stenosis with the development of collateral vessels [12, 30]. Therefore, these criteria only cover advanced stage MMD, and exclude cases at an earlier disease stage.
To date, five loci have been reported in Japanese MMD cases: 3p24–p26 [31], 6q25 [32], 8q23 [33], and 17q25/17q25.3 [34, 35]. Linkage analyses were applied to all loci, with the exception of 6q25, in which the association of HLA with MMD was conducted [32]. Loci variation is noteworthy because it argues against the epidemiological observation of a single major locus (17q25.3), and because it is linked with the default application of current diagnostic criteria. As the status of MMD is judged solely by the clinical diagnostic criteria, cases with stenosis only or unilateral MMD are eliminated and treated as “unaffected”, thereby rejecting the autosomal dominant mode of inheritance [31, 33]. Given that more than 80 % of Japanese patients with MMD are carriers of RNF213 R4810K, many researchers are skeptical about such versatility of genetic loci (3p24–p26 and 8q23) in Japanese pedigrees. In earlier studies, the dogmatic application of clinical diagnosis elicited the genetic problem known as “skipping of generations”. For example, when a grandparent and grandchild are affected with MMD but the grandparent’s daughter, i.e., the mother, only has stenosis, the “skipping generation phenomenon” occurs. Several examples can be found in the study by Liu et al. [12] (Fig. 1).
To overcome these genetic irregularities, Mineharu et al. [35] conducted a genome-wide linkage analysis by introducing a “carrier state”, which widened the clinical spectrum and included phenotypes, such as stenosis, unilateral cases, or the absence of abnormalities (Fig. 1). They analyzed 15 three-generation pedigrees and obtained a single and strong linkage signal at 17q25.3 (LOD score 8.11, p = 3.4 × 10−6)with an autosomal dominant mode of inheritance. The locus at 17q.25.3 has been confirmed repeatedly by different family sets [11–17, 36], and has led to the initial identification of the susceptibility gene, RNF213. However, confirmation is warranted for the other loci on 3p24–p26, 8q23, and 17q25.
Genetics of RNF213 mutations
R4810K and other mutations
Our previous studies showed that in East Asia, the founder variant RNF213 R4810K was much more frequently found in MMD patients (Japanese, 90.1 %; Korean, 78.9 %; Chinese, 23.1 %) than the general population (Japanese, 2.5 %; Korean, 2.7 %; Chinese, 0.9 %) [12, 18]. Following on from these studies, several groups also identified RNF213 R4810K in MMD patients from Taiwanese, Indian, Bangladeshi, and Filipino populations [14, 15, 37]. RNF213 R4810K was found to be absent from control individuals as well as Caucasian MMD cases [12, 15], which may explain their lower incidence of MMD. Indeed, the MMD incidence in Caucasians was estimated to be one-tenth of that in the Japanese population [7, 38].
Many non-R4810K mutations in RNF213 have, however, been identified in both Asian and Caucasian MMD cases (Fig. 2; Table 2) [11, 12, 14, 15, 37, 39]. These mutations have two characteristics: (1) they cluster at the C-terminal portion of RNF213, and (2) they do not fall into the category of null mutations resulting in a loss-of-function (nonsense or frame-shift mutations). Almost all RNF213 mutations, including R4810K (30 out of 32, expect for A529del and A1622V), are located within exons 41–68 (NM_001256071.2), corresponding to the region from the RING finger domain to the C-terminus of the RNF213 protein. Additionally, all 32 mutations are missense, in-frame deletions (A529del and K4115del), or in-frame insertions (E4950_F4951ins7). This suggests that the mutations have a dominant negative or gain-of-function effect. Indeed, mutations in the C-terminal portion of RNF213 would be predicted to cause functional alterations of the protein, which is more likely to be linked to a dominant negative or gain-of-function than a loss-of-function mechanism.
Fig. 2.
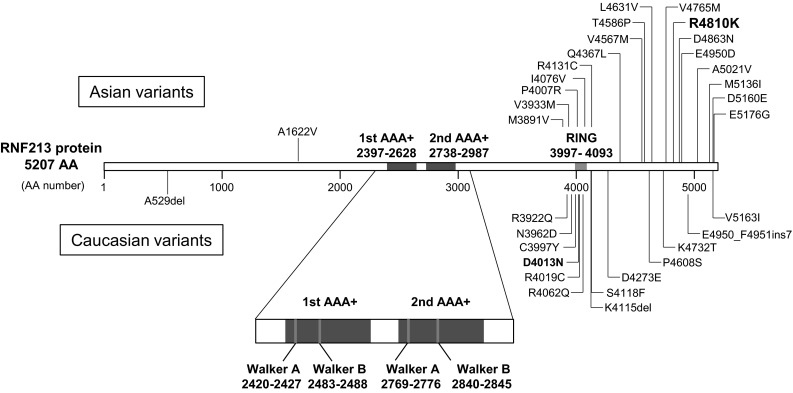
Variants shown are described previously [11, 12, 14, 15, 37, 39] (see details in Table 2). Variants in Asian and Caucasian patients are shown above and below the protein, respectively. The domain structure was based on [19]. AA amino acid, AAA+ ATPase associated with diverse cellular activities domain, RING ring finger domain
Table 2.
RNF213 mutations other than R4810K in MMD patients
| Mutation | Ethnicity | References |
|---|---|---|
| A1622V | Asian | [37] |
| M3891V | Asian | [11] |
| V3933M | Asian | [37] |
| P4007R | Asian | [14] |
| I4076V | Asian | [15] |
| R4131C | Asian | [37] |
| Q4367L | Asian | [14] |
| V4567M | Asian | [11] |
| T4586P | Asian | [14] |
| L4631V | Asian | [14] |
| V4765M | Asian | [11] |
| D4863N | Asian | [12] |
| E4950D | Asian | [12, 14] |
| A5021V | Asian | [12, 14] |
| M5136I | Asian | [14] |
| D5160E | Asian | [12] |
| E5176G | Asian | [12] |
| A529del | Caucasian | [15] |
| R3922Q | Caucasian | [15] |
| N3962D | Caucasian | [12] |
| D4013N | Caucasian | [12, 15] |
| R4019C | Caucasian | [15] |
| R4062Q | Caucasian | [12] |
| C3997Y | Caucasian | [15] |
| K4115del | Caucasian | [15] |
| S4118F | Caucasian | [39] |
| D4273E | Caucasian | [15] |
| P4608S | Caucasian | [12] |
| K4732T | Caucasian | [15] |
| E4950_F4951ins7 | Caucasian | [15] |
| V5163I | Caucasian | [15] |
Interestingly, five of these non-R4810K mutations are thought to be disease causing. D4013N in Caucasian patients and E4950D and A5021V in Chinese patients, originally identified by our group [12], have also been independently reported by others [14, 15]. D4013N segregation was confirmed both in European [12] and American [15] MMD pedigrees, raising the possibility that D4013N may have a founder effect in Caucasian populations worldwide. Furthermore, the two de novo mutations K4115del [15] and S4118F [39] have been identified in Caucasian cases. They are located in close proximity to each other, and were detected in early onset (<1-year-old) MMD patients, indicating that mutations in this region might have severe deleterious effects on RNF213 function.
Gene dosage effects
Gene dosage effects of RNF213 R4810K have been reported in a clinical genetics/epidemiological study and a case report by Miyatake et al. [13]. Homozygous RNF213 R4810K (AA) carriers with MMD were observed, but homozygosity was not seen in unaffected controls. Moreover, homozygosity was also associated with an earlier age of onset and greater disease severity compared with MMD cases harboring heterozygous RNF213 R4810K (GA) [13]. In the case report study, which described sibling MMD cases with homozygous and heterozygous RNF213 R4810K, the age of disease onset in the homozygote sibling was earlier than that of the heterozygote sibling, and the latter developed a milder clinical course [40]. The authors, therefore, claimed that the dosage of RNF213 R4810K alleles was strongly associated with clinical phenotype, even in family members sharing a similar genetic background. However, we have observed homozygous RNF213 R4810K carriers in an unaffected control population [18, 41], and also found sibling MMD cases, including identical twins, with the same dosage of RNF213 R4810K alleles but discordant phenotypes [12]. Therefore, it appears that heterogeneity of the MMD phenotype cannot be explained solely by gene dosage effects; indeed, environmental factors may play a critical role in phenotype variation.
Molecular characterization of RNF213
Molecular characterization of RNF213 as an AAA+ ATPAse (ATPase associated with diverse cellular activities)
The full-length cDNA of RNF213 was first cloned by Liu et al. [12]. It was found to code for a relatively large protein which functions both as an AAA+ ATPase and an E3 ligase (Fig. 2).
Various cell functions are mediated by AAA + ATPases, including membrane fusion/transport (NSF/Sec18p), proteolysis (ClpA), heat shock protein and protease Hsp78), motors (dyneins), protein disaggregation/refolding (Shp104/Hsp78/ClpB), DNA recombination/repair (RuvB, Rad17, Rfc2-5), and mitosis/meiosis (Cdc48p, Katanin) [42]. Morito et al. [19] demonstrated that RNF213 has two AAA+ modules and takes a hexamer form. Oligomerization is initiated by ATP binding in the Walker A motif of the first AAA+ module. This oligomer complex is then relaxed after ATP hydroxylation by the Walker B motif of the second AAA+. The cyclicity of ATP binding and ATP hydrolysis is required to generate a moving action for many AAA+ ATPases [42], which convert the chemical energy of ATP to physical energy (for example dyneins), but the role of Walker A and B motifs in maintaining ATP cyclicity is unknown.
Several diseases are known to be caused by AAA+ ATPase dysfunction, for example, PEX1/PEX6 mutations cause multiple organ degeneration such as Zellweger syndrome [43, 44], while mutations in Cdc48 cause amyotrophic lateral sclerosis [45, 46]. MMD is the only cerebrovascular or cardiovascular disease known to be associated with an AAA+ ATPase.
As RNF213 also has E3 ligase activity [12], it may additionally play a role in protein degradation or signaling processes. However, the complete physiological functions of RNF213 remain unknown as no investigations have been made into its dual AAA+ ATPase and E3 ligase activities, and its cofactors have not yet been identified.
Interferons as natural regulators of RNF213 expression
MMD patients have been shown to have elevated levels of several growth factors in their cerebrospinal fluid, including basic fibroblast growth factor [47], transforming growth factor-β [48], platelet-derived growth factor [49], hepatocyte growth factor [50], and an uncharacterized 4473 Da peptide [51]. Recently, two groups have independently found that RNF213 is induced by interferons (IFNs) [21, 52].
Kobayashi et al. [21] demonstrated that IFNβ and IFNγ induce RNF213 transcription in an endothelial cell (EC)-specific manner. This induction is mediated by the STAT box in the RNF213 promoter region. Ohkubo et al. [52] also found that IFNγ and tumor necrosis factor-α synergistically activate RNF213 transcription both in vitro and in vivo. They found that the AKT and PKR pathways contribute to the up-regulation of RNF213, although it remains to be determined what form of cellular signaling these are involved in. Further studies are needed to elucidate the complete signaling pathways associated with RNF213.
Lowered angiogenicity of endothelial cells (ECs) as a pathological effect of RNF213 R4810K
Kim et al. [53] reported that circulating endothelial progenitor cells obtained from patients with MMD are defective in angiogenic functions, as judged by the tube formation assay. This observation was unexpected because moyamoya vessels were thought to represent a hyperangiogenic phenomenon. This finding stimulated the following studies:
ECs derived from MMD patient iPSCs show unique EC-specific gene expression profiles
To obtain an MMD disease model, iPSCs were established from fibroblasts donated from six subjects [24]: two wild-type controls, two RNF213 R4810K heterozygotes (one affected and the other not affected with MMD), and two patients homozygous for RNF213 R4810K. iPSC ECs (iPSECs) were differentiated from iPSCs, and those derived from heterozygotes or homozygotes showed significantly decreased angiogenic activities compared with control iPSECs in accordance with the observation of Kim et al. [53]. In parallel, features of lowered angiogenic activity were recapitulated in human umbilical venous endothelial cells (HUVECs) overexpressing RNF213 R4810K, but not in those overexpressing WT RNF213. These authors also conducted expression array analyses in fibroblasts and counterpart iPSECs from the same donors. They observed differential expression profiles of mRNAs in iPSECs derived from controls and carriers of RNF213 R4810K, but none in the fibroblasts from the same donors. A total of 121 genes were down-regulated (Supplemental Table 1) and 36 genes were up-regulated (Supplemental Table 2) [24]. These expression profile differences were considered to be functionally related to the lowered angiogenic activities of ECs. These observations strongly indicated that differentiation from stem cells (i.e., iPSCs) to ECs induced a change of the gene expression profile by RNF213 R4810K.
Attention was focused on cell cycle-associated genes (Supplemental Table 1, asterisks), because they were enriched by gene ontology category analysis as down-regulated in iPSECs from RNF213 R4810K carriers. The expression of one of these genes, the key mitotic player Securin (PTTG1), which activates angiogenesis [54], was investigated in HUVECs and shown to be inhibited by RNF213 R4810K overexpression [24]. RNA silencing of Securin in HUVECs and wild-type iPSECs was found to inhibit angiogenesis, indicating that RNF213 R4810K lowers angiogenesis, at least in part, by the down-regulation of Securin. As this work only focused on a single gene out of the 128 identified, the biological implication of the expression profile differences found in iPSECs requires further investigation.
Tube formation, a comprehensive measure of angiogenic activity, is affected by various factors, such as EC proliferation rates and maturity [55]. As the overexpression of RNF213 R4810K inhibited HUVEC proliferation, Hitomi et al. [56] further investigated the effects of RNF213 R4810K on the cell cycle using HeLa cells, fibroblasts, and iPSECs. They found that overexpression of RNF213 R4810K, but not WT RNF213, delayed mitosis in HeLa cells, and that this was associated with abnormal mobilization of the metaphase–anaphase spindle checkpoint protein, mitotic arrest deficient 2 (MAD2). This abnormal mobilization was also seen in patient fibroblasts. Furthermore, both WT and mutant RNF213 could be co-immunoprecipitated with MAD2. Finally, iPSECs from MMD patients had higher mitotic failure rates than those from controls.
Collectively lowered angiogenic activity in vitro data suggest that RNF213 R4810K acts on EC signal production and proliferation/cell cycle. Deleterious cell proliferation/cell cycles are mediated by Securin and/or MAD2, which cross-talk with mutant, and probably WT, RNF213. As cell cycle abnormality is a common denominator for some monogenic diseases, such as Schimke immuno-osseous dysplasia, MOPDII, or Seckel syndrome (Table 1), further studies are warranted to explore this.
Lowered angiogenicity of RNF213 R4810K as an AAA + ATPase
Kobayashi et al. [21] investigated the effects of RNF213 R4810K induction on angiogenic activity, as measured by tube formation and by the migration assay. They confirmed that treatment with IFNβ, a cytokine that inhibits both angiogenesis and arteriogenesis [57, 58], inhibited angiogenesis in iPSECs (Fig. 3). This reduced angiogenesis could be rescued either by STAT box (Signal Transduction and Transcription) or RNF213 depletion in HUVECs. This led to the conclusion that the reduced anti-angiogenic activity of IFNβ is partially mediated by RNF213, which acts as a mediator downstream of the IFNβ signaling pathway. They also confirmed that overexpression of RNF213 R4810K, but not WT RNF213, can recapture the reduced angiogenicity induced by IFNβ, suggesting that RNF213 R4810K overexpression mimics IFNβ action.
Fig. 3.
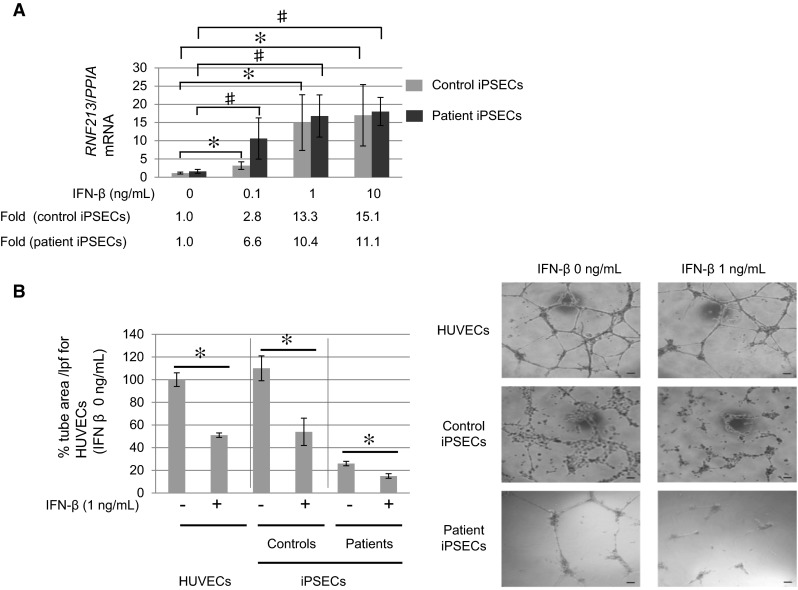
Inhibition of angiogenesis by INFβ and lowered angiogenic activity of iPSECs established from controls and patients. iPSCs were established from controls and patients with MMD. Mature iPSECs were developed from iPSCs as reported by Hitomi et al. [24]. a Treatment with INFβ induced mRNA of RNF213 significantly. b Tube formation was lowered in patients. Treatment with INFβ inhibited tube formation for iPSEECs from patients. Cited from Kobayashi et al. [21]
Morito et al. [19] demonstrated that disruption of Walker A or B motifs on the first or second AAA + modules decreases ATPase activity. However, while both motifs are necessary to maintain the oligomeric state, the Walker B motif has little impact on oligomerization. Furthermore, Morito et al. demonstrated that RNF213 R4810K forms a hexamer complex similar to the WT protein. Kobayashi et al. [21] further investigated the AAA+ ATPase mechanism by overexpressing various mutants in HUVECs: vector, RNF213 WT, RNF213 R4810K, a mutation of RNF213 Walker B motif (E2488Q) on the first AAA+ module (RNF213 WEQ), which disrupts ATP hydrolysis activity, and RNF213 first AAA+ module deletion mutant (RNF213 ΔAAA). They found that RNF213 R4810K and RNF213 WEQ, but neither RNF213 WT nor RNF213 ΔAAA, inhibited angiogenesis compared with the vector alone. They further showed that the ATPase activity was decreased in HUVECs transfected with RNF213 R4810K, RNF213 WEQ, and RNF213 ΔAAA. These results indicate that RNF213 R4810K is a molecular mimic of RNF213 WEQ. This also suggested that the Walker B motif in the first AAA+ module is functionally important in manifesting the function of ECs. A possible explanation for this is that disruption of the RNF213 first B motif disrupts ATP hydrolysis cyclicity, thereby inhibiting angiogenesis. As RNF213 R4810K is considered to have a similar mode of action to RNF213 WEQ, we speculate that it impairs the ATP hydrolysis cycle in the same way as the Walker B mutation.
RNF213 R4810K showed a reduced angiogenesis response to hypoxia in vivo
Kobayashi et al. [21] also focused their attention on the effects of RNF213 R4810K on angiogenesis after hypoxia exposure in vivo. They developed transgenic mouse (Tg) strains overexpressing RNF213 R4757K (the mouse homolog of human R4810K) in ECs or vascular smooth muscle cells (SMCs).
Hypoxia is known to induce angiogenesis in the cerebrum [59]. Mice were exposed to hypoxia (8 % O2) for 2 weeks from 3 weeks of age. Angiogenesis was found to be specifically reduced in Tg-ECs overexpressing RNF213 R4757K compared with other strains, i.e., Tg-SMCs overexpressing RNF213 R4757K or Tg WT RNF213 overexpressing RNF213 wild type specifically in ECs or RNF213 knock-out (KO) or WT mice (Fig. 4). The authors could recapture the lowered angiogenicity of Tg ECs in vivo, but magnetic resonance imaging failed to identify stenosis in the cerebral arteries or infarction.
Fig. 4.
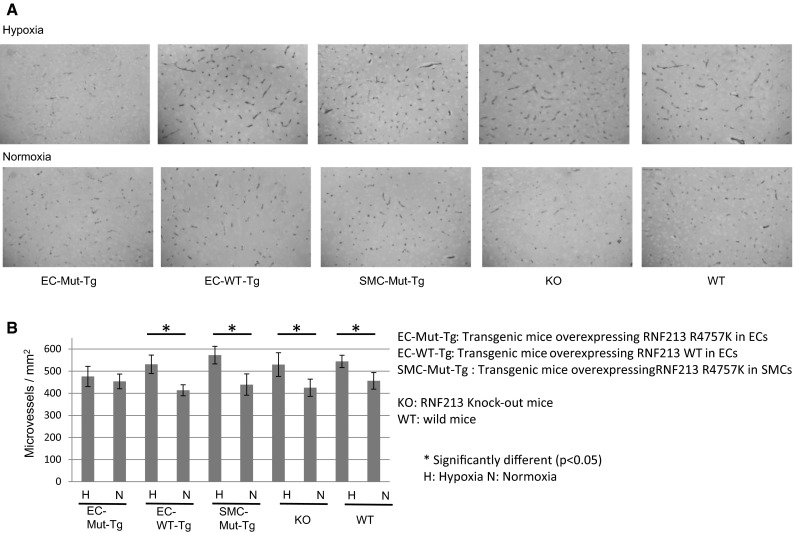
Lowered adaptive cerebral angiogenesis after exposure to hypoxia in transgenic mice expressing RNF213 R4757K in endothelial cells. a Several lines of transgenic or knockout or wild-type mice were exposed to hypoxia for 2 weeks at 8 % oxygen. Cerebral angiogenesis was evaluated by immunostaining Glut4. b Adaptive cerebral angiogenesis was abolished in transgenic mice overexpressing RNF213 R4757K (Human allelic ortholog of R4810K) in ECs, while in other mice adaptive angiogenesis was observed. Cited from Kobayashi et al. [21]
As lowered angiogenesis induced by RNF213 R4810K (R4757K in the mouse) observed in the in vitro ECs (HUVEC or iPSECs) could be successfully recaptured in the Tg mouse model overexpressing RNF213 R4757K, this suggested that ECs of RNF213 R4810K carriers may have a lowered angiogenicity and be particularly susceptible to hypoxia.
Other relevant studies
RNF213 KO mice were established by Kobayashi et al. [20], but did not induce abnormalities in the cardiovascular system. The effects of RNF213 ablation of diabetic progression were studied in the Akita mouse [60] which develops diabetes through an unfolded protein response of insulin 2. The authors tested whether RNF213 ablation (KO) influenced the development of diabetes and intracranial arteries around the circle of Willis. Although no stenosis was detected in the cerebral arteries of the RNF213 KO mouse, significant alleviation of endoplasmic reticulum (ER) stress was observed in pancreatic beta cells. Because ER stress enhances protein degradation and consequently depletes insulin levels in the Akita mouse, the authors speculated that RNF213 is involved in protein degradation as an E3 ligase in the proteasome.
Sonobe et al. [61] investigated the effects of RNF213 KO on vascular anatomy. They investigated cranial arteries using high-resolution magnetic resonance angiography, but found no abnormalities. They also investigated the effects on vascular remodeling after ligation of the carotid artery, but could not replicate the stenotic region, a hallmark of MMD. Conversely, Ito et al. [23] recently reported the recovery of blood flow after hind limb ischemia by femoral artery ligation in RNF213 KO mice. Recoveries were enhanced in RNF213 KO mice compared with WT counterparts. Although RNF213 KO animal models have yielded conflicting results in the cerebrum and hind limbs, Fujimura et al. [62] speculated that RNF213 influences vascular remodeling in chronic ischemia.
Inconsistencies necessitate additional experiments
Liu et al. [12] found that the inhibition of RNF213 expression in zebrafish induces abnormal arteriogenesis, but this is not observed in KO mouse models [20–22] despite the enhanced post-ischemic angiogenesis seen in the KO mouse [23]. This finding may be physiologically compatible with lowered angiogenesis in the cerebrum after hypoxic exposure in EC-specific R4757K Tg mice [21]; thus, the overexpression of RNF213 R4758K in ECs inhibits angiogenesis and conversely RNF213 depletion enhances angiogenesis.
Further discrepancies are noted between the observed inhibition of angiogenesis following RNF213 R4810K overexpression [21] and that in HUVECs following RNF213 depletion [52]. These differences are associated with the controversies in the reported RNF213 R4810K genetic mechanisms, involving loss-of-function, gain-of-function [52, 62], and dominant negative [21]. Alternatively, they could reflect species differences in innate immunity, e.g., of zebrafish and mice [63, 64], and further studies are needed to resolve these discrepancies.
Hypothetical pathological roles of RNF213 R4810K in MMD
Three major abnormalities: ECs, SMCs, and hemostasis
Several monogenic diseases have been reported to be complicated by MMD (Table 1). As various biological processes are involved in these diseases, including signal transduction, chromatin remodeling/DNA repair, DNA repair/angiogenesis, inflammation, vascular smooth muscle cell dysfunction, and coagulopathy, the pathological process of MMD cannot be explained in a consolidated signaling pathway. However, the diseases can be classified into three major abnormalities: (1) impaired functions of ECs, (2) SMC dysfunction, and (3) hemostasis abnormalities. For simplicity, we would like to propose an intuitive working hypothesis based on Table 1 and recent findings on RNF213. For analogy, we call this a three-route model (Fig. 5), in which MMD can occur through three different routes. These routes lead to the common outcome of SMC proliferation.
Fig. 5.

Three-route model of the hypothetical molecular pathology of moyamoya disease/syndrome. The model assumes that any of three independent abnormalities, endothelial dysfunction, smooth muscle cell dysfunction, and abnormal hemostasis, can lead to exaggerated proliferation of SMCs. Each abnormality can result in vascular stenosis. RNF213 R4810K is the major detrimental factor that elicits endothelial cell dysfunction. Pro-inflammatory signals such as IFNs can activate the transcription of RNF213
In the first route, RNF213 functions as a key mediator in ECs. Given that Type I IFN overproduction (Aicardi–Goutieres syndrome) [65] is complicated by MMD and RNF213 is highly activated by IFNs [21, 52], pro-inflammatory signals enhance IFN overproduction, which then activates RNF213 transcription. It should also be noted that pro-inflammatory signals can be induced by viral infections or by damaged, unrepaired DNA [65]. Amplified pro-inflammatory signals can lead to thrombosis, as seen in Sneddon’s syndrome [66–69].
In the second route, SMC dysfunction, which leads to exaggerated SMC proliferation, is a major outcome. Alpha-actin-2 (ACTA2) and guanylate cyclase 1 (GUCY1A3) are known to promote vascular SMC proliferation and induce MMD [70, 71]. While ACTA2 mutation causes moyamoya syndrome with thoracic aortic aneurysm and dissection by the mode of autosomal dominant, GUCY1A3 induced moyamoya syndrome with achalasia in the mode of autosomal recessive. It is of particular interest that GUCY1A3 encodes the major nitric oxide receptor. In Alagille syndrome (involving the Ras pathway), the impaired differentiation of both ECs and SMCs occurs [72].
The last route is associated with hemostasis. Several diseases in this category are known to induce hemostatic abnormalities, including Sickle-cell disease [73], mutations of protein S and protein C [74, 75], thrombotic thrombocytopenic purpura [76], and Noonan syndrome [77]. Furthermore, genes in the Ras signaling pathway [78] and Wnt signaling pathway [79] influence platelet activation. Activated thrombi formation results in ischemia and thereby causing hypoxia. In addition, Ras pathways may also trigger vascular inflammation [80] or SMC dysfunction [81] in a direct way.
Hypoxia, vascular injury, or chronic inflammation generate pro-inflammatory signals: stimulators of stenosis (neointimal formation)
It has been consistently demonstrated that RNF213 R4810K lowers angiogenic activities in ECs [21, 24, 53], but it remains unclear how this leads to stenosis (neointimal formation). This may be answered by examining pro-inflammatory signals, such as those involved in the JAK-STAT pathway.
Hypoxia [82], vascular injury [83], and chronic inflammation accompanied by elevated Type I IFNs [84] are known to activate EC mobilization for angiogenesis, which in turn leads to the production of adhesion molecules, cytokines, and chemokines. These pro-inflammatory signals stimulate SMC proliferation, migration, and secretion of extracellular matrix, causing neointimal formation [85]. Recently, IFN regulatory factors, activated by IFNα/β, were reported to modulate neointimal formation with sirtuin (SIRT)1 [86]. Given that RNF213 R4810K is a mimic of IFNβ, it may amplify the effects of IFNα/β, thereby magnifying neointimal formation by perturbing the IRF9/SIRT1 axis. The investigation of cytokine signaling in cells expressing RNF213 R4810K is expected to provide answers to several of these pending questions.
Future public health contributions to MMD
Since RNF213 was identified as a susceptible gene for MMD and as a founder mutation carried by 15 million people from the East Asian population [18], it has emerged as a key player in vascular disease. However, recent progress has also resulted in some unanswered questions, such as how can RNF213 R4810K describe the entire spectrum of the diseases associated with MMD? What is the health risk to RNF213 R4810K carriers? Can environmental factors explain the observed low penetrance of one in 150 carriers developing MMD?
Although in vitro and in vivo experimental approaches are expected to address many of these unresolved biomedical questions, well-designed human epidemiological studies are also essential. Given the large number of RNF213 R4810K carriers in the general population, there is an urgent need to evaluate their health risks. In parallel, ethical issues should be taken into account to avoid genetic stigmatization of these carriers.
A broader definition of MMD
The current diagnosis of MMD is based on the definition of the angiographic appearance of moyamoya vessels. However, RNF213 R410K genetics has unveiled different stages of disease progression. Given that 80 % of MMD cases are carriers of RNF213 R4810K, a definition of MMD based on this seems broader than one based on angiography. Another enigma is the difference between MMD and moyamoya syndrome. Chong et al. [87] recently reported a Down syndrome case with MMD, who is a carrier of RNF213 R4810K. This indicates that the interaction of RNF213 R4810K with other genes can lead to the manifestation of MMD. Within this context, the relationship between MMD syndrome and RNF213 should be examined and thereby definition of MMD being expanded. Indeed, a broader diagnostic criterion based on RNF213 is needed to illustrate the entire spectrum of MMD, as well as to delineate the natural course of carriers of RNF213 mutations in an epidemiological study.
Health risks associated with RNF213 R4810K
Prevalence of stenotic lesions or MMD was significantly higher (larger than 20 %) in the carriers of R4810K, if the carrier has family history of MMD [12]. The high risk among carriers in familial MMD shows a sharp contrast with the low risk of carriers in general population. Thus, carriers in the familial MMD is worthy for follow-up to ensure the early intervention.
Recently, Koizumi et al. [41] conducted a genetic epidemiological study with a case–control design (N = 4308) to investigate the association of RNF213 R4810K with blood pressure in the general Japanese population. They found 60 carriers (1.4 %). Regression analysis adjusted for age, sex, and body mass index based on the additive model demonstrated significant association with systolic blood pressure (mmHg/allele): β (Standard errors) 8.9 (2.0) (p = 10−5). In contrast, diastolic blood pressure did not show the association. Those data strongly indicate that RNF213 R4810K is a risk factor of blood pressure in free-living carriers in general population.
Monogenic diseases stochastically associated with MMD are often accompanied by coronary heart diseases (CHD) (BRCC3 [88] and ACTA2 [70]). Similarly, several case studies have reported the association between MMD and CHD [89–92]. Recently, Nam et al. [92] found that 4.6 % of 456 MMD patients were affected with CHD. Because these patients were young and lacked CHD risk factors, this suggests that CHD may be accelerated by the presence of RNF213 R4810K.
In early pathological studies [93–95], arterial stenosis was found to occur systematically, not only in the intracranial arteries but also in coronary, pulmonary, renal, and pancreatic arteries. Therefore, RNF213 R4810K carriers may have ischemic damage in these organs. These findings collectively imply that stenotic regions occur in various arteries and suggest the existence of both cardio- and cerebrovascular risks. Future large-scale genetic cohort studies should evaluate the risk of RNF213 R4810K on health outcomes of cardio- and cerebrovascular diseases, such as ischemic stroke, hemorrhagic stroke, myocardial infarction, and hypertension.
Environmental factors to explain low penetrance
The total number of registered MMD patients in 2012 was 15,177 in Japan (http://www.nanbyou.or.jp/entry/3664, Aug 5, 2015). Assuming that 80 % of these patients are carriers, the prevalence of MMD is 10−4. Carriers are estimated to be 2 % of the general population, resulting in only one out of ~150 carriers developing MMD. Therefore, another factor is needed to explain such a 1/150 low penetrance.
Kaku et al. [96] reported that vascular constrictive changes of affected arteries occur in MMD in comparison with other steno-occlusive diseases. It is uncertain whether such changes represent anatomical abnormalities involving narrowing of the cavernous sinus. Given that these are rare, they may increase the risk of MMD for RNF213 R4810K carriers. The constrictive remodeling hypothesis can be tested in animal models by introducing stenosis in the carotid artery. However, to date, ligation of the carotid artery has failed to replicate intimal hyperplasia when applied to the RNF213 ablation mouse [61]. Further studies are, therefore, warranted to test this hypothesis.
Inflammation is another possibility to explain such a low penetrance. Kobayashi et al. [21] and Ohkubo et al. [52] recently demonstrated that IFNs activate RNF213 transcription, so inflammation may induce MMD in association with RNF213 R4810K.
Yamashita et al. [25] reported that thrombi formation predominantly occurs in intracranial arteries of MMD patients, while Ikeda confirmed its presence in systemic arties [93]. Thrombi formation is often an opportunistic event precipitated by environmental factors, and we, therefore, speculate that this partially explains the 1/150 gap.
At present, it is highly probable that inflammatory signals trigger MMD. However, there has been no epidemiological evidence on the association of infection histories or other life-style or behavior factors with MMD in the carriers. Epidemiological evidence obtained through cohort studies focusing on carriers is deficient at present and is needed.
Therapeutic approach
Several studies have shown that the ablation of RNF213 does not cause deleterious effects on angiogenesis, except in zebrafish [12, 20, 61], suggesting that it might not affect mammalian species. A promising hypothesis is that RNF213 R4810K causes MMD by a dominant negative or gain-of-function mechanism. If this is the case, a pharmacological antagonist that inhibits ATP binding to the Walker A motif would be a suitable candidate as a drug target.
Ethical issues
RNF213 R4810K carriers have a very high prevalence in Japan and Korea (1–2 %) and are extremely likely to develop MMD. At present, however, there are insufficient data to predict the health risk of the carriers, except for subjects of familial MMD. It is, therefore, important to obtain carrier data and to elucidate the MMD risk attributable to RNF213 R4810K. In parallel, public health researchers should collaborate with genetic counsellors to facilitate genetic risk communication, not only to carriers, but also to society to avoid the social discrimination of carriers. A great deal of uncertainty currently surrounds application of genetic testing to the general population; indeed, it may have no benefit for the general population and only limited benefit for unaffected members in familial cases with MMD.
Conclusions
MMD was first described in 1957 by Takeuchi and Shimizu [97]. Although genetic factors had long been speculated, the angiographic definition of the MMD likely misled its genetic analysis. Recently, RNF213 R4810K was identified as the major susceptibility gene [10–12], and full-length cDNA cloning, iPS technology, and animal models have enabled the pathological roles of RNF213 R4810K to be investigated. Systemic biomedical and genetic epidemiology studies with specific emphasis on carriers will provide a deep understanding not only of MMD but also of associated health and environmental risk factors. Furthermore, such research will lead to a novel disease definition than the present one, and pave the way for new preventive strategies for cerebrovascular diseases, especially those in children.
Electronic supplementary material
Acknowledgments
This work was supported by a Grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (No. 25253047). It was partially supported by a Grant from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (No. 15K19243), and a Grant from the Research Committee on Spontaneous Occlusion of the Circle of Willis of the Ministry of Health and Welfare of Japan (No. H26-Nanjito-Ippan-078).
Compilance with ethical standards
Conflicts of interest
Prof. Koizumi and Dr. Hitomi have a patent JP2010068737 pending regarding with MMD. Other authors declare that they have no conflicts of interest.
Human and animal rights statement
This article does not contain any studies with human participants or animals performed by any of the authors.
References
- 1.Hosoda Y. A pathological study of so-called “spontaneous occulusion of the circle of Willis” (“cerebrovascular moyamoya disease”) Folia Angiol. 1976;14:85–86. [Google Scholar]
- 2.Oka K, Yamashita M, Sadoshima S, Tanaka K. Cerebral haemorrhage in moyamoya disease at autopsy. Virchows Arch A Pathol Anat Histol. 1981;392:247–261. doi: 10.1007/BF02155663. [DOI] [PubMed] [Google Scholar]
- 3.Suzuki J, Takaku A. Cerebrovascular, “moyamoya” disease. Disease showing abnormal net-like vessels in base of brain. Arch Neurol. 1969;20:288–299. doi: 10.1001/archneur.1969.00480090076012. [DOI] [PubMed] [Google Scholar]
- 4.Amlie-Lefond C, Bernard TJ, Sebire G, Friedman NR, Heyer GL, Lerner NB, et al. Predictors of cerebral arteriopathy in children with arterial ischemic stroke: results of the International Pediatric Stroke Study. Circulation. 2009;119:1417–1423. doi: 10.1161/CIRCULATIONAHA.108.806307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynch JK, Hirtz DG, DeVeber G, Nelson KB. Report of the National Institute of Neurological Disorders and Stroke workshop on perinatal and childhood stroke. Pediatrics. 2002;109:116–123. doi: 10.1542/peds.109.1.116. [DOI] [PubMed] [Google Scholar]
- 6.Phi JH, Wang KC, Cho BK, Lee MS, Lee JH, Yu KS, et al. Long-term social outcome in children with moyamoya disease who have reached adulthood. J Neurosurg Pediatr. 2011;8:303–309. doi: 10.3171/2011.6.PEDS10578. [DOI] [PubMed] [Google Scholar]
- 7.Kim T, Lee H, Bang JS, Kwon OK, Hwang G, Oh CW. Epidemiology of moyamoya disease in Korea: based on national health insurance service data. J Korean Neurosurg Soc. 2015;57:390–395. doi: 10.3340/jkns.2015.57.6.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuroda S, Houkin K. Moyamoya disease: current concepts and future perspectives. Lancet Neurol. 2008;7:1056–1066. doi: 10.1016/S1474-4422(08)70240-0. [DOI] [PubMed] [Google Scholar]
- 9.Guey S, Tournier-Lasserve E, Herve D, Kossorotoff M. Moyamoya disease and syndromes: from genetics to clinical management. Appl Clin Genet. 2015;8:49–68. doi: 10.2147/TACG.S42772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koizumi A. Genetic analysis of familial moyamoya. In: Annual report (2010) of the Research Committee on Spontaneous Occlusion of the Circle of Willis (Moyamoya Disease by Science Research Grants of Ministry of Health, Labor and Welfare, Japan (Chaired by N. Hashimoto) 2010. p. 25–26.
- 11.Kamada F, Aoki Y, Narisawa A, Abe Y, Komatsuzaki S, Kikuchi A, et al. A genome-wide association study identifies RNF213 as the first Moyamoya disease gene. J Hum Genet. 2011;56:34–40. doi: 10.1038/jhg.2010.132. [DOI] [PubMed] [Google Scholar]
- 12.Liu W, Morito D, Takashima S, Mineharu Y, Kobayashi H, Hitomi T, et al. Identification of RNF213 as a susceptibility gene for moyamoya disease and its possible role in vascular development. PLoS ONE. 2011;6:e22542. doi: 10.1371/journal.pone.0022542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miyatake S, Miyake N, Touho H, Nishimura-Tadaki A, Kondo Y, Okada I, et al. Homozygous c.14576G > A variant of RNF213 predicts early-onset and severe form of moyamoya disease. Neurology. 2012;78:803–810. doi: 10.1212/WNL.0b013e318249f71f. [DOI] [PubMed] [Google Scholar]
- 14.Wu Z, Jiang H, Zhang L, Xu X, Zhang X, Kang Z, et al. Molecular analysis of RNF213 gene for moyamoya disease in the Chinese Han population. PLoS ONE. 2012;7:e48179. doi: 10.1371/journal.pone.0048179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cecchi AC, Guo D, Ren Z, Flynn K, Santos-Cortez RL, Leal SM, et al. RNF213 rare variants in an ethnically diverse population with moyamoya disease. Stroke. 2014;45:3200–3207. doi: 10.1161/STROKEAHA.114.006244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bang OY, Ryoo S, Kim SJ, Yoon CH, Cha J, Yeon JY, et al. Adult moyamoya disease: a burden of intracranial stenosis in east asians? PLoS ONE. 2015;10:e0130663. doi: 10.1371/journal.pone.0130663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moteki Y, Onda H, Kasuya H, Yoneyama T, Okada Y, Hirota K, et al. Systematic validation of RNF213 coding variants in Japanese Patients With Moyamoya Disease. J Am Heart Assoc. 2015;4:e001862. doi: 10.1161/JAHA.115.001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu W, Hitomi T, Kobayashi H, Harada KH, Koizumi A. Distribution of moyamoya disease susceptibility polymorphism p.R4810K in RNF213 in East and Southeast Asian populations. Neurol Med Chir (Tokyo) 2012;52:299–303. doi:10.2176/nmc.52.299. [DOI] [PubMed]
- 19.Morito D, Nishikawa K, Hoseki J, Kitamura A, Kotani Y, Kiso K, et al. Moyamoya disease-associated protein mysterin/RNF213 is a novel AAA+ ATPase, which dynamically changes its oligomeric state. Sci Rep. 2014;4:4442. doi: 10.1038/srep04442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kobayashi H, Yamazaki S, Takashima S, Liu W, Okuda H, Yan J, et al. Ablation of Rnf213 retards progression of diabetes in the Akita mouse. Biochem Biophys Res Commun. 2013;432:519–525. doi: 10.1016/j.bbrc.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi H, Matsuda Y, Hitomi T, Okuda H, Shioi H, Matsuda T, et al. Biochemical and functional characterization of RNF213 (mysterin) R4810K, a susceptibility mutation of moyamoya disease, in angiogenesis in vitro and in vivo. J Am Heart Assoc. 2015;4:e002146. doi: 10.1161/JAHA.115.002146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sonobe S, Fujimura M, Niizuma K, Fujimura T, Furudate S, Nishijima Y, et al. Increased vascular MMP-9 in mice lacking RNF213: moyamoya disease susceptibility gene. NeuroReport. 2014;25:1442–1446. doi: 10.1097/WNR.0000000000000289. [DOI] [PubMed] [Google Scholar]
- 23.Ito A, Fujimura M, Niizuma K, Kanoke A, Sakata H, Morita-Fujimura Y, et al. Enhanced post-ischemic angiogenesis in mice lacking RNF213; a susceptibility gene for moyamoya disease. Brain Res. 2015;1594:310–320. doi: 10.1016/j.brainres.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 24.Hitomi T, Habu T, Kobayashi H, Okuda H, Harada KH, Osafune K, et al. Downregulation of Securin by the variant RNF213 R4810K (rs112735431, G > A) reduces angiogenic activity of induced pluripotent stem cell-derived vascular endothelial cells from moyamoya patients. Biochem Biophys Res Commun. 2013;438:13–19. doi: 10.1016/j.bbrc.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 25.Yamashita M, Oka K, Tanaka K. Histopathology of the brain vascular network in moyamoya disease. Stroke. 1983;14:50–58. doi: 10.1161/01.str.14.1.50. [DOI] [PubMed] [Google Scholar]
- 26.Yamashita M, Oka K, Tanaka K. Cervico-cephalic arterial thrombi and thromboemboli in moyamoya disease–possible correlation with progressive intimal thickening in the intracranial major arteries. Stroke. 1984;15:264–270. doi: 10.1161/01.str.15.2.264. [DOI] [PubMed] [Google Scholar]
- 27.Kudo T. Spontaneous occlusion of the circle of Willis. A disease apparently confined to Japanese. Neurology. 1968;18:485–496. doi: 10.1212/wnl.18.5.485. [DOI] [PubMed] [Google Scholar]
- 28.Nishimoto A, Takeuchi S. Abnormal cerebrovascular network related to the internal cartoid arteries. J Neurosurg. 1968;29:255–260. doi: 10.3171/jns.1968.29.3.0255. [DOI] [PubMed] [Google Scholar]
- 29.Research Committee on the Pathology and Treatment of Spontaneous Occlusion of the Circle of Willis Guidelines for diagnosis and treatment of moyamoya disease (spontaneous occlusion of the circle of Willis) Neurol Med Chir. 2012;52:245–266. doi: 10.2176/nmc.52.245. [DOI] [PubMed] [Google Scholar]
- 30.Miyawaki S, Imai H, Shimizu M, Yagi S, Ono H, Mukasa A, et al. Genetic variant RNF213 c.14576G > A in various phenotypes of intracranial major artery stenosis/occlusion. Stroke. 2013;44:2894–2897. doi: 10.1161/STROKEAHA.113.002477. [DOI] [PubMed] [Google Scholar]
- 31.Ikeda H, Sasaki T, Yoshimoto T, Fukui M, Arinami T. Mapping of a familial moyamoya disease gene to chromosome 3p24.2-p26. Am J Hum Genet. 1999;64:533–537. doi: 10.1086/302243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoue TK, Ikezaki K, Sasazuki T, Matsushima T, Fukui M. Linkage analysis of moyamoya disease on chromosome 6. J Child Neurol. 2000;15:179–182. doi: 10.1177/088307380001500307. [DOI] [PubMed] [Google Scholar]
- 33.Sakurai K, Horiuchi Y, Ikeda H, Ikezaki K, Yoshimoto T, Fukui M, et al. A novel susceptibility locus for moyamoya disease on chromosome 8q23. J Hum Genet. 2004;49:278–281. doi: 10.1007/s10038-004-0143-6. [DOI] [PubMed] [Google Scholar]
- 34.Yamauchi T, Tada M, Houkin K, Tanaka T, Nakamura Y, Kuroda S, et al. Linkage of familial moyamoya disease (spontaneous occlusion of the circle of Willis) to chromosome 17q25. Stroke. 2000;31:930–935. doi: 10.1161/01.str.31.4.930. [DOI] [PubMed] [Google Scholar]
- 35.Mineharu Y, Liu W, Inoue K, Matsuura N, Inoue S, Takenaka K, et al. Autosomal dominant moyamoya disease maps to chromosome 17q25.3. Neurology. 2008;70:2357–2363. doi: 10.1212/01.wnl.0000291012.49986.f9. [DOI] [PubMed] [Google Scholar]
- 36.Liu W, Hashikata H, Inoue K, Matsuura N, Mineharu Y, Kobayashi H, et al. A rare Asian founder polymorphism of Raptor may explain the high prevalence of Moyamoya disease among East Asians and its low prevalence among Caucasians. Environ Health Prev Med. 2010;15:94–104. doi: 10.1007/s12199-009-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee MJ, Chen YF, Fan PC, Wang KC, Wang K, Wang J, et al. Mutation genotypes of RNF213 gene from moyamoya patients in Taiwan. J Neurol Sci. 2015;353:161–165. doi: 10.1016/j.jns.2015.04.019. [DOI] [PubMed] [Google Scholar]
- 38.Scott RM, Smith ER. Moyamoya disease and moyamoya syndrome. N Engl J Med. 2009;360:1226–1237. doi: 10.1056/NEJMra0804622. [DOI] [PubMed] [Google Scholar]
- 39.Harel T, Posey JE, Graham BH, Walkiewicz M, Yang Y, Lalani SR, et al. Atypical presentation of moyamoya disease in an infant with a de novo RNF213 variant. Am J Med Genet A (online:2015.7.21), doi:10.1002/ajmg.a.37230. [DOI] [PMC free article] [PubMed]
- 40.Miyatake S, Touho H, Miyake N, Ohba C, Doi H, Saitsu H, et al. Sibling cases of moyamoya disease having homozygous and heterozygous c.14576G > A variant in RNF213 showed varying clinical course and severity. J Hum Genet. 2012;57:804–806. doi: 10.1038/jhg.2012.105. [DOI] [PubMed] [Google Scholar]
- 41.Koizumi A, Kobayashi H, Liu W, Fujii Y, Senevirathna ST, Nanayakkara S, et al. P.R4810K, a polymorphism of RNF213, the susceptibility gene for moyamoya disease, is associated with blood pressure. Environ Health Prev Med. 2013;18:121–129. doi: 10.1007/s12199-012-0299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ogura T, Wilkinson AJ. AAA+ superfamily ATPases: common structure–diverse function. Genes Cells. 2001;6:575–597. doi: 10.1046/j.1365-2443.2001.00447.x. [DOI] [PubMed] [Google Scholar]
- 43.Geisbrecht BV, Collins CS, Reuber BE, Gould SJ. Disruption of a PEX1-PEX6 interaction is the most common cause of the neurologic disorders Zellweger syndrome, neonatal adrenoleukodystrophy, and infantile Refsum disease. Proc Natl Acad Sci USA. 1998;95:8630–8635. doi: 10.1073/pnas.95.15.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reuber BE, Germain-Lee E, Collins CS, Morrell JC, Ameritunga R, Moser HW, et al. Mutations in PEX1 are the most common cause of peroxisome biogenesis disorders. Nat Genet. 1997;17:445–448. doi: 10.1038/ng1297-445. [DOI] [PubMed] [Google Scholar]
- 45.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864. doi: 10.1016/j.neuron.2010.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abramzon Y, Johnson JO, Scholz SW, Taylor JP, Brunetti M, Calvo A, et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2012;33:2231 e2231–2231 e2236. doi:10.1016/j.neurobiolaging.2012.04.005. [DOI] [PMC free article] [PubMed]
- 47.Hoshimaru M, Takahashi JA, Kikuchi H, Nagata I, Hatanaka M. Possible roles of basic fibroblast growth factor in the pathogenesis of moyamoya disease: an immunohistochemical study. J Neurosurg. 1991;75:267–270. doi: 10.3171/jns.1991.75.2.0267. [DOI] [PubMed] [Google Scholar]
- 48.Hojo M, Hoshimaru M, Miyamoto S, Taki W, Nagata I, Asahi M, et al. Role of transforming growth factor-beta1 in the pathogenesis of moyamoya disease. J Neurosurg. 1998;89:623–629. doi: 10.3171/jns.1998.89.4.0623. [DOI] [PubMed] [Google Scholar]
- 49.Aoyagi M, Fukai N, Sakamoto H, Shinkai T, Matsushima Y, Yamamoto M, et al. Altered cellular responses to serum mitogens, including platelet-derived growth factor, in cultured smooth muscle cells derived from arteries of patients with moyamoya disease. J Cell Physiol. 1991;147:191–198. doi: 10.1002/jcp.1041470202. [DOI] [PubMed] [Google Scholar]
- 50.Nanba R, Kuroda S, Ishikawa T, Houkin K, Iwasaki Y. Increased expression of hepatocyte growth factor in cerebrospinal fluid and intracranial artery in moyamoya disease. Stroke. 2004;35:2837–2842. doi: 10.1161/01.STR.0000148237.13659.e6. [DOI] [PubMed] [Google Scholar]
- 51.Maruwaka M, Yoshikawa K, Okamoto S, Araki Y, Sumitomo M, Kawamura A, et al. Biomarker research for moyamoya disease in cerebrospinal fluid using surface-enhanced laser desorption/ionization time-of-flight mass spectrometry. J Stroke Cerebrovasc Dis. 2015;24:104–111. doi: 10.1016/j.jstrokecerebrovasdis.2014.07.028. [DOI] [PubMed] [Google Scholar]
- 52.Ohkubo K, Sakai Y, Inoue H, Akamine S, Ishizaki Y, Matsushita Y, et al. Moyamoya disease susceptibility gene RNF213 links inflammatory and angiogenic signals in endothelial cells. Sci Rep. 2015;5:13191. doi: 10.1038/srep13191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JH, Jung JH, Phi JH, Kang HS, Kim JE, Chae JH, et al. Decreased level and defective function of circulating endothelial progenitor cells in children with moyamoya disease. J Neurosci Res. 2010;88:510–518. doi: 10.1002/jnr.22228. [DOI] [PubMed] [Google Scholar]
- 54.Kim CS, Ying H, Willingham MC, Cheng SY. The pituitary tumor-transforming gene promotes angiogenesis in a mouse model of follicular thyroid cancer. Carcinogenesis. 2007;28:932–939. doi: 10.1093/carcin/bgl231. [DOI] [PubMed] [Google Scholar]
- 55.Arnaoutova I, George J, Kleinman HK, Benton G. The endothelial cell tube formation assay on basement membrane turns 20: state of the science and the art. Angiogenesis. 2009;12:267–274. doi: 10.1007/s10456-009-9146-4. [DOI] [PubMed] [Google Scholar]
- 56.Hitomi T, Habu T, Kobayashi H, Okuda H, Harada KH, Osafune K, et al. The moyamoya disease susceptibility variant RNF213 R4810K (rs112735431) induces genomic instability by mitotic abnormality. Biochem Biophys Res Commun. 2013;439:419–426. doi: 10.1016/j.bbrc.2013.08.067. [DOI] [PubMed] [Google Scholar]
- 57.Schirmer SH, Bot PT, Fledderus JO, van der Laan AM, Volger OL, Laufs U, et al. Blocking interferon beta stimulates vascular smooth muscle cell proliferation and arteriogenesis. J Biol Chem. 2010;285:34677–34685. doi: 10.1074/jbc.M110.164350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Takano S, Ishikawa E, Matsuda M, Yamamoto T, Matsumura A. Interferon-beta inhibits glioma angiogenesis through downregulation of vascular endothelial growth factor and upregulation of interferon inducible protein 10. Int J Oncol. 2014;45:1837–1846. doi: 10.3892/ijo.2014.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tsipis CP, Sun X, Xu K, Lamanna JC. Hypoxia-induced angiogenesis and capillary density determination. Methods Mol Biol. 2014;1135:69–80. doi: 10.1007/978-1-4939-0320-7_6. [DOI] [PubMed] [Google Scholar]
- 60.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early-onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46:887–894. doi: 10.2337/diab.46.5.887. [DOI] [PubMed] [Google Scholar]
- 61.Sonobe S, Fujimura M, Niizuma K, Nishijima Y, Ito A, Shimizu H, et al. Temporal profile of the vascular anatomy evaluated by 9.4-T magnetic resonance angiography and histopathological analysis in mice lacking RNF213: a susceptibility gene for moyamoya disease. Brain Res. 2014;1552:64–71. doi: 10.1016/j.brainres.2014.01.011. [DOI] [PubMed] [Google Scholar]
- 62.Fujimura M, Sonobe S, Nishijima Y, Niizuma K, Sakata H, Kure S, et al. Genetics and biomarkers of moyamoya disease: significance of RNF213 as a susceptibility gene. J Stroke. 2014;16:65–72. doi: 10.5853/jos.2014.16.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kasher PR, Jenkinson EM, Briolat V, Gent D, Morrissey C, Zeef LA, et al. Characterization of samhd1 morphant zebrafish recapitulates features of the human type I interferonopathy Aicardi–Goutieres syndrome. J Immunol. 2015;194:2819–2825. doi: 10.4049/jimmunol.1403157. [DOI] [PubMed] [Google Scholar]
- 64.Rehwinkel J, Maelfait J, Bridgeman A, Rigby R, Hayward B, Liberatore RA, et al. SAMHD1-dependent retroviral control and escape in mice. EMBO J. 2013;32:2454–2462. doi: 10.1038/emboj.2013.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Crow YJ, Manel N. Aicardi–Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol. 2015;15:429–440. doi: 10.1038/nri3850. [DOI] [PubMed] [Google Scholar]
- 66.Aquino Gondim Fde A, Leacock RO, Subrammanian TA, Cruz-Flores S. Intracerebral hemorrhage associated with Sneddon’s syndrome: is ischemia-related angiogenesis the cause? Case report and review of the literature. Neuroradiology. 2003;45:368–372. doi: 10.1007/s00234-003-0990-4. [DOI] [PubMed] [Google Scholar]
- 67.Carhuapoma JR, D’Olhaberriague L, Levine SR. Moyamoya syndrome associated with Sneddon’s syndrome and antiphospholipid-protein antibodies. J Stroke Cerebrovasc Dis. 1999;8:51–56. doi: 10.1016/s1052-3057(99)80054-8. [DOI] [PubMed] [Google Scholar]
- 68.Fierini F, Barilaro A, Giambene B, Carlucci G, Grandi V, Maio V, et al. Moyamoya in a patient with Sneddon’s syndrome. Clin Neurol Neurosurg. 2015;129:34–36. doi: 10.1016/j.clineuro.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 69.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Stone DL, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med. 2014;370:911–920. doi: 10.1056/NEJMoa1307361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and moyamoya disease, along with thoracic aortic disease. Am J Hum Genet. 2009;84:617–627. doi: 10.1016/j.ajhg.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Herve D, Philippi A, Belbouab R, Zerah M, Chabrier S, Collardeau-Frachon S, et al. Loss of alpha1beta1 soluble guanylate cyclase, the major nitric oxide receptor, leads to moyamoya and achalasia. Am J Hum Genet. 2014;94:385–394. doi: 10.1016/j.ajhg.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13:357–378. doi: 10.1038/nrd4252. [DOI] [PubMed] [Google Scholar]
- 73.Merkel KH, Ginsberg PL, Parker JC, Jr, Post MJ. Cerebrovascular disease in sickle cell anemia: a clinical, pathological and radiological correlation. Stroke. 1978;9:45–52. doi: 10.1161/01.str.9.1.45. [DOI] [PubMed] [Google Scholar]
- 74.Park HW, Oh D, Kim N, Cho HY, Moon KC, Chae JH, et al. Congenital thrombotic thrombocytopenic purpura associated with unilateral moyamoya disease. Pediatr Nephrol. 2008;23:1555–1558. doi: 10.1007/s00467-008-0847-5. [DOI] [PubMed] [Google Scholar]
- 75.Salih M, Andeejani A, Gader A, Kolawole T, Palkar V. Moyamoya syndrome associated with protein C deficiency. Med Sci Res. 1995;23:573–575. [Google Scholar]
- 76.Hiyama H, Kusano R, Muragaki Y, Miura N. Moyamoya disease associated with thrombotic thrombocytopenic purpura (TTP) No Shinkei Geka. 1994;22:567–572. [PubMed] [Google Scholar]
- 77.Artoni A, Selicorni A, Passamonti SM, Lecchi A, Bucciarelli P, Cerutti M, et al. Hemostatic abnormalities in Noonan syndrome. Pediatrics. 2014;133:e1299–e1304. doi: 10.1542/peds.2013-3251. [DOI] [PubMed] [Google Scholar]
- 78.Tourdot BE, Conaway S, Niisuke K, Edelstein LC, Bray PF, Holinstat M. Mechanism of race-dependent platelet activation through the protease-activated receptor-4 and Gq signaling axis. Arterioscler Thromb Vasc Biol. 2014;34:2644–2650. doi: 10.1161/ATVBAHA.114.304249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lattanzio S, Santilli F, Liani R, Vazzana N, Ueland T, Di Fulvio P, et al. Circulating dickkopf-1 in diabetes mellitus: association with platelet activation and effects of improved metabolic control and low-dose aspirin. J Am Heart Assoc. 2014;3:e001000. doi: 10.1161/JAHA.114.001000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lasater EA, Li F, Bessler WK, Estes ML, Vemula S, Hingtgen CM, et al. Genetic and cellular evidence of vascular inflammation in neurofibromin-deficient mice and humans. J Clin Invest. 2010;120:859–870. doi: 10.1172/JCI41443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lasater EA, Bessler WK, Mead LE, Horn WE, Clapp DW, Conway SJ, et al. Nf1+/− mice have increased neointima formation via hyperactivation of a Gleevec sensitive molecular pathway. Hum Mol Genet. 2008;17:2336–2344. doi: 10.1093/hmg/ddn134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang J, Hu H, Palma NL, Harrison JK, Mubarak KK, Carrie RD, et al. Hypoxia-induced endothelial CX3CL1 triggers lung smooth muscle cell phenotypic switching and proliferative expansion. Am J Physiol Lung Cell Mol Physiol. 2012;303:L912–L922. doi: 10.1152/ajplung.00014.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davies MG, Hagen PO. Pathobiology of intimal hyperplasia. Br J Surg. 1994;81:1254–1269. doi: 10.1002/bjs.1800810904. [DOI] [PubMed] [Google Scholar]
- 84.George PM, Oliver E, Dorfmuller P, Dubois OD, Reed DM, Kirkby NS, et al. Evidence for the involvement of type I interferon in pulmonary arterial hypertension. Circ Res. 2014;114:677–688. doi: 10.1161/CIRCRESAHA.114.302221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buckley ML, Ramji DP. The influence of dysfunctional signaling and lipid homeostasis in mediating the inflammatory responses during atherosclerosis. Biochim Biophys Acta. 2015;1852:1498–1510. doi: 10.1016/j.bbadis.2015.04.011. [DOI] [PubMed] [Google Scholar]
- 86.Zhang SM, Zhu LH, Chen HZ, Zhang R, Zhang P, Jiang DS, et al. Interferon regulatory factor 9 is critical for neointima formation following vascular injury. Nat Commun. 2014;5:5160. doi: 10.1038/ncomms6160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chong PF, Ogata R, Kobayashi H, Koizumi A, Kira R. Early onset of moyamoya syndrome in a Down syndrome patient with the genetic variant RNF213 p.R4810K. Brain Dev (online:2014.12.26), doi:10.1016/j.braindev.2014.12.006. [DOI] [PubMed]
- 88.Miskinyte S, Butler MG, Herve D, Sarret C, Nicolino M, Petralia JD, et al. Loss of BRCC3 deubiquitinating enzyme leads to abnormal angiogenesis and is associated with syndromic moyamoya. Am J Hum Genet. 2011;88:718–728. doi: 10.1016/j.ajhg.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Akasaki T, Kagiyama S, Omae T, Ohya Y, Ibayashi S, Abe I, et al. Asymptomatic moyamoya disease associated with coronary and renal artery stenoses—a case report. Jpn Circ J. 1998;62:136–138. doi: 10.1253/jcj.62.136. [DOI] [PubMed] [Google Scholar]
- 90.Lee JH, Youn TJ, Yoon YE, Park JJ, Hong SJ, Chun EJ, et al. Coronary artery stenosis in moyamoya disease: tissue characterization by 256-slice multi-detector CT and virtual histology. Circulation. 2013;127:2063–2065. doi: 10.1161/CIRCULATIONAHA.112.136473. [DOI] [PubMed] [Google Scholar]
- 91.Choi W, Kim YN, Kim KH. Variant angina in moyamoya disease—a correlative etiology and different presentation: a case report. J Med Case Rep. 2015;9:86. doi: 10.1186/s13256-015-0537-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nam TM, Jo KI, Yeon JY, Hong SC, Kim JS. Coronary heart disease in moyamoya disease: are they concomitant or coincidence? J Korean Med Sci. 2015;30:470–474. doi: 10.3346/jkms.2015.30.4.470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikeda E. Systemic vascular changes in spontaneous occlusion of the circle of Willis. Stroke. 1991;22:1358–1362. doi: 10.1161/01.str.22.11.1358. [DOI] [PubMed] [Google Scholar]
- 94.Yamada I, Himeno Y, Matsushima Y, Shibuya H. Renal artery lesions in patients with moyamoya disease: angiographic findings. Stroke. 2000;31:733–737. doi: 10.1161/01.str.31.3.733. [DOI] [PubMed] [Google Scholar]
- 95.Togao O, Mihara F, Yoshiura T, Tanaka A, Kuwabara Y, Morioka T, et al. Prevalence of stenoocclusive lesions in the renal and abdominal arteries in moyamoya disease. AJR Am J Roentgenol. 2004;183:119–122. doi: 10.2214/ajr.183.1.1830119. [DOI] [PubMed] [Google Scholar]
- 96.Kaku Y, Morioka M, Ohmori Y, Kawano T, Kai Y, Fukuoka H, et al. Outer-diameter narrowing of the internal carotid and middle cerebral arteries in moyamoya disease detected on 3D constructive interference in steady-state MR image: is arterial constrictive remodeling a major pathogenesis? Acta Neurochir. 2012;154:2151–2157. doi: 10.1007/s00701-012-1472-4. [DOI] [PubMed] [Google Scholar]
- 97.Takeuchi K, Shimizu K. Hypoplasia of the bilateral internal carotid arteries. No Shinkei. 1957;9:37–43. [Google Scholar]
- 98.Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58:341–344. doi: 10.1034/j.1399-0004.2000.580501.x. [DOI] [PubMed] [Google Scholar]
- 99.Farmer JP, Khan S, Khan A, Ortenberg J, Freeman C, O’Gorman AM, et al. Neurofibromatosis type 1 and the pediatric neurosurgeon: a 20-year institutional review. Pediatr Neurosurg. 2002;37:122–136. doi: 10.1159/000064398. [DOI] [PubMed] [Google Scholar]
- 100.Rosser TL, Vezina G, Packer RJ. Cerebrovascular abnormalities in a population of children with neurofibromatosis type 1. Neurology. 2005;64:553–555. doi: 10.1212/01.WNL.0000150544.00016.69. [DOI] [PubMed] [Google Scholar]
- 101.Koss M, Scott RM, Irons MB, Smith ER, Ullrich NJ. Moyamoya syndrome associated with neurofibromatosis Type 1: perioperative and long-term outcome after surgical revascularization. J Neurosurg Pediatr. 2013;11:417–425. doi: 10.3171/2012.12.PEDS12281. [DOI] [PubMed] [Google Scholar]
- 102.Ganesan V, Kirkham FJ. Noonan syndrome and moyamoya. Pediatr Neurol. 1997;16:256–258. doi: 10.1016/s0887-8994(97)89980-8. [DOI] [PubMed] [Google Scholar]
- 103.Yamashita Y, Kusaga A, Koga Y, Nagamitsu S, Matsuishi T. Noonan syndrome, moyamoya-like vascular changes, and antiphospholipid syndrome. Pediatr Neurol. 2004;31:364–366. doi: 10.1016/j.pediatrneurol.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 104.Shiihara T, Kato M, Mitsuhashi Y, Hayasaka K. Costello syndrome showing moyamoya-like vasculopathy. Pediatr Neurol. 2005;32:361–363. doi: 10.1016/j.pediatrneurol.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 105.Choi JH, Oh MY, Yum MS, Lee BH, Kim GH, Yoo HW. Moyamoya syndrome in a patient with Noonan-like syndrome with loose anagen hair. Pediatr Neurol. 2015;52:352–355. doi: 10.1016/j.pediatrneurol.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 106.Rachmel A, Zeharia A, Neuman-Levin M, Weitz R, Shamir R, Dinari G. Alagille syndrome associated with moyamoya disease. Am J Med Genet. 1989;33:89–91. doi: 10.1002/ajmg.1320330112. [DOI] [PubMed] [Google Scholar]
- 107.Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. doi: 10.1161/01.CIR.0000121361.01862.A4. [DOI] [PubMed] [Google Scholar]
- 108.Al Kaissi A, Bieganski T, Baranska D, Chehida FB, Gharbi H, Ghachem MB, et al. Robinow syndrome: report of two cases and review of the literature. Australas Radiol. 2007;51:83–86. doi: 10.1111/j.1440-1673.2006.01668.x. [DOI] [PubMed] [Google Scholar]
- 109.Qaiser R, Scott RM, Smith ER. Identification of an association between Robinow syndrome and moyamoya. Pediatr Neurosurg. 2009;45:69–72. doi: 10.1159/000204907. [DOI] [PubMed] [Google Scholar]
- 110.Boerkoel CF, O’Neill S, Andre JL, Benke PJ, Bogdanovic R, Bulla M, et al. Manifestations and treatment of Schimke immuno-osseous dysplasia: 14 new cases and a review of the literature. Eur J Pediatr. 2000;159:1–7. doi: 10.1007/s004310050001. [DOI] [PubMed] [Google Scholar]
- 111.Lucke T, Clewing JM, Boerkoel CF, Hartmann H, Das AM, Knauth M, et al. Cerebellar atrophy in Schimke-immuno-osseous dysplasia. Am J Med Genet A. 2007;143A:2040–2045. doi: 10.1002/ajmg.a.31878. [DOI] [PubMed] [Google Scholar]
- 112.Rauch A, Thiel CT, Schindler D, Wick U, Crow YJ, Ekici AB, et al. Mutations in the pericentrin (PCNT) gene cause primordial dwarfism. Science. 2008;319:816–819. doi: 10.1126/science.1151174. [DOI] [PubMed] [Google Scholar]
- 113.Bober MB, Khan N, Kaplan J, Lewis K, Feinstein JA, Scott CI, Jr, et al. Majewski osteodysplastic primordial dwarfism type II (MOPD II): expanding the vascular phenotype. Am J Med Genet A. 2010;152A:960–965. doi: 10.1002/ajmg.a.33252. [DOI] [PubMed] [Google Scholar]
- 114.Codd PJ, Scott RM, Smith ER. Seckel syndrome and moyamoya. J Neurosurg Pediatr. 2009;3:320–324. doi: 10.3171/2008.12.PEDS08205. [DOI] [PubMed] [Google Scholar]
- 115.Charuvanij A, Laothamatas J, Torcharus K, Sirivimonmas S. Moyamoya disease and protein S deficiency: a case report. Pediatr Neurol. 1997;17:171–173. doi: 10.1016/s0887-8994(97)00072-6. [DOI] [PubMed] [Google Scholar]
- 116.Bonduel M, Hepner M, Sciuccati G, Torres AF, Tenembaum S. Prothrombotic disorders in children with moyamoya syndrome. Stroke. 2001;32:1786–1792. doi: 10.1161/01.str.32.8.1786. [DOI] [PubMed] [Google Scholar]
- 117.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, et al. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–849. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 118.Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, et al. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell. 1990;63:835–841. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- 119.Addissie YA, Kotecha U, Hart RA, Martinez AF, Kruszka P, Muenke M. Craniosynostosis and Noonan syndrome with KRAS mutations: expanding the phenotype with a case report and review of the literature. Am J Med Genet A. 2015 doi: 10.1002/ajmg.a.37259. [DOI] [PubMed] [Google Scholar]
- 120.Aoki Y, Niihori T, Kawame H, Kurosawa K, Ohashi H, Tanaka Y, et al. Germline mutations in HRAS proto-oncogene cause Costello syndrome. Nat Genet. 2005;37:1038–1040. doi: 10.1038/ng1641. [DOI] [PubMed] [Google Scholar]
- 121.Minami Y, Oishi I, Endo M, Nishita M. Ror-family receptor tyrosine kinases in noncanonical Wnt signaling: their implications in developmental morphogenesis and human diseases. Dev Dyn. 2010;239:1–15. doi: 10.1002/dvdy.21991. [DOI] [PubMed] [Google Scholar]
- 122.Boerkoel CF, Takashima H, John J, Yan J, Stankiewicz P, Rosenbarker L, et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat Genet. 2002;30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 123.Yigit G, Brown KE, Kayserili H, Pohl E, Caliebe A, Zahnleiter D, et al. Mutations in CDK5RAP2 cause Seckel syndrome. Mol Genet Genom Med. 2015 doi: 10.1002/mgg3.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu S, Xu Z, Liang H. Sneddon’s syndrome: a comprehensive review of the literature. Orphanet J Rare Dis. 2014;9:215–221. doi: 10.1186/s13023-014-0215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.