

Key points
Maternal hypoxia is a common perturbation that leads to growth restriction and may program vascular dysfunction in adult offspring.
An adverse prenatal environment may render offspring vulnerable to increased cardiovascular risk when challenged with a ‘second hit’, such as a high salt diet.
We investigated whether maternal hypoxia impaired vascular function, structure and mechanics in mouse offspring, and also whether this was exacerbated by excess dietary salt intake in postnatal life.
Maternal hypoxia predisposed adult male and female offspring to endothelial dysfunction.
The combination of prenatal hypoxia and high dietary salt intake caused significant stiffening of mesenteric arteries, and also altered structural characteristics of the aorta consistent with vascular stiffening.
The results of the present study suggest that prenatal hypoxia combined with a high salt diet in postnatal life can contribute to vascular dysfunction.
Abstract
Gestational hypoxia and high dietary salt intake have both been associated with impaired vascular function in adulthood. Using a mouse model of prenatal hypoxia, we examined whether a chronic high salt diet had an additive effect in promoting vascular dysfunction in offspring. Pregnant CD1 dams were placed in a hypoxic chamber (12% O2) or housed under normal conditions (21% O2) from embryonic day 14.5 until birth. Gestational hypoxia resulted in a reduced body weight for both male and female offspring at birth. This restriction in body weight persisted until weaning, after which the animals underwent catch‐up growth. At 10 weeks of age, a subset of offspring was placed on a high salt diet (5% NaCl). Pressurized myography of mesenteric resistance arteries at 12 months of age showed that both male and female offspring exposed to maternal hypoxia had significantly impaired endothelial function, as demonstrated by impaired vasodilatation to ACh but not sodium nitroprusside. Endothelial dysfunction caused by prenatal hypoxia was not exacerbated by postnatal consumption of a high salt diet. Prenatal hypoxia increased microvascular stiffness in male offspring. The combination of prenatal hypoxia and a postnatal high salt diet caused a leftward shift in the stress–strain relationship in both sexes. Histopathological analysis of aortic sections revealed a loss of elastin integrity and increased collagen, consistent with increased vascular stiffness. These results demonstrate that prenatal hypoxia programs endothelial dysfunction in both sexes. A chronic high salt diet in postnatal life had an additive deleterious effect on vascular mechanics and structural characteristics in both sexes.
Key points
Maternal hypoxia is a common perturbation that leads to growth restriction and may program vascular dysfunction in adult offspring.
An adverse prenatal environment may render offspring vulnerable to increased cardiovascular risk when challenged with a ‘second hit’, such as a high salt diet.
We investigated whether maternal hypoxia impaired vascular function, structure and mechanics in mouse offspring, and also whether this was exacerbated by excess dietary salt intake in postnatal life.
Maternal hypoxia predisposed adult male and female offspring to endothelial dysfunction.
The combination of prenatal hypoxia and high dietary salt intake caused significant stiffening of mesenteric arteries, and also altered structural characteristics of the aorta consistent with vascular stiffening.
The results of the present study suggest that prenatal hypoxia combined with a high salt diet in postnatal life can contribute to vascular dysfunction.
Abbreviations
- C
control
- CHS
control offspring fed high salt
- CNS
control offspring fed normal salt
- CSA
cross‐sectional area
- CVD
cardiovascular disease
- E
embryonic day
- H
hypoxia
- HS
high salt
- HHS
hypoxia offspring fed high salt
- HNS
hypoxia offspring fed normal salt
- KPSS
high potassium physiological salt solution
- KPSSmax
maximum vessel contraction to KPSS
- NO
nitric oxide
- NS
normal salt
- P
postnatal day
- PE
phenylephrine
- Rmax
maximum relaxation
- SNP
sodium nitroprusside
Introduction
Cardiovascular disease (CVD) is recognized as the leading cause of global mortality. The development of CVD is associated with risk factors such as smoking, age, sex and hypertension (Mozaffarian et al. 2008). Epidemiological evidence suggests that CVD can also be linked to poor development in early life (Barker, 2002). Adverse conditions during fetal life may require the fetus and placenta to make adaptations to organogenesis so as to remain viable in the short‐term, although these adaptations frequently lead to intrauterine growth restriction (Barker, 1998). If these adaptations occur during critical periods of development, permanent changes in organ structure and function can occur, leading to poor health outcomes and an increased susceptibility to disease in adulthood. This concept is known as developmental programming (Barker, 1995, 1998).
Impaired oxygen supply to the fetus is a common clinical complication during pregnancy and can arise from living at high altitude (Giussani et al. 1993), maternal smoking (Bulterys et al. 1990) and poor placentation leading to reduced fetoplacental perfusion (Krebs et al. 1996). Mild, acute forms of hypoxia initiate the centralization of blood flow away from the peripheral circulation to the heart and brain (Giussani et al. 1993; Baschat et al. 1997). Although this adaptation is necessary for the immediate preservation of critical organs, the decreased blood flow can impair development of peripheral organs resulting in significant adverse consequences in later life. If the hypoxic insult is prolonged, these immediate adaptations may be sustained and result in compromised growth in utero. Low birth weight and brain sparing following chronic in utero hypoxia have been observed across all species studied, including humans (Wladimiroff et al. 1986; Giussani et al. 2001), chicks (Mulder et al. 2002; Giussani et al. 2007) and the rat (Williams et al. 2005 a; Williams et al. 2005 b), and this is associated with an increased risk of CVD later in life.
Endothelial dysfunction and arterial stiffness are both associated with CVD in adulthood (Benetos et al. 2002). Interestingly, impaired peripheral vascular endothelial function has been demonstrated in low birth weight infants (Norman & Martin, 2003), children (Martin et al. 2000) and young adults (Goodfellow et al. 1998; Leeson et al. 2001), and increased arterial stiffness has been shown to contribute to elevated blood pressure in the human population (te Velde et al. 2004). There is now substantial evidence from animal models to show that perturbations in utero such as hypoxia (Williams et al. 2005 b), vitamin D deficiency (Tare et al. 2011) and undernutrition (Brawley et al. 2003) can also program vascular dysfunction in adulthood. Offspring born to rats that were exposed to chronic hypoxia during late pregnancy develop endothelial dysfunction (Williams et al. 2005 b; Giussani et al. 2012) and enhanced myogenic tone (Hemmings et al. 2005). These factors may contribute to the increased arterial blood pressure observed in middle‐aged rats exposed to chronic hypoxia in utero (Rook et al. 2014). Furthermore, ventricular (Camm et al. 2010) and aortic wall thickening (Rouwet et al. 2002; Giussani et al. 2012) and increased susceptibility to cardiac ischaemia‐reperfusion injury (Xu et al. 2006; Xue & Zhang, 2009) have also been reported in adult rodents prenatally exposed to hypoxia. Together, these studies demonstrate that prenatal hypoxia can significantly elevate the propensity to develop CVD in adulthood.
Importantly, prenatally‐programmed vulnerability to CVD may be exacerbated by a postnatal ‘second‐hit’ such as a chronic high salt (HS) diet (Ruta et al. 2010), a high fat diet (Rueda‐Clausen et al. 2012) and ageing. In a rat model of prenatal hypoxia, vascular alterations such as increased myogenic tone only arose in animals aged >6 months (Hemmings et al. 2005). In the same model, a combination of prenatal hypoxia and the postnatal consumption of a high fat diet increased susceptibility to cardiac and vascular pathologies (Rueda‐Clausen et al. 2012). In Western societies, diets are often not only high in fat, but also high in salt (Cordain et al. 2005). A chronic increase in dietary sodium intake has been shown to result in increased vascular stiffness in mice (Yu et al. 2004) and enhanced vasoconstrictor responsiveness in the rat (Sofola et al. 2002). Although a HS diet has been shown to exacerbate disease outcomes following maternal protein restriction (Woods et al. 2004) and a congenital nephron deficit (Ruta et al. 2010), no study has yet investigated whether the adverse vascular outcomes following prenatal hypoxia are exacerbated by a HS diet.
In the present study, we used our recently reported model of prenatal hypoxia in the mouse (Cuffe et al. 2014 a; Cuffe et al. 2014 b) to assess the impact of chronic late gestational hypoxia on microvascular structure and function in aged male and female offspring. In particular, we aimed to examine whether chronic high dietary sodium intake could exacerbate any deficits in microvascular structure and function. We hypothesized that arteries from hypoxia‐exposed offspring would have endothelial dysfunction and increased vascular stiffness. In addition, we hypothesized that these alterations would be exacerbated by high dietary sodium intake.
Methods
Ethics approval
All experiments were approved in advance by the University of Queensland animal ethics committee and were conducted in accordance with the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Animal treatment: maternal hypoxia
At embryonic day (E) 14.5 of pregnancy, time‐mated CD‐1 mice were randomly allocated to normoxic room conditions (control, C; n = 21) or housed inside a hypoxic chamber continuously flushed with nitrogen gas to maintain an oxygen concentration of 12% (hypoxia, H; n = 22) for the remainder of pregnancy. Food and water was provided ad libitum. Body weight, food and water consumption were monitored daily throughout the experimental protocol. This experimental protocol does not result in changes in maternal food intake and weight gain, and is thus not compromised by maternal undernutrition (Cuffe et al. 2014 a). The dams were removed from the hypoxic chamber upon littering down (postnatal day, P0). A subset of dams and offspring (C: n = 10; H: n = 11) were culled at P0. Pup body weight was recorded, and maternal and pup blood were collected for measurement of haematocrit. The remaining offspring remained with their mothers until weaning at P21, and were then weighed weekly for the duration of the study.
Dietary intervention: chronic HS diet
A subset of animals aged 10 weeks (n = 11 per sex per treatment, one or two animals from each litter) was randomly allocated to receive a HS (HS) diet (5% NaCl, wt/wt; modified AIN 93M, SF05‐023; Specialty Feeds, Memphis, TN, USA). The remaining animals (n = 11 per sex per treatment) were maintained on a matched control normal salt (NS) diet (0.26% NaCl, wt/wt; AIN 93M; Specialty Feeds). Food and water consumption were monitored for the first 5 days of diet administration to ensure the animals adjusted to the dietary change. Animals were maintained on the appropriate diet until post‐mortem at 12 months of age.
Urinalysis
Aged offspring (mean ± SEM: 12 ± 0.5 months) were housed in individual metabolic cages for 24 h (with food and water provided ad libitum) to measure urinary flow rates and electrolyte excretion. All animals were acclimatized to the metabolic cages on two consecutive days prior to the experiment. Urine was collected and stored at −20°C for subsequent analysis. Urinary sodium, chloride and potassium concentrations were measured using a COBAS Integra 400 Plus analyser (Roche Diagnostics Ltd, Rotkreuz, Switzerland).
Pressurized myography
Following completion of renal function measurements, animals were killed by CO2 inhalation (six to 10 per group). Second‐order mesenteric arteries were dissected from connective tissue, placed in physiological salt solution (composition in mmol l–1: 118 NaCl, 4.65 KCl, 1.18 MgSO4, 1.18 KH2PO4 and 2.5 CaCl2; 25 mm NaHCO3, 5.5 mm glucose and 0.026 mm EDTA), mounted in a pressurized myograph and the intraluminal pressure gradually increased to 45 mmHg. Maximal vessel contractility was determined using high potassium physiological salt solution (KPSS) containing an equimolar substitution of KCl for NaCl. Vessel contractility was assessed by cumulative concentration–response curves to phenylephrine (PE; 10−9 to 10−4 mol l−1). The contractile responses were expressed as a percentage of the maximum response to KPSS (KPSSmax). Endothelium‐dependent and endothelium independent relaxations were assessed in vessels submaximally constricted with PE (70% of KPSSmax), using ACh (10−9 to 10−4 mol l−1) and sodium nitroprusside (SNP; 10−10 to 10−4 mol l−1), respectively. Relaxation responses were expressed as the percentage relaxation from the PE‐induced contraction. Non‐linear regression (Prism 6; GraphPad Software Inc., San Diego, CA, USA) was used to determine the pEC50 and maximum response (Rmax) for each vessel. To measure vascular structure and mechanics, vessels were superfused with Ca2+‐free physiological salt solution containing 1 mmol l−1 of EGTA to remove intrinsic tone. Intraluminal pressure was increased stepwise from 3 to 140 mmHg and lumen diameter and wall thickness were measured at each pressure. Medial cross‐sectional area (CSA), distensibility, circumferential stress and strain were calculated as described previously (Virdis et al. 2002; Iglarz et al. 2003).
Histomorphometric analyses of the aorta
Thoracic aortas were fixed in 4% paraformaldehyde before processing to paraffin. Transverse aortic sections were taken at 5 μm. Sections were stained with Masson's trichrome to assess collagen content and Verhoeff's van Gieson elastin stain to detect elastin expression. Slides were imaged using a 20× objective on a BX61 microscope (Olympus, Tokyo, Japan). Masson's trichrome‐stained aortic sections were graded by a blinded observer as showing 0–10% media collagen content (grade 0), 10–25% media collagen content (grade 1), 25–50% media collagen content (grade 2) or 50–75% media collagen content (grade 3). Elastin content was evaluated by subtracting background, converting image to binary and calculating the percentage of black (elastin) and white pixels.
Statistical analysis
All data are presented as the mean ± SEM. Maternal haematocrit was analysed via a Student's t test. Offspring haematocrit and body weight at P0 were analysed by two‐way ANOVA examining the effects of prenatal hypoxia (P trt) and sex (P sex). Data for body and organ weights, urinalysis and vascular parameters of male and female offspring at 12 months of age were analysed separately. These data were analysed by two‐way ANOVA examining the effects of prenatal hypoxia (P trt) and postnatal HS diet (P diet). Bonferroni post hoc tests were used when necessary. P < 0.05 was considered statistically significant.
Results
Offspring weights and growth
Maternal (P = 0.007) (Fig. 1 A) and pup haematocrit (P trt < 0.0001) (Fig. 1 B) was elevated in hypoxia‐exposed animals. Litter size did not differ between treatment groups (C: 14 ± 1, H: 14 ± 1). At P0, body weights for both males and females (litter averages) were reduced by ∼12% in animals from hypoxia‐exposed dams (male C: 1.73 ± 0.5 g, male H: 1.52 ± 0.03 g, female C: 1.67 ± 0.04 g, female H: 1.46 ± 0.02 g; P trt < 0.0001) (Fig. 1 C). Hypoxia‐exposed animals remained lighter than control counterparts by ∼7% at the weaning age of P21 (male C: 11.9 ± 0.3 g, male H: 11.0 ± 0.3 g, female C: 11.2 ± 0.3 g, female H: 10.4 ± 0.4 g) (P trt = 0.01). There was no difference in body weights between treatment groups in male (Fig. 2 A) or female (Fig. 2 B) offspring from 3 to 12 months of age.
Figure 1. Maternal and offspring parameters at day of birth .
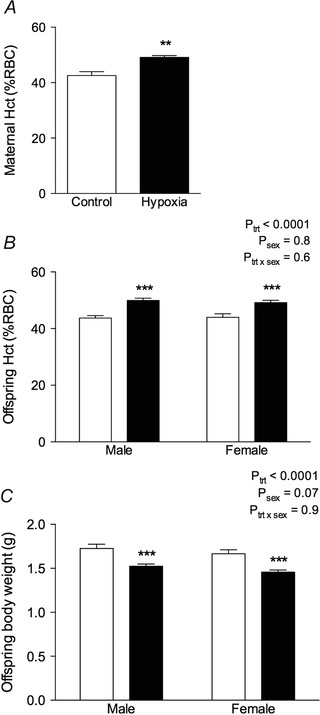
Maternal haematocrit (Hct, % packed red blood cell volume) (A), offspring haematocrit (B) and offspring body weight (C) were determined at P0. Control: white bars; hypoxia: black bars. Values are the mean ± SEM (n = 8–10 per group). **P < 0.001 from a t test, ***P < 0.0001 (from Bonferroni post hoc compared to same sex controls).
Figure 2. Growth curves of offspring .
Growth curves of male (A) and female (B) offspring either on a NS (0.26% NaCl) or HS diet (5% NaCl) from the age of 3–12 months. Values are the mean ± SEM (of n = 11 litters per group). Control: white points; hypoxia: black points.
Urinalysis
Urinary excretion of electrolytes did not differ between control and hypoxic‐exposed animals. However, sodium and chloride excretion was significantly elevated in male and female offspring (∼3‐ to 5‐fold) fed the HS diet compared to those on the control NS diet (data not shown). Potassium excretion did not differ between dietary groups.
Microvascular function
The maximal contractile response to high potassium physiological salt solution (KPSS) was not affected by maternal hypoxia but was enhanced in male offspring maintained on the HS diet compared to the NS diet (C, control; H, hypoxia) (CNS: 47.4 ± 1.0; HNS: 47.4 ± 1.0; CHS: 52.7 ± 0.9; HHS: 55.6 ± 2.2%; P diet < 0.0001). The maximum contractile response to phenylephrine was unaffected by prenatal hypoxia treatment or dietary HS intake (Fig. 3 A). However, sensitivity to phenylephrine was increased in animals maintained on the HS diet, irrespective of the maternal treatment (P = 0.008) (Fig. 3 A and Table 1). The maximum dilatator response to ACh was significantly attenuated in male hypoxia‐exposed offspring (Fig. 3 C and Table 1), whereas the sensitivity to ACh was similar in all groups. The reduced response to ACh indicates impairment in endothelium‐dependent relaxation because responsiveness to the nitric oxide donor SNP was statistically similar in all treatment groups (Fig. 3 E and Table 1). There was a trend towards reduced maximum relaxation to SNP in male offspring fed the HS diet, although this was not statistically significant (P = 0.07) (Table 1). In female offspring, the maximal response to KPSS did not differ between treatment and dietary groups (CNS: 52.2 ± 1.9; HNS: 50.2 ± 2.3; CHS: 48.4 ± 1.4; HHS: 51.0 ± 1.8%). Arteries from female offspring showed similar responsiveness to phenylephrine, regardless of treatment or dietary groups (Fig. 3 B and Table 1). Evidence of endothelial dysfunction was also seen in female hypoxia‐exposed offspring, which had reduced maximum responses to ACh (Fig. 3 D and Table 1). Sensitivity to ACh (Fig. 3 D and Table 1) and responsiveness to SNP (Fig. 3 F and Table 1) did not differ between groups.
Figure 3. Mesenteric arteries: functional responses .
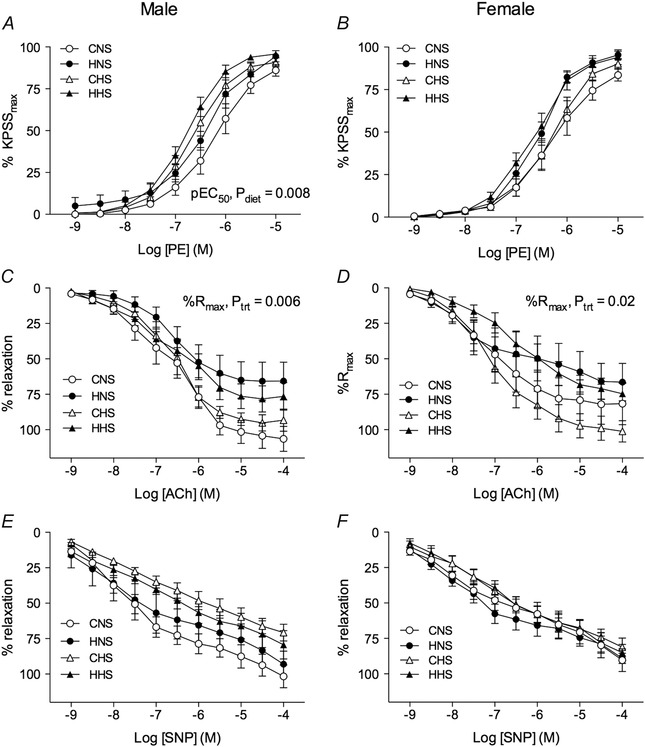
Mesenteric arteries were obtained from aged male (left) and female (right) control and hypoxia‐exposed offspring fed a NS or HS diet. Functional responses to cumulative additions of (A and B) phenylephrine, (C and D) ACh and (E and F) SNP were measured in arteries pressurized to 45 mmHg. Data in A and B are expressed as a percentage of the maximum contraction to high potassium physiological salt solution (KPSSmax). C–F, vessels were contracted to ∼70% of KPSSmax with phenylephrine, and relaxation responses to the vasodilatator agonists expressed as a percentage of the PE‐induced contraction. Values are the mean ± SEM (n = 6–10 per group). Control: white points; hypoxia: black points.
Table 1.
Vascular sensitivities (expressed as pEC50) and maximum responses to phenylephrine (% KPSSmax), ACh (% Rmax) and SNP (% Rmax)
| NS | HS | Two‐way ANOVA | ||||||
|---|---|---|---|---|---|---|---|---|
| Control | Hypoxia | Control | Hypoxia | P trt | P diet | P trt × diet | ||
| Males | ||||||||
| PE | pEC50 | –6.2 ± 0.2 | –6.4 ± 0.1 | –6.6 ± 0.1 | –6.8 ± 0.1 | 0.2 | 0.008 | 0.9 |
| % KPSSmax | 101.1 ± 6.2 | 102.7 ± 2.7 | 96.1 ± 1.9 | 100.0 ± 2.1 | 0.5 | 0.4 | 0.8 | |
| ACh | pEC50 | –6.6 ± 0.2 | –6.7 ± 0.2 | –6.6 ± 0.1 | –6.6 ± 0.2 | 0.6 | 0.8 | 0.9 |
| % Rmax | 107.3 ± 9.2 | 66.3 ± 13.2 | 95.4 ± 5.0 | 77.2 ± 9.5 | 0.006 | 0.96 | 0.3 | |
| SNP | pEC50 | –7.4 ± 0.1 | –6.9 ± 0.4 | –6.5 ± 0.4 | –6.6 ± 0.3 | 0.6 | 0.09 | 0.4 |
| % Rmax | 89.1 ± 7.2 | 93.7 ± 8.5 | 67.2 ± 5.5 | 74.2 ± 8.9 | 0.9 | 0.07 | 0.5 | |
| Females | ||||||||
| PE | pEC50 | –6.4 ± 0.2 | –6.5 ± 0.1 | –6.3 ± 0.1 | –6.7 ± 0.1 | 0.07 | 0.7 | 0.5 |
| % KPSSmax | 92.9 ± 4.3 | 105.2 ± 5.1 | 94.1 ± 2.2 | 97.6 ± 4.5 | 0.06 | 0.4 | 0.3 | |
| ACh | pEC50 | –7.0 ± 0.2 | –6.7 ± 0.5 | –7.0 ± 0.2 | –6.4 ± 0.2 | 0.2 | 0.6 | 0.8 |
| % Rmax | 89.4 ± 13.4 | 65.7 ± 12.4 | 98.5 ± 8.2 | 71.8 ± 6.9 | 0.02 | 0.5 | 0.9 | |
| SNP | pEC50 | –7.5 ± 0.3 | –7.8 ± 0.3 | –7.7 ± 0.1 | –7.7 ± 0.2 | 0.6 | 0.9 | 0.6 |
| % Rmax | 70.0 ± 6.9 | 76.3 ± 4.4 | 66.6 ± 5.3 | 68.0 ± 6.1 | 0.5 | 0.3 | 0.7 | |
Values are the mean ± SEM (n = 6–10 per group). The effect of treatment (trt), diet or their interaction (trt × diet) was evaluated by two‐way ANOVA.
Vascular structure
Mesenteric arteries from male offspring all had similar lumen diameters (Fig. 4 A) and media:lumen ratios (Fig. 4 E) across increasing intraluminal pressures. No differences in lumen diameters and media:lumen ratios were observed at 45 mmHg, which is the pressure at which the functional experiments were conducted (Table 2). Male animals fed the HS diet, had decreased medial CSA (P diet = 0.03) (Fig. 4 C) compared to those on a NS diet. CSA did not differ in offspring from the hypoxia and control groups. In female offspring, neither the maternal hypoxia, nor the HS diet had an effect on lumen diameter (Fig. 4 B), medial CSA (Fig. 4 D) or media:lumen ratio (Fig. 4 F) at any pressure (Table 2).
Figure 4. Structural properties of mesenteric arteries .
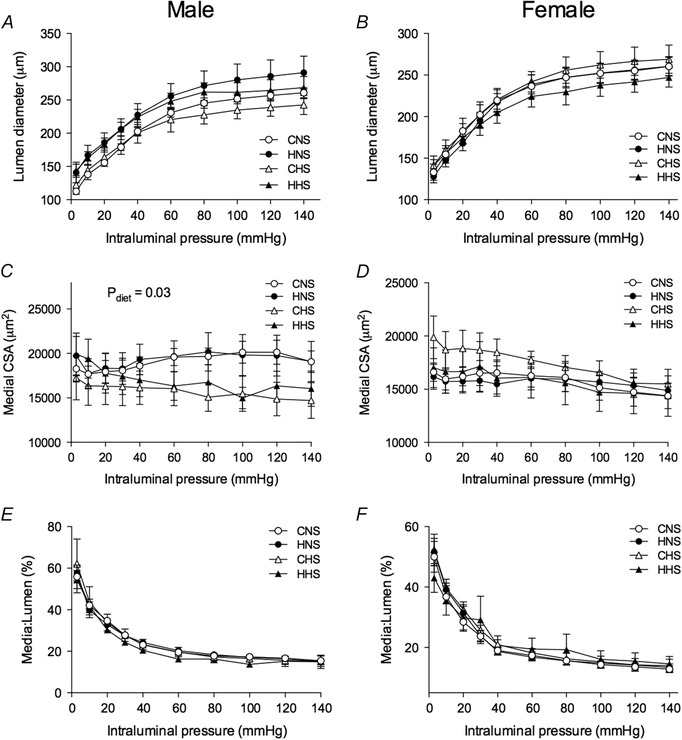
Arteries were obtained from aged male (left) and female (right) control and hypoxia‐exposed offspring fed a NS or HS diet. Lumen diameter in male (A) and female (B) offspring, medial CSA in male (C) and female (D) offspring, as well as media:lumen ratio in male (E) and female (F) offspring in response to increasing intraluminal pressure in deactivated mesenteric arteries. Values are the mean ± SEM (n = 6–10 per group). Control: white points; hypoxia: black points.
Table 2.
Structural parameters of mesenteric arteries from aged offspring when pressurized to 45 mmHg
| NS | HS | Two‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| Control | Hypoxia | Control | Hypoxia | P trt | P diet | P trt × diet | |
| Males | |||||||
| Lumen diameter (μm) | 212 ± 6 | 237 ± 18 | 207 ± 16 | 232 ± 12 | 0.1 | 0.8 | 0.97 |
| Medial CSA (μm2) | 18666 ± 1042 | 19577 ± 1814 | 15962 ± 1855 | 16143 ± 752 | 0.7 | 0.03 | 0.8 |
| Media:lumen | 21.5 ± 1.0 | 22.5 ± 1.0 | 21.8 ± 2.4 | 18.1 ± 1.2 | 0.3 | 0.1 | 0.09 |
| Females | |||||||
| Lumen diameter (μm) | 227 ± 14 | 225 ± 10 | 226 ± 12 | 210 ± 11 | 0.5 | 0.5 | 0.5 |
| Medial CSA (μm2) | 16472 ± 970 | 15992 ± 947 | 18206 ± 1123 | 15921 ± 1648 | 0.3 | 0.5 | 0.5 |
| Media:lumen | 19.0 ± 1.1 | 18.7 ± 1.2 | 21.0 ± 1.6 | 21.2 ± 3.6 | 0.98 | 0.4 | 0.9 |
Values are the mean ± SEM (n = 6–10 per group). The effect of treatment (trt), diet or their interaction (trt × diet) was evaluated by two‐way ANOVA.
Vascular mechanics
There was a leftward shift in the stress–strain curve in arteries from males fed the HS diet compared to those on the NS diet, indicating increased vessel stiffness (Fig. 5 A). This leftward shift was greatest in male HHS mesenteric arteries compared to NS arteries (Fig. 5 A). The rate constant of the stress–strain curve was greater in both male offspring fed a HS diet and hypoxia‐exposed male offspring (P trt = 0.03, P diet = 0.02) (Table 3). By contrast, in female offspring, the combination of hypoxia and a HS diet shifted the stress–strain curve to the left, indicating increased vessel stiffness. This was accompanied by a significant increase in the rate constant of the stress–strain curve in HHS female arteries compared to other treatment groups (P diet = 0.008, P trt × diet = 0.03) (Table 3).
Figure 5. Mechanical properties of mesenteric arteries .
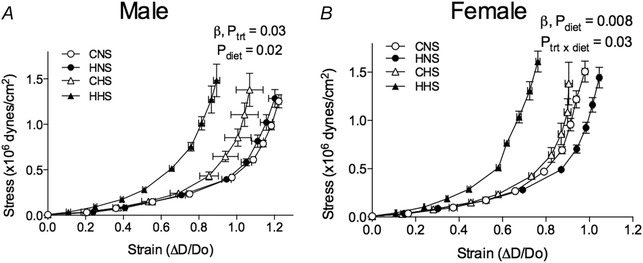
Arteries were obtained from aged male (left) and female (right) control and hypoxia‐exposed offspring fed a NS or HS diet. Stress–strain relationship in vessels of male (A) and female (B) offspring. Values are the mean ± SEM (n = 6–10 per group). Control: white points; hypoxia: black points.
Table 3.
Rate constant, ß, calculated from the exponential function fitted to stress‐strain curves of mesenteric arteries from aged offspring
| NS | HS | Two‐way ANOVA | |||||
|---|---|---|---|---|---|---|---|
| ß, rate constant | Control | Hypoxia | Control | Hypoxia | P trt | P diet | P trt × diet |
| Males | 4.1 ± 0.3 | 4.8 ± 0.5 | 4.9 ± 0.6 | 6.0 ± 0.3 | 0.03 | 0.02 | 0.6 |
| Females | 5.4 ± 0.4 | 5.0 ± 0.4 | 5.8 ± 0.5 | 7.9 ± 0.7* | 0.1 | 0.008 | 0.03 |
Values are the mean ± SEM (n = 6–10 per group). The effect of treatment (trt), diet or their interaction (trt × diet) was evaluated by two‐way ANOVA. *P < 0.01 (from Bonferroni post hoc compared to HS controls).
Aorta histology
Representative sections of male aortas from all treatment groups stained with Masson's trichrome to detect collagen and Verhoeff's Van Gieson to detect elastin are shown in Fig. 6. Significant collagen deposition can be observed in the media of aortas taken from control male offspring fed the HS diet. Equivalent levels of collagen deposition can be seen in aortas taken from hypoxia males fed NS and HS diets. When estimated semi‐quantitatively, the aortic collagen content score was increased ∼2‐fold in male (P diet = 0.02) (Fig. 7 A) and female (P diet = 0.007) (Fig. 7 B) offspring fed the HS diet (Fig. 6). Male but not female hypoxia‐exposed offspring fed a NS diet also showed increased aortic collagen content.
Figure 6. Microphotographs of aortic sections .
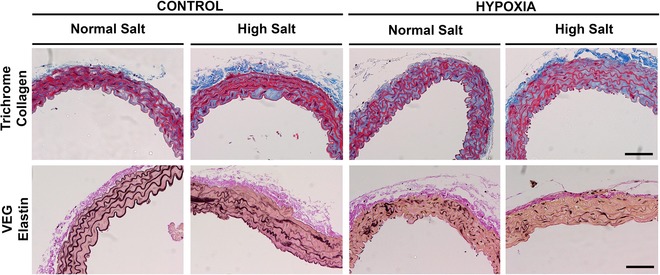
Sections were stained with Masson's trichrome to detect collagen (blue) and Verhoeff's Van Gieson to detect elastin (black). Aortas were obtained from aged male hypoxia‐exposed offspring fed a NS or HS diet. Scale bar = 50 μm.
Figure 7. Semi‐quantification of aortic histology .
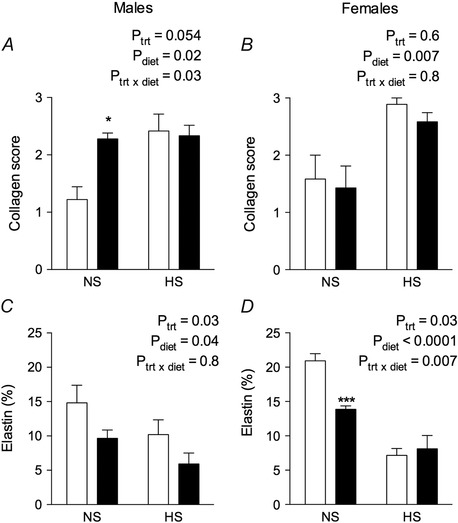
Collagen scores in aorta sections stained with Masson's trichrome to detect collagen in male (A) and female (B) control and hypoxia exposed offspring fed a NS or HS diet. Elastin (%) in aorta sections stained with Verhoeff's Van Gieson in male (C) and female (D) offspring. Values are the mean ± SEM (n = 6–8 per group). *P < 0.01, ***P < 0.0001 (from Bonferroni post hoc compared to control offspring fed the NS diet).
Aortas of male CNS offspring presented elastin fibres laid down as concentric layers and occupying the greatest proportion of media compared to other treatment groups (Figs 6 and 7 C). Modest degeneration of elastin fibre integrity was observed in male CHS aortas (Fig. 6). Elastin content was ∼30% lower in aortas from male CHS aortas compared to male CNS aortas (P diet = 0.04) (Fig. 7 C). Prenatal hypoxia caused marked fragmentation and disorganization of the elastin fibres (Fig. 6) and reduced elastin content in male (P trt = 0.03) (Fig. 7 C) and female offspring (P trt = 0.03) (Fig. 7 D). Degeneration of the elastin fibres was most pronounced in aortas of the HHS male, where disorganization and discontinuity of the elastin fibre segments can clearly be observed in Fig. 6. Elastin content in aortas of male HHS offspring was ∼42% lower compared to elastin content in aortas of CHS offspring (Fig. 7 C). The HS diet reduced aortic elastin significantly in both female control and hypoxia‐exposed offspring (P diet < 0.0001) (Fig. 7 D).
Discussion
The present study examined the effects of maternal hypoxia during late gestation, and determined how this gestational insult interacted with the postnatal consumption of a chronic HS diet to affect vascular function, structure and mechanics in adult offspring. We show for the first time that, although prenatal hypoxia exposure alone caused mild endothelial dysfunction in mesenteric vessels, the combined insult of prenatal hypoxia and a chronic postnatal HS diet caused significant stiffening of the mesenteric vasculature and altered extracellular matrix composition in the aorta; most notably a loss of elastin fibre integrity. Our results suggest that, although prenatal hypoxia may be unavoidable, consuming a healthy diet, particularly with respect to sodium intake, may prevent or at least limit adverse vascular outcomes in adulthood.
During early life, suboptimal influences such as maternal low protein diet (Brawley et al. 2003), maternal high fat diet (Khan et al. 2004) and vitamin D insufficiency (Tare et al. 2011) can program vascular dysfunction. In particular, endothelial dysfunction as a result of impaired responsiveness to endothelium‐dependent vasodilatators such as ACh is commonly reported in models of developmental programming (Mcmillen & Robinson, 2005). In our model of prenatal hypoxia, there was reduced ACh‐induced vasodilatation in the offspring of both sexes, although there were no differences in vasodilatator responses to the NO donor SNP. This suggests that the impaired vascular response to ACh in hypoxia‐exposed offspring is a result of impaired endothelial function rather than decreased vascular smooth muscle sensitivity to NO‐induced relaxation. This is consistent with previous reports using a model of prenatal hypoxia in the rat showing impaired endothelial function in the resistance vasculature of hypoxia‐exposed adult offspring (Hemmings et al. 2005; Williams et al. 2005 b; Morton et al. 2011). Notably, Morton et al. (2011) used Sprague–Dawley rats at 12 months (the same age used in the present study) to show that prenatal hypoxia impaired flow‐mediated vasodilatation in both sexes. Although, in the present study, we did not directly determine the mechanisms underlying this endothelial dysfunction, it may be a result of reduced NO bioavailability associated with increased superoxide anion production, as previously reported by Williams et al. (2005 b). Increased oxidative stress in the fetal rat vasculature has been associated with endothelial dysfunction (Giussani et al. 2012), suggesting that hypoxia‐induced oxidative stress during fetal development can have adverse cardiovascular outcomes in later life.
In both the human population and animal models, endothelial dysfunction is associated with low birth weight (Leeson et al. 2001). Our model of chronic hypoxia in the mouse led to low birth weight, and resulted in endothelial dysfunction in aged offspring. Importantly, our model of gestational hypoxia using the mouse is not confounded by maternal undernutrition (Cuffe et al. 2014 a). Similar models of maternal hypoxia during late gestation in the rat have reported a significant reduction in maternal food intake during the time spent inside the hypoxia chamber (Williams et al. 2005 b; Camm et al. 2010). Undernourishment during pregnancy alone has been shown to alter vascular function (Brawley et al. 2003; Torrens et al. 2003). Therefore, the combination of reduction in maternal oxygen supply and reduced food consumption provides a dual insult to the developing fetus. Similarly, the vascular dysfunction seen in rat models of uteroplacental insufficiency may be attributable to a reduction in both oxygen and nutrient supply to the fetus (Tare et al. 2012). Our findings suggest that fetal hypoxia alone is able to result in vascular dysfunction in offspring. Growth restriction was sustained in our model during lactation but substantial catch‐up growth was observed post‐weaning, both of which are predictors of adult‐onset cardiovascular dysfunction. Reduced body weight pre‐weaning may signify an impaired lactational environment, which has previously has been reported in a model of uteroplacental insufficiency in the rat (O'Dowd et al. 2008). However, in our model, absolute growth during lactation was similar in the control and prenatal hypoxia groups. Sensitivity to vasodilatators and resistance artery stiffness was improved when these growth‐restricted offspring were cross‐fostered onto healthy mothers to improve the lactational environment (Tare et al. 2012). These studies highlight the importance of a healthy diet throughout lactation for growth‐restricted offspring.
Vulnerability to CVD programmed in utero may be exacerbated by a postnatal ‘second‐hit’, such as a HS (Ruta et al. 2010) or high fat diet (Rueda‐Clausen et al. 2012). The chronic HS diet produced modest alterations to microvascular function in aged control and hypoxia‐exposed male (but not female) offspring. To our knowledge, the present study is the first to examine the impact of a chronic HS diet in the CD‐1 mouse strain and, importantly, to examine these effects in both sexes. Our findings therefore warrant further investigation. Dietary salt loading to assess microvascular function has overwhelmingly been performed in the rat (Liu et al. 1999; Sofola et al. 2002). Sprague–Dawley rats fed a HS diet have increased small artery reactivity to vasoconstrictors but no overall impairment in endothelial function. However, the mechanism of ACh‐induced dilatation was altered (Sofola et al. 2002). Reduced endothelium‐dependent responses associated with HS intake have been reported in skeletal muscle arterioles (Boegehold, 1995), cerebral arterioles and small feed arteries of rat gracilis muscle (Liu et al. 1997). However, many of these vascular function studies were performed following exposure to extremely high levels of sodium (7–10% NaCl) in the diet over an acute period, which has limited relevance to the human population. In the present study, we have employed a more modest increase in dietary sodium (from 0.26% to 5%) across the adult lifespan of the mouse, which makes our studies of greater relevance. However, we cannot discount the possibility that more overt vascular deficits may be observed with a further increase in dietary sodium.
Vascular stiffening is a major risk factor for cardiovascular dysfunction. Increased vascular stiffness is not only associated with a low birth weight in humans, but also increases with age (Martin et al. 2000; te Velde et al. 2004) and other cardiovascular risk factors such as hypertension, diabetes and tobacco smoking (Benetos et al. 2002). A striking finding to emerge from the present study was that vascular stiffness was markedly exacerbated by the combination of prenatal hypoxia and a postnatal HS diet. Importantly, these changes in the stress–strain relationship of the microvasculature were geometry independent because no changes were observed in media:lumen ratios. Prenatal hypoxia alone had a limited effect on vascular stiffness, which is consistent with observations in aged rats exposed to hypoxia during late gestation (Morton et al. 2011). Given that vascular stiffening occurred in the absence of geometric changes, we examined the deposition and organization of aortic extracellular matrix proteins, particularly collagen and elastin, to determine the mechanical properties of vessel walls. We found reduced elastin content and significant disorganization and fragmentation of the elastin layers in hypoxia‐exposed offspring. The synthesis of elastin fibres responsible for vessel elasticity is restricted to fetal and early postnatal life and is therefore vulnerable to influences early in life such as chronic hypoxia (Davis, 1995). However, during the postnatal period, elastin fibres are sensitive to degradation by matrix metalloproteinases and elastases produced by inflammatory cells during the pathogenesis of vascular lesions such as aneurisms and arteriosclerosis (Galis & Khatri, 2002). Although the HS diet reduced elastin content in both treatment groups, the effect was significantly greater in hypoxia‐exposed offspring, particularly males. The significant disorganization and loss of elastin fibres may be the result of a prenatal impairment in elastin synthesis and enhanced postnatal degradation of fibres, consistent with vascular injury. Substantial collagen accumulation was observed in hypoxia‐exposed male offspring, although it was most prominent in offspring fed the HS diet irrespective of prenatal treatment. Dietary salt intake is a known contributor to fibrosis in the kidney, heart and vasculature (Yu et al. 2004), which in turn leads to vascular stiffening and increased cardiovascular risk. We observed slight differences between males and females in the histological changes in response to prenatal hypoxia and a postnatal HS diet. Most notably, hypoxia alone did not increase aortic collagen deposition in females, although it did in male offspring. Because the female hormonal milieu contains oestrogen, a hormone with vasoprotective effects, female offspring may be protected against a mild prenatal insult such as hypoxia (Mendelsohn & Karas, 1999). The postnatal HS diet increased aortic collagen deposition in both sexes, suggesting that the protective effects of oestrogen on the vasculature may be diminished in the ageing female mouse. This is observed in the human population where rates of CVD are higher in postmenopausal women than premenopausal women, associated with the declining plasma oestrogen levels with age (Khalil, 2010). Because the loss of elastin fibre integrity and increased media collagen content were observed in a compliance vessel, we cannot discount the possibility that the pathophysiology could present differently in the resistance vasculature. However, these changes are indicative of vascular stiffening and are consistent with the reduced vascular compliance observed in the microvasculature of hypoxia‐exposed offspring fed the HS diet.
In conclusion, we report for the first time that vascular stiffening in aged offspring exposed to gestational hypoxia is markedly exacerbated by a postnatal HS diet. The changes in aortic extracellular matrix composition probably underlie the increase in vascular stiffness in hypoxia‐exposed offspring fed the chronic HS diet. Prenatal hypoxia alone impaired endothelial function in adult male and female mice, demonstrating that the endothelium, a vital regulator of vascular tone, is susceptible to adverse environmental conditions in utero. Because high dietary salt intake is endemic in Western societies, such adverse changes in vascular function and wall stiffness may be inadvertently enhanced by suboptimal dietary choices. This may have serious consequences for individuals predisposed to CVD because of their in utero environments. It is advised that individuals born with a low birth weight take precautions to ensure that dietary sodium intake in adult life is not excessive so as to limit adverse cardiovascular outcomes.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
SLW, RRS, TMP and KMM were responsible for conception and design of the experiments. SLW and TT were responsible for the collection, analysis and interpretation of data. All authors were involved in drafting the article and revising it critically for intellectual content. All authors approved the final version of the manuscript submitted for publication.
Funding
This project was funded by the National Health and Medical Research Council (NHMRC‐APP1009338) of Australia. KMM was supported by fellowships provided by the NHMRC. SLW was supported by an Australian Postgraduate Award.
References
- Barker DJ (1995). Intrauterine programming of adult disease. Mol Med Today 1, 418–423. [DOI] [PubMed] [Google Scholar]
- Barker DJ (1998). In utero programming of chronic disease. Clin Sci 95, 115–128. [PubMed] [Google Scholar]
- Barker DJP (2002). Fetal programming of coronary heart disease. Trends Endocrinol Metabol 13, 364–368. [DOI] [PubMed] [Google Scholar]
- Baschat AA, Gembruch U, Reiss I, Gortner L & Diedrich K (1997). Demonstration of fetal coronary blood flow by Doppler ultrasound in relation to arterial and venous flow velocity waveforms and perinatal outcome – the ‘heart‐sparing effect’. Ultrasound Obstet Gynecol 9, 162–172. [DOI] [PubMed] [Google Scholar]
- Benetos A, Waeber B, Izzo J, Mitchell G, Resnick L, Asmar R & Safar M (2002). Influence of age, risk factors, and cardiovascular and renal disease on arterial stiffness: clinical applications. Am J Hyperten 15, 1101–1108. [DOI] [PubMed] [Google Scholar]
- Boegehold MA (1995). Flow‐dependent arteriolar dilation in normotensive rats fed low‐ or high‐salt diets. Am J Physiol Heart Circ Physiol 269, H1407–H1414. [DOI] [PubMed] [Google Scholar]
- Brawley L, Itoh S, Torrens C, Barker A, Bertram C, Poston L & Hanson M (2003). Dietary protein restriction in pregnancy induces hypertension and vascular defects in rat male offspring. Pediatr Res 54, 83–90. [DOI] [PubMed] [Google Scholar]
- Bulterys MG, Greenland S & Kraus JF (1990). Chronic fetal hypoxia and sudden infant death syndrome: interaction between maternal smoking and low hematocrit during pregnancy. Pediatrics 86, 535–540. [PubMed] [Google Scholar]
- Camm EJ, Hansell JA, Kane AD, Herrera EA, Lewis C, Wong S, Morrell NW & Giussani DA (2010). Partial contributions of developmental hypoxia and undernutrition to prenatal alterations in somatic growth and cardiovascular structure and function. Am J Obstetr Gynecol 203, 495.e424–495.e434. [DOI] [PubMed] [Google Scholar]
- Cordain L, Eaton SB, Sebastian A, Mann N, Lindeberg S, Watkins BA, O'Keefe JH & Brand‐Miller J (2005). Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr 81, 341–354. [DOI] [PubMed] [Google Scholar]
- Cuffe J, Walton S, Singh R, Spiers J, Bielefeldt‐Ohmann H, Wilkinson L, Little M & Moritz K (2014. a). Mid‐ to late term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways and nutrient transporters in a sex‐specific manner. J Physiol 592, 3127–3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuffe J, Walton S, Steane S, Singh R, Simmons D & Moritz K (2014. b). The effects of gestational age and maternal hypoxia on the placental renin angiotensin system in the mouse. Placenta 35, 953–961. [DOI] [PubMed] [Google Scholar]
- Davis EC (1995). Elastic lamina growth in the developing mouse aorta. J Histochem Cytochem 43, 1115–1123. [DOI] [PubMed] [Google Scholar]
- Galis ZS & Khatri JJ (2002). Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 90, 251–262. [PubMed] [Google Scholar]
- Giussani DA, Camm EJ, Niu Y, Richter HG, Blanco CE, Gottschalk R, Blake EZ, Horder KA, Thakor AS, Hansell JA, Kane AD, Wooding FBP, Cross CM & Herrera EA (2012). Developmental programming of cardiovascular dysfunction by prenatal hypoxia and oxidative stress. PLoS ONE 7, e31017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Phillips PS, Anstee S & Barker DJ (2001). Effects of altitude versus economic status on birth weight and body shape at birth. Pediatr Res 49, 490–494. [DOI] [PubMed] [Google Scholar]
- Giussani DA, Salinas CE, Villena M & Blanco CE (2007). The role of oxygen in prenatal growth: studies in the chick embryo. J Physiol 585, 911–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giussani DA, Spencer JA, Moore PJ, Bennet L & Hanson MA (1993). Afferent and efferent components of the cardiovascular reflex responses to acute hypoxia in term fetal sheep. J Physiol 461, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodfellow J, Bellamy MF, Gorman ST, Brownlee M, Ramsey MW, Lewis MJ, Davies DP & Henderson AH (1998). Endothelial function is impaired in fit young adults of low birth weight. Cardiovasc Res 40, 600–606. [DOI] [PubMed] [Google Scholar]
- Hemmings DG, Williams SJ & Davidge ST (2005). Increased myogenic tone in 7‐month‐old adult male but not female offspring from rat dams exposed to hypoxia during pregnancy. Am J Physiology Heart Circ Physiol 289, H674–H682. [DOI] [PubMed] [Google Scholar]
- Iglarz M, Touyz RM, Amiri F, Lavoie M‐F, Diep QN & Schiffrin EL (2003). Effect of peroxisome proliferator – activated receptor‐α and ‐γ activators on vascular remodeling in endothelin‐dependent hypertension. Arterioscler Thromb Vasc Biol 23, 45–51. [DOI] [PubMed] [Google Scholar]
- Khalil RA (2010). Potential approaches to enhance the effects of estrogen on senescent blood vessels and postmenopausal cardiovascular disease. Cardiovasc Hematol Agents Med Chem 8, 29–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan I, Dekou V, Hanson M, Poston L & Taylor P (2004). Predictive adaptive responses to maternal high‐fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation 110, 1097–1102. [DOI] [PubMed] [Google Scholar]
- Krebs C, Macara LM, Leiser R, Bowman AW, Greer IA & Kingdom JC (1996). Intrauterine growth restriction with absent end‐diastolic flow velocity in the umbilical artery is associated with maldevelopment of the placental terminal villous tree. Am J Obstet Gynecol 175, 1534–1542. [DOI] [PubMed] [Google Scholar]
- Leeson C, Kattenhorn M, Morley R, Lucas A & Deanfield J (2001). Impact of low birth weight and cardiovascular risk factors on endothelial function in early adult life. Circulation 103, 1264–1268. [DOI] [PubMed] [Google Scholar]
- Liu Y, Fredricks KT, Roman RJ & Lombard JH (1997). Response of resistance arteries to reduced PO2 and vasodilators during hypertension and elevated salt intake. Am J Physiol Heart Circ Physiol 273, H869–H877. [DOI] [PubMed] [Google Scholar]
- Liu Y, Rusch NJ & Lombard JH (1999). Loss of endothelium and receptor‐mediated dilation in pial arterioles of rats fed a short‐term high salt diet. Hypertension 33, 686–688. [DOI] [PubMed] [Google Scholar]
- Martin H, Hu J, Gennser G & Norman M (2000). Impaired endothelial function and increased carotid stiffness in 9‐year‐old children with low birthweight. Circulation 102, 2739–2744. [DOI] [PubMed] [Google Scholar]
- Mcmillen IC & Robinson JS (2005). Developmental origins of the metabolic syndrome: prediction, plasticity, and programming. Physiol Rev 85, 571–633. [DOI] [PubMed] [Google Scholar]
- Mendelsohn ME & Karas RH (1999). The protective effects of estrogen on the cardiovascular system. New Engl J Med 340, 1801–1811. [DOI] [PubMed] [Google Scholar]
- Morton JS, Rueda‐Clausen CF & Davidge ST (2011). Flow‐mediated vasodilation is impaired in adult rat offspring exposed to prenatal hypoxia. J Appl Physiol 110, 1073–1082. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Wilson PWF & Kannel WB (2008). Beyond established and novel risk factors: lifestyle risk factors for cardiovascular disease. Circulation 117, 3031–3038. [DOI] [PubMed] [Google Scholar]
- Mulder ALM, Miedema A, De Mey JGR, Giussani DA & Blanco CE (2002). Sympathetic control of the cardiovascular response to acute hypoxemia in the chick embryo. Am J Physiol Regul Integr Comp Physiol 282, R1156–R1163. [DOI] [PubMed] [Google Scholar]
- Norman M & Martin H (2003). Preterm birth attenuates association between low birth weight and endothelial dysfunction. Circulation 108, 996–1001. [DOI] [PubMed] [Google Scholar]
- O'Dowd R, Kent JC, Moseley JM & Wlodek ME (2008). Effects of uteroplacental insufficiency and reducing litter size on maternal mammary function and postnatal offspring growth. Am J Physiol Regul Integr Comp Physiol 294, R539–R548. [DOI] [PubMed] [Google Scholar]
- Rook W, Johnson CD, Coney AM & Marshall JM (2014). Prenatal hypoxia leads to increased muscle sympathetic nerve activity, sympathetic hyperinnervation, premature blunting of neuropeptide y signaling, and hypertension in adult life. Hypertension 64, 1321–1327. [DOI] [PubMed] [Google Scholar]
- Rouwet EV, Tintu AN, Schellings MWM, van Bilsen M, Lutgens E, Hofstra L, Slaaf DW, Ramsay G & le Noble FAC (2002). Hypoxia induces aortic hypertrophic growth, left ventricular dysfunction, and sympathetic hyperinnervation of peripheral arteries in the chick embryo. Circulation 105, 2791–2796. [DOI] [PubMed] [Google Scholar]
- Rueda‐Clausen CF, Morton JS, Dolinsky VW, Dyck JR & Davidge ST (2012). Synergistic effects of prenatal hypoxia and postnatal high‐fat diet in the development of cardiovascular pathology in young rats. Am J Physiol Regul Integr Comp Physiol 303, R418–R426. [DOI] [PubMed] [Google Scholar]
- Ruta L‐AM, Dickinson H, Thomas MC, Denton KM, Anderson WP & Kett MM (2010). High‐salt diet reveals the hypertensive and renal effects of reduced nephron endowment. Am J Physiol Renal Physiol 298, F1384–F1392. [DOI] [PubMed] [Google Scholar]
- Sofola OA, Knill A, Hainsworth R & Drinkhill M (2002). Change in endothelial function in mesenteric arteries of Sprague–Dawley rats fed a high salt diet. J Physiol 543, 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Emmett SJ, Coleman HA, Skordilis C, Eyles DW, Morley R & Parkington HC (2011). Vitamin D insufficiency is associated with impaired vascular endothelial and smooth muscle function and hypertension in young rats. J Physiol 589, 4777–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Bubb KJ & Wlodek ME (2012). Uteroplacental insufficiency and lactational environment separately influence arterial stiffness and vascular function in adult male rats. Hypertension 60, 378–386. [DOI] [PubMed] [Google Scholar]
- te Velde SJ, Ferreira I, Twisk JW, Stehouwer CD, van Mechelen W & Kemper HC (2004). Birthweight and arterial stiffness and blood pressure in adulthood – results from the Amsterdam growth and health longitudinal study. Int J Epidemiol 33, 154–161. [DOI] [PubMed] [Google Scholar]
- Virdis A, Neves MF, Amiri F, Viel E, Touyz RM & Schiffrin EL (2002). Spironolactone improves angiotensin‐induced vascular changes and oxidative stress. Hypertension 40, 504–510. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Campbell ME, McMillen IC & Davidge ST (2005. a). Differential effects of maternal hypoxia or nutrient restriction on carotid and femoral vascular function in neonatal rats. Am J Physiol Regul Integr Comp Physiol 288, R360–R367. [DOI] [PubMed] [Google Scholar]
- Williams SJ, Hemmings DG, Mitchell JM, McMillen IC & Davidge ST (2005. b). Effects of maternal hypoxia or nutrient restriction during pregnancy on endothelial function in adult male rat offspring. J Physiol 565, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladimiroff JW, Tonge HM & Stewart PA (1986). Doppler ultrasound assessment of cerebral blood flow in the human fetus. Br J Obstet Gynaecol 93, 471–475. [PubMed] [Google Scholar]
- Woods LL, Weeks DA & Rasch R (2004). Programming of adult blood pressure by maternal protein restriction: role of nephrogenesis. Kidney Int 65, 1339–1348. [DOI] [PubMed] [Google Scholar]
- Xu Y, Williams SJ, O'Brien D & Davidge ST (2006). Hypoxia or nutrient restriction during pregnancy in rats leads to progressive cardiac remodeling and impairs postischemic recovery in adult male offspring. FASEB J 20, 1251–1253. [DOI] [PubMed] [Google Scholar]
- Xue Q & Zhang L (2009). Prenatal hypoxia causes a sex‐dependent increase in heart susceptibility to ischemia and reperfusion injury in adult male offspring: role of protein kinase Cϵ. J Pharmacol Exp Ther 330, 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Larson D, Slayback D, Lundeen T, Baxter J & Watson R (2004). Characterization of high‐salt and high‐fat diets on cardiac and vascular function in mice. Cardiovasc Toxicol 4, 37–46. [DOI] [PubMed] [Google Scholar]