

Key points
It is generally accepted that mitochondrial volume density in human skeletal muscle diminishes with chronic high altitude exposure.
All data supporting this concept were collected during mountaineering expeditions, which are associated with the confounding effects of whole body negative energy balance.
Here we examine the effect of 28 days of exposure to 3454 m on skeletal muscle mitochondrial volume density in a setting where whole body weight, whole body composition, leg lean mass, skeletal muscle fibre area and maximal power output were preserved.
Our results demonstrate that total skeletal muscle mitochondrial volume density increases in response to high altitude exposure secondary to a preferential increase in intermyofibrillar mitochondrial populations.
This study provides direct evidence contradicting the notion that high altitude exposure diminishes skeletal muscle mitochondrial volume density, highlighting an inconsistent understanding of the role of hypoxia on skeletal muscle mitochondria.
Abstract
The role of hypoxia on skeletal muscle mitochondria is controversial. Studies superimposing exercise training on hypoxic exposure demonstrate an increase in skeletal muscle mitochondrial volume density (MitoVD) over equivalent normoxic training. In contrast, reductions in both skeletal muscle mass and MitoVD have been reported following mountaineering expeditions. These observations may, however, be confounded by negative energy balance, which may obscure the results. Accordingly we sought to examine the effects of high altitude hypoxic exposure on mitochondrial characteristics, with emphasis on MitoVD, while minimizing changes in energy balance. For this purpose, skeletal muscle biopsies were obtained from nine lowlanders at sea level (Pre) and following 7 and 28 days of exposure to 3454 m. Maximal ergometer power output, whole body weight and composition, leg lean mass and skeletal muscle fibre area all remained unchanged following the altitude exposure. Transmission electron microscopy determined that intermyofibrillar (IMF) MitoVD was augmented (P = 0.028) by 11.5 ± 9.2% from Pre (5.05 ± 0.9%) to 28 Days (5.61 ± 0.04%). In contrast, there was no change in subsarcolemmal (SS) MitoVD. As a result, total MitoVD (IMF + SS) was increased (P = 0.031) from 6.20 ± 1.5% at Pre to 6.62 ± 1.4% at 28 Days (7.8 ± 9.3%). At the same time no changes in mass‐specific respiratory capacities, mitochondrial protein or antioxidant content were found. This study demonstrates that skeletal muscle MitoVD may increase with 28 days acclimation to 3454 m.
Key points
It is generally accepted that mitochondrial volume density in human skeletal muscle diminishes with chronic high altitude exposure.
All data supporting this concept were collected during mountaineering expeditions, which are associated with the confounding effects of whole body negative energy balance.
Here we examine the effect of 28 days of exposure to 3454 m on skeletal muscle mitochondrial volume density in a setting where whole body weight, whole body composition, leg lean mass, skeletal muscle fibre area and maximal power output were preserved.
Our results demonstrate that total skeletal muscle mitochondrial volume density increases in response to high altitude exposure secondary to a preferential increase in intermyofibrillar mitochondrial populations.
This study provides direct evidence contradicting the notion that high altitude exposure diminishes skeletal muscle mitochondrial volume density, highlighting an inconsistent understanding of the role of hypoxia on skeletal muscle mitochondria.
Abbreviations
- au
arbitrary units
- C/F ratio
capillary‐to‐fibre ratio
- CI + CIIP
maximal state 3 respiration–oxidative phosphorylation capacity
- CIIP
submaximal state 3 respiration through mitochondrial complex II
- CIP
submaximal state 3 respiration through mitochondrial complex I
- COXIV
cytochrome c oxidase IV
- CS
citrate synthase
- ETFP
maximal fatty acid oxidation
- ETS
electron transfer system capacity
- HIF
hypoxia‐inducible transcription factor
- HXII
hexokinase II
- IMF
intermyofibrillar
- Lactate
submaximal state‐3 lactate stimulated respiration
- LCR
mitochondrial leak coupling ratio
- LDH
lactate dehydrogenase
- LDVD
lipid droplets volume density
- LLTL
live low–train low
- MFN2
mitofusin‐2
- MitoVD
mitochondrial volume density
- PGC‐1α
peroxisome proliferator‐activated receptor γ coactivator 1 alpha
- RCR
respiratory control ratio
- RT
room temperature
- SOD2
superoxide dismutase 2
- SS
subsarcolemmal
- TEM
transmission electron microscopy
- TMPD
N,N,N',N'‐tetramethyl‐p‐phenylenediamine dihydrochloride
- ww
wet weight
Introduction
As oxidative phosphorylation is the primary bioenergetic pathway in which ATP is generated, a drop in cellular oxygen concentration threatens energetic balance and thus physiological function. Accordingly, hypoxia serves as a stimulus to induce adaptations that help maintain metabolic homeostasis in spite of a reduction in cellular oxygen availability. While these global metabolic alterations have been identified (Ratcliffe et al. 1998; Semenza et al. 2006; Vigano et al. 2008), hypoxic regulation of human skeletal muscle mitochondria, specifically, remains controversial.
Exercise training increases skeletal muscle mitochondria and attendant oxidative capacity (Holloszy, 1967; Coyle et al. 1984). These adaptations to exercise are governed by peroxisome proliferator‐activated receptor γ coactivator 1‐α (PGC‐1α), a master regulator of mitochondrial adaptation referred to as mitochondrial biogenesis (Wu et al. 1999; Baar et al. 2002), and are partially dependent on hypoxia sensing through hypoxia‐inducible transcription factors (HIF) 1‐ and 2‐α (Taylor, 2008; Rasbach et al. 2010). PGC‐1α overexpression upregulates HIF‐1α regulated genes (O'Hagan et al. 2009) as well as HIF‐2α (Rasbach et al. 2010). It is believed that exercise per se transiently simulates local hypoxic/ischaemic conditions within the working muscle and this stimulus partially facilitates exercise‐induced mitochondrial alterations (O'Hagan et al. 2009). Indeed, acute normoxic exercise training transiently increases HIF‐1α and HIF‐2α mRNA in human skeletal muscle (Lundby et al. 2006) and conditions resembling ischaemia induce PGC‐1 gene expression in cultured skeletal myotubes (Arany et al. 2008).
Superimposing exercise and hypoxia reduces intramuscular oxygen partial pressure beyond that observed with normoxic exercise (Richardson et al. 2006). Consequently, some investigators theorized that the more pronounced hypoxia and attendant metabolic stresses on the skeletal muscle with hypoxic exercise may facilitate a greater adaptive response in the skeletal muscle and selectively enhance mitochondrial characteristics (Hoppeler et al. 2008). In support of this theory, living near sea level but training in hypoxic conditions, effectively increased mitochondrial volume density (MitoVD) more than the complementary live low–train low (LLTL) training (Desplanches et al. 1993, 2014; Vogt et al. 2001; Schmutz et al. 2010). However, an ostensibly contradictory effect of hypoxia on mitochondria is provided by mountaineering studies, which show that chronic hypoxic exposure significantly reduces MitoVD, (Hoppeler et al. 1990 a; Levett et al. 2012), but in these conditions it cannot be ruled out that, in particular, fluctuations in energy balance may be confounding when attempting to examine mitochondrial physiology. Given these data, the role of hypoxic exposure on skeletal muscle MitoVD in humans is unclear.
Therefore, in this study we investigate the role of terrestrial hypoxic exposure (3454 m) on skeletal muscle mitochondrial characteristics. Unlike previous studies that have demonstrated a loss of MitoVD following high altitude exposure (Hoppeler et al. 1990 a; Levett et al. 2012), in our study we minimized confounding factors by ensuring adequate housing and were thereby able to maintain maximal power output, whole body mass and composition, total leg fat‐free mass and skeletal muscle fibre cross‐sectional area to examine how hypoxic exposure, specifically, regulates mitochondrial characteristics, with emphasis on MitoVD. We hypothesized that acclimatization to high altitude exposure will increase MitoVD, as has been demonstrated in studies examining the effects of hypoxic exercise on skeletal muscle mitochondria (Desplanches et al. 1993, 2014; Vogt et al. 2001; Schmutz et al. 2010).
Methods
The present experimental protocols were approved by the ethical committee of the Eidgenössische Technische Hochschule Zürich (ETH, EK 2011‐N‐51) and were conducted in accordance with the Declaration of Helsinki. Participants were fully informed about the purposes, benefits and risks associated with this study and gave their written informed consent prior to the initiation of the experiments.
A total of nine young and healthy individuals, eight male and one female, voluntarily participated in this study (mean ± SD: age, 26.9 ± 3.7 years; height, 178.8 ± 8.9 cm; body mass, 75.2 ± 10.3 kg). All participants were recreationally active; they were neither sedentary nor highly trained.
Experimental design
Subjects reported to the laboratory in Zurich (432 m) several times prior to high altitude ascent for acquisition of preliminary (Pre) measurements. Subsequently, subjects were transported by train to the Jungfraujoch Research Station in the Swiss Alps located at 3454 m. The ascent profile was uniform across all subjects. Here, skeletal muscle biopsies were sampled following 7 and 28 days of acclimatization to study mitochondrial characteristics. For a detailed description the reader is referred to (Siebenmann et al. 2015).
During 14 days prior to the sea level studies all subjects were given physical activity meters that they wore 24 h a day for those 14 days and the plan was that the subjects should have mimicked these at the research‐station. However many of the devices failed at altitude and we do not have complete recordings. However all subjects were well aware of the importance of this aspect of the study and to the best of their ability replicated normal sea level activities while at the research station. For example, those (n = 2) who used to bike to and from school did similar ‘exercise’ on a Monark ergometer (Monark E839, Varberg, Sweden). The remaining subjects walked to a nearby mountain hut (30 min each way) on a road 5 days week–1 which should resemble their sea level transportation to and from school. One subject did two sessions (30–35 min) of upper body strength training per week at sea level, and to be able to continue this habit we specifically transported his six private dumbbells up to the research station. Thus, we did our best to replicate sea level physical activity levels while residing at the research station. We did not record the subjects’ dietary intake but they were free to order groceries from a nationwide shop at sea level which were then brought to the research station on a daily basis. The subjects prepared all meals themselves and were asked to consume their ‘normal diet’. Since no weight gain or loss was observed we are confident that neither activity level nor dietary intake could be regarded as a confounder in the present study.
Body composition
A densitometer (Lunar iDXATM, GE Healthcare, Madison, WI, USA) was used for the determination of lean and fat tissue prior to high altitude ascent (Pre) and again 2 days after return to sea level (Post).
Exercise testing
Each participant conducted an incremental exercise test on a braked cycle ergometer (Monark E839) until voluntary exhaustion to determine peak oxygen uptake (). Pulmonary gas exchange and ventilation were continuously recorded using an online gas collection system (Innocor M400, Innovision, Odense, Denmark), where O2 and CO2 concentration were continuously measured and monitored as breath‐by‐breath values. The gas analysers and the flowmeter of the applied spirometer were calibrated prior to each test according to the manufacturer's instructions. All subjects began with three consecutive submaximal 5 min workloads, starting at 50 W which was then increased to 100 and 150 W. Thereafter, the workload was increased by 30 W every 90 s until voluntary exhaustion. was determined as the highest mean over a 10 s period.
Skeletal muscle sampling
Skeletal muscle biopsies were obtained under standardized conditions from the m. vastus lateralis under local anaesthesia (1% lidocaine) of the skin and superficial muscle fascia, using the Bergström technique with a needle modified for suction. The biopsy was immediately dissected macroscopically free of fat and connective tissue and divided into sections for ex vivo measurements of mitochondrial respiration and for later transmission electron microscopy (TEM), Western blotting, immunohistochemistry and assay analysis. The part of the biopsy for the determination of mitochondrial respiration capacity was immediately placed in ice‐cold biopsy preservation solution. TEM specimens were processed as described below. The samples for skeletal muscle lysates were snap frozen in liquid nitrogen and stored at −80°C until processing. Tissue for immunohistochemistry was instantly mounted in an embedding medium (Tissue‐Tek, Sakura Finetek, Torrance, CA, USA), snap frozen in isopentane cooled to −160°C with liquid nitrogen, and subsequently stored at −80°C until cryosectioning.
Transmission electron microscopy
Four 1 mm3 pieces of each muscle biopsy were fixed in 2.5% glutaraldehyde at room temperature and processed as previously described (Montero et al. 2015). TEM images were obtained in a FEI Tecnai G2 Spirit electron microscope (FEI, Hillsboro, OR, USA) mounted with an Orius SC1000 CCD camera (Gatan, Pleasanton, CA, USA). A total of 216 images (3840 × 2528 pixels, each pixel being 4.14 × 4.14 nm) per biopsy were acquired in a random systematic order from 24 meshes distributed on 8 grids from 4 blocks. In each of the 24 meshes, 9 images were acquired using automated image capturing by the TEM photomontage software. A random starting point was selected for the first micrograph, whereafter further micrographs were taken at fixed x‐, y‐intervals of −400%. The Cavalieri feature in the Stereo‐Investigator software (MBF Bioscience, USA) was used to estimate skeletal muscle volume density of lipid droplets (LDVD) and mitochondria (MitoVD) by point counting (West, 2012). The grid spacing was 1 μm along both x‐ and y‐axes. Each point was assigned as either intermyofibrillar (IMF) mitochondria, subsarcolemmal (SS) mitochondria, lipid droplet (LD), skeletal muscle or ‘nothing’. SS mitochondria were defined as the mitochondria that were not separated by myofibrils from the sarcolemma.
Cryosections
Tissue‐Tek‐embedded muscle samples were cut at an optimal cutting temperature (−22°C) using a cryostat (Leica CM 1850, Leica Biosystems, Germany). Three serial transverse sections (8 μm thick) were obtained from each muscle sample and mounted on glass slides (Thermo Scientific, Superfrost Plus). The sections were set to air dry and stored at –20°C until further processing. Prior to immunohistochemical analysis the sections were fixed in acetone for 30 s and left for air drying at room temperature (RT) for 10 min.
Skeletal muscle fibre cross‐sectional area (FCSA)
Fixed sections were blocked with 5% goat serum in PBS (pH = 7.4) for 20 min at RT for elimination of non‐specific binding. The sarcolemma of the muscle fibres was stained using anti‐laminin (1:80) mouse monoclonal primary antibody (NCL‐Laminin, Novocastra). Alexa Flour 488 (1:600) (ab150117, Abcam) was used as secondary antibody. Antibody incubations were performed at 37°C for 30 min in a humidified chamber. Sections were mounted with cover slips using a mounting medium (Vectashield H‐1000). All muscle sections were digitally captured on a Leica fluorescence microscopy (Leica DM500 B, Leica Microsystems, Germany). Digital imaging was performed at ×10 magnification. Semi‐automated image processing and computation of FCSA was performed using the software ilastik (version 1.1.5, ilastik: Integrative Learning and Segmentation Toolkit) for automated simple segmentation of the images and the FIJI software (NIH, USA) for determination of the FCSA using a custom‐made plug‐in that analysing the areas obtained from the simple segmentation as intensity maps. The FCSA was determined for an average of 202 ± 54 fibres per sample (N = 27), with a minimum of 100 and a maximum of 300 fibres analysed.
Muscle capillarization
The 8 μm serial transverse sections were also used for immunohistochemical analysis of capillary density. The fixed sections were blocked with 1% BSA prior to incubation with primary antibodies against caveolin‐1 (Cat 610060, BD bioscience) and collagen IV (M0785, Dako) in conjunction with biotinylated secondary antibodies (E032 and E033, Dako) and the VECTASTAIN ABC‐AP KIT (Vector laboratories, USA) for detection of capillaries and muscle fibres. A slide scanner (Zeiss Mirax Midi, Germany) connected to a 3‐CCD camera (Hitachi HV‐F22, 1360 × 1024 pixels, Japan) was used for digitizing the cryo‐sections at ×20 magnification. Capillary density was determined by counting the number of capillaries (250 ± 58 capillaries; mean ± SD) surrounding an average of 133 (minimum 99; maximum 215) coherent fibres and expressed as the capillary‐to‐fibre ratio (C/F ratio). Counting was performed manually using Photoshop CS6 and Panoramic Viewer (3DHISTECH Ltd, Hungary). To adjust the C/F ratio for capillaries located in the outer circumference of the counted area, the number of capillaries in the outer circumference of each section was divided by 2, before being added to the number of capillaries in the inner section. The total number of capillaries was then divided by the number of counted fibres.
Muscle lysate preparation
Snap frozen muscle sections (15–20 mg) were freeze dried for 15 h at −55°C (ScanVac CoolSafe55‐4, Denmark), homogenized (Precellys 24 Tissue Homogenizer, Bertin Technologies, France) in freshly made homogenization buffer, whereafter muscles lysates were prepared as described in detail elsewhere (Nordsborg et al. 2012). Total protein concentrations were determined by BCA assay (Pierce, USA). For Western blotting, samples were diluted to the same protein concentration with 6 × modified Laemmli buffer (Nordsborg et al. 2012) and stored at −80°C until analysis. The remaining lysates were separated into aliquots and stored at −80°C.
Citrate synthase activity
Citrate synthase activity was determined in muscle lysates using a commercially available citrate synthase assay kit (C3260, Sigma‐Aldrich) according to the manufacturer. All activities were normalized to milligrams of total protein.
SDS–PAGE and Western blotting
Standard Western blotting procedures (Jacobs et al. 2013 c) were applied for quantification of catalase (ab16731, Abcam), superoxide dismutase 2 (SOD2) (ab13534, Abcam), hexokinase II (HXII) (2867S, Cell Signalling Technology), lactate dehydrogenase (LDH) (ab135396, Abcam), mitofusin 2 (MFN2) (H00009927‐M03, Abnova), cytochrome c oxidase IV (COXIV) (Ab33985, Abcam) and skeletal muscle specific alpha‐actin (actin) (A2066, Sigma‐Aldrich). Anti‐mouse or anti‐rabbit IgG HRP‐conjugated antibodies (W4021 and W4011, Promega) were used for labelling of primary antibodies. Protein bands were detected with Immobilon Western Chemiluminescent HRP Substrate (Merck‐Millipore) using the Las‐4000 image analyser system (Fujifilm Life Science). Quantification of band intensity was performed using FIJI software. All samples from each individual subject were loaded on the same gel together with human pool control samples. Signal intensity from each sample was adjusted according to the mean signal intensity of the all samples on the same gel and normalized to alpha‐actin. All Western blots were run in duplicate and the average values were subsequently calculated.
Skeletal muscle mitochondrial respiration
Samples were prepared as described in detail previously (Jacobs et al. 2012 a). In short, after mechanical fibre separation, chemical permeabilization in an ice‐cold biopsy preservation solution (2.77 mm CaK2EGTA buffer, 7.23 mm K2EGTA buffer, 0.1 μm free calcium, 20 mm imidazole, 20 mm taurine, 50 mm 2‐(N‐morpholino) ethanesulfonic acid hydrate (K‐MES), 0.5 mm dithiothreitol (DTT), 6.56 mm MgCl2.6H2O, 5.77 mm ATP, 15 mm phosphocreatine, and 50 μg ml−1 of saponin; pH 7.1) and washing (10 min at 4°C) in mitochondrial respiration medium (0.5 mm EGTA, 3 mm MgCl2.6H2O, 60 mm potassium lactobionate, 20 mm taurine, 10 mm KH2PO4, 20 mm Hepes, 110 mm sucrose, and 1 mg ml−1 BSA; pH 7.1), respectively, muscle bundles were blotted dry and measured for wet weight (ww) in a balance‐controlled scale (XS205 DualRange Analytical Balance; Mettler‐Toledo AG, Greifensee, Switzerland). Respiration measurements were then performed in the mitochondrial respiration medium plus catalase (u ml−1). Oxygen consumption of skeletal muscle samples was measured at 37°C using the high‐resolution Oxygraph‐2k (Oroboros, Innsbruck, Austria) with the titration of each substrate, uncoupler and inhibitor in series. Standardized instrumental and chemical calibrations were performed as recommended by the manufacturer and described previously (Jacobs et al. 2012 a). Oxygen flux was computed by the software, accounting for non‐linear changes in the negative time derivative of the oxygen concentration signal (DatLab, Oroboros, Innsbruck, Austria). All samples were measured in duplicate in a hyperoxygenated environment to control for any potential differences in oxygen diffusion capacity. Oxygen concentration within the chambers ranged between 200 and 450 nmol ml−1.
Respiratory titration protocol
The titration protocol utilized has been described in detail previously (Jacobs et al. 2012 a). Leak respiration in the absence of adenylates was induced with the addition of malate (2 mm) and octanoyl carnitine (0.2 mm). Maximal electron flow through electron‐transferring‐flavoprotein (ETF) and fatty acid oxidative capacity (ETFP) was determined following the addition of ADP (5 mm). In the ETFP state, the ETF‐linked transfer of electrons requires the metabolism of acetyl‐CoA, hence the addition of malate, in order to facilitate convergent electron flow into the Q‐junction from both mitochondrial complex I (CI) and ETF, allowing β‐oxidation to proceed. The contribution of electron flow through CI is far below capacity and so here the rate‐limiting metabolic branch is electron transport through ETF such that malate + octanoyl carnitine + ADP stimulated respiration is representative of, rather than specific to, electron capacity through ETF (Eaton et al. 1996; Saks et al. 1998; Gnaiger, 2009; Pesta & Gnaiger, 2011; Jacobs et al. 2013 b). Submaximal state 3 respiratory capacity specific to CI (NADH dehydrogenase; CIP) was first induced following the additions of lactate (30 mm), pyruvate (5 mm) and glutamate (10 mm). Maximal state 3 respiration–oxidative phosphorylation capacity (CI + CIIP), was then induced with the addition of succinate (19 mm). This maximal state 3 state represents respiration that is resultant on saturating concentrations of ADP and substrate supply for both CI and mitochondrial complex II (succinate dehydrogenase; CII). Convergent electron input to CI and CII provides higher respiratory values compared to the isolated respiration of either CI (pyruvate/glutamate + malate or glutamate + malate) or CII (succinate + rotenone) (Rasmussen & Rasmussen, 2000; Gnaiger, 2009). Consequently CI + CIIP is more physiologically relevant to the study of mitochondrial function (Brand & Nicholls, 2011) and is necessary to establish confirmation of a complete and intact electron transport system. CI + CIIP demonstrates a naturally intact electron transport system's capacity to catalyse a sequential set of redox reactions that are partially coupled to the production of ATP via ATP synthase. Compared to a corresponding leak state with an equivalent substrate supply, CI + CIIP maintains a lower electrochemical gradient across the inner mitochondrial membrane. That gradient is dictated by the degree of coupling to the phosphorylation system (Gnaiger, 2009; Pesta & Gnaiger, 2011). As an internal control for compromised integrity of the mitochondrial preparation, the mitochondrial outer membrane was assessed with the addition of cytochrome c (10 μm). The addition of exogenous cytochrome c suggested complete mitochondrial membrane integrity across samples measured at sea level (110.77 ± 31.65 to 109.87 ± 30.74 pmol O2 min−1 (mg ww)−1), following 7 days of exposure (115.13 ± 36.62 to 116.75 ± 36.48 pmol O2·min−1 (mg ww)−1) and this was also the case after 28 days of exposure (124.38 ± 18.50 to 124.03 ± 18.30 pmol O2·min−1 (mg ww)−1). Oligomycin (2.5 μm) was added to inhibit ATP synthase. Phosphorylative restraint of electron transport was assessed by uncoupling ATP synthase (complex V) from the electron transport system with the titration of the proton ionophore carbonyl cyanide p‐(trifluoromethoxy) phenylhydrazone (FCCP; 1–2.5 μm) reaching electron transport system (ETS) capacity. The inner mitochondrial membrane potential is completely collapsed with an open transmembrane proton circuit in the ETS respiratory state. The uninhibited flow of electrons through the respiratory system can therefore directly serve as an indication of maximal electron flow through the ETS. Rotenone (0.5 μm) and antimycin A (2.5 μm) were added, in sequence, to terminate respiration by inhibiting CI and complex III (cytochrome bc1 complex), respectively. With CI inhibited, electron flow specific to CII (CIIP) can be measured. Inhibition of respiration with antimycin A then allows for the determination and correction of residual oxygen consumption (ROX), indicative of non‐mitochondrial oxygen consumption in the chamber. Respiration measurements were concluded by simultaneous titration of ascorbate (2 mm) and TMPD (0.5 mm) to assess complex IV (COXIV) activity. TMPD and ascorbate are redox substrates that donate electrons directly to COXIV.
Indices of mitochondrial efficiency
There are various measures of mitochondrial efficiency analysed. Here, leak control and coupling control ratios were calculated as indirect indications of mitochondrial efficiency. Leak control ratios (LCRs) are produced between two respiratory states, a leak state (i.e. low respiration, state 4) to a higher respiratory state (i.e. state 3 respiration). These corresponding states are paired by an identical substrate supply. The reference state is defined by the leak state, i.e. flux in the P state is greater than in the LOMY state given the same substrate supply (detailed above). LCRs help describe mitochondrial coupling efficiency with a theoretical minimum of 0.0 indicating a fully coupled system to a value of 1.0 representing a fully non‐coupled (dyscoupled) system (Gnaiger, 2009). Alternatively, coupling control ratios are based on the theory that a tightly coupled electron transport system is distinguished from a dyscoupled system by the magnitude of difference between two steady respiratory states, with the reference state being the stated defined by the minimum oxygen flux (Chance & Williams, 1955). The most common coupling control ratio is the respiratory control ratio (RCR; state 3 respiration/state 4 respiration).
Results
Whole body and skeletal muscle mass
No differences in whole body mass before altitude exposure and following 28 days exposure to 3454 m were observed (P = 0.867; Fig. 1), suggesting that subjects remained weight stable throughout the duration of the study. Dual‐energy X‐ray absorptiometry (DEXA) results confirm these findings as percentage body fat and lean body mass remained unchanged in measurements taken before and again after 28 days of exposure to 3454 m (data not shown). Moreover, neither leg lean mass (P = 0.918) nor fibre cross‐sectional area (FCSA; P = 0.646) were altered in response to altitude exposure (Fig. 1).
Figure 1. Body weight, leg lean mass and fibre cross‐sectional area .
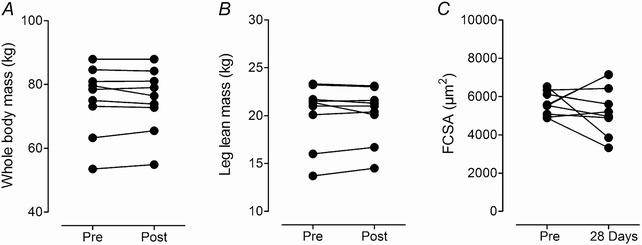
Individual whole body mass (A), leg lean mass (B), and skeletal muscle fibre cross‐sectional area (FCSA; C), before (Pre) and after (Post) the 28 days sojourn at 3454 m or on the 28th day at 3454 m.
Maximal exercise capacity () and power output ()
and data are presented in Table 1. Measures of , both absolute and relative to body weight, remained unchanged in response to altitude exposure, suggesting that overall fitness remained stable throughout the course of the study. However, following the altitude exposure period tended (P = 0.055) to increase by 140 ml, which, assuming a 4 ml increase in per gram increase in Hb mass (Prommer & Schmidt, 2010), may be attributed entirely to the concomitantly occurring 5.26 % increase in Hb mass (Siebenmann et al. 2015).
Table 1.
Maximal exercise capacity and power output
| Pre | Post | P values | |
|---|---|---|---|
| (l min−1) | 3.89 ± 0.88 | 4.03 ± 0.87 | 0.055 |
| (ml kg−1 min−1) | 51.6 ± 8.3 | 53.7 ± 8.1 | 0.055 |
| (W) | 336.3 ± 62.1 | 327.8 ± 62.9 | 0.150 |
| (W kg−1) | 4.49 ± 0.66 | 4.36 ± 0.55 | 0.136 |
Relative and absolute measures of oxygen consumption () and maximal power output () are presented. W; watts. Values are means ± SD.
Skeletal muscle mitochondrial volume density (MitoVD)
The skeletal muscle mitochondria were distinguished according to their location in the muscle fibre as either intermyofibrillar (IMF) or subsarcolemmal (SS) mitochondria. Exposure to hypoxia led to changes in IMF MitoVD (Pre, 5.05 ± 0.89%; 7 days at 3454 m, 4.98 ± 0.99%; 28 days at 3454 m, 5.61 ± 0.04%; P = 0.028)). On average, IMF MitoVD increased by 11.5 ± 9.2% after 28 days at 3454 m compared to sea level (Fig. 2 A). In contrast, there was no change in the SS MitoVD (Pre, 1.15 ± 0.65; 7 days at 3454 m, 0.93 ± 0.40; 28 days at 3454 m, 1.01 ± 0.47%; P = 0.233; Fig. 2 B). The total MitoVD (IMF + SS) increased by 7.8 ± 9.3% (P = 0.031) with 28 days of high altitude exposure, i.e. from 6.20 ± 1.49% to 6.62 ± 1.35% (Fig. 2 C).
Figure 2. Skeletal muscle mitochondrial volume density (MitoVD) .
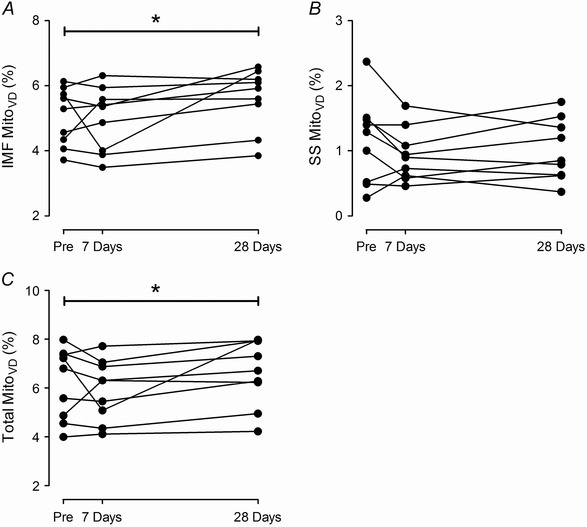
Stereological estimations of intermyofibrillar (IMF) MitoVD (A), subsacrolemmal (SS) MitoVD (B), and total MitoVD (C) in individuals before (Pre), after 7 days and after 28 days sojourn at 3454 m. Note the different scaling of the y‐axis. *P < 0.05 vs. Pre.
Skeletal muscle lipid droplet volume density (LDVD)
The skeletal muscle LDVD was assessed in the same way as MitoVD and there were no changes in LDVD in response to either 7 or 28 days exposure to hypoxia, on average (Pre, 0.38 ± 0.14%; 7 days at 3454 m, 0.49 ± 0.38%; 28 days at 3454 m, 0.36 ± 0.15%; P = 0.513; Fig. 3 A).
Figure 3. Lipid droplets volume density (LDVD) and capillary‐to‐fibre ratio (C/F ratio).
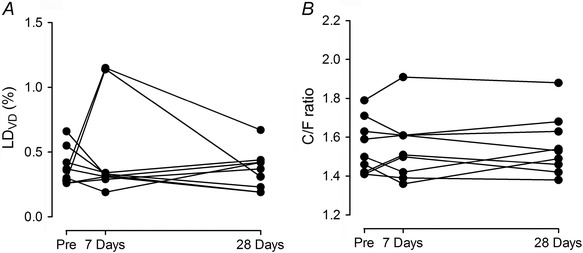
Stereological estimation of LDVD (A) and C/F ratios (B) for individual subjects before (Pre), after 7 days and after 28 days sojourn at 3454 m.
Capillary‐to‐fibre ratio (C/F ratio)
The C/F ratio did not change in response to exposure to 3454 m (Pre, 1.55 ± 0.1; 7 days at 3454 m, 1.56 ± 0.2; 28 days at 3454 m, 1.56 ± 0.2; P = 0.907; Fig. 3 B).
Skeletal muscle citrate synthase (CS) and cytochrome c oxidase (COXIV)
Skeletal muscle CS (P = 0.303) and COXIV (P = 0.885) activity did not change in response to 28 days high altitude exposure (Fig. 4 A and B). There was also no change in protein expression for COXIV in response to hypoxic exposure (Fig. 5 F). While there was a significant correlation (P ≤ 0.05) between both CS and COXIV activities and total MitoVD when examining all data sets (Pre, 7 days and 28 days) combined (Fig. 4 C and D), there was no correlation between the absolute changes (Δ) in CS activities (ΔCS) with those observed for MitoVD (ΔMitoVD) (Fig. 4 E) or between ΔCOXIV and ΔMitoVD (Fig. 4 F).
Figure 4. Citrate synthase (CS) and cytochrome c oxidase (COXIV) activities and correlation to total MitoVD .
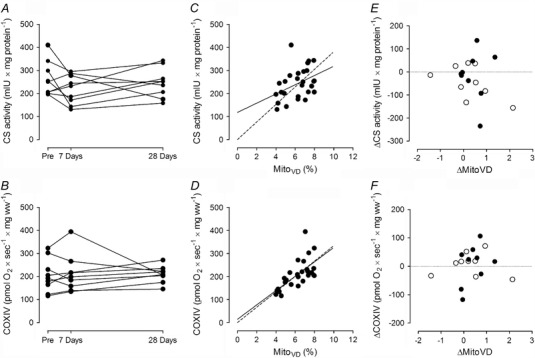
CS activity (A) and COXIV activity (B) before (Pre), after 7 days and after 28 days sojourn at 3454 m for individual subjects. C and D, correlation between total MitoVD and CS activity (C; Pearson r = 0.400, P = 0.040); E, COXIV activity (D; Pearson r = 0.660, P < 0.001). Lines represent linear regression and dashed lines linear regression through zero. E and F, correlation between absolute changes (Δ) of total MitoVD (ΔMitoVD) and CS activity (ΔCS; E) and COXIV activity (ΔCOXIV; F), Pre to 7 days (open circles), Pre to 28 days (filled circles) (all P values >> 0.1).
Figure 5. Protein expression levels evaluated by Western blot .
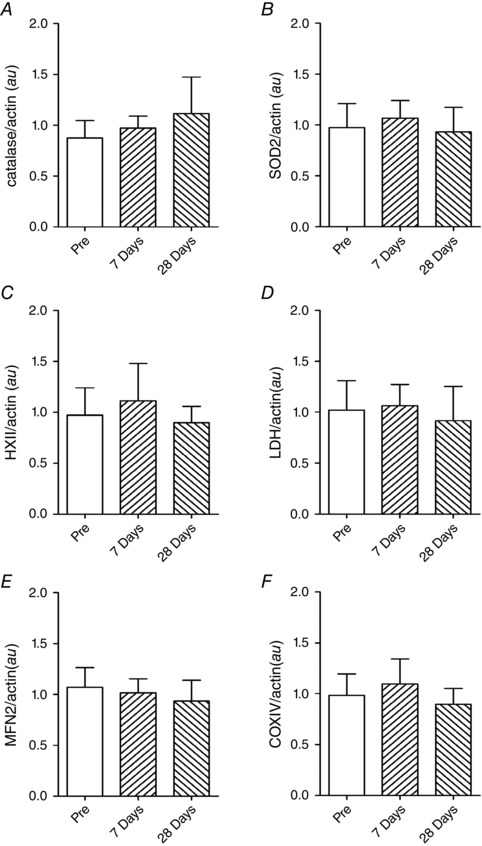
A, catalase; B, superoxide dismutase2 (SOD2); C, hexokinase II (HXII); D, lactate dehydrogenase (LDH); E, mitofusin 2 (MFN2); F, cytochrome c oxidase IV (COXIV). Data determined in samples taken before (Pre), and after 7 and 28 days at 3454 m (means ± SEM, N = 9). au, arbitrary units.
Skeletal muscle mass‐specific respiration
All respiratory states analysed are reported in Table 2. There were no changes in skeletal muscle mitochondrial mass‐specific respiration (pmol O2 s−1 (mg ww)−1), at any respiratory state, throughout the course of the study.
Table 2.
Skeletal muscle respirometric analysis
| Pre | 7 days | 28 days | P values | ||
|---|---|---|---|---|---|
| ETFP | 34.3 ± 11.80 | 31.52 ± 9.3 | 29.7 ± 5.7 | 0.903 | |
| Lactate | 64.6 ± 23.8 | 66.6 ± 19.2 | 67.8 ± 16.2 | 0.309 | |
| CIP | 82.4 ± 23.0 | 90.1 ± 30.8 | 94.0 ± 16.70 | 0.329 | |
| CI + CIIP | 110.8 ± 31.7 | 115.1 ± 36.6 | 124.4 ± 18.5 | 0.070 | |
| ETS | 121.8 ± 33.3 | 125.5 ± 44.6 | 139.8 ± 22.6 | 0.289 | |
| CIIP | 70.6 ± 21.6 | 68.1 ± 22.3 | 70.8 ± 9.2 | 0.863 | |
| COXIV | 204.0 ± 71.8 | 212.1 ± 80.2 | 209.6 ± 35.5 | 0.928 | |
|
|
0.38 ± 0.11 | 0.47 ± 0.15 | 0.43 ± 0.11 | 0.109 | |
| LCRCI + CII | 0.47 ± 0.07 | 0.43 ± 0.04 | 0.47 ± 0.08 | 0.379 | |
| LCRETS | 0.42 ± 0.06 | 0.40 ± 0.05 | 0.42 ± 0.05 | 0.635 | |
| RCR | 2.17 ± 0.32 | 2.34 ± 0.23 | 2.16 ± 0.34 | 0.423 |
Skeletal muscle mitochondrial mass‐specific respiration (pmol O2 s−1 (mg ww)−1) is reported for samples collected before altitude exposure (Pre), following 7 days exposure to 3454 m, and after 28 days exposure to 3454 m. ETFP, maximal fatty acid oxidation; Lactate, submaximal state‐3 lactate stimulated respiration; CIP, submaximal state 3 respiration through mitochondrial complex I; CI + CIIP, maximal state 3 respiration–oxidative phosphorylation capacity; ETS, electron transport system capacity; CIIP, submaximal state 3 respiration through mitochondrial complex II; LCR, leak control ratios of ETFP, CI + CIIP, and ETS, respectively; and respiratory control ratio (RCR; state 3). Values are means ± SD.
Skeletal muscle mitochondrial efficiency
Mitochondrial leak coupling ratios (LCR) and the respiratory control ratio (RCR), as indices of respiratory efficiency, are reported in Table 2. There was no indication suggesting a change in electron coupling control when comparing measures prior to and following 28 days exposure to 3454 m.
Mitochondrial dynamics
In an attempt to better understand and explain the observed changes in MitoVD, we measured an essential protein regulating in mitochondrial fusion, mitofusin‐2 (MFN2). There was, however, no discernible change in MFN2 following 7 or 28 days exposure to high altitude (Fig. 5 E).
Skeletal muscle non‐aerobic/glycolytic enzyme protein content
Protein content of two fundamental regulating enzymes of glycolysis, hexokinase II (HKII) and lactate dehydrogenase (LDH) were analysed. Neither HKII nor LDH changed in response to high altitude exposure (Fig. 5 C and D).
Skeletal muscle antioxidants
High altitude exposure results in increased oxidative stress. Acclimatization is believed to involve adaptation to increased concentration of oxidants by either diminishing respiratory capacity and/or increasing antioxidant concentrations. We analysed the protein content of two antioxidants, catalase and mitochondrial‐specific superoxide dismutase 2 (SOD2). Skeletal muscle antioxidant protein concentrations did not change in response to high altitude exposure (Fig. 5 A and B).
Discussion
This study examined skeletal muscle mitochondrial adaptation in nine individuals exposed to terrestrial hypoxia (3454 m) while maintaining whole body mass and composition, total leg fat‐free mass and skeletal muscle fibre cross‐sectional area. The novel findings of this study are: (i) total mitochondrial volume density (MitoVD) increases with acclimatization to 3454 m; and (ii) the increase in MitoVD is delineated by a preferential increase in intermyofibrillar (IMF) mitochondrial volume density, with no change in subsarcolemmal (SS) populations. These changes in MitoVD were observed as the principal mitochondrial adaptation to high altitude exposure, as several additional complementary characteristics (i.e. mitochondrial enzyme activity and protein content, antioxidant concentrations, mass‐specific respiratory control, and indices of electron coupling control) remained unchanged.
An assessment of literature reviews discussing the effect of hypoxic exposure on MitoVD collectively suggest that while intermittent hypoxic exposure superimposed on exercise may enhance MitoVD, chronic exposure to environmental hypoxia induces a loss of total MitoVD in human skeletal muscle (Hoppeler & Desplanches, 1992; Hoppeler et al. 2003; Howald & Hoppeler, 2003; Murray & Horscroft, 2015). Indeed, a review published in this issue (Murray & Horscroft, 2015) affirms this theory by concluding that extended exposure to extreme high altitude appears to result in a loss of MitoVD in human skeletal muscle with a specific loss of subsarcolemmal mitochondrial populations. A limitation to this theory, however, as also addressed in the aforementioned review (Murray & Horscroft, 2015), is that the collection of data supporting these concepts are derived from mountaineering studies in extreme hypoxia where particular fluctuations in energy balance may be confounding when attempting to examine mitochondrial physiology.
Hypoxic exposure commonly results in negative energy balance and an attendant loss of body mass (Westerterp et al. 1992, 1994,2000; Wing‐Gaia et al. 2013), predominantly (60–70%) consisting of a loss of fat‐free mass (Rose et al. 1988; Butterfield et al. 1992; Pulfrey & Jones, 1996; Armellini et al. 1997) and a reduction of energy intake (Westerterp et al. 2000). The degree of hypoxia‐associated weight loss appears to be influenced by both absolute altitude and the duration of exposure (Kayser, 1994). Additionally, energy expenditure in hypoxia also fluctuates, with either a decrease reported when subjects are restricted to a hypobaric chamber (Westerterp et al. 2000) or an increase when hypoxic exposure coincides with mountaineering (Reynolds et al. 1999). Given this information it seems hasty to conclude that high altitude exposure per se results in diminished MitoVD when the sum total of studies exploring this topic in human subjects have also observed losses in total body, total fat‐free mass, and/or skeletal muscle cross‐sectional area (Hoppeler et al. 1990 a; MacDougall et al. 1991; Levett et al. 2012).
Hypoxia has been identified as a stimulus for regulated in development and DNA damage response 1 (REDD1, also referred to as Rtp801; Ellisen et al. 2002), which inhibits mammalian target of rapamycin (mTOR) a central regulator of protein synthesis (Brugarolas et al. 2004). REDD1 is thought to contribute to skeletal muscle wasting associated with more severe chronic obstructive pulmonary disease (Favier et al. 2010; Langen et al. 2013), a disease state characterized by upper respiratory airflow limitation and lung inflammation that can result in chronic systemic hypoxaemia (Pauwels et al. 2001), ergo one theory is that high‐altitude‐associated skeletal muscle atrophy may be induced by an upregulation of REDD1 expression. Although we have previously observed a slight decrease in total mTORC1 following a 7‐ to 9‐day sojourn at 4559 m (Vigano et al. 2008), data that would support this theory, skeletal muscle functional respiratory capacity was, however, unaffected (Jacobs et al. 2013 a). Moreover, in human subjects, acute hypoxic exposure modified the mTORC1 pathway 4 h after feeding, which would suggest conditions that are favourable for muscle protein accretion, higher insulin concentration and attendant PKB and S6K1 phosphorylation, despite an increase REDD1 transcription (D'Hulst et al. 2013). Lastly, any effect of hypoxia‐stimulated REDD1 expression on exercise‐induced increases in mean protein synthesis (MPS) appears transient (Etheridge et al. 2011; Chaillou et al. 2012), suggesting that REDD1‐associated inhibition of mTOR1 may be silenced by concurrent exercise or activity during chronic exposure to hypoxia. In this respect, high‐altitude studies that have reported loss of skeletal muscle mass, most likely to be those in which the subjects were sedentary or had a decrease in activity thermogenesis with high altitude exposure, may also have been influenced by an upregulation of REDD1. Much less is known regarding the regulation of REDD1 on mitochondrial protein synthesis, a process that is maintained despite extreme metabolic stress such as a 40% reduction in caloric restriction (Miller et al. 2012).
Previous data suggest that a net loss of total skeletal muscle protein with acclimatization (Holm et al. 2010) may account for any downregulation of mitochondrial proteins reported following high altitude exposure (Mizuno et al. 1990; Vigano et al. 2008). In vitro HIF‐1α upregulation and activation has been shown to suppress mitochondrial DNA (Zhang et al. 2008), mitochondrial protein expression and mitochondrial mass (Jensen et al. 2011). However, in the current study there was no change in mitochondrial enzyme activity, protein expression, or mass‐specific oxidative capacity. The lack of change in CS or COXIV with high altitude exposure is supported by our previous findings (Jacobs et al. 2012 a). Moreover, not all studies, even those involving exposure to extreme high altitude, have demonstrated a loss in MitoVD. Levett et al. (2012) failed to see a change in MitoVD in non‐climbing base‐camp staff after 19 days at 5300 m terrestrial altitude and MacDougall et al. (1991) reported no changed to MitoVD following a 40 day progressive decompression ultimately reaching an inspired partial pressure of oxygen of 43 Torr, an equivalent to 8848 m. In the present study we demonstrate that 28 days exposure to 3454 m of terrestrial hypoxia, when maintaining maximal power output, body weight, fat‐free mass and skeletal muscle fibre cross‐sectional area, results in an increase in MitoVD, with a particular increase in IMF mitochondrial density (Fig. 2). Interestingly, we have previously shown the intermyofibrillar protein synthesis rate to be increased in healthy volunteers with 7–9 days exposure to 4559 m, whereas subsarcolemmal protein synthesis rate remained unchanged (Holm et al. 2010).
Two biochemically and positionally distinct mitochondrial populations exist within skeletal muscle, intermyofibrillar (IMF) and the subsarcolemmal (SS) mitochondria. The IMF populations are situated in‐between myofibrils and principally support the energetic requirements of contraction while SS populations exist immediately beneath the sarcolemma and support energetic requirements specific to maintaining membrane potential, active membrane transport, and various cytoplasmic processes (Krieger et al. 1980; Cogswell et al. 1993; Iossa et al. 2002). In the current study the IMF MitoVD preferentially increased, explaining the increase in total MitoVD, whereas SS MitoVD remained unchanged (Fig. 2).
An earlier, and seemingly overlooked, hypothesis posited that hypoxic exposure actually enhances aerobic bioenergetics (Hochachka et al. 1983). In line with this theory, animals that are consistently exposed to hypoxic conditions have evolved with measures of enhanced skeletal muscle oxidative capacity (Valdivia, 1958; Scott, 2011). The high altitude native bar‐headed goose has skeletal muscle mitochondria that appear redistributed closer to adjacent capillaries (Scott et al. 2009) and hypobaric hypoxia increased mitochondrial density in murine cerebral subcortex (Gutsaeva et al. 2008). In human subjects, studies that superimpose hypoxia on exercise training also resulted in an increase in MitoVD compared with normoxic training (Desplanches et al. 1993, 2014; Vogt et al. 2001; Schmutz et al. 2010). Given the results of the present study in combination with past literature, it is clear that there is an inconsistent understanding of the role of hypoxia on skeletal muscle mitochondria and a need for further studies to elucidate these findings.
In an attempt to explain the increases MitoVD following 28 days exposure to 3454 m, various enzymatic and functional mitochondrial characteristics were analysed. High altitude exposure is commonly associated with elevated oxidative stress (Moller et al. 2001) and perhaps attendant increases in skeletal muscle antioxidant defence would explain the increase in MitoVD. However, there were no alterations in enzymatic concentrations of catalase or mitochondrial‐specific SOD2 with acclimatization (Fig. 5). Additionally, common biomarkers serving as a proxy for mitochondrial content (Larsen et al. 2012), CS activity, COXIV activity (Fig. 4), and COXIV protein content, also did not change with high altitude exposure (Fig. 5). These data support earlier reports where 28 days of terrestrial exposure to 3454 m also failed to increase CS and COXIV (Jacobs et al. 2012 a). Though cellular measures of endogenous antioxidants did not change, the increase in MitoVD in combination with the maintenance of mass‐specific respiratory capacity and measures of both TCA cycle and ETS proteins following hypoxic exposure may still result in a lower oxidant production by means of expanding the inner mitochondrial area responsible for maintaining mitochondrial membrane potential (electrochemical gradient). This may effectively diminish the amount of mitochondrial proton leak, electron slip, cation cycling, and overall dyscoupling at any given location along the mitochondrial reticulum, helping to regulate hypoxia‐induced increases in oxidative stress in skeletal muscle (Clanton, 2007). Our previous data demonstrating diminished skeletal muscle respiratory capacity per unit of mitochondrial content following chronic high altitude exposure support this theory (Jacobs et al. 2012 b).
Non‐aerobic metabolic pathways complement aerobic metabolism in human bioenergetics to maintain energetic homeostasis. Therefore potential hypoxia‐induced alterations in aerobic metabolism are theorized to be accompanied by divergent changes in non‐aerobic metabolic pathways (Murray & Horscroft, 2015). Mostly this theory was not confirmed in the present study. In addition to an increase in total MitoVD and none in skeletal muscle oxidative capacity, neither hexokinase nor lactate dehydrogenase increased in response to 28 days high altitude exposure (Fig. 5). These findings are supported by data showing no change in the capillary‐to‐fibre ratio, which is HIF‐1 regulated (Arany et al. 2008), negligible changes in intramuscular triglyceride, LDVD, and previous data which also demonstrated no increase in capillarity with high altitude exposure (Lundby et al. 2004). There did appear to be transient increases in LDVD in two subjects measured after 7 days of high altitude exposure compared, to preliminary and data collected after 28 days (Fig. 3 A). These data may indicate a brief alteration in nutrient partitioning, reflecting an increased reliance on carbohydrates and a reciprocal decrease in fat oxidation, as has been observed when humans sojourn to high altitude (Brooks et al. 1991; Sawhney et al. 1991; Roberts et al. 1996). However, LDVD for the remaining seven subject either did not change or diminished slightly and thus we failed to find any significant changes in nutrient partitioning throughout 28 days exposure to 3454 m.
The data from this current study demonstrating a MitoVD expansion in skeletal muscle after 28 days of 3454 m were collected at a lower elevation and over a shorter time period than in those studies showing a loss of skeletal muscle MitoVD (Hoppeler et al. 1990 b; Levett et al. 2012), so a potential dose and/or time response of skeletal muscle mitochondria adaptation to high‐altitude cannot be ruled out and further study is necessary to validate or refute this theory.
In summary, this study demonstrates that 28 days of exposure to 3454 m results in an increase in skeletal muscle mitochondrial volume density, with a specific increase in intermyofibrillar MitoVD and no change in subsarcolemmal populations. These data refute the idea that high altitude exposure per se diminishes skeletal muscle mitochondrial density. The explanation for this increase remains unclear, as TCA cycle, electron transport system enzymes and mitochondrial‐specific superoxide dehydrogenase 2 all remained unchanged. This study demonstrates the necessity to revisit the role of hypoxia on mitochondrial physiology, especially while maintaining energetic balance.
Additional information
Competing interests
None of the authors have any competing interests to declare in relation to this manuscript.
Author contributions
All data were collected in the laboratory of Carsten Lundby at the University of Zürich or at the Jungfraujoch Research Station. Conception and design of the experiments: C.L., A.K.M.L., S.G., D.F. and C.S. Collection, assembly, analysis and interpretation of data: C.L., A.K.M.L., S.G., D.F., C.S., S.F., N.K., M.H. and R.A.J. Drafting the article or revising it critically for important intellectual content: C.L., A.K.M.L. and R.A.J. All authors read and approved the final version of the manuscript.
Funding
Grants from the Swiss National Science Foundation (grant 320030_143745) and the Zürich Centre for Integrative Human Physiology to Carsten Lundby made the experiments possible.
Acknowledgements
The authors acknowledge the assistance and support of the Centre for Microscopy and Image Analysis, University of Zurich for performing scanning electron microscopy experiments.
References
- Arany Z, Foo S‐Y, Ma Y, Ruas JL, Bommi‐Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J et al (2008). HIF‐independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC‐1α. Nature 451, 1008–1012. [DOI] [PubMed] [Google Scholar]
- Armellini F, Zamboni M, Robbi R, Todesco T, Bissoli L, Mino A, Angelini G, Micciolo R & Bosello O (1997). The effects of high altitude trekking on body composition and resting metabolic rate. Horm Metab Res 29, 458–461. [DOI] [PubMed] [Google Scholar]
- Baar K, Wende AR, Jones TE, Marison M, Nolte LA, Chen M, Kelly DP & Holloszy JO (2002). Adaptations of skeletal muscle to exercise: rapid increase in the transcriptional coactivator PGC‐1. FASEB J 16, 1879–1886. [DOI] [PubMed] [Google Scholar]
- Brand Martin D & Nicholls David G (2011). Assessing mitochondrial dysfunction in cells. Biochem J 435, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks GA, Butterfield GE, Wolfe RR, Groves BM, Mazzeo RS, Sutton JR, Wolfel EE & Reeves JT (1991). Increased dependence on blood glucose after acclimatization to 4,300 m. J Appl Physiol (1985) 70, 919–927. [DOI] [PubMed] [Google Scholar]
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW & Kaelin WG Jr (2004). Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18, 2893–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield GE, Gates J, Fleming S, Brooks GA, Sutton JR & Reeves JT (1992). Increased energy intake minimizes weight loss in men at high altitude. J Appl Physiol 72, 1741–1748. [DOI] [PubMed] [Google Scholar]
- Chaillou T, Koulmann N, Simler N, Meunier A, Serrurier B, Chapot R, Peinnequin A, Beaudry M & Bigard X (2012). Hypoxia transiently affects skeletal muscle hypertrophy in a functional overload model. Am J Physiol Regul Integr Comp Physiol 302, R643–654. [DOI] [PubMed] [Google Scholar]
- Chance B & Williams GR (1955). Respiratory enzymes in oxidative phosphorylation. III. The steady state. J Biol Chem 217, 409–427. [PubMed] [Google Scholar]
- Clanton TL (2007). Hypoxia‐induced reactive oxygen species formation in skeletal muscle. J Appl Physiol (1985) 102, 2379–2388. [DOI] [PubMed] [Google Scholar]
- Cogswell AM, Stevens RJ & Hood DA (1993). Properties of skeletal muscle mitochondria isolated from subsarcolemmal and intermyofibrillar regions. Am J Physiol Cell Physiol 264, C383–C389. [DOI] [PubMed] [Google Scholar]
- Coyle EF, Martin WH 3rd, Sinacore DR, Joyner MJ, Hagberg JM & Holloszy JO (1984). Time course of loss of adaptations after stopping prolonged intense endurance training. J Appl Physiol 57, 1857–1864. [DOI] [PubMed] [Google Scholar]
- Desplanches D, Amami M, Dupré‐Aucouturier S, Valdivieso P, Schmutz S, Müller M, Hoppeler H, Kreis R & Flück M (2014). Hypoxia refines plasticity of mitochondrial respiration to repeated muscle work. Eur J Appl Physiol 114, 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplanches D, Hoppeler H, Linossier MT, Denis C, Claassen H, Dormois D, Lacour JR & Geyssant A (1993). Effects of training in normoxia and normobaric hypoxia on human muscle ultrastructure. Pflügers Arch 425, 263–267. [DOI] [PubMed] [Google Scholar]
- D'Hulst G, Jamart C, Van Thienen R, Hespel P, Francaux M & Deldicque L (2013). Effect of acute environmental hypoxia on protein metabolism in human skeletal muscle. Acta Physiol (Oxf) 208, 251–264. [DOI] [PubMed] [Google Scholar]
- Eaton S, Bartlett K & Pourfarzam M (1996). Mammalian mitochondrial beta‐oxidation. Biochem J 320, 345–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellisen LW, Ramsayer KD, Johannessen CM, Yang A, Beppu H, Minda K, Oliner JD, McKeon F & Haber DA (2002). REDD1, a developmentally regulated transcriptional target of p63 and p53, links p63 to regulation of reactive oxygen species. Mol Cell 10, 995–1005. [DOI] [PubMed] [Google Scholar]
- Etheridge T, Atherton PJ, Wilkinson D, Selby A, Rankin D, Webborn N, Smith K & Watt PW (2011). Effects of hypoxia on muscle protein synthesis and anabolic signaling at rest and in response to acute resistance exercise. Am J Physiol Endocrinol Metab 301, E697–E702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier FB, Costes F, Defour A, Bonnefoy R, Lefai E, Bauge S, Peinnequin A, Benoit H & Freyssenet D (2010). Downregulation of Akt/mammalian target of rapamycin pathway in skeletal muscle is associated with increased REDD1 expression in response to chronic hypoxia. Am J Physiol Regul Integr Comp Physiol 298, R1659–R1666. [DOI] [PubMed] [Google Scholar]
- Gnaiger E ( 2009). Capacity of oxidative phosphorylation in human skeletal muscle: New perspectives of mitochondrial physiology. Int J Biochem Cell Biol 41, 1837–1845. [DOI] [PubMed] [Google Scholar]
- Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H & Paintadosi CA (2008). Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase‐dependent mechanism. J Neurosci 28, 2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Stanley C, Maerkt P & Sumar‐Kalinowski J (1983). Metabolic meaning of elevated levels of oxidative enzymes in high altitude adapted animals: an interpretive hypothesis. Respir Physiol 52, 303–313. [DOI] [PubMed] [Google Scholar]
- Holloszy JO (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242, 2278–2282. [PubMed] [Google Scholar]
- Holm L, Haslund ML, Robach P, van Hall G, Calbet JA, Saltin B & Lundby C (2010). Skeletal muscle myofibrillar and sarcoplasmic protein synthesis rates are affected differently by altitude‐induced hypoxia in native lowlanders. PLoS One 5, e15606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppeler H & Desplanches D (1992). Muscle structural modifications in hypoxia. Int J Sports Med 13, S166–S168. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR & Cerretelli P (1990. a). Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med 11, S3–S9. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Kleinert E, Schlegel C, Claassen H, Howald H, Kayar SR & Cerretelli P (1990. b). Morphological adaptations of human skeletal muscle to chronic hypoxia. Int J Sports Med 11(Suppl. 1), S3–S9. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Klossner S & Vogt M (2008). Training in hypoxia and its effects on skeletal muscle tissue. Scand J Med Sci Sports 18, 38–49. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Vogt M, Weibel ER & Flück M (2003). Response of skeletal muscle mitochondria to hypoxia. Exp Physiol 88, 109–119. [DOI] [PubMed] [Google Scholar]
- Howald H & Hoppeler H (2003). Performing at extreme altitude: muscle cellular and subcellular adaptations. Eur J Appl Physiol 90, 360–364. [DOI] [PubMed] [Google Scholar]
- Iossa S, Mollica M, Lionetti L, Crescenzo R, Botta M, Samec S, Solinas G, Mainieri D, Dulloo A & Liverini G (2002). Skeletal muscle mitochondrial efficiency and uncoupling protein 3 in overeating rats with increased thermogenesis. Pflugers Arch 445, 431–436. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Boushel R, Wright‐Paradis C, Calbet JA, Robach P, Gnaiger E & Lundby C (2013. a). Mitochondrial function in human skeletal muscle following high altitude exposure. Exp Physiol 98, 245–255. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Boushel R, Wright‐Paradis C, Calbet JAL, Robach P, Gnaiger E & Lundby C (2013. b). Mitochondrial function in human skeletal muscle following high altitude exposure. Exp Physiol 98, 245–255. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Díaz V, Soldini L, Haider T, Thomassen M, Nordsborg NB, Gassmann M & Lundby C (2013. c). Fast‐twitch glycolytic skeletal muscle is predisposed to age‐induced impairments in mitochondrial function. J Gerontol A Biol Sci Med Sci 68, 1010–1022. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Siebenmann C, Hug M, Toigo M, Meinild A‐K & Lundby C (2012. a). Twenty‐eight days at 3454‐m altitude diminishes respiratory capacity but enhances efficiency in human skeletal muscle mitochondria. FASEB J 26, 5192–5200. [DOI] [PubMed] [Google Scholar]
- Jacobs RA, Siebenmann C, Hug M, Toigo M, Meinild AK & Lundby C (2012. b). Twenty‐eight days at 3454‐m altitude diminishes respiratory capacity but enhances efficiency in human skeletal muscle mitochondria. FASEB J 26, 5192–5200. [DOI] [PubMed] [Google Scholar]
- Jensen KS, Binderup T, Jensen KT, Therkelsen I, Borup R, Nilsson E, Multhaupt H, Bouchard C, Quistorff B, Kjaer A et al (2011). FoxO3A promotes metabolic adaptation to hypoxia by antagonizing Myc function. EMBO J 30, 4554–4570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayser B (1994). Nutrition and energetics of exercise at altitude. Theory and possible practical implications. Sports Med 17, 309–323. [DOI] [PubMed] [Google Scholar]
- Krieger DA, Tate CA, McMillin‐Wood J & Booth FW (1980). Populations of rat skeletal muscle mitochondria after exercise and immobilization. J Appl Physiol 48, 23–28. [DOI] [PubMed] [Google Scholar]
- Langen RC, Gosker HR, Remels AH & Schols AM (2013). Triggers and mechanisms of skeletal muscle wasting in chronic obstructive pulmonary disease. Int J Biochem Cell Biol 45, 2245–2256. [DOI] [PubMed] [Google Scholar]
- Larsen S, Nielsen J, Hansen CN, Nielsen LB, Wibrand F, Stride N, Schroder HD, Boushel R, Helge JW, Dela F & Hey‐Mogensen M (2012). Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol 590, 3349–3360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levett DZ, Radford EJ, Menassa DA, Graber EF, Morash AJ, Hoppeler H, Clarke K, Martin DS, Ferguson‐Smith AC, Montgomery HE, Grocott MPW, Murray AJ & Group tCXER (2012). Acclimatization of skeletal muscle mitochondria to high‐altitude hypoxia during an ascent of Everest. FASEB J 26, 1431–1441. [DOI] [PubMed] [Google Scholar]
- Lundby C, Gassmann M & Pilegaard H (2006). Regular endurance training reduces the exercise induced HIF‐1alpha and HIF‐2alpha mRNA expression in human skeletal muscle in normoxic conditions. Eur J Appl Physiol 96, 363–369. [DOI] [PubMed] [Google Scholar]
- Lundby C, Pilegaard H, Andersen JL, van Hall G, Sander M & Calbet JAL (2004). Acclimatization to 4100 m does not change capillary density or mRNA expression of potential angiogenesis regulatory factors in human skeletal muscle. J Exp Biol 207, 3865–3871. [DOI] [PubMed] [Google Scholar]
- MacDougall JD, Green HJ, Sutton JR, Coates G, Cymerman A, Young P & Houston CS (1991). Operation Everest II: structural adaptations in skeletal muscle in response to extreme simulated altitude. Acta Physiol Scand 142, 421–427. [DOI] [PubMed] [Google Scholar]
- Miller BF, Robinson MM, Bruss MD, Hellerstein M & Hamilton KL (2012). A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno M, Juel C, Bro‐Rasmussen T, Mygind E, Schibye B, Rasmussen B & Saltin B (1990). Limb skeletal muscle adaptation in athletes after training at altitude. J Appl Physiol 68, 496–502. [DOI] [PubMed] [Google Scholar]
- Moller P, Loft S, Lundby C & Olsen NV (2001). Acute hypoxia and hypoxic exercise induce DNA strand breaks and oxidative DNA damage in humans. FASEB J 15, 1181–1186. [DOI] [PubMed] [Google Scholar]
- Montero DB, Cathomen A, Jacobs RA, Flück D, de Leur J, Keiser S, Bonne TC, Kirk N, Lundby A‐KM & Lundby C (2015). Haematological rather than skeletal muscle adaptations contribute to the increase in peak oxygen uptake induced by moderate endurance training. J Physiol 593, 4677–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AJ & Horscroft JA (2015). Mitochondrial function at extreme high altitude. J Physiol 594, 1137–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordsborg NB, Siebenmann C, Jacobs RA, Rasmussen P, Diaz V, Robach P & Lundby C (2012). Four weeks of normobaric “live high‐train low” do not alter muscular or systemic capacity for maintaining pH and K+ homeostasis during intense exercise. J Appl Physiol 112, 2027–2036. [DOI] [PubMed] [Google Scholar]
- O'Hagan KA, Cocchiglia S, Zhdanov AV, Tambuwala MM, Cummins EP, Monfared M, Agbor TA, Garvey JF, Papkovsky DB, Taylor CT & Allan BB (2009). PGC‐1α is coupled to HIF‐1α‐dependent gene expression by increasing mitochondrial oxygen consumption in skeletal muscle cells. Proc Natl Acad Sci USA 106, 2188–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauwels RA, Buist AS, Calverley PM, Jenkins CR & Hurd SS (2001). Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am J Respir Crit Care Med 163, 1256–1276. [DOI] [PubMed] [Google Scholar]
- Pesta D & Gnaiger E (2011). High‐resolution respirometry. OXPHOS protocols for human cell cultures and permeabilized fibres from small biopsies of human muscle. Methods Mol Biol 810, 25–58. [DOI] [PubMed] [Google Scholar]
- Prommer N & Schmidt W (2010). Impact of alterations in total hemoglobin mass on . Med Sci Sports Med 38, 68–75. [DOI] [PubMed] [Google Scholar]
- Pulfrey SM & Jones PJ (1996). Energy expenditure and requirement while climbing above 6,000 m. J Appl Physiol 81, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Rasbach KA, Gupta RK, Ruas JL, Wu J, Naseri E, Estall JL & Spiegelman BM (2010). PGC‐1alpha regulates a HIF2alpha‐dependent switch in skeletal muscle fibre types. Proc Natl Acad Sci USA 107, 21866–21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen UF & Rasmussen HN (2000). Human skeletal muscle mitochondrial capacity. Acta Physiol Scand 168, 473–480. [DOI] [PubMed] [Google Scholar]
- Ratcliffe PJ, O'Rourke JF, Maxwell PH & Pugh CW (1998). Oxygen sensing, hypoxia‐inducible factor‐1 and the regulation of mammalian gene expression. J Exp Biol 201, 1153–1162. [DOI] [PubMed] [Google Scholar]
- Reynolds RD, Lickteig JA, Deuster PA, Howard MP, Conway JM, Pietersma A, deStoppelaar J & Deurenberg P (1999). Energy metabolism increases and regional body fat decreases while regional muscle mass is spared in humans climbing Mt. Everest. J Nutr 129, 1307–1314. [DOI] [PubMed] [Google Scholar]
- Richardson RS, Duteil S, Wary C, Wray DW, Hoff J & Carlier PG (2006). Human skeletal muscle intracellular oxygenation: the impact of ambient oxygen availability. J Physiol 571, 415–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Butterfield GE, Cymerman A, Reeves JT, Wolfel EE & Brooks GA (1996). Acclimatization to 4,300‐m altitude decreases reliance on fat as a substrate. J Appl Physiol 81, 1762–1771. [DOI] [PubMed] [Google Scholar]
- Rose MS, Houston CS, Fulco CS, Coates G, Sutton JR & Cymerman A (1988). Operation Everest. II: Nutrition and body composition. J Appl Physiol 65, 2545–2551. [DOI] [PubMed] [Google Scholar]
- Saks GA, Veksler VI, Kuznetsov VA, Kay L, Sikk P, Tiivel T, Tranqui L, Olivares J, Winkler K, Widemann F & Kunz WS (1998). Permeabilized cell and skinned fibre techniques in studies of mitochondrial function in vivo. Mol Cell Biochem 184, 81–100. [PubMed] [Google Scholar]
- Sawhney RC, Malhotra AS & Singh T (1991). Glucoregulatory hormones in man at high altitude. Eur J Appl Physiol Occup Physiol 62, 286–291. [DOI] [PubMed] [Google Scholar]
- Schmutz S, Däpp C, Wittwer M, Durieux A‐C, Mueller M, Weinstein F, Vogt M, Hoppeler H & Flück M (2010). A hypoxia complement differentiates the muscle response to endurance exercise. Exp Physiol 95, 723–735. [DOI] [PubMed] [Google Scholar]
- Scott GR ( 2011). Elevated performance: the unique physiology of birds that fly at high altitudes. J Exp Biol 214, 2455–2462. [DOI] [PubMed] [Google Scholar]
- Scott GR, Egginton S, Richards JG & Milsom WK (2009). Evolution of muscle phenotype for extreme high altitude flight in the bar‐headed goose. Proc Biol Sci 276, 3645–3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL, Shimoda LA & Prabhakar NR (2006). Regulation of gene expression by HIF‐1. Novartis Found Symp 272, 2–8. [PubMed] [Google Scholar]
- Siebenmann C, Cathomen A, Hug M, Keiser S, Lundby AK, Hilty MP, Goetze JP, Rasmussen P & Lundby C (2015). Hemoglobin mass and intravascular volume kinetics during and after exposure to 3,454 m altitude. J Appl Physiol 15, 1194–1201. [DOI] [PubMed] [Google Scholar]
- Taylor CT ( 2008). Mitochondria and cellular oxygen sensing in the HIF pathway. Biochem J 409, 19–26. [DOI] [PubMed] [Google Scholar]
- Valdivia E ( 1958). Total capillary bed in striated muscles of guinea pigs native to the Peruvian mountains. Am J Physiol 194, 585–589. [DOI] [PubMed] [Google Scholar]
- Vigano A, Ripamonti M, De Palma S, Capitanio D, Vasso M, Wait R, Lundby C, Cerretelli P & Gelfi C (2008). Proteins modulation in human skeletal muscle in the early phase of adaptation to hypobaric hypoxia. Proteomics 8, 4668–4679. [DOI] [PubMed] [Google Scholar]
- Vogt M, Puntschart A, Geiser J, Zuleger C, Billeter R & Hoppeler H (2001). Molecular adaptations in human skeletal muscle to endurance training under simulated hypoxic conditions. J Appl Physiol 91, 173–182. [DOI] [PubMed] [Google Scholar]
- West MJ ( 2012). Estimating volume in biological structures. Cold Spring Harbor Protocols 2012, pdb.top071787. [DOI] [PubMed] [Google Scholar]
- Westerterp KR, Kayser B, Brouns F, Herry JP & Saris WH (1992). Energy expenditure climbing Mt. Everest. J Appl Physiol 73, 1815–1819. [DOI] [PubMed] [Google Scholar]
- Westerterp KR, Kayser B, Wouters L, Le Trong JL & Richalet JP (1994). Energy balance at high altitude of 6,542 m. J Appl Physiol 77, 862–866. [DOI] [PubMed] [Google Scholar]
- Westerterp KR, Meijer EP, Rubbens M, Robach P & Richalet JP (2000). Operation Everest III: Energy and water balance. Pflügers Arch 439, 483–488. [DOI] [PubMed] [Google Scholar]
- Wing‐Gaia SL, Gershenoff DC, Drummond MJ & Askew EW (2013). Effect of leucine supplementation on fat free mass with prolonged hypoxic exposure during a 13‐day trek to Everest Base Camp: a double‐blind randomized study. Appl Physiol Nutr Metab 39, 318–323. [DOI] [PubMed] [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC & Spiegelman BM (1999). Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC‐1. Cell 98, 115–124. [DOI] [PubMed] [Google Scholar]
- Zhang H, Bosch‐Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ & Semenza GL (2008). Mitochondrial autophagy is an HIF‐1‐dependent adaptive metabolic response to hypoxia. J Biol Chem 283, 10892–10903. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]